Transforming growth factor-β (TGF-β) signaling is
widely known to serve an important role in the extracellular
microenvironment and numerous cellular processes, including cell
proliferation, differentiation, apoptosis and migration (1). Based on a significant amount of evidence
in the literature, TGF-β signaling is currently considered to have
paradoxical impacts on cancer. TGF-β functions as a tumor
suppressor in normal epithelial cells or in the early stages of
oncogenesis. However, as a tumor develops, TGF-β becomes a potent
tumor promoter in the epithelium at a later stage and even
increases the production of TGF-β, supporting tumor progression and
metastasis (2,3).
TGF-β is a regulatory cytokine that is secreted by
tumor and stromal cells in the tumor microenvironment (TME).
Members of the TGF-β superfamily include TGF-βs, bone morphogenic
proteins, growth and differentiation factors, activins, inhibins
and the anti-Müllerian hormone (4).
Inactive TGF-β cytokines, known as latent TGF-βs, are located in
the extracellular matrix. Upon activation, the ligand binds to
TGF-β receptor type II (TβRII), which is constitutively activated,
and then interacts with TβRI (also termed activin receptor-like
kinase), resulting in the formation of hetero-tetrameric complex
and phosphorylation of TβRI (5,6). The
co-receptors (known as TβRIII) may modulate the access of ligands
to TβRI and TβRII, rather than being directly involved in the
pathway (7). The hetero-tetrameric
complex of active receptors initiates downstream signaling through
canonical or non-canonical TGF-β signaling. In the canonical
signaling, active TβRI recruits and phosphorylates
receptor-regulated Smad (R-Smad) proteins, including Smad1-3, Smad5
and Smad8. Activated R-Smads associate with common-mediator Smad
proteins (Smad4 in mammals) to form hetero-trimers, which
subsequently translocate to the nucleus, bind to Smad-binding
elements and regulate TGF-β-responsive genes in collaboration with
cofactors, such as zinc-finger and forkhead (8–10). The
inhibitory Smad proteins (including Smad6 and Smad7) compete with
R-Smads for binding to the receptors and recruit ubiquitin ligases
to degrade TβRI and R-Smads, thus modulating the intensity and
duration of Smad-dependent signaling (11,12). In
the non-canonical TGF-β signaling pathway, phosphorylated
hetero-tetrameric receptors activate phosphoinositide
3-kinase/protein kinase B (PI3K/Akt), Ras homolog gene family
member A and mitogen-activated protein kinase (MAPK) among others
(13,14).
Dysregulated TGF-β signaling is common in several
types of cancer, including head and neck squamous cell carcinoma
(HNSCC) (15), and serves a crucial
role in tumor prevention and progression. HNSCC accounts for ~90%
of head and neck cancer cases, and common risk factors include
tobacco exposure, alcohol use, human papillomavirus infection and
areca nut consumption. A series of therapies have been applied in
the treatment of HNSCC, including surgery, radiotherapy,
neoadjuvant chemotherapy and a combination of these methods
(16). However, the 5-year survival
rate of this disease has not evidently increased in last 30 years
and remains at ≤50% (17–21). Accumulating evidence suggested that
deregulation of TGF-β signaling is of great importance in HNSCC and
may be the result of defected TGF-β signaling (22–24).
Previous studies have demonstrated that the expression of Smad4 and
Smad2 is frequently lost in HNSCC, while increased TGF-β1
expression has been reported in the majority of these tumors.
Mutations of TβRII have been reported to occur in 21% of oral
squamous cell carcinoma (OSCC) (25–27). In
addition, TβRII mRNA exhibited a >50% loss in HNSCC and adjacent
tissue samples as compared with the levels in normal tissue samples
(28,29). However, the exact role of TGF-β in
HNSCC is not completely understood. The present study reviews the
current understanding on the TGF-β signaling pathway and its impact
on cells in the HNSCC microenvironment.
The tumor suppressive effect of TGF-β was supported
by several animal models with defected TGF-β signaling. For
example, TβRI/phosphatase and tensin homolog (PTEN) knockout mice
developed full-penetrance HNSCC while mice with PTEN deletion
presented with hyperproliferation in the head and neck epithelium
(30). Similarly, Smad4 deletion in
head and neck epithelium also exhibited spontaneous HNSCC in mice
(26). In normal epithelial cells,
TGF-β may maintain homeostasis through the regulation of
proliferation and apoptosis. TGF-β signaling arrests the cell cycle
in phase G1, and mechanisms underlying normal cell growth
inhibition include upregulation of cyclin-dependent kinase (CDK)
inhibitors and downregulation of Myc expression (31). Smad3/Smad4 complexes interact with
forkhead box O (FoxO) to increase the expression of CDK inhibitors,
namely p15 and p21 (1,3,32). In the
presence of co-repressors, Smad3/Smad4 complex also interact with
regulatory elements of the Myc promoter, a cell cycle regulator
gene (33,34). As a consequence, the mRNA and protein
expression of Myc are reduced. Smads induce apoptosis in epithelial
cells through the activation of P53, Bcl-2-like protein 11 and
death-associated protein kinase, and the repression of Akt. TβRI
also induces apoptosis in normal epithelial cells through
TGF-β-activated kinase 1 (TAK1)-p38/c-Jun N-terminal kinase,
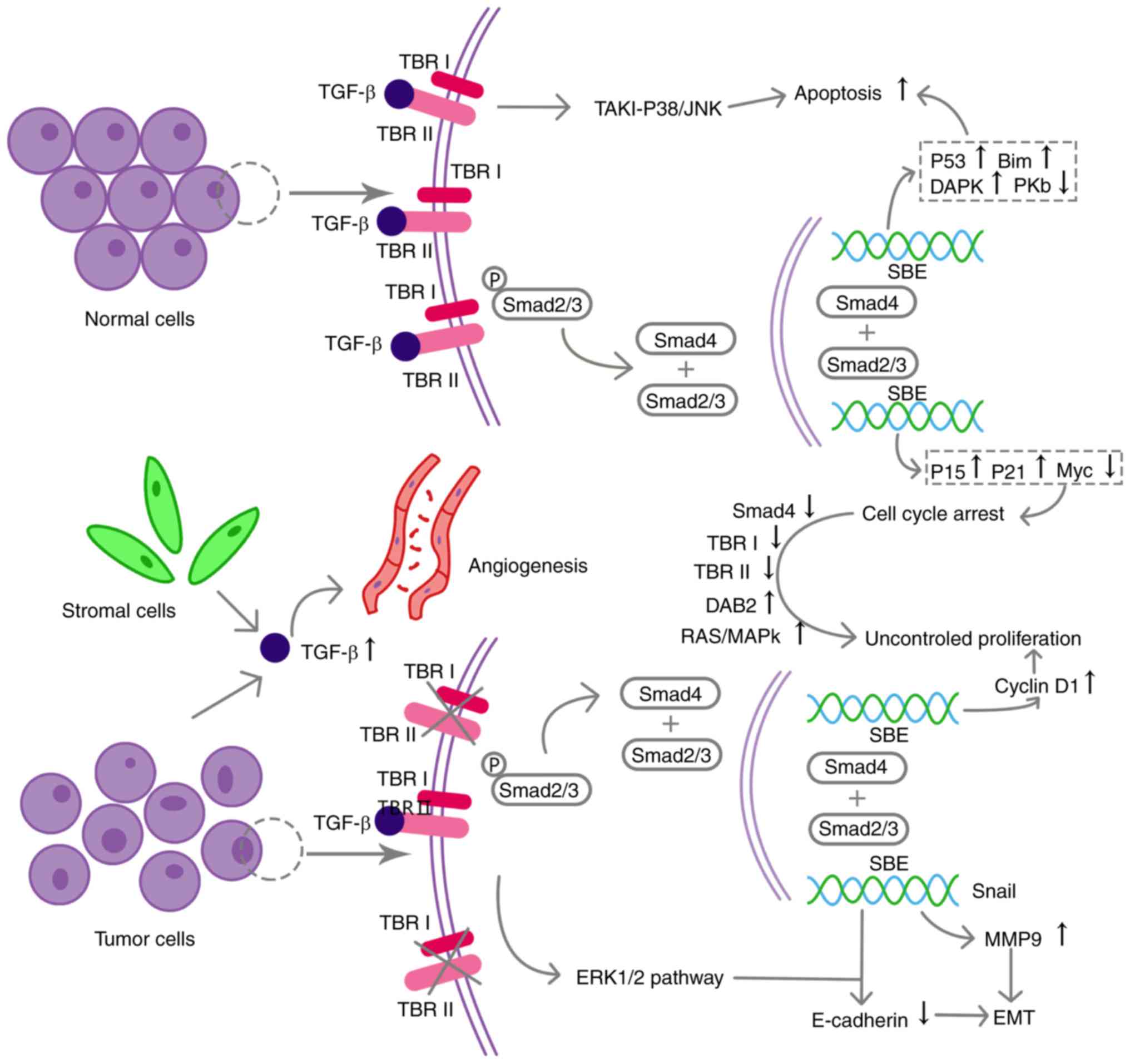
independent of Smads (Fig. 1)
(1,35–40).
However, there is no direct evidence to support that the same
molecular mechanisms are involved in HNSCC. Inhibitor of
DNA-binding/differentiation (ID) proteins are known to negatively
regulate cell differentiation by interfering with basic
helix-loop-helix transcription factors. It has been reported that
in keratinocytes TGF-β upregulated the expression of cyclic
AMP-dependent transcription factor-3, which served as a cofactor
assisting the binding of Smad3/Smad4 complexes to the ID1 promoter;
consequently, ID1 expression was downregulated and cell
differentiation was promoted in vitro (41,42).
Although TGF-β signaling is widely known to mediate
cell cycle arrest and enhance apoptosis in normal epithelium or in
the early stage of tumor formation, it also induces epithelial cell
overproliferation and inhibits apoptosis at a later stage of
oncogenesis. For instance, Lu et al (27) demonstrated that tumor cells and
epithelial cells from adjacent tissues expressed increased levels
of TGF-β1, as compared with those in epithelial cells from normal
control human tissues. However, in transgenic mice, overexpression
of TGF-β1 was reported to result in the hyperproliferation of cells
at the head and neck epithelium, and to enhance inflammation and
angiogenesis (27). This suggested
that TGF-β1 promoted cell proliferation by the formation of an
extracellular microenvironment in favor of tumor formation, even at
early stage of carcinogenesis (Fig.
1) (27). Although, TGF-β1
functions as a potent chemotactic molecule for leukocytes, it has
been reported that inflammatory cytokines and growth factors
secreted by infiltrated leukocytes may counteract the negative
effects of TGF-β1 on the cell cycle (27). In fact, loss of TβRI or TβRII partly
subverts TGF-β1-induced cell cycle arrest, and this effect along
with the increased production of TGF-β1 may result in its
accumulation in the extracellular microenvironment. The loss of
TβRI or TβRII has also been proven to lead to increased cell
proliferation and inhibit the apoptosis of HNSCC cells,
respectively (29,30). Additionally, a previous study
identified that improved proliferation and inhibited apoptosis due
to decreased TβRI levels were alternatively associated with the
activation of the PI3K/Akt signaling pathway (43).
TGF-β signaling disruptions are associated with poor
prognosis partly due to the induction of epithelial-mesenchymal
transition (EMT). EMT is a cellular process during which a cell
with epithelial characteristics, such as cell polarity and
cell-cell conjunction, translates to a cell with mesenchymal
characteristics, such as motility (44). Decreased E-cadherin and increased
vimentin levels are hallmarks of the EMT, while Snail and Twist are
important factors negatively regulating E-cadherin (45). In OSCC, TGF-β signaling has been
implicated in EMT through Snail and upregulation of matrix
metalloprotease 9 (MMP-9) levels (46,47). Yu
et al (48) demonstrated that
TGF-β1 expression in HNSCC was correlated with decreased E-cadherin
level through the phosphorylation of Smad2/3 and subsequent
involvement of Smad4, which bound to the Snail promoter (49). Independently of Smads, TGF-β1 also
regulates the Snail family proteins via the extracellular
signal-regulated kinase (ERK)1/2 pathway in HNSCC (45). In addition, MMP-9 degrades the
extracellular matrix components and basement membrane, and is
regulated by TGF-β1 through Smad2/3 and myosin light chain kinase
in human HNSCC cell lines (Fig. 1)
(50). Notably, Sun et al
(46) demonstrated a reciprocal
interaction between MMP-9 and Snail regulation. MMP-9 induced EMT
partly through the expression of Snail, while Snail was involved in
TGF-β1-modulated MMP-9 expression by increasing Ets-1 (46). Another study indicated that TGF-β1
promoted MMP-9 expression by Slug (Snail2) (51). Furthermore, TGF-β1 may enhance EMT in
cooperation with other growth factors in HNSCC. Compared with
TGF-β1 or epithelial growth factor (EGF) alone, long-term
co-stimulation with TGF-β1 and EGF in an OSCC cell culture model
induced a phenotype transition, displaying upregulation of vimentin
and downregulation of E-cadherin at the protein level, as well as
markedly enhanced invasiveness (52).
It was also reported that these observations may be associated with
TGF-β1/EGF causing extracellular matrix remodeling by a
plasmin/MMP-10/MMP-1-dependent collagen remodeling axis (52).
The TGF-β signaling pathway serves as a tumor
suppressor at an early stage, whereas it serves as a tumor promoter
in transformed epithelial cells at a later stage (31). Accordingly, the potential mechanisms
underlying this conversion of the role of TGF-β have been widely
discussed due to their important effect on the balance of normal
and transformed cells, and these mechanisms may serve as
therapeutic targets in malignancies of an epithelial origin. Among
the numerous possible mechanisms, investigation of mutations in the
Smad-dependent pathway and disruption of the balance between this
and other pathways may be of great importance.
In keratinocytes, the defected Smad4 pathway or
alternative activated pathways (such as Erk) may abrogate growth
inhibition and enable the pro-oncogenic effects of TGF-β. Defective
Smads-dependent pathways overturn the effect of TGF-β that induces
cell cycle arrest and apoptosis and at the same time reciprocally
interact with alternative pathways facilitating cancer invasion
(53). Smad4 is a gatekeeper gene in
HNSCC that directly controls cell proliferation, and decreased
Smad4 expression has been verified to be correlated with
irresponsiveness to TGF-β-induced growth inhibition (53). In addition, oncogenic Ras
downregulates Smad4 by promoting the process of degradation
(54); in return, low levels of Smad4
activate Ras-dependent ERK signaling, which is then involved in the
progression to undifferentiated carcinoma in keratinocytes
(55).
Disabled homolog 2 (DAB2), a putative tumor
suppressor gene, suppresses Smad2 phosphorylation and activation.
Hannigan et al (56) reported
that, in SCC cell lines, the epigenetically downregulated DAB2 has
been verified to negatively regulate Smad2 and its downstream
pathway. By contrast, upon enhanced expression of DAB2, the cell
lines presented growth prohibitive responses to TGF-β again
(56). Therefore, downregulated DAB2
contributes to converting TGF-β into a tumor promoter, facilitating
cell proliferation and anchorage-independent growth (Fig. 1).
Other mechanisms have also been suggested to be
involved in this role conversion. For instance, TβRII is reportedly
significantly decreased in primary HNSCC (57), resulting in TGF-β1 accumulation in the
TME. In place of suppressing overproliferation in epithelial cells,
excessive TGF-β1 levels may directly affect the tumor stroma,
inducing effects such as promotion of angiogenesis, myofibroblast
formation and exertion of a chemotactic effect on neutrophils and
macrophages (29). Notably, TβRII
mutation in the epithelium is also a classic mechanism that
subverts growth arrest and contributes to a tumor-friendly
microenvironment by increasing inflammation and angiogenesis
(Fig. 1). Additionally, the
suppressive effect of Smad7 on the canonical TGF-β signaling
pathway is greater than that on non-canonical pathways involving
TAK1 signaling, such as TAK1-NF-κB signaling, which favors
malignant progression. Freudlsperger et al (58) demonstrated that, in HNSCC cell lines,
the relatively preferential suppression by Smad7 on Smad-dependent
growth inhibition may favor the conversion. The development of
cancer cell tolerance to the TGF-β-mediated growth inhibition is
currently an important focus of carcinogenesis, progression and
treatment research; however, the intricate conversion mechanism
remains unclear to date.
The concept of tumor immune surveillance was
initially described in 1967 by Burnet (59). This refers to an organism discerning
and eliminating cancer cells via the innate and adaptive immune
system. At the same time, cancer cells are able to evade
discernment and attack from the immune system, thus promoting tumor
progression. Over the last 50 years, TGF-β has been demonstrated to
abrogate tumor-suppressing immune cell functions and to support
tumor-promoting functions in certain types of cancer (23,24,60). This
section outlines how TGF-β signaling promotes tumor progression in
HNSCC by affecting several types of immune cells.
Previous studies have indicated that
tumor-associated macrophages are mainly of the M2 phenotype and are
positively correlated with the histopathological grades of HNSCC
bearing hosts (67–69). Exposure to enhanced expression of
TGF-β results in an M2 macrophage phenotype, which typically
expressed CD206 (70). Mechanisms of
TGF-β inducing M2 include the negative regulation of Toll-like
receptor (TLR) signaling that causes induction of anti-tumor
cytokines, such as tumor necrosis factor (TNF)-α, interleukin
(IL)-12 and interferon (IFN)-γ, in order to participate in
macrophage responses (71,72). Furthermore, Standiford et al
(73) demonstrated that activated
TGF-β signaling is required in IL receptor-associated kinase
induction, which is a critically negative regulator of TLR
signaling. Besides participating in macrophages polarization, TGF-β
recruits macrophages to the TME, where they further produce TGF-β
and thus a vicious cycle is formed (60).
Myeloid-derived suppressor cells (MDSCs), first
detected in cancer patients in 1984 (80), consist of a heterogeneous population
of immature myeloid cells, including the precursors of DCs and
macrophages. One of the most notable characteristics of MDSCs is
that they suppress the activity of T cells (81,82).
Previous studies have reported the presence of MDSCs in the
peripheral blood of HNSCC patients, and that high infiltration of
MDSCs promoted an immune-suppressive TME (83–85). Bian
et al (30) demonstrated that
decreased TGF-β signaling upregulated C-X-C motif chemokine ligand
(CXCL)1, CXCL5, prostaglandin-endoperoxide synthase 2 and CXC
receptor 3, contributing to the recruitment of MDSCs in an HNSCC
model in TβRI/phosphatase and tensin homolog (PTEN) 2cKO mice.
However, further research is required to support whether the
recruitment of MDSCs in an HNSCC model is associated with a TGF-β
signaling defect alone, or simultaneous TGF-β signaling defect and
PTEN loss.
T lymphocytes can be classified into four subtypes,
including cytotoxic T, helper T (Th), regulatory T (Treg) and
memory T (Tm) cells. With the exception of Tm cells, all these
subtypes have been verified to serve an important role in tumor
escape from immune surveillance (86). Similarly to epithelial cells, TGF-β
induces primary T lymphocyte growth inhibition in G1 phase by
downregulating CDK4 (87). In
addition, the trigger of the conversion of local primary T cells to
Treg or Th cells in mice is partly associated with the
transcription factor FoxP3 and retinoic acid receptor-related
orphan receptor γt, respectively, whose transcription activity can
be modulated by TGF-β (88,89).
Besides tumor and immune cells, stromal cells are
another important component of the TME. HNSCC has a relatively low
survival rate, which may be the consequence of the complex
symbiotic association among tumor cells, surrounding stromal cells
(including fibroblasts) and the neoplastic extracellular matrix
(97). TGF-β interferes with these
cells in a paracrine or autocrine manner, leading to increased
inflammation and angiogenesis, and consequently to tumorigenesis
and tumor invasiveness, as reported in a study by Lu et al
(27). A full discussion of how TGF-β
signaling regulates stromal cells in the TME is beyond the scope of
the present review, and the attention of this section will focus on
the effects of TGF-β signaling on cancer-associated fibroblasts
(CAFs).
Not applicable.
The present study was supported by National Natural
Science Foundation of China grants (grant nos. 81672672, 81772891,
81572650 and 81621062), and by National Program on Key Research
Project of China (grant no. 2016YFC0902700).
Not applicable.
XP was mainly responsible for the manuscript
writing. YLT and XHL provided suggestions on the ideas and
performed the final corrections.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Meulmeester E and Ten Dijke P: The dynamic
roles of TGF-β in cancer. J Pathol. 223:205–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pickup M, Novitskiy S and Moses HL: The
roles of TGFβ in the tumour microenvironment. Nat Rev Cancer.
13:788–799. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Drabsch Y and ten Dijke P: TGF-β
signalling and its role in cancer progression and metastasis.
Cancer Metastasis Rev. 31:553–568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wakefield LM and Hill CS: Beyond TGFβ:
Roles of other TGFβ superfamily members in cancer. Nat Rev Cancer.
13:328–341. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cantelli G, Crosas-Molist E, Georgouli M
and Sanz-Moreno V: TGFβ-induced transcription in cancer. Semin
Cancer Biol. 42:60–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
López-Casillas F, Wrana JL and Massagué J:
Betaglycan presents ligand to the TGF beta signaling receptor.
Cell. 73:1435–1444. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gatza CE, Sun YO and Blobe GC: Roles for
the type III TGF-β receptor in human cancer. Cell Signal.
22:1163–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmierer B and Hill CS: Kinetic analysis
of Smad nucleocytoplasmic shuttling reveals a mechanism for
transforming growth factor beta-dependent nuclear accumulation of
Smads. Mol Cell Biol. 25:9845–9858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakao A, Imamura T, Souchelnytskyi S,
Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH,
Miyazono K and ten Dijke P: TGF-beta receptor-mediated signalling
through Smad2, Smad3 and Smad4. EMBO J. 16:5353–5362. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koinuma D, Tsutsumi S, Kamimura N, Imamura
T, Aburatani H and Miyazono K: Promoter-wide analysis of Smad4
binding sites in human epithelial cells. Cancer Sci. 100:2133–2142.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itoh S and ten Dijke P: Negative
regulation of TGF-beta receptor/Smad signal transduction. Curr Opin
Cell Biol. 19:176–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kavsak P, Rasmussen RK, Causing CG, Bonni
S, Zhu H, Thomsen GH and Wrana JL: Smad7 binds to Smurf2 to form an
E3 ubiquitin ligase that targets the TGF beta receptor for
degradation. Mol Cell. 6:1365–1375. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ohkawara B, Shirakabe K, Hyodo-Miura J,
Matsuo R, Ueno N, Matsumoto K and Shibuya H: Role of the
TAK1-NLK-STAT3 pathway in TGF-beta-mediated mesoderm induction.
Genes Dev. 18:381–386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Massagué J and Gomis RR: The logic of TGFβ
signaling. FEBS Lett. 580:2811–2820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malkoski SP and Wang XJ: Two sides of the
story? Smad4 loss in pancreatic cancer versus head-and-neck cancer.
FEBS Lett. 586:1984–1992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krstevska V: Evolution of treatment and
high-risk features in resectable locally advanced Head and Neck
squamous cell carcinoma with special reference to extracapsular
extension of nodal disease. J BUON. 20:943–953. 2015.PubMed/NCBI
|
|
17
|
Moutsopoulos NM, Wen J and Wahl SM:
TGF-beta and tumors-an ill-fated alliance. Curr Opin Immunol.
20:234–240. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edwards BK, Ward E, Kohler BA, Eheman C,
Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I,
Seeff LC, et al: Annual report to the nation on the status of
cancer, 1975–2006, featuring colorectal cancer trends and impact of
interventions (risk factors, screening, and treatment) to reduce
future rates. Cancer. 116:544–573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grsic K, Opacic IL, Sitic S, Milkovic PM,
Suton P and Sarcevic B: The prognostic significance of estrogen
receptor β in head and neck squamous cell carcinoma. Oncol Lett.
12:3861–3865. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bae WJ, Lee SH, Rho YS, Koo BS and Lim YC:
Transforming growth factor β1 enhances stemness of head and neck
squamous cell carcinoma cells through activation of Wnt signaling.
Oncol Lett. 12:5315–5320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Honjo Y, Bian Y, Kawakam K, Molinolo A,
Longenecker G, Boppana R, Larsson J, Karlsson S, Gutkind JS, Puri
RK and Kulkarni AB: TGF-β receptor I conditional knockout mice
develop spontaneous squamous cell carcinoma. Cell Cycle.
6:1360–1366. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Connolly EC and Akhurst RJ: The
complexities of TGF-β action during mammary and squamous cell
carcinogenesis. Curr Pharm Biotechnol. 12:2138–2149. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pring M, Prime S, Parkinson EK and
Paterson I: Dysregulated TGF-beta1-induced Smad signalling occurs
as a result of defects in multiple components of the TGF-beta
signalling pathway in human head and neck carcinoma cell lines. Int
J Oncol. 28:1279–1285. 2006.PubMed/NCBI
|
|
25
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bornstein S, White R, Malkoski S, Oka M,
Han G, Cleaver T, Reh D, Andersen P, Gross N, Olson S, et al: Smad4
loss in mice causes spontaneous head and neck cancer with increased
genomic instability and inflammation. J Clin Invest. 119:3408–3419.
2009.PubMed/NCBI
|
|
27
|
Lu SL, Reh D, Li AG, Woods J, Corless CL,
Kulesz-Martin M and Wang XJ: Overexpression of transforming growth
factor β1 in head and neck epithelia results in inflammation,
angiogenesis, and epithelial hyperproliferation. Cancer Res.
64:4405–4410. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Snijders AM, Schmidt BL, Fridlyand J,
Dekker N, Pinkel D, Jordan RC and Albertson DG: Rare amplicons
implicate frequent deregulation of cell fate specification pathways
in oral squamous cell carcinoma. Oncogene. 24:4232–4242. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu SL, Herrington H, Reh D, Weber S,
Bornstein S, Wang D, Li AG, Tang CF, Siddiqui Y, Nord J, et al:
Loss of transforming growth factor-beta type II receptor promotes
metastatic head-and-neck squamous cell carcinoma. Genes Dev.
20:1331–1342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bian Y, Hall B, Sun ZJ, Molinolo A, Chen
W, Gutkind JS, Waes CV and Kulkarni AB: Loss of TGF-β signaling and
PTEN promotes head and neck squamous cell carcinoma through
cellular senescence evasion and cancer-related inflammation.
Oncogene. 31:3322–3332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White RA, Malkoski SP and Wang XJ: TGFβ
signaling in head and neck squamous cell carcinoma. Oncogene.
29:5437–5446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alexandrow MG and Moses H: Transforming
growth factor b and cell cycle regulation. Cancer Res.
55:1452–1457. 1995.PubMed/NCBI
|
|
33
|
Kim T, Cui R, Jeon YJ, Fadda P, Alder H
and Croce CM: MYC-repressed long noncoding RNAs antagonize
MYC-induced cell proliferation and cell cycle progression.
Oncotarget. 6:18780–18789. 2015.PubMed/NCBI
|
|
34
|
Chen CR, Kang Y, Siegel PM and Massagué J:
E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to
c-myc repression. Cell. 110:19–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Massague J: TGFbeta in Cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pardali K and Moustakas A: Actions of
TGF-beta as tumor suppressor and pro-metastatic factor in human
cancer. Biochim Biophys Acta. 1775:21–62. 2007.PubMed/NCBI
|
|
37
|
Sorrentino A, Thakur N, Grimsby S,
Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH and
Landström M: The type I TGF-beta receptor engages TRAF6 to activate
TAK1 in a receptor kinase-independent manner. Nat Cell Biol.
10:1199–1207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamashita M, Fatyol K, Jin C, Wang X, Liu
Z and Zhang YE: TRAF6 mediates smad-independent activation of JNK
and p38 by TGF-beta. Mol Cell. 31:918–924. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang S, Ekman M, Thakur N, Bu S,
Davoodpour P, Grimsby S, Tagami S, Heldin CH and Landström M:
TGFbeta1-induced activation of ATM and p53 mediates apoptosis in a
Smad7-dependent manner. Cell Cycle. 5:2787–2795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jang CW, Chen CH, Chen CC, Chen JY, Su YH
and Chen RH: TGF-beta induces apoptosis through Smad-mediated
expression of DAP-kinase. Nat Cell Biol. 4:51–58. 2001. View Article : Google Scholar
|
|
41
|
Korchynskyi O and ten Dijke P:
Identification and functional characterization of distinct
critically important bone morphogenetic protein-specific response
elements in the Id1 promoter. J Biol Chem. 277:4883–4891. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kang Y, Chen CR and Massagué J: A
self-enabling TGFbeta response coupled to stress signaling: Smad
engages stress response factor ATF3 for Id1 repression in
epithelial cells. Mol Cell. 11:915–926. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bian Y, Terse A, Du J, Hall B, Molinolo A,
Zhang P, Chen W, Flanders KC, Gutkind JS, Wakefield LM and Kulkarni
AB: Progressive tumor formation in mice with conditional deletion
of TGF-beta signaling in head and neck epithelia is associated with
activation of the PI3K/Akt pathway. Cancer Res. 69:5918–5926. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu S, Ye D, Guo W, Yu W, He Y, Hu J, Wang
Y, Zhang L, Liao Y, Song H, et al: G9a is essential for
EMT-mediated metastasis and maintenance of cancer stem cell-like
characters in head and neck squamous cell carcinoma. Oncotarget.
6:6887–6901. 2015.PubMed/NCBI
|
|
45
|
Smith A, Teknos TN and Pan Q: Epithelial
to mesenchymal transition in head and neck squamous cell carcinoma.
Oral Oncol. 49:287–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun L, Diamond ME, Ottaviano AJ, Joseph
MJ, Ananthanarayan V and Munshi HG: Transforming growth factor-beta
1 promotes matrix metalloproteinase-9-mediated oral cancer invasion
through snail expression. Mol Cancer Res. 6:10–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qiao B, Johnson NW and Gao J:
Epithelial-mesenchymal transition in oral squamous cell carcinoma
triggered by transforming growth factor-beta1 is Snail
family-dependent and correlates with matrix metalloproteinase-2 and
−9 expressions. Int J Oncol. 37:663–668. 2010.PubMed/NCBI
|
|
48
|
Yu C, Liu Y, Huang D, Dai Y, Cai G, Sun J,
Xu T, Tian Y and Zhang X: TGF-β1 mediates epithelial to mesenchymal
transition via the TGF-β/Smad pathway in squamous cell carcinoma of
the head and neck. Oncol Rep. 25:15812011.PubMed/NCBI
|
|
49
|
Hoot KE, Lighthall J, Han G, Lu SL, Li A,
Ju W, Kulesz-Martin M, Bottinger E and Wang XJ:
Keratinocyte-specific Smad2 ablation results in increased
epithelial-mesenchymal transition during skin cancer formation and
progression. J Clin Invest. 118:2722–2732. 2008.PubMed/NCBI
|
|
50
|
Sinpitaksakul SN, Pimkhaokham A,
Sanchavanakit N and Pavasant P: TGF-beta1 induced MMP-9 expression
in HNSCC cell lines via Smad/MLCK pathway. Biochem Biophys Res
Commun. 371:713–718. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Joseph MJ, Dangi-Garimella S, Shields MA,
Diamond ME, Sun L, Koblinski JE and Munshi HG: Slug is a downstream
mediator of transforming growth factor-beta1-induced matrix
metalloproteinase-9 expression and invasion of oral cancer cells. J
Cell Biochem. 108:726–736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Richter P, Umbreit C, Franz M and Berndt
A, Grimm S, Uecker A, Böhmer FD, Kosmehl H and Berndt A: EGF/TGFβ1
co-stimulation of oral squamous cell carcinoma cells causes an
epithelial-mesenchymal transition cell phenotype expressing laminin
332. J Oral Pathol Med. 40:46–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Korc M: Smad4: Gatekeeper gene in head and
neck squamous cell carcinoma. J Clin Invest. 119:3208–3211.
2009.PubMed/NCBI
|
|
54
|
Saha D, Datta PK and Beauchamp RD:
Oncogenic ras represses transforming growth factor-beta/Smad
signaling by degrading tumor suppressor Smad4. J Biol Chem.
276:29531–29537. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Iglesias M, Frontelo P, Gamallo C and
Quintanilla M: Blockade of Smad4 in transformed keratinocytes
containing a Ras oncogene leads to hyperactivation of the
Ras-dependent Erk signalling pathway associated with progression to
undifferentiated carcinomas. Oncogene. 19:4134–4145. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hannigan A, Smith P, Kalna G, Lo Nigro C,
Orange C, O'Brien DI, Shah R, Syed N, Spender LC, Herrera B, et al:
Epigenetic downregulation of human disabled homolog 2 switches
TGF-beta from a tumor suppressor to a tumor promoter. J Cli Invest.
120:2842–2857. 2010. View Article : Google Scholar
|
|
57
|
Wang D, Song H, Evans JA, Lang JC,
Schuller DE and Weghorst CM: Mutation and downregulation of the
transforming growth factor beta type II receptor gene in primary
squamous cell carcinomas of the head and neck. Carcinogenesis.
18:2285–2290. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Freudlsperger C, Bian Y, Contag Wise S,
Burnett J, Coupar J, Yang X, Chen Z and Van Waes C: TGF-β and NF-κB
signal pathway cross-talk is mediated through TAK1 and SMAD7 in a
subset of head and neck cancers. Oncogene. 32:1549–1559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Burnet FM: Immunological aspects of
malignant disease. Lancet. 1:1171–1174. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang L: TGFbeta and cancer metastasis: An
inflammation link. Cancer Metastasis Rev. 29:263–271. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Steinman RM and Cohn ZA: Identification of
a novel cell type in peripheral lymphoid organs of mice. I.
Morphology, quantitation, tissue distribution. J Exp Med.
137:1142–1162. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Banchereau J and Steinman RM: Dendritic
cells and the control of immunity. Nature. 392:245–252. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Duray A, Demoulin S, Hubert P, Delvenne P
and Saussez S: Immune suppression in head and neck cancers: A
review. Clin Dev Immunol. 2010:7016572010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wrzesinski SH, Wan YY and Flavell RA:
Transforming growth factor-beta and the immune response:
implications for anticancer therapy. Clin Cancer Res. 13:5262–5270.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Khazaie K and von Boehmer H: The impact of
CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and
cancer. Semin Cancer Biol. 16:124–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Curry JM, Sprandio J, Cognetti D,
Luginbuhl A, Bar-ad V, Pribitkin E and Tuluc M: Tumor
microenvironment in head and neck squamous cell carcinoma. Semin
Oncol. 41:217–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
El-Rouby DH: Association of macrophages
with angiogenesis in oral verrucous and squamous cell carcinomas. J
Oral Pathol Med. 39:559–564. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu SY, Chang LC, Pan LF, Hung YJ, Lee CH
and Shieh YS: Clinicopathologic significance of tumor cell-lined
vessel and microenvironment in oral squamous cell carcinoma. Oral
Oncol. 44:277–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Marcus B, Arenberg D, Lee J, Kleer C,
Chepeha DB, Schmalbach CE, Islam M, Paul S, Pan Q, Hanash S, et al:
Prognostic factors in oral cavity and oropharyngeal squamous cell
carcinoma. Cancer. 101:2779–2787. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Flavell R, Sanjabi S, Wrzesinski S and
Licona-Limón P: The polarization of immune cells in the tumour
environment by TGFbeta. Nat Rev Immunol. 10:554–567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Seya T, Akazawa T, Uehori J, Matsumoto M,
Azuma I and Toyoshima K: Role of toll-like receptors and their
adaptors in adjuvant immunotherapy for cancer. Anticancer Res.
23:4369–4376. 2003.PubMed/NCBI
|
|
73
|
Standiford TJ, Kuick R, Bhan U, Chen J,
Newstead M and Keshamouni VG: TGF-β-induced IRAK-M expression in
tumor-associated macrophages regulates lung tumor growth. Oncogene.
30:2475–2484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Schantz SP, Shillitoe EJ, Brown B and
Campbell B: Natural killer cell activity and head and neck cancer:
A clinical assessment. J Natl Cancer Inst. 77:869–875.
1986.PubMed/NCBI
|
|
75
|
Schantz SP and Goepfert H: Multimodality
therapy and distant metastases. The impact of natural killer cell
activity. Arch Otolaryngol Head Neck Surg. 113:1207–1213. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wahl SM, Wen J and Moutsopoulos NM: The
kiss of death: Interrupted by NK-cell close encounters of another
kind. Trends Immunol. 27:161–164. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zwirner NW, Fuertes MB, Girart MV, Domaica
CI and Rossi LE: Cytokine-driven regulation of NK cell functions in
tumor immunity: Role of the MICA-NKG2D system. Cytokine Growth
Factor Rev. 18:159–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Klöss S, Chambron N, Gardlowski T, Weil S,
Koch J, Esser R, Pogge von Strandmann E, Morgan MA, Arseniev L,
Seitz O and Köhl U: Cetuximab reconstitutes pro-inflammatory
cytokine secretions and tumor-infiltrating capabilities of
sMICA-inhibited NK cells in HNSCC tumor spheroids. Front Immunol.
6:5432015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ghiringhelli F, Menard C, Martin F and
Zitvogel L: The role of regulatory T cells in the control of
natural killer cells: Relevance during tumor progression. Immunol
Rev. 214:229–238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Buessow SC, Paul RD and Lopez DM:
Influence of mammary tumor progression on phenotype and function of
spleen and in situ lymphocytes in mice. J Natl Cancer Inst.
73:249–255. 1984.PubMed/NCBI
|
|
81
|
Chen WC, Lai CH, Chuang HC, Lin PY and
Chen MF: Inflammation-induced myeloid-derived suppressor cells
associated with squamous cell carcinoma of the head and neck. Head
Neck. 39:347–355. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mao L, Deng WW, Yu GT, Bu LL, Liu JF, Ma
SR, Wu L, Kulkarni AB, Zhang WF and Sun ZJ: Inhibition of SRC
family kinases reduces myeloid-derived suppressor cells in head and
neck cancer. Int J Cancer. 140:1173–1185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pyzer AR, Cole L, Rosenblatt J and Avigan
DE: Myeloid-derived suppressor cells as effectors of immune
suppression in cancer. Int J Cancer. 139:1915–1926. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Russell SM, Lechner MG, Gong L, Megiel C,
Liebertz DJ, Masood R, Correa AJ, Han J, Puri RK, Sinha UK, et al:
USC-HN2, a new model cell line for recurrent oral cavity squamous
cell carcinoma with immunosuppressive characteristics. Oral Oncol.
47:810–817. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Filipazzi P, Huber V and Rivoltini L:
Phenotype, function and clinical implications of myeloid-derived
suppressor cells in cancer patients. Cancer Immunol Immunother.
61:255–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tu E, Chia PZ and Chen W: TGFβ in T cell
biology and tumor immunity: Angel or devil? Cytokine Growth Factor
Rev. 25:423–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wolfraim LA, Walz TM, James Z, Fernandez T
and Letterio JJ: p21Cip1 and p27Kip1 act in synergy to alter the
sensitivity of naive T cells to TGF-beta-mediated G1 arrest through
modulation of IL-2 responsiveness. J Immunol. 173:3093–3102. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tone Y, Furuuchi K, Kojima Y, Tykocinski
ML, Greene MI and Tone M: Smad3 and NFAT cooperate to induce Foxp3
expression through its enhancer. Nat Immunol. 9:194–202. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ivanov II, McKenzie BS, Zhou L, Tadokoro
CE, Lepelley A, Lafaille JJ, Cua DJ and Littman DR: The orphan
nuclear receptor RORgammat directs the differentiation program of
proinflammatory IL-17+ T helper cells. Cell.
126:1121–1133. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Schaefer C, Kim GG, Albers A, Hoermann K,
Myers EN and Whiteside TL: Characteristics of CD4+CD25+ regulatory
T cells in the peripheral circulation of patients with head and
neck cancer. Br J Cancer. 92:913–920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Boucek J, Mrkvan T, Chovanec M, Kuchar M,
Betka J, Boucek V, Hladikova M, Betka J, Eckschlager T and Rihova
B: Regulatory T cells and their prognostic value for patients with
squamous cell carcinoma of the head and neck. J Cell Mol Med.
14:426–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Strauss L, Bergmann C, Szczepanski M,
Gooding W, Johnson JT and Whiteside TL: A unique subset of
CD4+CD25highFoxp3+ T cells secreting interleukin-10 and
transforming growth factor-beta1 mediates suppression in the tumor
microenvironment. Clin Cancer Res. 13:4345–4354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bergmann C, Strauss L, Wang Y, Szczepanski
MJ, Lang S, Johnson JT and Whiteside TL: T regulatory type 1 cells
in squamous cell carcinoma of the head and neck: Mechanisms of
suppression and expansion in advanced disease. Clin Cancer Res.
14:3706–3715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Li C, Zhao Y and Zhang W and Zhang W:
Increased prevalence of T(H)17 cells in the peripheral blood of
patients with head and neck squamous cell carcinoma. Oral Surg Oral
Med Oral Pathol Oral Radiol Endod. 112:81–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Stockinger B, Veldhoen M and Martin B:
Th17 T cells: Linking innate and adaptive immunity. Semin Immunol.
19:353–361. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Laad A, Kode J, Chavan S, Rao R, Fakih AR
and Chiplunkar S: Limiting dilution analysis of proliferating and
cytotoxic lymphocytes in the peripheral blood and tumours of oral
cancer patients. Eur J Cancer B Oral Oncol. 32B:1–342. 1996.
|
|
97
|
Sweeny L, Liu Z, Lancaster W, Hart J,
Hartman YE and Rosenthal EL: Inhibition of fibroblasts reduced head
and neck cancer growth by targeting fibroblast growth factor
receptor. Laryngoscope. 122:1539–1544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wheeler SE, Shi H, Lin F, Dasari S,
Bednash J, Thorne S, Watkins S, Joshi R and Thomas SM: Enhancement
of head and neck squamous cell carcinoma proliferation, invasion,
and metastasis by tumor-associated fibroblasts in preclinical
models. Head Neck. 36:385–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lim KP, Cirillo N, Hassona Y, Wei W,
Thurlow JK, Cheong SC, Pitiyage G, Parkinson EK and Prime SS:
Fibroblast gene expression profile reflects the stage of tumour
progression in oral squamous cell carcinoma. J Pathol. 223:459–469.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Takahashi H, Sakakura K, Kawabata-Iwakawa
R, Rokudai S, Toyoda M, Nishiyama M and Chikamatsu K:
Immunosuppressive activity of cancer-associated fibroblasts in head
and neck squamous cell carcinoma. Cancer Immunol Immunother.
64:1407–1417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Rosenthal E, McCrory A, Talbert M, Young
G, Murphy-Ullrich J and Gladson C: Elevated expression of TGF-beta1
in head and neck cancer-associated fibroblasts. Mol Carcinog.
40:116–121. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Xu BJ, Yan W, Jovanovic B, An AQ, Cheng N,
Aakre ME, Yi Y, Eng J, Link AJ and Moses HL: Quantitative analysis
of the secretome of TGF-beta signaling-deficient mammary
fibroblasts. Proteomics. 10:2458–2470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Meng W, Xia Q, Wu L, Chen S, He X, Zhang
L, Gao Q and Zhou H: Downregulation of TGF-beta receptor types II
and III in oral squamous cell carcinoma and oral
carcinoma-associated fibroblasts. BMC Cancer. 11:882011. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bhowmick NA, Chytil A, Plieth D, Gorska
AE, Dumont N, Shappell S, Washington MK, Neilson EG and Moses HL:
TGF-beta signaling in fibroblasts modulates the oncogenic potential
of adjacent epithelia. Science. 303:848–851. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Nema R, Vishwakarma S, Agarwal R, Panday
RK and Kumar A: Emerging role of sphingosine-1-phosphate signaling
in head and neck squamous cell carcinoma. Onco Targets Ther.
9:3269–3280. 2016.PubMed/NCBI
|