Introduction
Lung cancer is a leading cause of cancer-associated
mortality worldwide (1). Non-small
cell lung carcinoma (NSCLC) accounts for ~85% of lung
cancer-associated mortalities worldwide (2,3). Despite
recent improvements in chemo- and radiotherapy, the 5-year overall
survival rate of patients with late-stage NSCLC is <15% due to
cancer recurrence and metastasis (4).
Elucidating the molecular mechanisms of NSCLC progression may help
to identify novel therapeutic targets, as well as novel treatment
approaches.
The polo-like kinase (Plk) family in eukaryotes
comprises highly conserved serine/threonine kinases (5). Plk1 controls mitotic entry, centrosome
maturation, bipolar spindle formation and chromosome condensation
and segregation (6). Plk1 has been
linked to tumor aggressiveness and patient prognosis in numerous
types of cancer, including NSCLC (7–9). Plk1
expression is upregulated in various cancer types (10–15), and
this induces cell proliferation and malignant transformation
(16). Previous studies have shown
that Plk1 overexpression is involved in the invasion and migration
of tumor cells in renal (17),
colorectal (18) and bladder
(19) cancer.
Signal transducer and activator of transcription
(STAT)3 of the Janus kinase-STAT signaling pathway is a latent
transcription factor with a variety of functions (20). Constitutively-activated
(phosphorylated) STAT3 protein contributes to tumor formation and
metastasis in the majority of human cancer types (21,22).
Aberrant expression of Plk1 and STAT3 has been implicated in the
migration of cancer cells (23–25). Plk1
functions in a transcriptional self-regulatory loop that involves
the phosphorylation of forkhead box (Fox)M1 (26), while STAT3 and Plk1 exhibit
cross-regulatory interactions in esophageal cancer (25). Based on these findings, it was
speculated that STAT3 and Plk1 interact in a manner similar to Plk1
and FoxM1 to modulate the migration of tumor cells. To test this
hypothesis, the present study investigated the effect of Plk1
inhibition on the migration of human lung adenocarcinoma epithelial
A549 cells and the interaction between Plk1 and STAT3.
Materials and methods
Cell lines and culture conditions
A549 cells were isolated in the Laboratory of
nuclear and radiation damage, The General Hospital of the Second
Artillery Corps of Chinese PLA (Beijing, China), HeLa and 293T
cells were donated by Professor Cheng Cao of the Academy of
Military Medical Sciences (Beijing, China). All cells were cultured
in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA), 100 U/ml penicillin and 100 µg/ml streptomycin (Gibco;
Thermo Fisher Scientific, Inc.). The cell cultures were incubated
at 37°C in a humidified atmosphere of 5% CO2.
Wound-healing assay
A549 cells (2×105 cells/well) were seeded
in 6-well dishes at 80% confluence. After 16 h, a scratch mark
(wound) was made to the central area of confluent cells with a
pipette tip. The cells were washed with serum-free DMEM, and the
Plk1 inhibitor BI2536 (0.1 µM) was added to the dishes. The
cultures were incubated in serum-free medium at 37°C in a
humidified atmosphere of 5% CO2. The scratch area was
captured under an inverted microscope (Olympus CX31; Olympus
Corporation, Tokyo, Japan) and magnified by a computer-based
microscopy imaging system (magnification, ×200; Olympus
Corporation) at 0, 24 and 48 h and the area of each wound was
evaluated using Image-Pro Plus software (version 6.0, Media
Cybernetics, Bethesda, MD, USA).
Small-interfering (si)RNA
transfection
Plk1 and the control siRNA were purchased from
GenePharma (GenePharma, Shanghai, China), and the sequences were
5′-AGATTGTGCCTAAGTCTCT-3′ and 5′-UUCUCCGAACGUGUCACGUTT-3′,
respectively. The cells (1×105 cells/well) were seeded
on a 6-well plate and transfected using Lipofectamine®
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) following
the manufacturer's protocols when 60% confluence was reached. The
cells were harvested at 0, 48 and 72 h following transfection. The
cells were washed with cold phosphate buffered saline (PBS),
centrifuged at 1,000 × g for 5 min at 4°C, and then the supernatant
was discarded, and the cell pellets were washed with PBS twice for
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) and western blot analysis. The experiments were repeated
three times.
Total RNA isolation and RT-qPCR
Total RNA was isolated from cells using the RNeasy
Mini kit (Qiagen GmbH, Hilden, German) according to the
manufacturer's protocol, and then reverse-transcribed with random
hexamers using the All-in-One First-Strand cDNA Synthesis kit
(GeneCopoeia, Rockville, MD, USA). The expression of the
transcripts was detected by RT-qPCR using the Real-time Premix EX
TaqMan kit (Takara Biotechnology Co., Ltd., Dalian, China). All the
experiments were carried out according to the manufacturers'
protocols. The cycle threshold (Cq) value for each target gene was
normalized to the level of β-actin, and relative expression was
calculated using the 2−∆∆Cq method (27). Each sample was analyzed in triplicate,
and the experiment was repeated three times. The reaction
conditions were as follows: 50°C for 2 min and 95°C for 5 min,
followed by 40 cycles of 95°C for 12 sec and 60°C for 40 sec. The
primer and probe sequences were as follows: Matrix
metalloproteinase (MMP)2 forward, 5′-AATCCATGATGGAGAGGCAGAC-3′,
reverse, 5′-CAGTCCGTCCTTACCGTCAAAG-3′ and probe,
5′-CTTTGGCCGCTGGGGAGCATGG-3′; vascular endothelial growth factor
(VEGF)A forward, 5′-ACATCACCATGCAGATTATGC-3′, reverse,
5′-GTCTTGCTCTATCTTTCTTTGGTC-3′ and probe,
5′-CACCAAGGCCAGCACATAGGAGAGA-3′; and β-actin forward,
5′-TCTGGCAACGGTGAAGGTGACA-3′, reverse, 5′-CACCTCCCCTGTGTGGACTT-3′
and probe, 5′-AGCGAGCATCCCCCAAAGTTCACA-3′.
Co-immunoprecipitation (Co-IP)
Cells were lysed in lysis buffer (50 mM Tris-HCl; 1
mM phenylmethylsulfonyl fluoride; 1 mM dithiothreitol; 10 mM sodium
fluoride; 10 µg/ml aprotinin; 10 µg/ml leupeptin; 10 µg/ml
pepstatin A; and 1% NonidetP-40). The cell lysates were kept on ice
for 30 min, and then centrifuged at 12,000 × g for 15 min at 4°C
and the soluble supernatants were collected.
For tagged Flag protein IP, cell supernatants were
incubated with anti-Flag M2 beads (15 µl; cat. no. A8592;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 4 h at 4°C.
Normal mouse IgG-Agarose (15 µl; cat. no. A0910; Sigma-Aldrich;
Merck KGaA) was used as the control and the condition were
identical. Additionally, 15 µl of the total supernatants (5%, v/v)
was included as a control. Precipitates were then washed three
times with lysis buffer and immunoblotted with horseradish
peroxidase-conjugated anti-Flag (cat. no. A8592; 1:5,000 dilution;
Sigma-Aldrich; Merck KGaA) or rabbit anti-STAT3 (cat. no. 4904;
1:1,000 dilution; Cell Signaling Technology, Inc., Danvers, MA,
USA).
For STAT3 protein IP, cell supernatants were
incubated with anti-STAT3 antibody (8 µl; cat. no. 4904; Cell
Signaling Technology, Inc.) for 2 h at 4°C, and then incubated with
Protein A/G agarose (20 µl; cat. no. 2336; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 12 h at 4°C. The normal
rabbit IgG (cat. no. A2909, 4 µl; rabbit; Santa Cruz Biotechnology,
Inc.) was used as the control and the conditions were identical.
Additionally, 15 µl of the total supernatants (5%, v/v) was
included as a control. Precipitates were then washed three times
with lysis buffer and immunoblotted with the indicated antibodies
(anti-Plk1 and anti-STAT3) for western blot analysis.
Western blot analysis
To isolate the protein, the cells were first washed
twice in PBS at 48 h after transferation and lysed in RIPA lysis
buffer (ProMab Biotechnologies, Inc., Richmond, CA, USA; 50 mM
Tris-HCI pH 7.6, 150 mM sodium chloride, 1.0% NP-40, 1% sodium
deoxychlorate, and 0.1% SDS) plus protease inhibitors. The lysates
were kept on ice for 30 min, and then centrifuged at 14,000 × g for
15 min. The surpernatant was collected and then 20 µg of each of
the proteins was separated by SDS-PAGE on 10% gels and transferred
onto nitrocellulose membranes The proteins were electrotransferred
to polyvinylidene difluoride membranes, blocked in 5% non-fat milk
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at room temperature
for 1 h and incubated with primary antibodies overnight at 4°C.
Primary antibodies against the following targets were used:
Anti-Plk1 (cat. no. J3108, mouse; 1:1,000; Santa Cruz
Biotechnology, Inc.); anti-STAT3 and anti-phosphorylated (p-)STAT3
Y705 (cat. no. 9145, rabbit; 1:1,000; Cell Signaling Technology,
Inc.); horseradish peroxidase (HRP)-conjugated anti-β-actin (cat.
no. A3584, 1:10,000; Sigma-Aldrich; Merck KGaA); anti-MMP2 (cat.
no. BS1236, rabbit; 1:500; Bioworld Technology, Inc., St. Louis
Park, MN, USA) and anti-VEGFA (cat. no. BS2431, rabbit; 1:500;
Bioworld Technology, Inc.). Subsequently, the membranes were washed
four times for 5 min each with PBS containing 0.05% Tween-20 and
incubated with anti rabbit or anti mouse horseradish peroxidase
conjugated secondary antibodies (HRP-conjugated anti-mouse and
HRP-conjugated rabbit antibodies (GE Healthcare; dilution, 1:2,000)
for 1 h at room temperature. Finally, the protein bands were
visualized using an enhanced chemiluminescence detection reagent
(GE Healthcare, Little Chalfont, UK) and detected using a ChemiDoc
imager (Bio-Rad Laboratories, Inc.).
Double luciferase reporter assay
HEK 293T cells (5×104 cells/well) were
seeded on 24-well plates and transfected with STAT3-flag (0.15 µg)
and Plk1-flag (0, 0.15 and 0.25 µg) using Lipofectamine®
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.), along
with a luciferase reporter gene (0.15 µg) and pRL-TK plasmid
(0.0003 µg). The relative firefly luciferase activity was
normalized to Renilla luciferase activity. Following
incubation for 24 h, the cells were lysed and the luciferase
activity was analyzed by the dual luciferase assay kit (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocol. The data are presented as the mean ± standard deviation
(SD) of three independent experiments performed in triplicate.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 19.0; IBM SPSS, Armonk, NY, USA). The data are
presented as the mean ± SD. A two-tailed Student's t-test for
paired data was used to compare the means of two groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
Inhibition of Plk1 suppresses the
migration of A549 cells
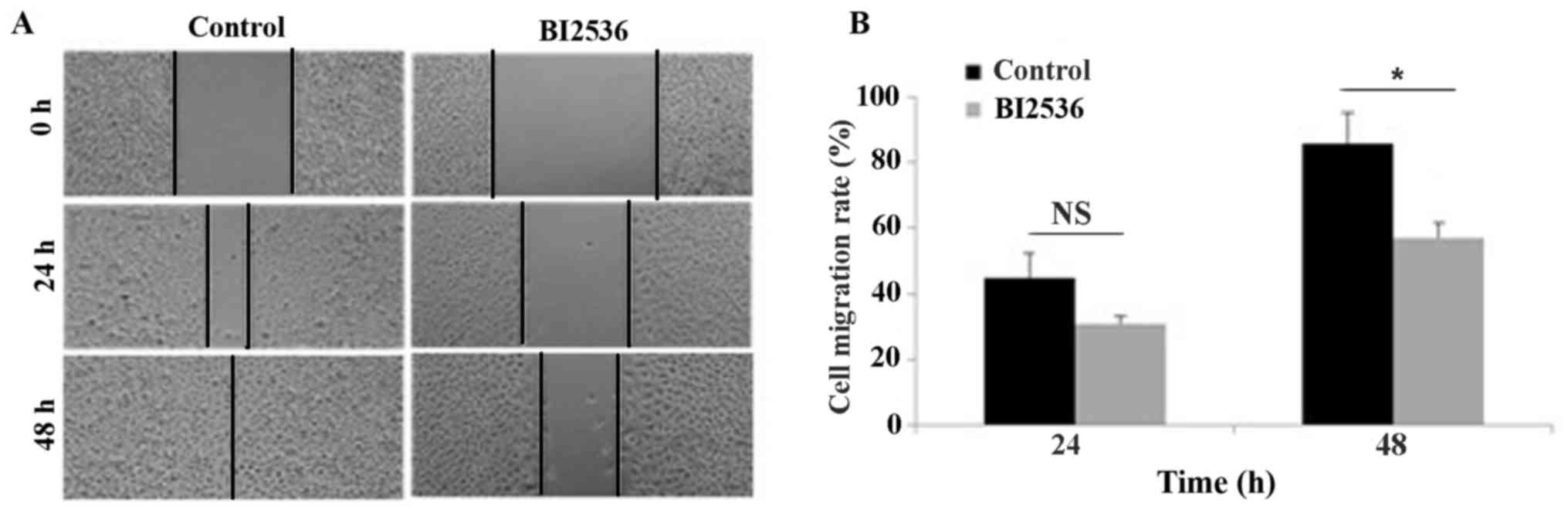
The effect of Plk1 inhibition on the migration of
A549 cells was evaluated with the wound-healing assay (Fig. 1A). Treatment with BI2536, which is an
inhibitor of Plk1, for 48 h decreased migration in control cells
from 85.60±9.50 to 57.10±٤.٤٨٪, indicating that the inhibition of
Plk١ is able to block the migration of human lung adenocarcinoma
epithelial cancer cells (Fig.
1B).
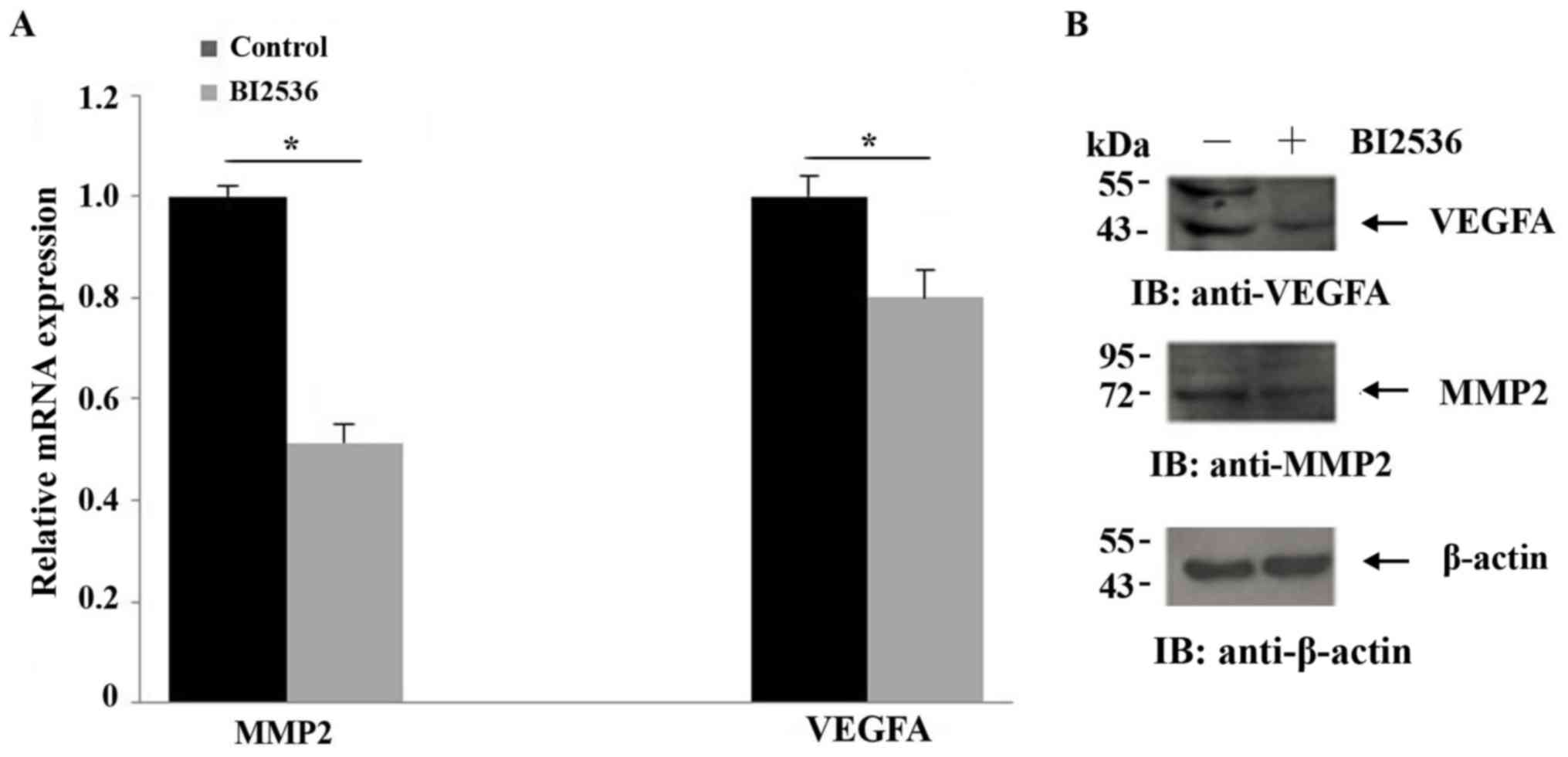
Inhibition of Plk1 suppresses MMP2 and
VEGFA expression
MMP2 and VEGFA have been implicated in the invasion
and migration of tumor cells (28–30). The
effect of Plk1 inhibition on the mRNA and protein expression of
these factors in BI2536-treated cells were analyzed with RT-qPCR
and western blotting, respectively. The expression of MMP2 and
VEGFA was decreased in the cells that were treated with BI2536
compared with control cells (Fig. 2),
providing additional evidence that Plk1 modulates the migration of
tumor cells.
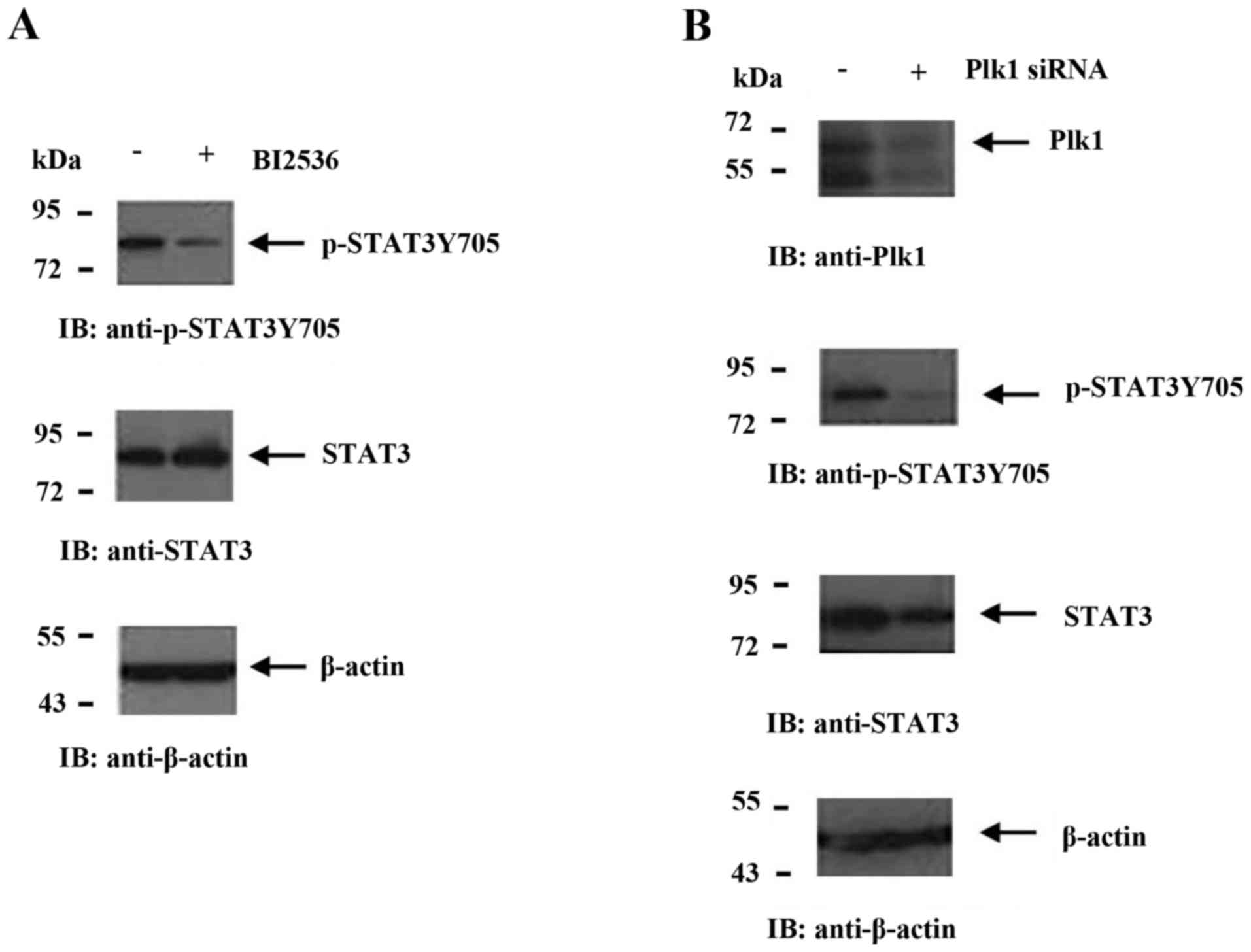
Suppression of Plk1 blocks the
phosphorylation of STAT3
MMP2 and VEGFA are downstream target genes of STAT3
(31,32). Therefore, the effect of Plk1
inhibition on STAT3 and p-STAT3 expression was investigated.
Treatment with BI2536 decreased the level of p-STAT3 without
altering the level of STAT3 (Fig.
3A). siRNA-mediated knockdown of Plk1 resulted in the
downregulation of STAT3 and p-STAT3 expression (Fig. 3B) and activity, as determined by the
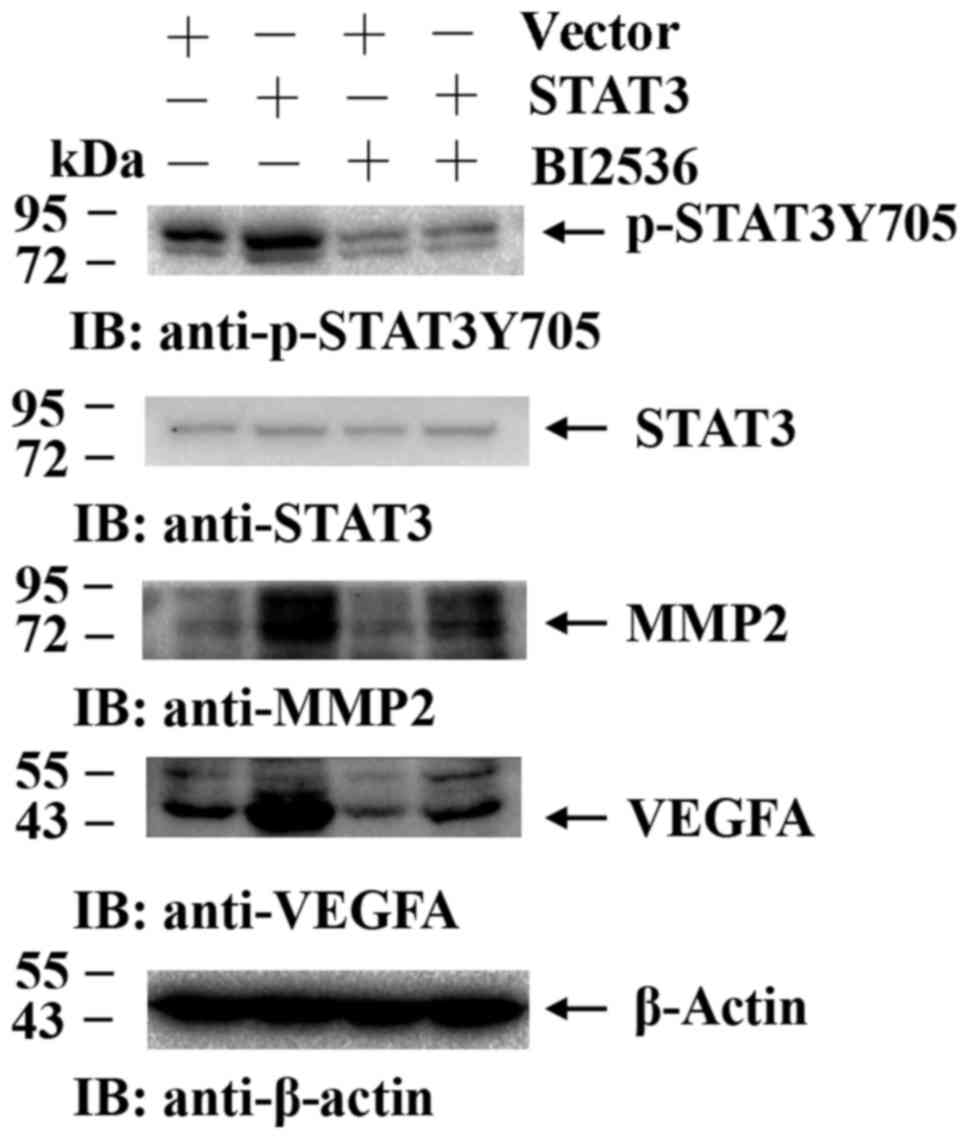
luciferase assay. This was confirmed by overexpressing STAT3 in
BI2536-treated A549 cells, which reversed the downregulation of
p-STAT3, VEGFA and MMP2 mediated by Plk1 inhibition compared with
the cells that were transfected with the control plasmid (Fig. 4). These results suggested that Plk1
modulates the migration of tumor cells via STAT3 signaling.
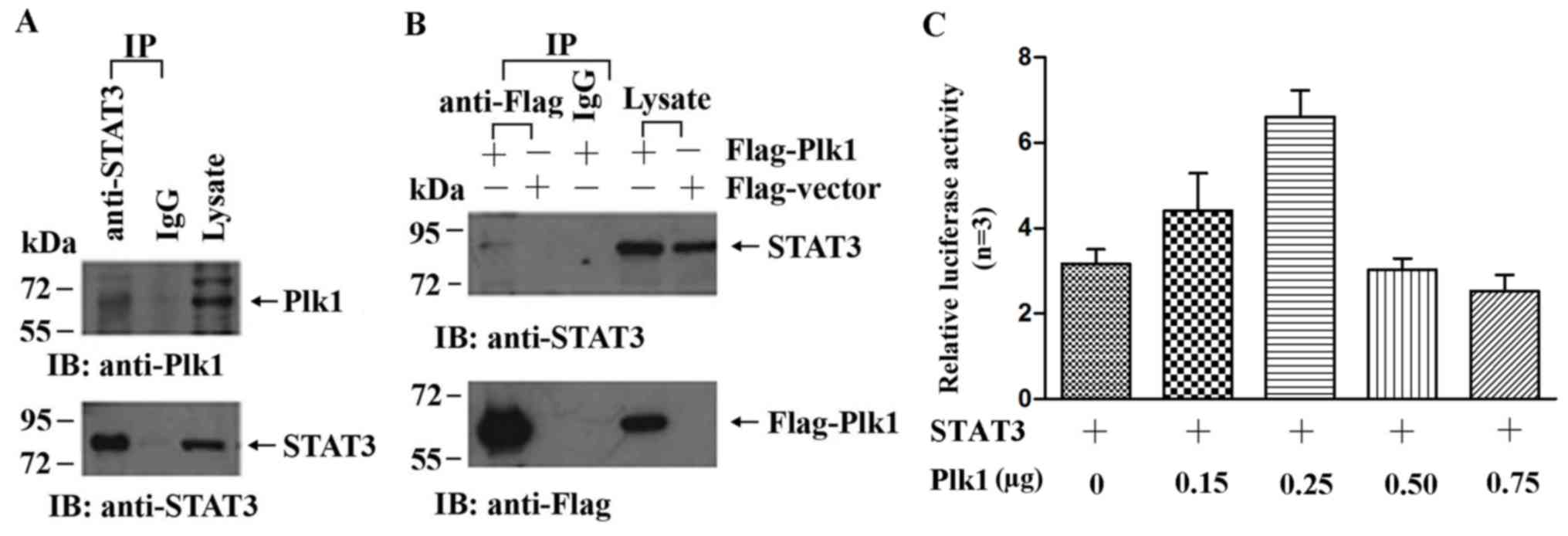
Plk1 interacts with STAT3
The inhibition of Plk1 blocked the phosphorylation
of STAT3, while the level of STAT3 was unchanged, suggesting a
possible interaction between the two proteins. To investigate this
possibility, the interaction between Plk1 and STAT3 was examined by
co-IP in HeLa cells, which exhibit higher endogenous expression of
Plk1 compared with A549 cells. HeLa cell lysates immunoprecipitated
with anti-STAT3 antibody were analyzed by western blotting using an
anti-Plk1 antibody. Plk1 was detected in the immunoprecipitate
obtained using anti-STAT3 antibody, but not that obtained using
rabbit IgG (Fig. 5A). To confirm the
interaction between STAT3 and Plk1, 293T cells were transfected
with plasmids expressing flag-Plk1 or flag-vector, and cell lysates
precipitated with anti-flag antibody were probed with anti-STAT3
antibody. The western blot analysis revealed that STAT3 is able to
interact with Plk1 (Fig. 5B). In
addition, the co-transfection of Plk1 and STAT3-luciferase reporter
plasmids increased the relative luciferase activity compared with
the control vectors (Fig. 5C). These
results suggested that Plk1 enhances STAT3 activity in tumor
cells.
Discussion
Cancer metastasis is complex and regulated by
multiple factors (33–35). Plk1 has been shown to promote invasion
and migration in several types of cancer (13,14), For
example, Han et al (18)
reported that Polo-like kinase 1 was over-expressed in colorectal
cancer and participated in the invasion and migration of colorectal
cancer cells. However, the underlying mechanism is unclear. The
present study indicates that the inhibition of Plk1 suppressed the
migration of A549 cells and decreased the expression of MMP2 and
VEGFA and phosphorylation of STAT3. Furthermore, to the best of our
knowledge, the present study presents for the first time the
interaction of Plk1 and STAT3 to regulate cell migration.
The overexpression of Plk1 is associated with a low
survival rate and poor prognosis in various types of cancer
(16,17,19), which
can be attributed to its role in promoting the invasion and
migration of cancer cells (17–19).
Consistent with these findings, in the present study, it was
observed that the migration of A549 cells was blocked by treatment
with BI2536, which is a small-molecular inhibitor of Plk1.
MMP2 belongs to a family of structurally-related
enzymes that degrade the components of the extracellular matrix
during tumor invasion and metastasis (36). The overexpression of MMP2 is observed
in solid tumors of different origins, including metastatic lesions
(37–40), this is positively associated with
tumor size, depth of invasion, lymphatic and venous invasion and
lymph node metastasis in gastric carcinoma (41). In the present study, MMP2 expression
was downregulated upon the inhibition of Plk1. VEGFA is a potent
inducer of angiogenesis, a process that allows solid tumors to
acquire nutrients for continuous growth and metastasis (42). VEGFA has been shown to be upregulated
in the majority of malignancies and promotes the progression and
metastasis of various types of cancer (43). Conversely, the knockdown of VEGFA
suppressed lymph node metastasis in a murine metastatic mammary
cancer model (44). In the present
study, the level of VEGFA was observed to be markedly reduced by
Plk1 inhibition. These results indicated that the inhibition of
Plk1 may suppress the migration of A549 cells by negatively
regulating the expression of MMP2 and VEGFA.
The activation of STAT3 leads to the upregulation of
MMP2 and VEGFA, thereby enhancing the migration and invasion of
tumor cells (36). STAT3 is
constitutively activated in numerous types of human cancer, usually
by phosphorylation of a conserved tyrosine residue in response to
extracellular signals from cytokines and growth factors (45). Activated p-STAT3 forms homo- or
heterodimers that translocate into the nucleus and induce the
transcription of target genes, including MMP2 and VEGFA. The
constitutive activation of STAT3 has also been shown to modulate
cancer invasion and metastasis (46–48), which
is an effect that is mitigated by the inhibition of STAT3
signaling. Therefore, it was hypothesized that the inhibition of
Plk1 suppresses MMP2 and VEGFA by negatively regulating STAT3
expression or activation. This was supported by the observations
that the levels of phosphorylated STAT3 but not total protein
expression, decreased upon pharmacological inhibition of Plk1, and
that the overexpression of STAT3 countered the effects of Plk1
inhibition in A549 cells. The present results are consistent with
those of a previous study (26) on
esophageal cancer cells, which reported that Plk1 knockdown or
pharmacological inhibition suppressed STAT3 activity. However, one
difference between this earlier study and the present study is that
BI2536 treatment reduced STAT3 expression in esophageal cancer
cells, whereas no change in STAT3 level was observed in A549 cells.
This may be attributable to intrinsic differences between the two
cell types.
A previous study (49)
reported an association between Plk1 and regulation of the G2/M
transcriptional network in mammalian cells. Plk1 has been shown to
be involved in a positive-feedback loop with its binding partner
FoxM1, a transcription factor that controls the expression of
various M-phase genes, including Plk1. The Plk1-FoxM1 complex
allows the direct phosphorylation of FoxM1 by Plk1 at the G2/M
phase. The activation of FoxM1 is required for Plk1 expression,
which is enhanced by FoxM1. STAT3 was reported to directly activate
Plk1 transcription in esophageal cancer cells (25,50).
Therefore, it was speculated that Plk1 and STAT3 interact in a
manner similar to Plk1 and FoxM1. Indeed, binding between Plk1 and
STAT3 was observed between Plk1 and STAT3 in co-IP experiments in
the present study. Furthermore, the luciferase reporter assay
revealed that Plk1 enhanced the activity of STAT3. However, Plk1 is
a Ser/Thr kinase, and it promotes tyrosine phosphorylation of STAT3
(51,52), therefore the interaction between Plk1
and STAT3 may be indirect. Additional experiments such as protein
microarrays or cross-linking are required to determine whether the
interaction between the two proteins is direct or indirect.
The results of the present study demonstrated that
Plk1 inhibition suppresses the migration of A549 cell via the
downregulation of STAT3 signaling and the target genes MMP2 and
VEGFA. These findings suggested that the inhibition of Plk1 may
prevent the migration and metastasis of cancer cells and can
therefore be an effective therapeutic approach for the treatment of
NSCLC.
Acknowledgements
The authors thank Professor Cheng Cao from the
Institute of Biotechnology of Academy of Military Medical Sciences
for providing antibodies against Plk1 and β-actin, and Dr Quanbin
Xu from the Institute of Biotechnology of Academy of Military
Medical Sciences and Professor Ling Gao from Key Laboratory of
Radiological Protection and Nuclear Emergency, National Institute
for Radiological Protection, China Centers for Disease Control for
providing the Plk1 inhibitor BI2536, A549 cells and the
overexpression vector.
Funding
The present study was supported by National Nature
Science Foundation of China (grant nos. 81602796 and 31770914) and
Major Technology Project of Military Logistics (grant no.
AEP17J001).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
WL and QJ designed the experiments and checked the
manuscript. WY performed the experiments, prepared the data and
wrote and edited the manuscript. HY performed the experiments and
programmed the software. FL designed and performed the experiments.
SW and NY performed the experiments.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kanwal M, Ding XJ and Cao Y: Familial risk
for lung cancer. Oncol Lett. 13:535–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stinchcombe TE and Socinski MA: Current
treatments for advanced stage non-small cell lung cancer. Proc Am
Thorac Soc. 6:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu Z, Chen X, Zhao Y, Tian T, Jin G, Shu
Y, Chen Y, Xu L, Zen K, Zhang C and Shen H: Serum microRNA
signatures identified in a genome-wide serum microRNA expression
profiling predict survival of non-small-cell lung cancer. J Clin
Oncol. 28:1721–1726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Combes G, Alharbi I, Braga LG and Elowe S:
Playing polo during mitosis: PLK1 takes the lead. Oncogene.
34:4819–4827. 2017. View Article : Google Scholar
|
|
6
|
Song B, Liu XS and Liu X: Polo-like kinase
1 (Plk1): An unexpected player in DNA replication. Cell Div.
7:32012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Z, Sun Q and Wang X: PLK1, a potential
target for cancer therapy. Transl Oncol. 10:22–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu C, Li S, Chen T, Hu H, Ding C, Xu Z,
Chen J, Liu Z, Lei Z, Zhang HT, et al: miR-296-5p suppresses cell
viability by directly targeting PLK1 in non-small cell lung cancer.
Oncol Rep. 35:497–503. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang XH, Lu Y, Liang JJ, Cao JX, Jin YQ,
An GS, Ni JH, Jia HT and Li SY: MiR-509-3-5p causes aberrant
mitosis and anti-proliferative effect by suppression of PLK1 in
human lung cancer A549 cells. Biochem Biophys Res Commun.
478:676–682. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang H, Tian C, Xu Y, Xie WL, Zhang J,
Zhang BY, Ren K, Wang K, Chen C, Wang SB, et al: Abortive cell
cycle events in the brains of scrapie-infected hamsters with
remarkable decreases of PLK3/Cdc25C and increases of PLK1/cyclin
B1. Mol Neurobiol. 48:655–668. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
King SI, Purdie CA, Bray SE, Quinlan PR,
Jordan LB, Thompson AM and Meek DW: Immunohistochemical detection
of Polo-like kinase-1 (PLK1) in primary breast cancer is associated
with TP53 mutation and poor clinical outcom. Breast Cancer Res.
14:R402012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takahashi T, Sano B, Nagata T, Kato H,
Sugiyama Y, Kunieda K, Kimura M, Okano Y and Saji S: Polo-like
kinase 1 (PLK1) is overexpressed in primary colorectal cancers.
Cancer Sci. 94:148–152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Medema RH, Lin CC and Yang JC: Polo-like
kinase 1 inhibitors and their potential role in anticancer therapy,
with a focus on NSCLC. Clin Cancer Res. 17:6459–6466. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo J and Liu X: Polo-like kinase 1, on
the rise from cell cycle regulation to prostate cancer development.
Protein Cell. 3:182–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun W, Su Q, Cao X, Shang B, Chen A, Yin H
and Liu B: High expression of polo-like kinase 1 is associated with
early development of hepatocellular carcinoma. Int J Genomics.
2014:3121302014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ito Y, Yoshida H, Matsuzuka F, Matsuura N,
Nakamura Y, Nakamine H, Kakudo K, Kuma K and Miyauchi A: Polo-like
kinase 1 (PLK1) expression is associated with cell proliferative
activity and cdc2 expression in malignant lymphoma of the thyroid.
Anticancer Res. 24:259–263. 2004.PubMed/NCBI
|
|
17
|
Zhang G, Zhang Z and Liu Z: Polo-like
kinase 1 is overexpressed in renal cancer and participates in the
proliferation and invasion of renal cancer cells. Tumour Biol.
34:1887–1894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han DP, Zhu QL, Cui JT, Wang PX, Qu S, Cao
QF, Zong YP, Feng B, Zheng MH and Lu AG: Polo-like kinase 1 is
over-expressed in colorectal cancer and participates in the
migration and invasion of colorectal cancer cells. Med Sci Monit.
18:BR237–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Zhang G and Kong C: High
expression of polo-like kinase 1 is associated with the metastasis
and recurrence in urothelial carcinoma of bladder. Urol Oncol.
31:1222–1230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao M, Gao FH, Wang JY, Liu F, Yuan HH,
Zhang WY and Jiang B: JAK2/STAT3 signaling pathway activation
mediates tumor angiogenesis by upregulation of VEGF and bFGF in
non-small-cell lung cancer. Lung Cancer. 73:366–374. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu Y, Zhao Q, Wang Z and Liu XY: Activated
STAT3 correlates with prognosis of non-small cell lung cancer and
indicates new anticancer strategies. Cancer Chemother Pharmacol.
75:917–922. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-beta-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morry J, Ngamcherdtrakul W, Gu S, Reda M,
Castro DJ, Sangvanich T, Gray JW and Yantasee W: Targeted treatment
of metastatic breast cancer by PLK1 siRNA delivered by an
antioxidant nanoparticle platform. Mol Cancer Ther. 4:763–772.
2017. View Article : Google Scholar
|
|
24
|
Xu L, Zhou R, Yuan L, Wang S, Li X, Ma H,
Zhou M, Pan C, Zhang J and Huang N: IGF1/IGF1R/STAT3
signaling-inducible IFITM2 promotes gastric cancer growth and
metastasis. Cancer Lett. 393:76–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y: Reciprocal Activation between
PLK1 and Stat3 contributes to survival and proliferation of
esophageal cancer cells. Gastroenterology. 142:5212012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pomerening JR: Positive-feedback loops in
cell cycle progression. FEBS Lett. 583:3388–3396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fossey SL, Bear MD, Kisseberth WC, Pennell
M and London CA: Oncostatin M promotes STAT3 activation, VEGF
production, and invasion in osteosarcoma cell lines. BMC Cancer.
11:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Song X, Zhu J, Li M, Ji Y, Wu F,
Chen Y, Cui X, Hu J, Wang L, et al: Tumor suppressor microRNA-34a
inhibits cell migration and invasion by targeting
MMP-2/MMP-9/FNDC3B in esophageal squamous cell carcinoma. Int J
Oncol. 51:3782017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong J, Zhu S, Zhang Y and Wang J:
Interplay of VEGFa and MMP2 regulates invasion of glioblastoma.
Tumor Biol. 35:11879–11885. 2014. View Article : Google Scholar
|
|
31
|
Kesanakurti D, Chetty C, Dinh DH, Gujrati
M and Rao JS: Role of MMP-2 in the regulation of IL-6/Stat3
survival signaling via interaction with α٥β١ integrin in glioma.
Oncogene. 32:327–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen X, Tang J, Hu J, Guo L, Xing Y and Xi
T: MiR-424 regulates monocytic differentiation of human leukemia
U937 cells by directly targeting CDX2. Biotechnol Lett.
35:1799–1806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsu F, Caluwe AD, Anderson D, Nichol A,
Toriumi T and Ho C: Patterns of spread and prognostic implications
of lung cancer metastasis in an era of driver mutations. Curr
Oncol. 24:228–233. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Su SC, Hsieh MJ, Yang WE, Chung WH, Reiter
RJ and Yang SF: Cancer metastasis: Mechanisms of inhibition by
melatonin. J Pineal Res. 62:e123702017. View Article : Google Scholar
|
|
35
|
Scott BJ, Oberheimbush NA and Kesari S:
Leptomeningeal metastasis in breast cancer-a systematic review.
Oncotarget. 4:3740–3747. 2016.
|
|
36
|
Roomi MW, Monterrey JC, Kalinovsky T, Rath
M and Niedzwiecki A: Inhibition of invasion and MMPs by a nutrient
mixture in human cancer cell lines: A correlation study. Exp Oncol.
32:243–248. 2010.PubMed/NCBI
|
|
37
|
Groblewska M, Mroczko B, Gryko M, Kedra B
and Szmitkowski M: Matrix metalloproteinase 2 and tissue inhibitor
of matrix metalloproteinases 2 in the diagnosis of colorectal
adenoma and cancer patients. Folia Histochem Cytobiol. 48:564–571.
2010.PubMed/NCBI
|
|
38
|
Duan W, Li R, Ma J, Lei J, Xu Q, Jiang Z,
Nan L, Li X, Wang Z, Huo X, et al: Overexpression of Nodal induces
a metastatic phenotype in pancreatic cancer cells via the Smad2/3
pathway. Oncotarget. 6:1490–1506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Pan T, Zhong X and Cheng C:
Androgen receptor promotes esophageal cancer cell migration and
proliferation via matrix metalloproteinase 2. Tumour Biol.
36:5859–5864. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zheng H, Takahashi H, Murai Y, Cui Z,
Nomoto K, Niwa H, Tsuneyama K and Takano Y: Expressions of MMP-2,
MMP-9 and VEGF are closely linked to growth, invasion, metastasis
and angiogenesis of gastric carcinoma. Anticancer Res.
26:3579–3583. 2006.PubMed/NCBI
|
|
41
|
Zhu Y, Yu X, Fu H, Wang P, Zheng X and
Wang Y: MicroRNA-21 is involved in ionizing radiation-promoted
liver carcinogenesis. Int J Clin Exp Med. 3:2112010.PubMed/NCBI
|
|
42
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nature Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shibata MA, Morimoto J, Shibata E and
Otsuki Y: Combination therapy with short interfering RNA vectors
against VEGF-C and VEGF-A suppresses lymph node and lung metastasis
in a mouse immunocompetent mammary cancer model. Cancer Gene Ther.
15:776–786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miao JW, Liu LJ and Huang J:
Interleukin-6-induced epithelial-mesenchymal transition through
signal transducer and activator of transcription 3 in human
cervical carcinoma. Int J Oncol. 45:165–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yan LI, Li LI, Li Q, DI W, Shen W, Zhang L
and Guo H: Expression of signal transducer and activator of
transcription 3 and its phosphorylated form is significantly
upregulated in patients with papillary thyroid cancer. Exp Ther
Med. 9:2195–2201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li C, Wang Z, Liu Y, Wang P and Zhang R:
STAT3 expression correlates with prognosis of thymic epithelial
tumors. J Cardiothoracic Surg. 8:922013. View Article : Google Scholar
|
|
47
|
Park JK, Hong R, Kim KJ, Lee TB and Lim
SC: Significance of p-STAT3 expression in human colorectal
adenocarcinoma. Oncol Rep. 20:597–604. 2008.PubMed/NCBI
|
|
48
|
Zhang M, Zhu GY, Gao HY, Zhao SP and Xue
Y: Expression of tissue levels of matrix metalloproteinases and
tissue inhibitors of metalloproteinases in gastric adenocarcinoma.
J Surg Oncol. 103:243–247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng F, Malureanu L, Huang J, Wang W, Li
H, van Deursen JM, Tindall DJ and Chen J: Plk1-dependent
phosphorylation of FoxM1 regulates a transcriptional programme
required for mitotic progression. Nat Cell Biol. 9:1076–1082.
2008.
|
|
50
|
Dibb M, Han N, Choudhury J, Hayes S,
Valentine H, West C, Ang YS and Sharrocks AD: The FOXM1-PLK1 axis
is commonly upregulated in oesophageal adenocarcinoma. Brit J
Cancer. 107:1766–1775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guo K, Yin G, Zi X-H and Yan W-G: A study
on expression and Tyrosine 705 phosphorylation of STAT3 in focal
cerebral ischemia-reperfusion rat model and its role in neuronal
apoptosisTrop J Pharma Res. 2. pp. 2672016, View Article : Google Scholar
|
|
52
|
Shin SB, Woo SU, Chin YW, Jang YJ and Yim
H: Sensitivity of TP53-Mutated cancer cells to the Phytoestrogen
Genistein is associated with direct inhibition of Plk1 activity. J
Cell Physiol. 232:2818–2828. 2017. View Article : Google Scholar : PubMed/NCBI
|