Introduction
Gliomas are the most common and lethal type of
primary intracranial tumor of the central nervous system and the
median survival time for patients with glioma is ~15 months
(1,2).
Despite current advancements in diagnosis and therapeutic
modalities, surgery remains the primary mode of treatment, as the
majority of gliomas largely develop resistance to chemotherapy or
radiotherapy (3). Due to the high
levels of aggressiveness and inducible resistance to chemotherapy
and radiotherapy associated with this disease, conventional
treatments generally fail to produce positive outcomes (4). As a result, efforts have been redirected
toward identifying a novel molecular target to glioma cells, for
the future development of more effective therapies (5).
Apoptosis is a form of programmed cell death that
functions to eliminate abnormal or transformed cells (6). Consequently, studies involving the
utilization of the apoptotic pathway to target over-proliferating
tumor cells have gained attention. It has been established that
resistance to apoptotic signals and the inhibition of apoptosis are
associated with glioma genesis (7).
Dysregulation of apoptosis may not only promote the survival and
proliferation of glioma cells, but also antagonize the effects of
apoptotic inducers (e.g. Tumor necrosis factor-related
apoptosis-inducing ligand) thereby developing resistance to
chemotherapy and radiotherapy and increasing the risk of malignant
phenotypes in cancer cells (8).
However, much remains unknown regarding the underlying molecular
mechanism of how glioma cells develop an apoptosis-resistant
phenotype.
Nuclear factor κ-light-chain-enhancer of B cells
(NF-κB) has been established as a pro-survival and anti-apoptotic
oncogenic factor in the majority of the tumor types, and various
oncogenes and growth factors may be additionally upregulated by
NF-κB activation in tumor cells (8–10).
Evidence has revealed that NF-κB activity is commonly elevated in
glioma, and that tumor grading and prognosis are associated with
its level of expression (11,12). Additional studies have also suggested
the involvement of NF-κB activation in the anti-apoptotic responses
of glioma cells (13,14). Therefore, it is important to identify
novel molecular mechanisms that are associated with the aberrant
activation of NF-κB in glioma that facilitate the anti-apoptotic
responses.
In non-dividing cells, NF-κB is sequestered in the
cytosol by the inhibitor of κB (IκB) family proteins, B-cell
lymphoma (Bcl) 3 protein, p105 and p100 (15). However, the presence of certain
antigens, cytokines and environmental stress signals may lead to
the phosphorylation and degradation of IκBα through the
26S-proteosome-dependent system (16). Following the release of NF-κB and its
nuclear translocation, NF-κB-targeted genes are activated (17). The phosphorylation of IκBα is
accomplished by the IκB kinase (IKK) complex. Studies in breast
cancer have previously identified IKK as being an oncogene and
activator of the NF-κB pathway to facilitate cell transformation
and to enable cancer cells to maintain a malignant phenotype
(16). Elevated NF-κB expression has
been detected in human ovarian and prostate cancer, which
contributes to tumorigenesis, drug resistance and the promotion of
cancer progression (18–20). However, a comprehensive understanding
of what controls NF-κB expression and activity in cancer cells is
lacking and, to the best of our knowledge, the upstream regulator
of NF-κB signaling has yet to defined.
Ubiquitination of proteins is of key importance in
post-translational modification, targeting proteins for degradation
via the proteasome, membrane trafficking, DNA repair, protein
kinase activation and chromatin modification (21). In an attempt to elucidate how glioma
cells usurp the NF-κB signaling cascade to adopt an
apoptosis-resistant phenotype, the present study aimed to
investigate the involvement of E3 ubiquitin-protein ligase MIB2
(MIB2), an E3 ligase that catalyzes the addition of single or short
chains of ubiquitin to lysine (22).
The association between NF-κB activation and MIB2 expression is not
well understood. The differential expression of MIB2 in glioma
cells and its function in the NF-κB pathway may serve a role in the
anti-apoptotic responses of glioma cells. The present study
demonstrated a novel interaction between NF-κB and MIB2, and
investigated the effects of these proteins on the induction of
resistance to apoptosis. The presence of aberrant MIB2 expression
in tumor specimens from patients with glioma also supports the
hypothesis that the MIB2/NF-κB interaction serves an essential role
in the development of drug resistance.
Materials and methods
Clinical specimens
Tissues used in the present study were obtained from
69 patients with glioma who had undergone craniotomy and tumor
extirpation between March 2008 and December 2010 at the Shenzhen
People's Hospital (Shenzhen, China). The present study was approved
by the Ethics Committee of Shenzhen People's Hospital. The
pathological diagnosis and grading of glioma were assessed
according to World Health Organization (WHO) grade II–IV
classification, which is based on the histological features of a
heterogeneous population of tumors with varying prognoses and
treatments (23). Clinical
information of the samples is summarized in Table I. In addition, 4 normal brain tissues
were obtained via donations from road traffic accident fatalities
without any prior pathologically-detectable conditions. Written
informed consent was obtained from all patients.
 | Table I.Clinicopathological characteristics
of patient samples and expression of MIB2 in glioma. |
Table I.
Clinicopathological characteristics
of patient samples and expression of MIB2 in glioma.
| Patient
characteristic | n | % |
|---|
| Sex |
|
Male | 39 | 56.5 |
|
Female | 30 | 43.5 |
| Age, years |
|
≤50 | 53 | 76.8 |
|
>50 | 16 | 23.2 |
| Grade |
| I | 8 | 11.6 |
| II | 24 | 34.8 |
|
III | 21 | 30.4 |
| IV | 16 | 23.2 |
| MIB2 score |
| 0 | 3 | 4.3 |
|
1-4 | 15 | 21.7 |
|
5-8 | 23 | 33.3 |
|
9-12 | 28 | 40.7 |
Cell lines
Primary normal human astrocytes (NHA) were purchased
from Lonza Group Ltd. (Basel, Switzerland) and maintained in
Clonetics™ Astrocyte Growth Medium (Lonza Group Ltd.) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Glioma T98G, LN-18 and A172 cell lines were
purchased from the American Type Culture Collection (Manassas, VA,
USA), and maintained in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.).
Plasmids and small interfering
(si)RNA
The pNF-κB-Luc plasmids were purchased from the
Clontech Laboratories, Inc. (Mountainview, CA, USA), and the
Flag-MIB2 plasmids used for MIB2 overexpression were purchased from
Addgene, Inc. (Cambridge, MA, USA). Human MIB2 siRNA was obtained
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Glioma T98G
cells were transfected with siRNAs targeting MIB2 (10 nM) or with
control siRNA (10 nM) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). The transfection was
performed at 37°C in a humidified incubator with 5% CO2.
After 24 h, cells were harvested for subsequent
experimentation.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from T98G, LN-18 and A172 cells or
patient tissues using TRIzol® reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. A total
of 5 µg RNA from each sample was reverse transcribed to
complementary cDNA (at 42°C for 1 hr) using the SMART®
MMLV Reverse Transcriptase kit (cat. no. 639523; Takara Bio, Inc.,
Otsu, Japan). qPCR was performed using QuantiTect SYBR Green PCR
Master mix (Qiagen GmbH, Hilden, Germany) and was analyzed on an
ABI Prism 7700 analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). PCR was performed under the following
conditions: Initial denaturation at 95°C for 5 min, followed by 35
cycles of 95°C for 30 sec, 55°C for 1 min and 72°C for 1 min, with
a final extension step at 72°C for 5 min. GAPDH served as the
internal control. The mRNA expression levels of MIB2 were
normalized to those of GAPDH, and relative quantification was
performed using the comparative cycle threshold method
(2−ΔΔCq) (24). All PCR
experiments were repeated ≥3 times. The sequences of the primers
were as follows: MIB2 forward, 5′-CATCGGCGACCTTGACACA-3′ and
reverse, 5′-CGACTGCTACACGCAGGTT-3′; and GAPDH forward,
5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse,
5′-GCCATCACGCCACAGTTTC-3′.
Western blot (WB) analysis
T98G, LN-18 and A172 cells were plated in tissue
culture dishes overnight. Following harvest, adherent cells were
washed with cold PBS twice and resuspended in lysis buffer (150 mM
NaCl, 50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 1% NP-40) containing
protease inhibitor cocktail (Amresco, LLC, Solon, OH, USA). Tissues
were dissociated using the Brain Tumor Dissociation kit (cat. no.
130-095-942; Miltenyi Biotec, Inc., Auburn, CA, USA). Protein
levels in the extracts were quantified using a BCA assay (Pierce;
Thermo Fisher Scientific, Inc.). Equal amounts of 100 µg total
protein extracts were resolved by 10% standard sodium dodecyl
sulfate polyacrylamide gel electrophoresis and transferred onto a
polyvinylidene fluoride membrane (0.45 mm; EMD Millipore,
Billerica, MA, USA). Membranes were blocked with 5% fat-free dry
milk/Tris-buffered saline with 5% Tween-20 (cat. no. 9997; Cell
Signaling Technology Inc., Danvers, MA, USA) at room temperature
for 1 h, then incubated with the following anti-human primary
antibodies overnight at 4°C: Anti-MIB2 (1:1,000; cat. no. sc-55149;
Santa Cruz Biotechnology, Inc.), anti-α-tubulin (1:5,000; cat. no.
T6074; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
anti-phosphorylated (p)-NF-κB p65 subunit (1:1,000; NF-κB p65; cat.
no. sc-33020; Santa Cruz Biotechnology, Inc.), anti-caspase-3
(1:2,000; cat. no. sc-1225; Santa Cruz Biotechnology, Inc.) and
anti-Bcl-2 (1:2,000; cat. no. sc-783 Santa Cruz Biotechnology,
Inc.). Membranes were subsequently incubated with a horseradish
peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) (cat
no. sc-2005; 1:5,000; Santa Cruz Biotechnology, Inc.) or goat
anti-rabbit IgG (cat no. sc-2004; 1:5,000; Santa Cruz
Biotechnology, Inc.) secondary antibody for 1 h at room temperature
and visualized using an enhanced chemiluminescence reagent
(PerkinElmer, Inc., Waltham, MA, USA). ImageJ software (1.46p;
National Institutes of Health, Bethesda, MD, USA) was used for
densitometry analysis.
Immunohistochemistry (IHC)
Immunohistochemical staining of MIB2 protein was
performed to examine changes in protein expression in 69 cases of
human glioma samples. An anti-MIB2 antibody (anti-MIB2; 1:100; cat.
no. sc-55149; Santa Cruz Biotechnology, Inc.) was used as the
primary antibody to detect MIB2, and the procedure was performed as
previously described. Immunohistochemistry was performed on 10%
formalin-fixed (for 24-48 h at room temperature), paraffin-embedded
tissue sections using the Envision peroxidase detection method
(25). Sections (4-µm) were
deparaffinized in xylene, dehydrated through a graded ethanol
series at 10 min intervals (100, 70, 40 and 0%) and subsequently
treated with 0.3% hydrogen peroxide in methanol for 30 min at room
temperature to eliminate the endogenous peroxidase activity. For
antigen retrieval, the sections were placed in 10 mM sodium citrate
buffer (pH 6.0) and heated to near boiling (95-98°C) in a water
bath for 20 min (or an oven) followed by cooling for 20 min at room
temperature. The primary antibodies (anti-MIB2; 1:100; cat. no.
sc-55149; Santa Cruz Biotechnology, Inc.) were incubated overnight
at 4°C subsequent to blocking non-specific binding with 10% normal
goat serum in PBS for 30 min at room temperature. For negative
controls, PBS was used as a substitute for the primary antibodies.
Sections were incubated with the secondary biotinylated antibody
(Envision peroxidase complex; horseradish peroxidase conjugated;
1:200; cat. no. ZK-9600; OriGene Technologies, Inc., Rockville, MD,
USA) for 20 min at room temperature. Sections were counterstained
with Mayer's hematoxylin after immunostaining prior to mounting for
30 sec at room temperature (26).
Immunohistochemical staining for protein expression in tumor
lesions and normal tissues was assessed by 3 pathologists (1 chief
pathologist and 2 associate chief pathologists; Pathology
Department of Shenzhen People's Hospital, Shenzhen, China) blinded
to the origin of the samples using a semi-quantitative method. An
upright light microscope (Positive-ECLIPSE E200-LED; Nikon
Corporation, Tokyo, Japan) was used at a magnification of ×200.
H-scores were calculated for each sample according to the intensity
of the nucleic and cytoplasmic staining. The expression levels of
MIB2 were scored based on the staining intensity and distribution
using the immunoreactive score (IRS), as follows: IRS = staining
intensity (SI) × percentage of positive cells (PP). The SI was
determined as follows: Absent, 0; weak, 1; moderate, 2; and strong,
3. The PP was scored as follows: 0%, 0; 0-25%, 1; 25-50%, 2;
50-75%, 3; and 75-100%, 4 (27,28).
Cell viability assay
Cells were plated in 6-well plates at a density of
5×105 cells per well and incubated for 24 h at 37°C,
followed by irradiation with ultraviolet (UV; 20 or 40
J/m2) and an additional 12-h incubation at 37°C. The
numbers of viable cells were determined by the trypan blue dye
exclusion test. Briefly, the Glioma T98G cells were seeded in
six-well plates (1×105 cells/ml). A 10 µl cell
suspension was mixed with 10 µl 0.4% trypan blue for 3-5 min at
room temperature (Nacalai Tesque, Inc., Kyoto, Japan), and live
cells were counted manually using a hemacytometer (Erma, Inc.,
Tokyo, Japan). The results were expressed as the percentage of the
values obtained when the cells were grown in the absence of
reagents.
Annexin V binding assay
The ApopNexin Annexin V-fluorescein isothiocyanate
apoptosis kit (cat. no. APT750; Merck KGaA) was used to
quantitatively examine apoptotic cells using a flow cytometer using
a BD FACSCanto™ II apparatus (BD Biosciences, Franklin Lakes, NJ,
USA) with BD FACSChorus™ software (FCS 3.1) for analysis according
to the manufacturer's protocol.
NF-κB-dependent reporter gene
transcription assay
A pNF-κB-Luc plasmid for NF-κB luciferase reporter
assay was obtained from Clontech Laboratories, Inc. NF-κB-dependent
luciferase activity was measured using a Dual-Luciferase Reporter
Assay system (Promega Corporation, Madison, WI, USA) according to
the manufacturer's protocol. Briefly, Glioma T98G cells
(1×105 cells/well) were seeded in a 96-well plate for 24
h. The cells were then transfected with plasmids using
Lipofectamine® 2000 in each well and then incubated for
a transfection period of 24 h at 37°C. Subsequently, the Dulbecco's
modified Eagle's medium was removed and replaced with fresh medium.
Luciferase activity was determined in a MicroLumat plus luminometer
(Berthold Technologies GmbH Co., Bad Wildbad, Germany) by injecting
100 µl of assay buffer containing luciferin and measuring light
emission for 10 sec. Co-transfection with pRL-CMV (Promega
Corporation), which expresses Renilla luciferase, was
performed to enable normalization of data for transfection
efficiency.
Statistical analysis
All statistical analyses were performed using the
SPSS 10.0 statistical software package (SPSS, Inc., Chicago, IL,
USA) and data were expressed as the mean ± standard deviation. The
differences between experimental conditions were compared
individually using Student's t-tests. Comparisons within groups
underwent P-values were calculated using one-way analysis of
variance (ANOVA) followed by Tukey's post hoc test, two-way ANOVA
followed by Tukey's post hoc test or Bonferroni's tests, or paired
t-tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
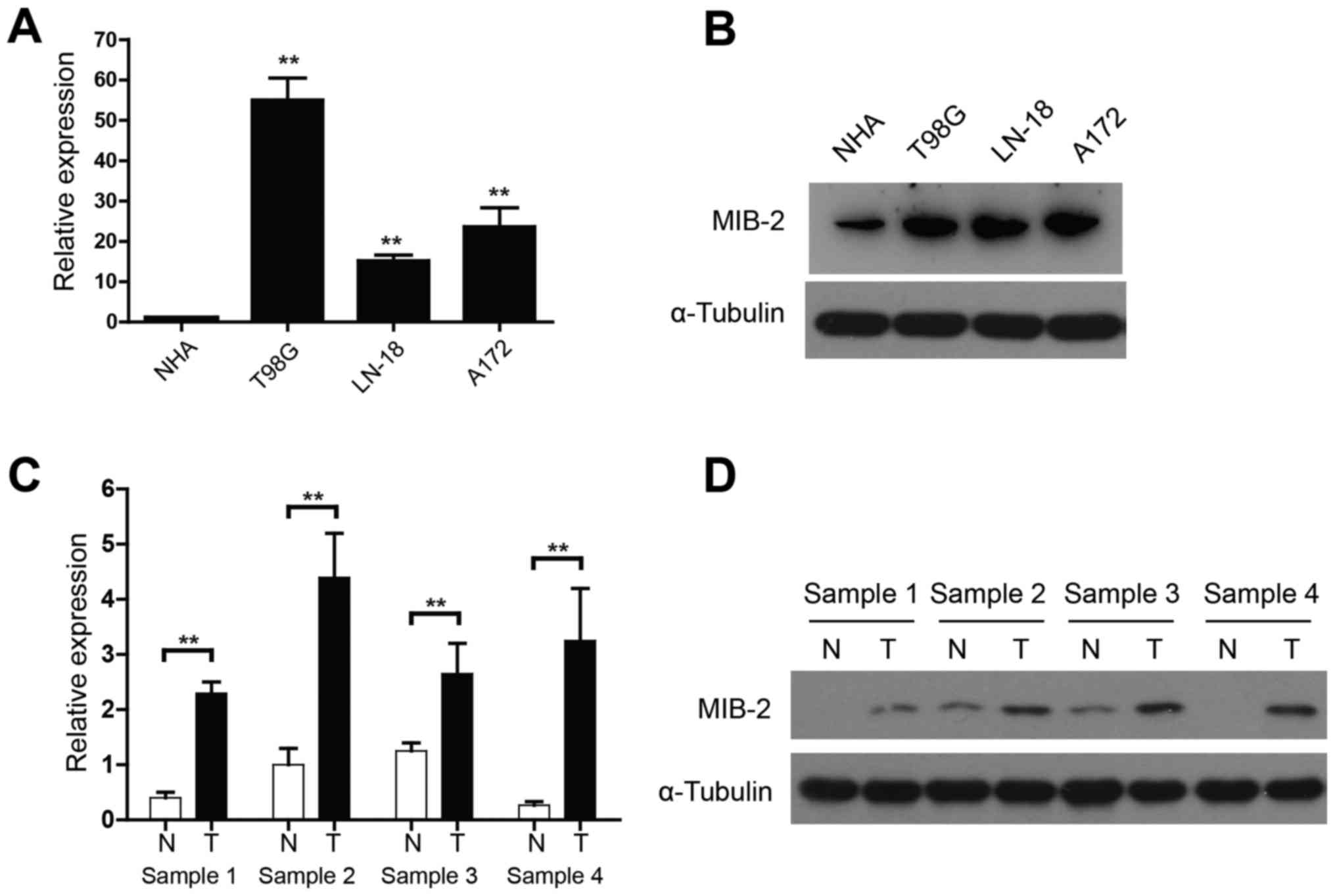
Elevated MIB2 expression in glioma
cell lines and human glioma specimens
The functional and clinical relevance of MIB2 in
human glioma remains to be investigated. The present study assessed
the MIB2 expression in NHA and various human glioma cell lines
using qPCR and WB analyses. When compared with NHA, mRNA expression
of MIB2 was significantly increased in all glioma T98G, LN-18 and
A172 cell lines (P<0.01; Fig. 1A).
WB analysis also confirmed the upregulated MIB2 expression in all
glioma cell lines, compared with NHA (Fig. 1B). In addition, four pairs of primary
glioma samples and adjacent non-cancerous brain tissues were used
to conduct a comparative analysis of MIB2 expression in human
gliomas. While the adjacent non-cancerous brain tissue expressed a
relatively low level of MIB2, the mRNA (Fig. 1C) and protein expression (Fig. 1D) levels of MIB2 indicated a
significant elevation in all four sets of human primary glioma
samples (P<0.01).
Additional confirmation of MIB2 expression in glioma
was performed using immunohistochemical staining of tumor sections.
Abundant MIB2 was detected and positively stained in all primary
gliomas, while its expression in normal brain tissues was absent or
only limited to a marginally measurable state (Fig. 2A). Additional analysis confirmed that
the average scores of MIB2 staining in primary glioma clinical
samples were significantly (P<0.01) increased compared with
those of adjacent normal brain tissues (Fig. 2B). These data demonstrated that MIB2
was highly expressed in human glioma, suggesting the possibility of
a specific role for MIB2 in glioma pathology.
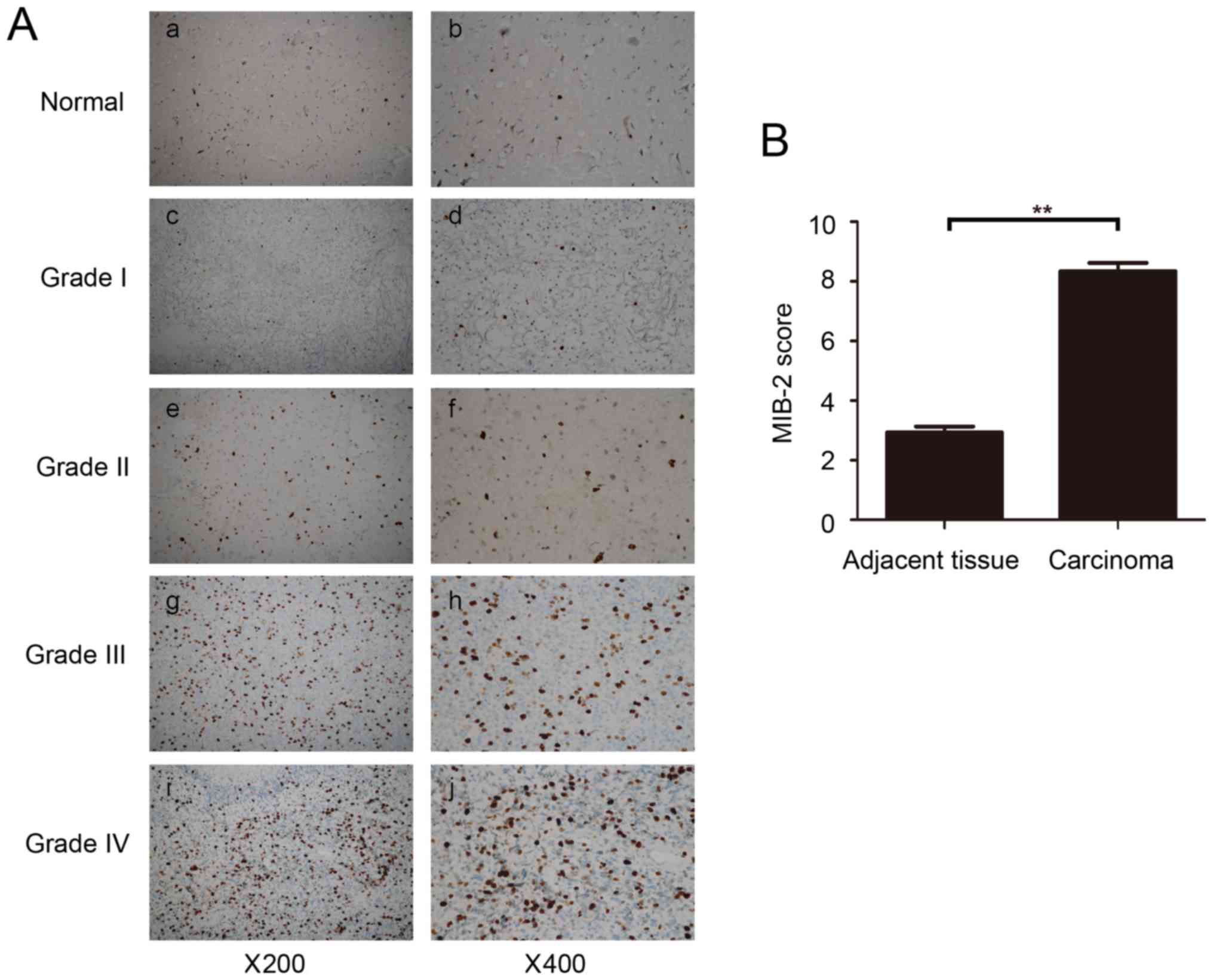 | Figure 2.Immunohistochemical analysis of MIB2
expression in normal brain tissues and primary glioma tumors. (A)
Representative images from IHC assays of normal brain tissues and
specimens of 69 archived glioma cases. (a,b) Normal brain tissue;
(c,d) WHO grade 1 pilocytic astrocytoma; (e,f) WHO grade 2 diffuse
astrocytoma; (g,h) WHO grade 3 anaplastic astrocytoma; (i,j) WHO
grade 4 glioblastoma multiforme. (a,c,e,g,i) Magnification, ×200.
(b,d,f,h and j) Magnification, ×400. (B) Comparative statistical
quantification of the mean values of MIB2 staining between normal
brain tissues (n=3) and glioma specimens of different WHO grades.
Student's t-test was used for statistical analysis. **P<0.01.
IHC, immunohistochemistry; MIB2, E3 ubiquitin-protein ligase; WHO,
World Health Organization. |
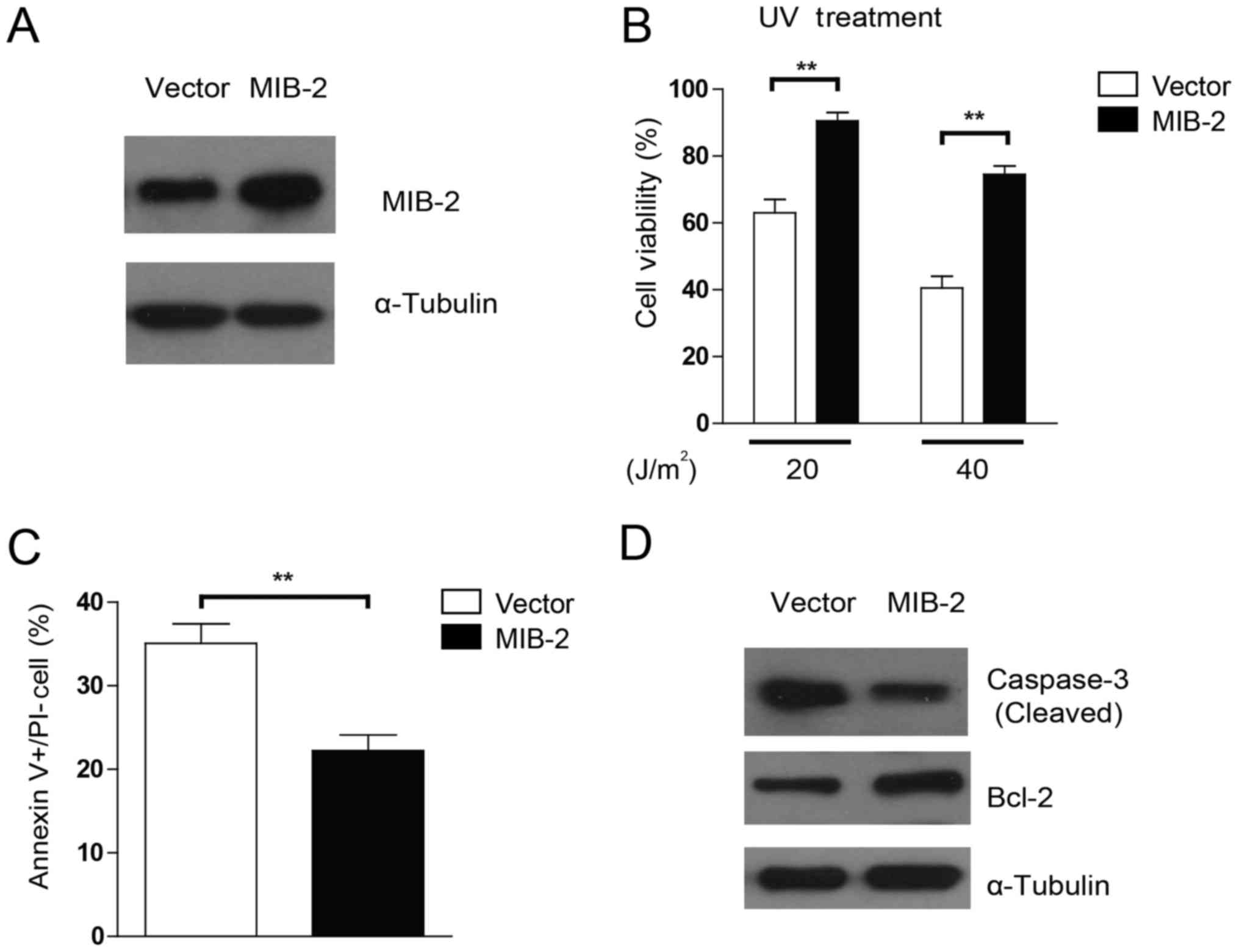
UV-induced apoptosis was repressed in
MIB2-overexpressing glioma cells
To examine whether the overexpression of MIB2 may
affect the sensitivity of glioma cells to apoptosis inducers,
stable transduction of MIB2 overexpression was achieved in T98G
cells (Fig. 3A). As UV irradiation
was administered to induce cell death, marked resistance toward
different dosages of UV irradiation was observed in
MIB2-overexpressing cell lines when compared with their
vector-carrying control cells, in a dose-dependent manner (Fig. 3B). To investigate the specific effects
of MIB2 overexpression in apoptosis, Annexin V binding assays were
conducted. As indicated in Fig. 3C,
T98G cells with MIB2 overexpression exhibited lower numbers of
apoptotic cells post-UV irradiation compared with those of the
vector control group, implying that MIB2 overexpression may confer
protection in glioma cells against pro-apoptotic agents.
Additionally, a pro-apoptotic protein, caspase-3, and an
anti-apoptotic protein, Bcl-2, were assessed for their differential
activities in cells with MIB2 overexpression. It was indicated that
MIB2-overexpressing cells exhibited less activation and cleavage of
caspase-3, but increased Bcl-2 expression, when compared with the
vector control cells (Fig. 3D). Taken
together, these results indicated that the functions of high MIB2
expression in glioma cells manifest in promoting cell survival and
resistance to apoptosis.
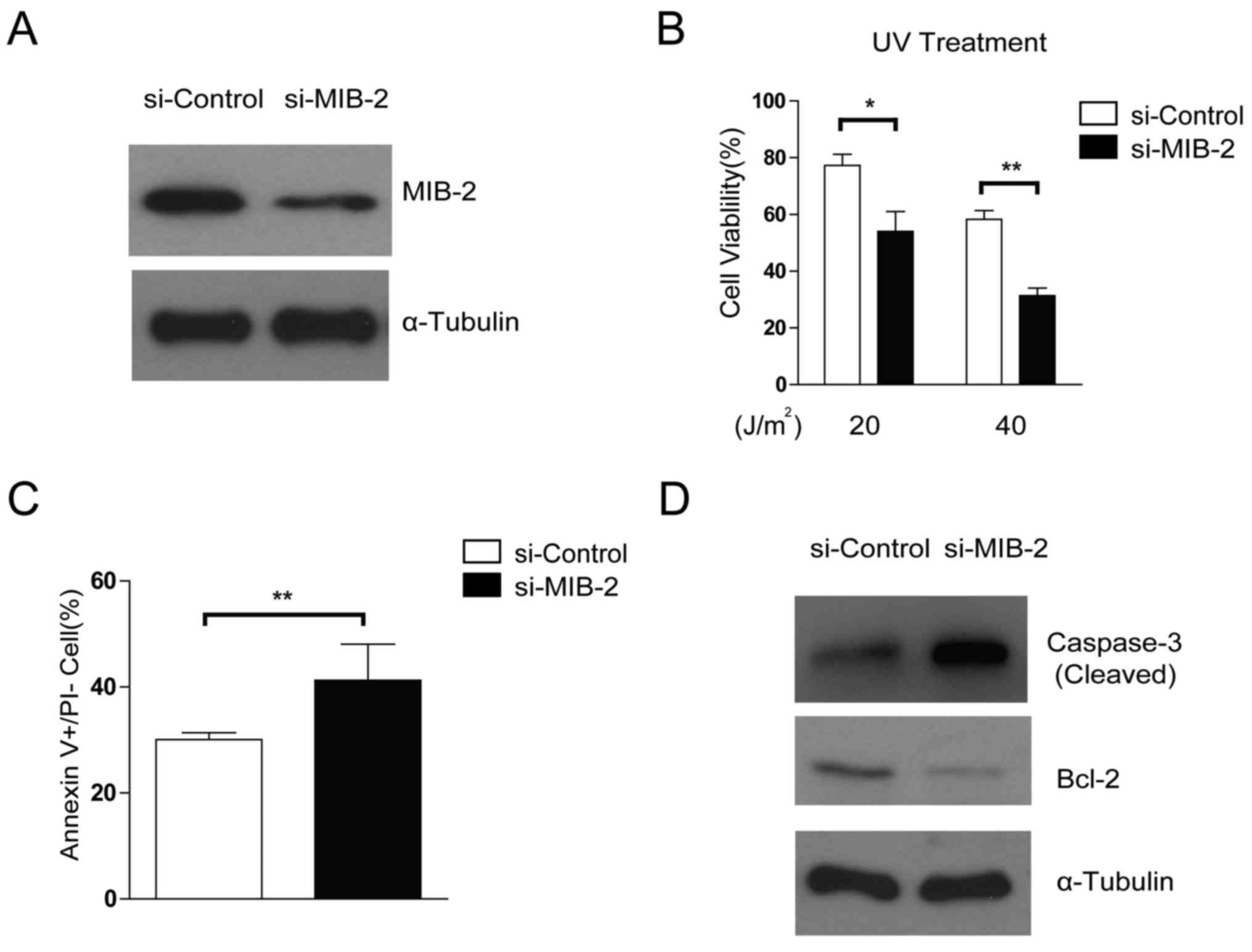
Depletion of MIB2 increases the
susceptibility of glioma cells toward apoptosis
The effects of specific MIB2 knockdown on glioma
cell survival and apoptosis were then compared using treatment with
RNA interference. The glioma T98G cell line with MIB2 depletion
exhibited increased rates of apoptosis following UV irradiation
(Fig. 4A and B), compared with the
control. Knockdown of MIB2 increased the rates of cellular
apoptosis in glioma cells compared with the control cells, as
confirmed by Annexin V binding assays (Fig. 4C). In addition, promoted activation
and cleavage of caspase-3 and a decreased level of Bcl-2 was
observed when MIB2 was depleted in T98G cell lines (Fig. 4D). These data suggested that MIB2 may
be a key controller in the adaptation of the anti-apoptotic
phenotype in glioma cells.
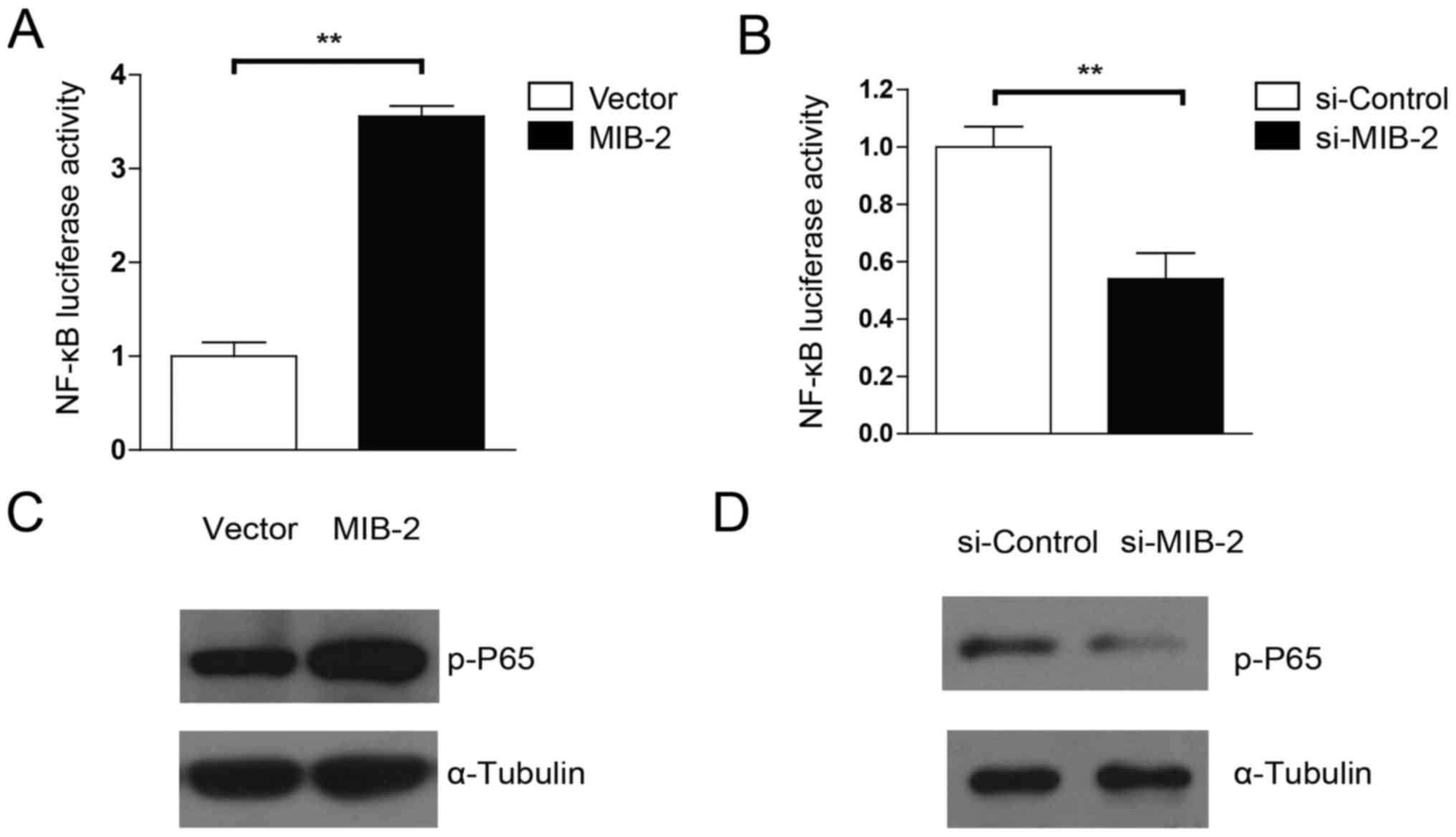
MIB2 is capable of activating the
NF-κB pathway
Evidence has suggested that Bcl-2 expression is
controlled by NF-κB activity, and that activation of NF-κB promotes
drug resistance in glioma cells (9,29).
Therefore, the present study examined whether the anti-apoptotic
activity of MIB2 in human glioma cells was associated with NF-κB
activation. MIB2-overexpressing T98G cells exhibited significantly
(P<0.01) more marked luciferase activity driven by the NF-κB
inducible promoter, compared with the vector control cells
(Fig. 5A). By contrast, cells with
MIB2 knockdown exhibited significantly decreased NF-κB activity
(Fig. 5B). The WB analysis
substantiated these results. NF-κB p65 phosphorylation was
upregulated in glioma T98G cells with MIB2 overexpression (Fig. 5C), whereas a significant decrease in
NF-κB p65 phosphorylation levels was observed in MIB2 knockdown
glioma cells (Fig. 5D). Taken
together, these results supported our hypothesis that MIB2 mediates
an increase in NF-κB signaling.
Discussion
In the present study, MIB2, an E3 ligase, was
identified as a key contributor that enabled glioma cells to
acquire anti-apoptotic properties through the activation of the
NF-κB pathway. Previous data have implicated NF-κB activation in
malignant transformation and resistance to apoptosis in human
glioma cells (15). However, the
underlying molecular mechanism of how NF-κB activity is regulated
remains unknown. The present study demonstrated that MIB2
expression was highly upregulated in glioma cells compared with
control cells. As a result, the cellular function of MIB2 was
suggested to be the activation of NF-κB signaling and the induction
of an anti-apoptotic phenotype. Furthermore, the overexpression of
MIB2 was significantly associated with the development of
resistance to UV irradiation in the primary human glioma samples
examined, while the downregulation of MIB2 restored the
pro-apoptotic responses toward apoptotic inducers. These data
indicated that MIB2 is an activator of NF-κB, and that activation
of the NF-κB pathway by MIB2 is a key mechanism for promoting
glioma cell survival.
Protein degradation via ubiquitination is a
catalytic mechanism that targets various cellular proteins, and is
therefore pivotal in regulating a wide variety of cellular
signaling pathways and cell survival (30). As neurons are particularly vulnerable
to changes in cellular proteins and alterations in signaling
processes (31), the ubiquitination
system is a key regulator of neuronal physiology, pathology and
tumorigenesis. At present, MIB2 has been elucidated to be an
important modulator of the Notch and glutamate receptor signaling
pathway, and to have an essential role in mammalian development
(32,33). Although suggestions that MIB2 may be
potent in inducing NF-κB targeted gene activation have been made,
the primary question remaining is how MIB2 expression in glioma is
associated with NF-κB activity, and how these interactions affect
cancer cell survival (34). MIB2 was
first characterized as an actin-binding, cytoskeleton-associated
protein in melanoma (35). In
Drosophila, MIB2 is responsible for proper muscle
development and maintenance of muscle integrity by preventing their
apoptotic degradation (36,37). The roles of MIB2 in immunity have been
studied extensively: Matsuda et al (31) presented the first study on the
involvement of MIB2 in cytokine signaling as being an activator of
NF-κB and a mitogen-activated protein kinase promoter (38). Additionally, Stempin et al
(32) revealed that the complex of
Bcl-10 and MIB2 promoted auto-ubiquitination and ubiquitination of
IKKα, and led to the activation of NF-κB. The results of these
studies supported the data indicating that MIB2 is specifically
involved in NF-κB activation.
In addition to its roles in causing cancer through
inflammation, NF-κB is known to regulate a range of downstream
anti-apoptotic genes, including cellular inhibitor of apoptosis 2
and Bcl-extra large (38,39). Owing to this anti-apoptotic function,
NF-κB has been demonstrated to actively participate in the
development of resistance to chemotherapy and radiotherapy in
numerous neoplasms (40–42). Notably, it has been established that
NF-κB is activated in response to treatment with chemotherapeutic
drugs or irradiation (43,44). More specifically, inhibiting NF-κB
activation enhances the cytotoxic effects of apoptotic inducers
(26). This association has been
described in various cancer cell types, including glioma (45). Therefore, a number of preclinical
studies have been conducted on agents that inhibit the activation
of the NF-κB signaling pathway. At present, the only NF-κB
inhibitors used clinically are proteasome inhibitors that function
to prevent the degradation of IκB, but the uses of these strategies
are limited due to their lack of efficacy and high cytotoxicity
(46). Additional analysis of the
mechanisms behind the induction of NF-κB may identify more targets
for NF-κB inhibition strategies. In light of this, the present
study aimed to clarify the roles of MIB2 in the activation of NF-κB
in response to UV irradiation, a DNA-damage agent known to induce
NF-κB activity (47,48). By first examining the expression of
MIB2 in glioma cell lines and clinical specimens, it was
demonstrated that MIB2 was expressed in a high abundance in tumors.
In addition, MIB2 was identified to mediate anti-apoptotic
responses via activating NF-κB. Depleting MIB2 resulted in enhanced
susceptibility of glioma cells to UV-induced cell death, suggesting
that the key downstream role for MIB2 is NF-κB-driven mediators of
cell survival and anti-apoptosis.
In conclusion, the results of the present study
propose a novel and important role for MIB2 in NF-κB-mediated
resistance to apoptosis in glioma cells. This suggests the
potential for developing a therapeutic strategy of inhibiting MIB2
to overcome anti-apoptotic responses.
Acknowledgements
Not applicable.
Funding
This research is supported by the 309th Hospital of
Chinese People's Liberation Army (grant no. 2015ZD-002) and
Liaoning Natural Funding (grant no. 2015020714).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JB, LX, YH, WF, XK, XM, YG, LB, WC and BS planned
the experiment, collected the data and wrote the paper. ZY and BC
analyzed the data. XL collected the samples.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Shenzhen People's Hospital (Shenzhen, China). Written
informed consent was obtained from all patients.
Consent for publication
Consent for publication was obtained from all
patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nieder C, Mehta MP and Jalali R: Combined
radio- and chemotherapy of brain tumours in adult patients. Clin
Oncol (R Coll Radiol). 21:515–524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fidoamore A, Cristiano L, Antonosante A,
d'Angelo M, Di Giacomo E, Astarita C, Giordano A, Ippoliti R,
Benedetti E and Cimini A: Glioblastoma stem cells microenvironment:
The paracrine roles of the niche in drug and radioresistance. Stem
Cells Int. 2016:68091052016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Argyriou AA and Kalofonos HP: Molecularly
targeted therapies for malignant gliomas. Mol Med. 15:115–122.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kasibhatla S and Tseng B: Why target
apoptosis in cancer treatment? Mol Cancer Therap. 2:573–580.
2003.
|
|
7
|
Wong ML, Kaye AH and Hovens CM: Targeting
malignant glioma survival signalling to improve clinical outcomes.
J Clin Neurosci. 14:301–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun SY, Hail N Jr and Lotan R: Apoptosis
as a novel target for cancer chemoprevention. J Natl Cancer Inst.
96:662–672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Biswas DK, Shi Q, Baily S, Strickland I,
Ghosh S, Pardee AB and Iglehart JD: NF-kappa B activation in human
breast cancer specimens and its role in cell proliferation and
apoptosis. Proc Natl Acad Sci USA. 101:10137–10142. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen HM and Tergaonkar V: NFkappaB
signaling in carcinogenesis and as a potential molecular target for
cancer therapy. Apoptosis. 14:348–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guan H, Zhang H, Cai J, Wu J, Yuan J, Li
J, Huang Z and Li M: IKBKE is over-expressed in glioma and
contributes to resistance of glioma cells to apoptosis via
activating NF-κB. J Pathol. 223:436–445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumar DM, Patil V, Ramachandran B, Nila
MV, Dharmalingam K and Somasundaram K: Temozolomide-modulated
glioma proteome: Role of interleukin-1 receptor-associated kinase-4
(IRAK4) in chemosensitivity. Proteomics. 13:2113–2124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamagishi N, Miyakoshi J and Takebe H:
Enhanced radiosensitivity by inhibition of nuclear factor kappa B
activation in human malignant glioma cells. Int J Radiat Biol.
72:157–162. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Angileri FF, Aguennouz M, Conti A, La
Torre D, Cardali S, Crupi R, Tomasello C, Germanò A, Vita G and
Tomasello F: Nuclear factor-kappaB activation and differential
expression of survivin and Bcl-2 in human grade 2-4 astrocytomas.
Cancer. 112:2258–2266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong ET and Tergaonkar V: Roles of
NF-kappaB in health and disease: Mechanisms and therapeutic
potential. Clin Sci (Lond). 116:451–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sizemore N, Lerner N, Dombrowski N,
Sakurai H and Stark GR: Distinct roles of the Ikappa B kinase alpha
and beta subunits in liberating nuclear factor kappa B (NF-kappa B)
from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B.
J Biol Chem. 277:3863–3869. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mankan AK, Lawless MW, Gray SG, Kelleher D
and McManus R: NF-kappaB regulation: The nuclear response. J Cell
Mol Med. 13:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Antoon JW, White MD, Slaughter EM, Driver
JL, Khalili HS, Elliott S, Smith CD, Burow ME and Beckman BS:
Targeting NFκB mediated breast cancer chemoresistance through
selective inhibition of sphingosine kinase-2. Cancer Biol Ther.
11:678–689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santini D, Schiavon G, Vincenzi B, Gaeta
L, Pantano F, Russo A, Ortega C, Porta C, Galluzzo S, Armento G, et
al: Receptor activator of NF-kB (RANK) expression in primary tumors
associates with bone metastasis occurrence in breast cancer
patients. PLoS One. 6:e192342011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoshiya Y, Gupta V, Segev DL, Hoshiya M,
Carey JL, Sasur LM, Tran TT, Ha TU and Maheswaran S: Mullerian
inhibiting substance induces NFkB signaling in breast and prostate
cancer cells. Mol Cell Endocrinol. 211:43–49. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mani A and Gelmann EP: The
ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol.
23:4776–4789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rajan N, Elliott RJ, Smith A, Sinclair N,
Swift S, Lord CJ and Ashworth A: The cylindromatosis gene product,
CYLD, interacts with MIB2 to regulate notch signalling. Oncotarget.
5:12126–12140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vigneswaran K, Neill S and Hadjipanayis
CG: Beyond the world health organization grading of infiltrating
gliomas: Advances in the molecular genetics of glioma
classification. Ann Transl Med. 3:952015.PubMed/NCBI
|
|
24
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nature Protoc.
3:1101–1108. 2008. View Article : Google Scholar
|
|
25
|
Kämmerer U, Kapp M, Gassel AM, Richter T,
Tank C, Dietl J and Ruck P: A new rapid immunohistochemical
staining technique using the EnVision antibody complex. J Histochem
Cytochem. 49:623–630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bednarski BK, Ding X, Coombe K, Baldwin AS
and Kim HJ: Active roles for inhibitory kappaB kinases alpha and
beta in nuclear factor-kappaB-mediated chemoresistance to
doxorubicin. Mol Cancer Therap. 7:1827–1835. 2008. View Article : Google Scholar
|
|
27
|
McClelland RA, Finlay P, Walker KJ,
Nicholson D, Robertson JF, Blamey RW and Nicholson RI: Automated
quantitation of immunocytochemically localized estrogen receptors
in human breast cancer. Cancer Res. 50:3545–3550. 1990.PubMed/NCBI
|
|
28
|
Jung Y, Joo KM, Seong DH, Choi YL, Kong
DS, Kim Y, Kim MH, Jin J, Suh YL, Seol HJ, et al: Identification of
prognostic biomarkers for glioblastomas using protein expression
profiling. Int J Oncol. 40:1122–1132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pitlick FA: Binding of bovine brain tissue
factor to concanavalin A-Sepharose. Partial purification of
coagulant, arylamidase, and alkaline phosphatase activities.
Biochim Biophys Acta. 428:27–34. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tu Y, Chen C, Pan J, Xu J, Zhou ZG and
Wang CY: The ubiquitin proteasome pathway (UPP) in the regulation
of cell cycle control and DNA damage repair and its implication in
tumorigenesis. Int J Clin Exp Pathol. 5:726–738. 2012.PubMed/NCBI
|
|
31
|
Wang X and Michaelis EK: Selective
neuronal vulnerability to oxidative stress in the brain. Front
Aging Neurosci. 2:122010.PubMed/NCBI
|
|
32
|
Koo BK, Yoon KJ, Yoo KW, Lim HS, Song R,
So JH, Kim CH and Kong YY: Mind bomb-2 is an E3 ligase for Notch
ligand. J Biol Chem. 280:22335–22342. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koo BK, Yoon MJ, Yoon KJ, Im SK, Kim YY,
Kim CH, Suh PG, Jan YN and Kong YY: An obligatory role of mind
bomb-1 in notch signaling of mammalian development. PLoS One.
2:e12212007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ye JS, Kim N, Lee KJ, Nam YR, Lee U and
Joo CH: Lysine 63-linked TANK-binding kinase 1 ubiquitination by
mindbomb E3 ubiquitin protein ligase 2 is mediated by the
mitochondrial antiviral signaling protein. J Virol. 88:12765–12776.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takeuchi T, Heng HH, Ye CJ, Liang SB,
Iwata J, Sonobe H and Ohtsuki Y: Down-regulation of a novel
actin-binding molecule, skeletrophin, in malignant melanoma. Am J
Pathol. 163:1395–1404. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nguyen HT, Voza F, Ezzeddine N and Frasch
M: Drosophila mind bomb2 is required for maintaining muscle
integrity and survival. J Cell Biol. 179:219–227. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barsi JC, Rajendra R, Wu JI and Artzt K:
Mind bomb1 is a ubiquitin ligase essential for mouse embryonic
development and Notch signaling. Mech Dev. 122:1106–1117. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Q, Wang X and Evers BM: Induction of
cIAP-2 in human colon cancer cells through PKC delta/NF-kappa B. J
Biol Chem. 278:51091–51099. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hinz M, Loser P, Mathas S, Krappmann D,
Dörken B and Scheidereit C: Constitutive NF-kappaB maintains high
expression of a characteristic gene network, including CD40, CD86,
and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells.
Blood. 97:2798–2807. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baldwin AS: Control of oncogenesis and
cancer therapy resistance by the transcription factor NF-kappaB. J
Clin Invest. 107:241–246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakshatri H, Bhat-Nakshatri P, Martin DA,
Goulet RJ Jr and Sledge GW Jr: Constitutive activation of NF-kappaB
during progression of breast cancer to hormone-independent growth.
Mol Cell Biol. 17:3629–3639. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cusack JC, Liu R and Baldwin AS: NF- kappa
B and chemoresistance: Potentiation of cancer drugs via inhibition
of NF-kappa B. Drug Resist Updat. 2:271–273. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cusack JC Jr, Liu R, Houston M, Abendroth
K, Elliott PJ, Adams J and Baldwin AS Jr: Enhanced chemosensitivity
to CPT-11 with proteasome inhibitor PS-341: Implications for
systemic nuclear factor-kappaB inhibition. Cancer Res.
61:3535–3540. 2001.PubMed/NCBI
|
|
44
|
Lu TP, Lai LC, Lin BI, Chen LH, Hsiao TH,
Liber HL, Cook JA, Mitchell JB, Tsai MH and Chuang EY: Distinct
signaling pathways after higher or lower doses of radiation in
three closely related human lymphoblast cell lines. Int J Radiat
Oncol Biol Phys. 76:212–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion O6-methylguanine.
Oncogene. 26:186–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Alberts SR, Foster NR, Morton RF, Kugler
J, Schaefer P, Wiesenfeld M, Fitch TR, Steen P, Kim GP and Gill S:
PS-341 and gemcitabine in patients with metastatic pancreatic
adenocarcinoma: A north central cancer treatment group (NCCTG)
randomized phase II study. Ann Oncol. 16:1654–1661. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ali F and Sultana S: Repeated short-term
stress synergizes the ROS signalling through up regulation of NFkB
and iNOS expression induced due to combined exposure of
trichloroethylene and UVB rays. Mol Cell Biochem. 360:133–145.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Triscott J, Rose Pambid M and Dunn SE:
Concise review: Bullseye: Targeting cancer stem cells to improve
the treatment of gliomas by repurposing disulfiram. Stem cells.
33:1042–1046. 2015. View Article : Google Scholar : PubMed/NCBI
|