Introduction
Epigenetic therapy (EGT), specifically with
hypomethylating agents (HMA) either alone or in combination,
continues to be successfully used in the treatment of high risk
myelodysplastic syndrome (MDS) and elderly acute myeloid leukemia
(AML) although resistance is a frequent and ultimately near
universal outcome (1). Mechanisms of
de-novo resistance to EGT in the setting of MDS and AML are
incompletely understood however recent studies have identified
diverse molecular mechanisms including integrin α5-mediated
hematopoietic progenitor cell quiescence (2), increased RNA 5-methylcytosine active
chromatin (3), elevated BCL2L10
expression (4) and Stat3/5 signalling
(5) as contributing factors.
MDS and AML are often associated with endogenous
immune compromise and overexpression of immune checkpoint molecule
expression (6). Identification of
EGT-mediated upregulation of genes associated with tumour immune
evasion including immune checkpoint pathway-related molecules
programmed cell death protein 1 (PD-1), programmed death-ligand 1
(PD-L1) and programmed death ligand 2 (PD-L2), may contribute to
the acquired EGT resistance phenotype through T cell exhaustion
(6,7).
In addition EGT-mediated induction of tumour cell PD-L1 expression
may contribute to enhanced PD-L1 reverse signalling facilitating
anti-apoptotic effects in tumour cells and emergence of disease
resistance (8).
The recent development of immune checkpoint
inhibitor therapy, successfully trialled in solid tumours and more
recently in lymphoid haematological malignancies (6,7), may
afford therapeutic benefit in the setting of EGT-mediated immune
evasion based resistance should these patients be identifiable.
Despite identification of several putative markers
of early response to EGT (3,5,9,10) characterisation of biomarkers of
acquired resistance to EGT in myeloid malignancies are required
together with delineation of the associated molecular
mechanisms.
Our study aimed to correlate the in vivo
effects of EGT on expression of PD-1, PD-L1 and orphan nuclear
receptor NUR77, previously identified as a key molecule in the
response to EGT (11), with clinical
response in patients with myeloid malignancies. In addition, in
vitro and in vivo characterisation of the effects of EGT
on NF-κB and Bcl-xL expression, potential downstream targets of
PD-L1 reverse signalling, was evaluated to delineate possible
components of the molecular mechanism responsible for these effects
which may inform on novel treatment strategies in the setting of
resistance to EGT.
Materials and methods
Cell culture
KG-1 leukemia cells were cultured in IMDM (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 20%
heat inactivated fetal calf serum (Gibco; Thermo Fisher Scientific,
Inc.) and kept in a 5% CO2 incubator at 37°C. Cells were
split into 6-well plates with 2×106 cells per well.
Agents: Azacytidine (Aza) (Sigma, Australia) and the histone
deacetylase inhibitors (HDACi) Panobinostat (LBH-589) (LBH)
(Sapphire Bioscience Pty Ltd., Sydney, Australia) and MCT-3
(12) were added to plates for 24 h.
Aza was dissolved in H2O with 0.2% acetic acid and used
at a final concentration of 1.0 µM. LBH-589 and MCT-3 were
dissolved in 1% DMSO and used at a final concentration of 20 nM and
0.5 µM respectively.
Patient and healthy control blood
samples
Mono-nuclear cell fractions were obtaining by ficoll
density gradient centrifugation of peripheral blood samples taken
from patients enrolled in low risk studies 357/14 and LR63/2015
with MDS/AML treated with Aza alone (n=9), 75 mg/m2 for
7 days of a 28 day cycle between February 2015 and January 2017 and
from a phase Ib/II clinical trial (n=4) of a 5 day schedule of Aza,
75 mg/m2 followed by LBH in high-risk MDS or AML
patients between January 2010 and January 2012 (13) and healthy controls aged 25–50 years
old) (n=5). Peripheral blood samples were collected from patients
at screening (prior to treatment) and at time points (days 5–28)
during the first cycle of treatment. The open-label, phase Ib/II
study (13) was conducted at three
centres: Alfred Hospital, Melbourne, Australia; Princess Alexandra
Hospital, Brisbane, Australia; and Austin Hospital, Melbourne,
Australia. Low risk studies 357/14 and LR63/2015 were conducted at
the Alfred Hospital, Melbourne, Australia and Box Hill Hospital,
Melbourne, Australia.
The open-label, phase Ib/II study (13) was approved by the Alfred Hospital
Ethics Committee, Melbourne, Australia on 15/07/2009, ethics
approval number AH189/09, and performed in accordance with the
principles of independent Human Research and Ethics Committees, and
registered with the Australian and New Zealand Clinical Trials
Registry (ACTRN12610000924055). Low risk study 357/14 was approved
by Alfred Health Ethics Committee on 20/8/2014 and low risk study
LR63/2015 was approved by Eastern Health Ethics Committee,
Melbourne, Australia on 27/07/2015. All participating patients and
healthy controls were required to provide written informed consent
for participation in the studies.
Growth inhibition assay
KG-1 cells in log phase were plated at a density of
0.2×106 in 2 ml of medium. Cells were harvested at 24 h.
Cell viability was assessed using 0.4% trypan blue staining
immediately after culture. Black staining cells were considered as
non-viable cells and unstained bright cells as viable. Cell growth
(%)=(the total number of viable cells at 24 h-the total number of
viable cells at the beginning of the experiment/the total number of
viable cells at the beginning of the experiment) as previously
described (9). All experiments were
repeated a minimum of 3 times with averages displayed
graphically.
RNA extraction
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.)
was used to extract RNA from KG-1 cells treated with Aza, LBH or
MCT-3 or a combination of Aza+LBH. TRIzol was also used to extract
RNA from the mono-nuclear cell fractions of blood samples from
patients with MDS/AML treated with Aza alone, 75 mg/m2
for 7 days of a 28 day cycle or from a phase Ib/II clinical trial
of a 5 day schedule of Aza followed by LBH in high-risk MDS or AML
patients (13) and healthy controls.
A single mRNA sample for each time point per patient was available
for extraction.
RT-qPCR
Reverse Transcription of KG-1 cell and patient and
healthy control mRNA was performed using a 20 µl reaction mix
containing dNTPs (100 mM), MultiScribe Reverse Transcriptase (50
U/µl), RT buffer (10X), RNase Inhibitor (20 U/µl), nuclease free
water (Invitrogen; Thermo Fisher Scientific, Inc.), primer and KG-1
cell, patient or healthy control RNA samples. RT-qPCR analysis of
KG-1 cell, patient and healthy control PD-1, PD-L1, NUR77, Bcl-xL
and NF-кB mRNA expression was performed in duplicate using
Sensifast SYBR No-Rox kit (Bioline, London, UK). Reaction volumes
of 20 µl contained 2X Sensifast SYBR no-rox mix, nuclease-free
water and RT reaction product (either KG-1 cell or patient/healthy
control sample). Each PCR run also included wells of no template
control (NTC). A melting point dissociation curve generated by the
instrument (Applied Biosystems 7500 Real-Time PCR System; Thermo
Fisher Scientific, Inc.) was used to confirm that only a single
product was present. The fluorescence data were quantitated using
the threshold cycle (Ct) value (14). Data was normalized to actin and
presented as the fold change compared with the pre-treatment screen
sample.
PCR primers
Forward and reverse primer sequences for: PD-1:
Forward 5′-GGAAACCCCTCCACCTTTA-3′ and reverse
5′-TCTGCCTGCCCGCTTACT-3′; PD-L1: Forward
5′-TGGCATTTGCTGAACGCATTT-3′ and reverse
5′-TGCAGCCAGGTCTAATGTTTT-3′; NUR77: Forward
5′-GCTGCAGAATGACTCCACC-3′ and reverse 5′-ACAGCACTGGGCTTA-3′; NF-κB:
Forward 5′-CAGGAAGATGTGGAGGAT-3′ and reverse
5′-TGTCGTGCTCCACAGCCAGGT-3′; Bcl-xL: Forward
5′-TTGGACAATGGACTGGTTGA-3′ and reverse 5′-GTAGAGTGGATGGTCAGTG-3′;
and actin: Forward 5′-GACAGGATGCAGAAGGAGATTACT-3′ and reverse
5′-TGATCCACATCTGCTGGAAGGT-3′.
Statistical methods
Results of in vitro studies and combined
patient in vivo studies were expressed as means ± standard
error of the mean (SEM), and analyzed using GraphPad Prism 5
software (GraphPad Software, Inc., La Jolla, CA, USA), using
unpaired t-tests for two-group comparisons and one-way analysis of
variance and Dunnett's post hoc (ANOVA) for three or more group
comparisons. P-value of <0.05 was considered to be statistically
significant.
Methodological evaluation of results of duplicate
RT-qPCR analysis of individual patient, single time point mRNA
samples were presented as mean fold change compared to screen
utilizing the 2−∆∆Cq method (14).
Results
Effect of Aza, LBH-589 and MCT-3 on
leukemic cell growth, PD-1 and PD-L1 mRNA expression in KG-1
cells
Single agent treatment with Aza, MCT-3 and LBH-589
decreased KG-1 leukemic cell growth with LBH-589 showing
significant inhibition of cell growth. Combination Aza+LBH-589
treatment also resulted in significant attenuation of cell growth
(Fig. 1A). Single agent treatment
with Aza, MCT-3 and LBH-589 resulted in significant induction of
PD-1 mRNA expression whilst combination treatment with Aza+LBH-589
also demonstrated significant induction of PD-1 mRNA expression
(Fig. 1B). Single agent treatment
with Aza demonstrated modest induction of PD-L1 mRNA expression
whilst LBH-589 treatment resulted in significant induction of PD-L1
mRNA expression and combination treatment with Aza+LBH-589 also
demonstrated significant induction of PD-L1 mRNA expression
(Fig. 1C).
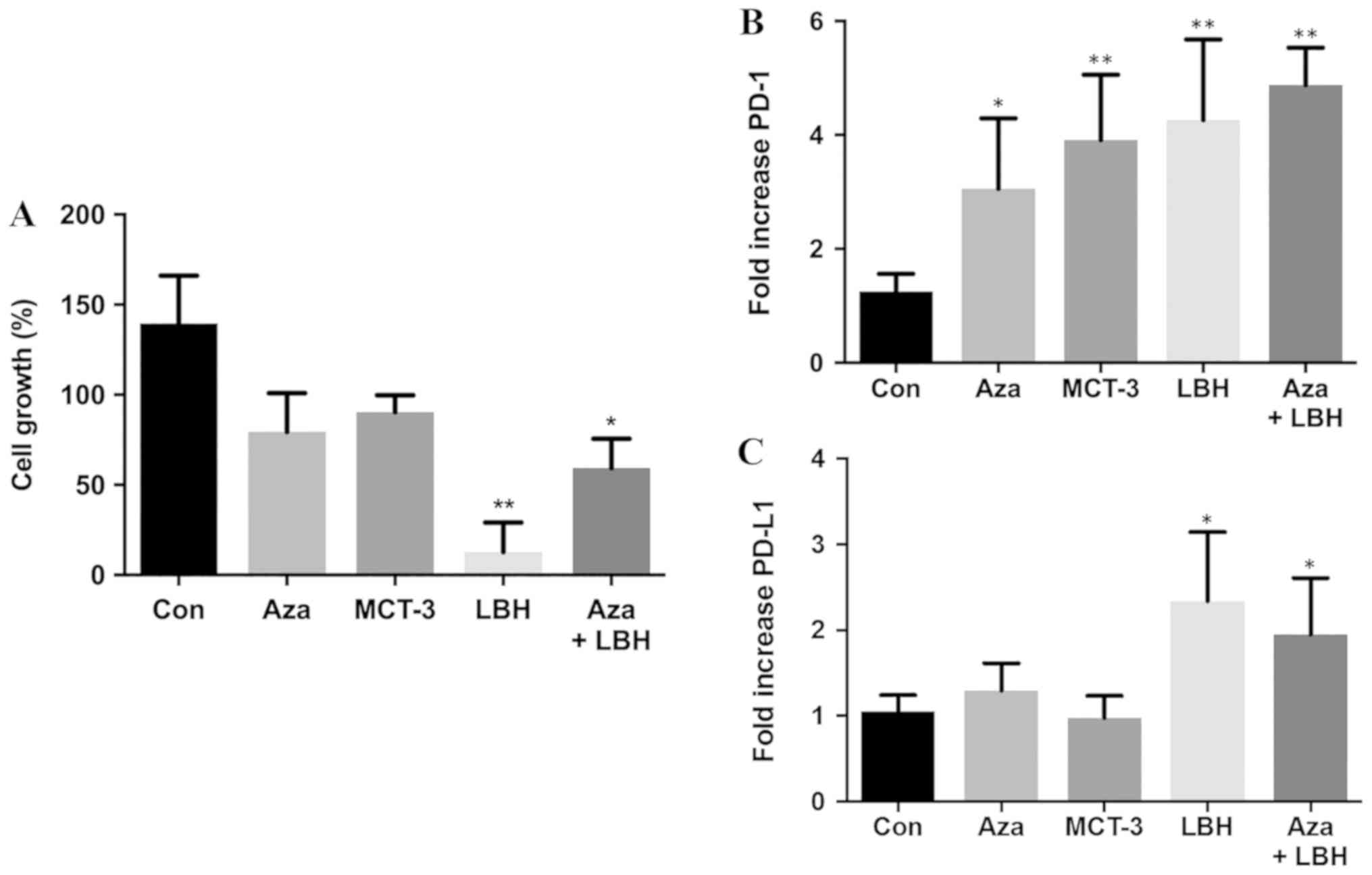 | Figure 1.In vitro effects on cell
growth, mRNA expression levels of PD-1 and PD-L1 of Aza, LBH-589,
MCT-3 or a combination of Aza and LBH589 in KG-1 cells. (A) In
vitro effects on cell growth in KG-1 cells untreated (Con) or
treated for 24 h with Aza, LBH-589 (LBH), MCT-3 or Aza+LBH as
assessed by trypan blue staining. n=3. In vitro mRNA
expression of (B) PD-1 and (C) PD-L1 mRNA in KG-1 cells untreated
(Con) and treated for 24-h treatment with Aza, MCT-3, LBH or a
combination of Aza+LBH. *P<0.05 vs. con, **P<0.01 vs. con,
n=3-4. PD-1, programmed cell death protein 1; PD-L1, programmed
death-ligand 1; Aza, azacytidine; Con, control. |
PD-1, PD-L1 and NUR77 mRNA expression
profiles from PBMC's of patients treated with Aza alone or Aza in
combination with LBH589
Expression levels of PD-1, PD-L1 and NUR77 mRNA were
determined at screening (prior to treatment commencement, day 0)
and at designated time points during 25–28 day first cycle of
treatment. Non-responders demonstrated a >2 fold increase in
PD-L1 expression over any other time point after treatment
commenced during the first cycle of treatment (Fig. 2A and Table
I) whilst responders, apart from patients 001 and 006,
demonstrated no increase in PD-L1 expression (Fig. 2B and Table
I). No correlation of PD-1 expression with response was
identified in individual patient analysis whilst paradoxically 4 of
6 non-responders also had induction of NUR77 expression, a
previously identified putative marker of response to EGT in MDS
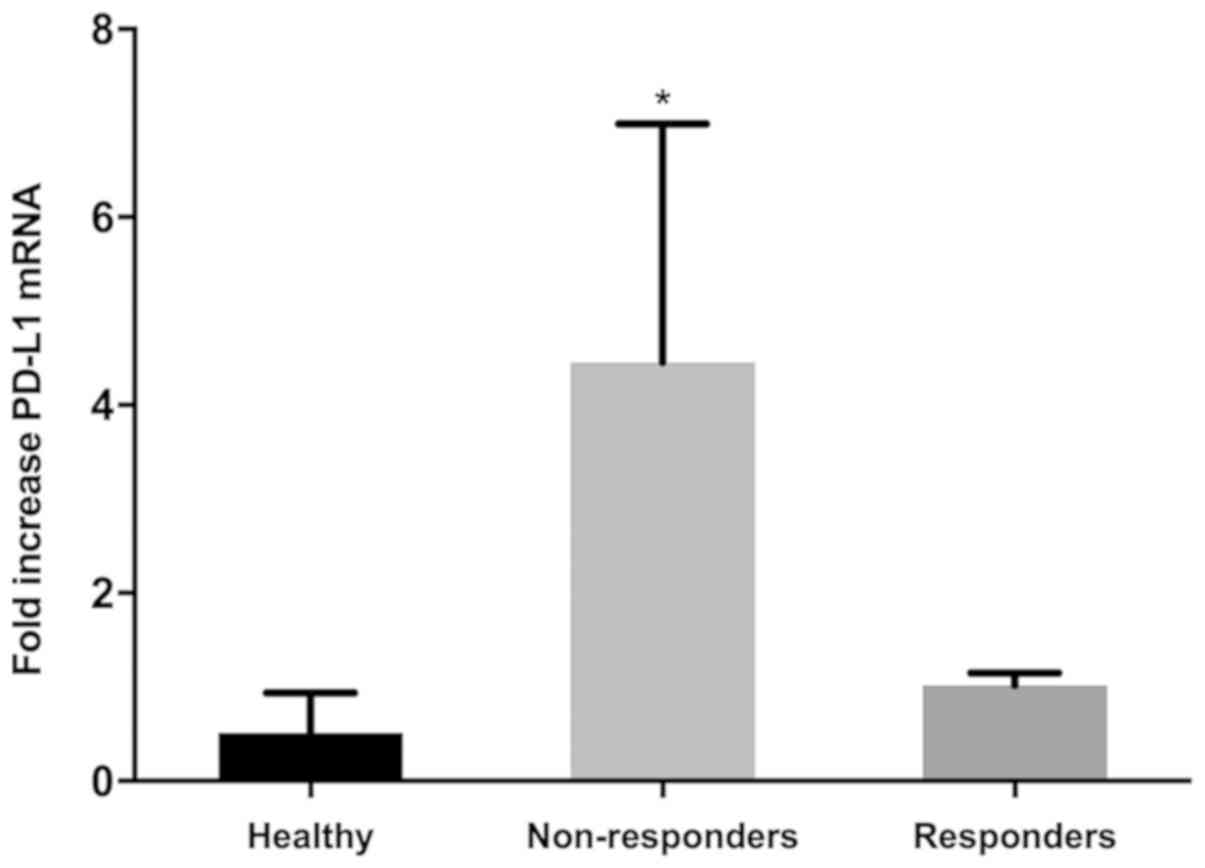
(Fig. 2A and Table I). Comparison of PD-L1 expression from
combined first cycle time points (screen, day 14, 21 and 28) of
healthy controls, responders and non-responders demonstrated
significant induction of PD-L1 expression in non-responders over
both healthy controls and responders (Fig. 3). Responses were defined according to
international working group criteria for AML and MDS (15).
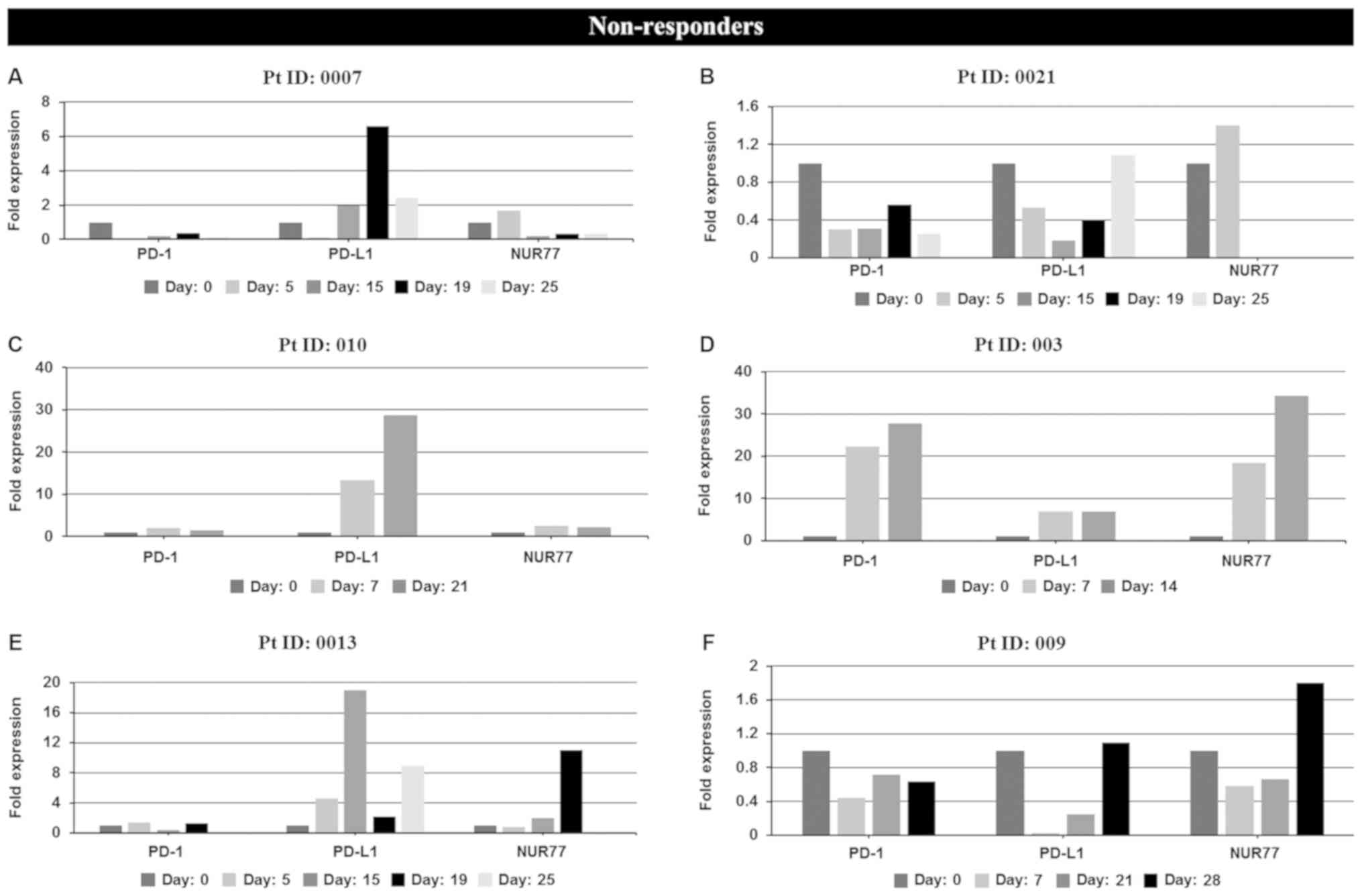 | Figure 2.In vivo mRNA expression
profiles at screening and during cycle 1 of PD-1, PD-L1 and NUR77
from PBMC's of individual non-responder and responder patients
treated with Aza alone or a combination of Aza and LBH589.
Non-responders: (A) patient 0007 (resistant), (B) patient 0021
(resistant), (C) patient 010 (resistant), (D) patient 003
(resistant), (E) patient 0013 [stable disease (SD)], (F) patient
009 [progressive disease (PD)]. Day: 0=screening sample, Days
5–28=samples taken on specific day post screening. Responders: (G)
patient 12 [partial response (PR)], (H) patient 002 (PR), (I)
patient 001 [complete response (CR)], (J) patient 005 (CR), (K)
patient 006 (CR), (L) patient 007 (CR), and (M) patient 015 [not
evaluated (NE)]. Clinical response to treatment determined at 1, 3
and 6 months. Responses were defined according to international
working group criteria for AML and MDS (15). Results for patient 006 expression
levels are presented using a log scale. PD-1, programmed cell death
protein 1; PD-L1, programmed death-ligand 1; PBMC, peripheral blood
mononuclear cells; Aza, azacytidine; AML, acute myeloid leukemia;
MDS, myelodysplastic syndrome. |
 | Table I.Clinical response data and correlation
with PD-1, PD-L1 and NUR77 mRNA expression at days 5–28. |
Table I.
Clinical response data and correlation
with PD-1, PD-L1 and NUR77 mRNA expression at days 5–28.
| Patient ID | Sex/age
(years) |
Diagnosis/treatment | Best response | PD-1
inductiona | PD-L1
inductiona | NUR77
inductiona |
|---|
| 0007 | M 58 | AMLb | Resistant | No | Yes | No |
| 0021 | F 78 | AMLb | Resistant | No | Yes | No |
| 003 | M 75 | AMLc | Resistant | Yes | Yes | Yes |
| 010 | F 65 | AMLc | Resistant | Yes | Yes | Yes |
| 0013 | M 79 | MDSb | SD | Yes | Yes | Yes |
| 009 | F 89 | MDSc | PD | No | Yes | Yes |
| 12 | F 59 | MDSc | PR | No | No | Yes |
| 002 | M 60 | AMLc | PR | Yes | No | Yes |
| 001 | M 45 | MDSc | CR | Yes | Yes | No |
| 005 | M 78 | MDSc | CR | No | No | No |
| 006 | F 90 | MDSc | CR | No | Yes | Yes |
| 007 | M 64 | AMLc | CR | No | No | No |
| 15 | M 77 | MDSb | NE | No | No | Yes |
In vitro and in vivo expression of
NF-кB and Bcl-xL in Aza, LBH-589, MCT-3 or a combination of Aza and
LBH-589 treated KG-1 cells and in patients non-responsive and
responsive to EGT
PD-L1 reverse signaling has recently been identified
as a potential mechanism of EGT resistance in the setting of MDS
treatment (8,16–19). We
investigated the in vitro and in vivo effect of EGT
on potential down-stream effector molecules of reverse PD-L1
signaling. In vitro studies identified 2–8 fold induction of
NF-κB and Bcl-xL mRNA expression in KG-1 cells treated with single
agent or combination EGT (Fig. 4A and
B). Characterisation of Bcl-xL expression from two patients,
one identified as non-responsive to EGT (Pt ID:010) and one
identified as responding to EGT (Pt ID:007) identified significant
upregulation of Bcl-xL mRNA expression in the patient resistant to
EGT (Fig. 4C) together with unchanged
or reduced levels of Bcl-xL and NF-κB mRNA in the patient who
responded to EGT (Fig. 4D and E).
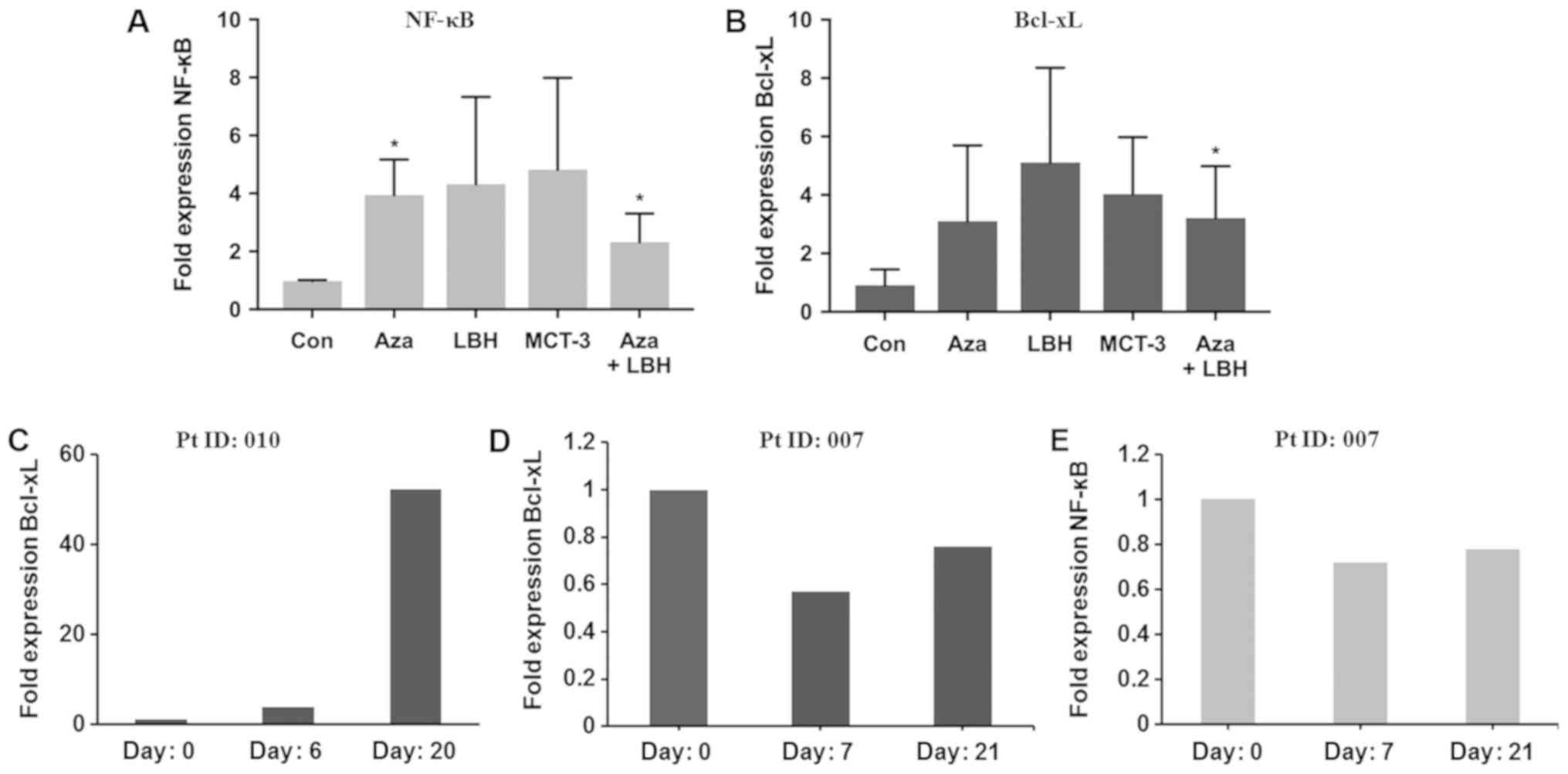 | Figure 4.In vitro and in vivo mRNA
expression levels of NF-κB and Bcl-xL in KG-1 cells treated with
Aza, LBH-589, MCT-3 or a combination of Aza+LBH-589 and from PBMC's
from a non-responder and responder patient. (A) NF-κB and (B)
Bcl-xL mRNA expression in KG-1 cells untreated (Con) or treated for
24 h with Aza, LBH-589 (LBH), MCT-3 or Aza+LBH *P<0.05 Con vs.
Aza and Aza+LBH (n=3) and (C) Bcl-xL mRNA expression in PBMC's from
non-responsive patient 010 at day 0, 6 and day 20 (n=1). (D) Bcl-xL
and (E) NF-κB mRNA expression in PBMC's from an EGT-responsive
patient 007 at day 0, 7 and day 21 (n=1). Responses were defined
according to international working group criteria for AML and MDS
(15). Aza, azacytidine; PBMC,
peripheral blood mononuclear cells; EGT, epigenetic therapy; Con,
control; AML, acute myeloid leukemia; MDS, myelodysplastic
syndrome. |
Discussion
Treatment of myeloid malignancies with single or
combination agent EGT has proven to be a significant advance in the
management of these conditions (20).
Response rates in the order of 50%, delayed time to response,
together with primary and acquired resistance to EGT have made
identification of biomarkers of response and resistance, together
with associated molecular mechanisms, increasingly important in the
management of patients treated with EGT (20).
Recent evidence identifies EGT-mediated upregulation
of genes associated with tumour immune evasion, including immune
checkpoint pathway-related molecules PD-1, PD-L1 and PD-L2 in the
setting of treatment of myeloid malignancies, which may contribute
to the acquired EGT resistance phenotype through T cell exhaustion
and enhanced PD-L1 reverse signaling (6–8,18).
Our current study investigated the expression of
immune checkpoint molecules PD-1 and PD-L1 together with the
putative EGT response marker orphan nuclear receptor NUR77
(9,11)
in PBMC's from patients with myeloid malignancies treated with EGT
and healthy controls and correlated these findings with clinical
response. In addition we investigated the in vitro and in
vivo effects of EGT on expression of potential downstream
molecules of reverse PD-L1 signalling as a possible molecular
mechanism for acquired resistance to EGT.
PD-L1 expression was increased in vitro in
response to EGT and correlated with resistance to EGT irrespective
of increased expression of the previously postulated marker of
response to EGT, NUR77 suggesting enhanced PD-L1 expression and
associated downstream molecular mechanisms may be a more potent
phenotypic contributor to EGT outcomes than potential response
biomarkers including NUR77. Whilst previous studies have
demonstrated EGT is able to increase PD-L1 expression in MDS
(16–18) and may correlate with poor responses to
treatment (18) our study is the
first to identify this effect independent of potential response
marker expression. Whilst our in vivo observations support
our in vitro findings a potential limitation of our study
exists due to logistical constraints in obtaining chronologically
identical patient blood samples for analysis during cycle 1.
Ongoing studies to evaluate more complete data sets over multiple
cycles of EGT will be undertaken to further refine the observations
made in this pilot study.
The molecular mechanisms responsible for EGT
resistance and loss of response in the setting of EGT-mediated
upregulation of inhibitory checkpoint molecules may include
enhanced immune evasion by leukemic cells through classical
PD-1-PD-L1 interactions (6–8) together with recently postulated
augmentation of tumour cell PD-L1 reverse signaling (8).
The contribution of tumour cell-intrinsic PD-L1
reverse signalling to disease pathogenesis has been recently
investigated. Initial reports identified B7-H1 or PD-L1 as a
ubiquitous anti-apoptotic receptor on cancer cells (19) whilst comprehensive evaluation of
tumour intrinsic PD-L1 signalling in ovarian and melanoma models
have demonstrated significant stimulatory effects on inflammation
and tumour cell growth (21). Our
studies evaluated potential downstream targets of PD-L1 reverse
signalling including the apoptosis and inflammation associated
genes Bcl-xL and NF-κB. EGT-mediated induction of Bcl-xL and NF-κB
gene expression both in vitro and in vivo in the
setting of treatment resistance demonstrates that enhanced PD-L1
expression may correlate with induction of PD-L1 reverse signalling
and upregulation of downstream targets which may contribute to
tumour cell resistance to EGT. In support of these observations
recent studies identify single or combination agent EGT treatment
of the HL-60 leukemia cell line, known not to express PD-L1,
as able to attenuate Bcl-xL expression, while concurrently
enhancing expression of the putative response marker NUR77
(9) suggesting induction of PD-L1
expression together with enhanced PD-L1 reverse signalling and
downstream effectors may be significant contributors to treatment
resistance and tumour cell persistence. It is intriguing to
speculate that whilst EGT in the setting of treatment for myeloid
malignancies induces malignant cell apoptosis, as has been
previously extensively documented in KG-1 cells (22,23) and
possibly supported by our observations of reduced KG-1 cell growth
with in vitro EGT, simultaneous anti-apoptotic effects
mediated by induction of PD-L1 expression together with enhanced
PD-L1 reverse signalling and upregulation of Bcl-xL may contribute
to malignant cell persistence and ultimately EGT failure.
EGT has recently been postulated to potentially
enhance and/or re-sensitize solid and haematological malignancies
to the effects of novel anti-PD-1/PD-L1 immunotherapeutics via
enhanced tumour neoantigen expression (7) and/or via upregulation of expression of
double stranded endogenous retroviral elements (dsERV's),
subsequent viral defense gene expression and interferon (IFN)
production, leading to cellular apoptosis and reduced cellular
proliferation (24–26).
A so-called ‘double edged sword effect’ of EGT
however arises from potentially concurrent therapy-mediated
induction of inhibitory checkpoint molecule expression such as
PD-L1 which may contribute to enhanced secondary resistance to EGT
in leukemia and MDS via mechanisms including reverse PD-L1
signaling which we have investigated during this study. Whilst
potentially problematic, EGT-mediated upregulation of inhibitory
immune checkpoint molecule expression may be able to be exploited
though identification of patients with EGT-mediated upregulation of
inhibitory immune checkpoint molecules including PD-L1 and
therapeutic intervention with anti-PD-1/PD-L1 therapy; a theory
currently the subject of several clinical trials (7). In addition characterization of PD-L1
expression may be an important marker of acquired EGT resistance
and anti-PD-1/PD-L1 therapy response in this patient
population.
Together our observations suggest enhanced PD-L1
expression may herald resistance to EGT over known markers of
response to EGT in myeloid malignancies and provide a potential
molecular mechanism involving modulation of effectors of PD-L1
reverse signaling which may, in-part, be responsible for these
effects. Larger prospective studies designed to formally evaluate
the contribution of PD-L1 expression to acquired EGT resistance
will establish the potential application of this molecule as a
clinical biomarker and therapeutic target in the setting EGT
resistance.
Acknowledgements
The abstract was presented as a poster presentation
at the 30th Lorne Cancer Conference 2018 Feb 8–11 2018 in Lorne,
Victoria, Australia.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL, YH, RR, CvH, EWCK, NL, and SF assisted with
analysis, interpretation and presentation of data. AS was
instrumental in conceiving and designing critical elements of the
clinical trial utilized in this study. AED assisted with analysis
and interpretation of data and made a significant intellectual
contribution to the study. All authors contacted read and approved
the final manuscript.
Ethics approval and consent to
participate
The open-label, phase Ib/II study (13) was approved by the Alfred Hospital
Ethics Committee on 15/07/2009, ethics approval number AH189/09 and
was performed in accordance with the principles of independent
human research and ethics committees, and registered with the
Australian and New Zealand Clinical Trials Registry
(ACTRN12610000924055). Low risk study 357/14 was approved by Alfred
Health Ethics Committee (AHEC) on 20/8/2014 and low risk study
LR63/2015 was approved by Eastern Health Ethics Committee (EHEC) on
27/07/2015. All participating patients and healthy controls were
required to provide written informed consent for participation in
the studies.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yun S, Vincelette ND, Abraham I, Robertson
KD, Fernandez-Zapico ME and Patnaik MM: Targeting epigenetic
pathways in acute myeloid leukemia and myelodysplastic syndrome: A
systematic review of hypomethylating agents trials. Clin
Epigenetics. 8:682016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Unnikrishnan A, Papaemmanuil E, Beck D,
Deshpande NP, Verma A, Kumari A, Woll PS, Richards LA, Knezevic K,
Chandrakanthan V, et al: Integrative genomics identifies the
molecular basis of resistance to azacitidine therapy in
myelodysplastic syndromes. Cell Rep. 20:572–585. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei
J, Watanabe KA, Shammo JM, Anastasi J, Shen QJ, et al: RNA cytosine
methylation and methyltransferases mediate chromatin organization
and 5-azacytidine response and resistance in leukaemia. Nat Commun.
9:11632018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vidal V, Robert G, Goursaud L, Durand L,
Ginet C, Karsenti JM, Luciano F, Gastaud L, Garnier G, Braun T, et
al: BCL2L10 positive cells in bone marrow are an independent
prognostic factor of azacitidine outcome in myelodysplastic
syndrome and acute myeloid leukemia. Oncotarget. 8:47103–47109.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miltiades P, Lamprianidou E,
Vassilakopoulos TP, Papageorgiou SG, Galanopoulos AG, Kontos CK,
Adamopoulos PG, Nakou E, Vakalopoulou S, Garypidou V, et al: The
Stat3/5 signaling biosignature in hematopoietic stem/progenitor
cells predicts response and outcome in myelodysplastic syndrome
patients treated with azacitidine. Clin Cancer Res. 22:1958–1968.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daver N, Boddu P, Garcia-Manero G, Yadav
SS, Sharma P, Allison J and Kantarjian H: Hypomethylating agents in
combination with immune checkpoint inhibitors in acute myeloid
leukemia and myelodysplastic syndromes. Leukemia. 32:1094–1105.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wolff F, Leisch M, Greil R, Risch A and
Pleyer L: The double-edged sword of (re)expression of genes by
hypomethylating agents: From viral mimicry to exploitation as
priming agents for targeted immune checkpoint modulation. Cell
Commun Signal. 15:132017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Granier C, De Guillebon E, Blanc C,
Roussel H, Badoual C, Colin E, Saldmann A, Gey A, Oudard S and
Tartour E: Mechanisms of action and rational for the use of
checkpoint inhibitors in cancer. ESMO Open. 2:e0002132017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu HB, Urbanavicius D, Tan P, Spencer A
and Dear AE: Mechanisms and potential molecular markers of early
response to combination epigenetic therapy in patients with myeloid
malignancies. Int J Oncol. 45:1742–1748. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lübbert M, Ihorst G, Sander PN, Bogatyreva
L, Becker H, Wijermans PW, Suciu S, Bissé E and Claus R: Elevated
fetal haemoglobin is a predictor of better outcome in MDS/AML
patients receiving 5-aza-2′-deoxycytidine (Decitabine). Br J
Haematol. 176:609–617. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou L, Ruvolo VR, McQueen T, Chen W,
Samudio IJ, Conneely O, Konopleva M and Andreeff M: HDAC inhibition
by SNDX-275 (Entinostat) restores expression of silenced
leukemia-associated transcription factors Nur77 and Nor1 and of key
pro-apoptotic proteins in AML. Leukemia. 27:1358–1368. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dear AE, Liu HB, Mayes PA and Perlmutter
P: Conformational analogues of oxamflatin as histone deacetylase
inhibitors. Org Biomol Chem. 4:3778–3784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan P, Wei A, Mithraprabhu S, Cummings N,
Liu HB, Perugini M, Reed K, Avery S, Patil S, Walker P, et al: Dual
epigenetic targeting with panobinostat and azacitidine in acute
myeloid leukemia and high-risk myelodysplastic syndrome. Blood
Cancer J. 4:e1702014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheson BD, Greenberg PL, Bennett JM,
Lowenberg B, Wijermans PW, Nimer SD, Pinto A, Beran M, de Witte TM,
Stone RM, et al: Clinical application and proposal for modification
of the International Working Group (IWG) response criteria in
myelodysplasia. Blood. 108:419–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dail M, Yang L, Green C, Ma C, Robert A,
Kadel EE, Koeppen H, Adamkewicz J, Byon J, Woodard J, et al:
Distinct patterns of PD-L1 and PD-L2 expression by tumor and
non-tumor cells in patients with MM, MDS and AML. Blood.
128:13402016.
|
|
17
|
Coats T, Smith AE, Mourikis TP, Irish JM,
Kordasti S and Mufti GJ: Mass cytometry reveals PD1 upregulation is
an early step in MDS disease progression. Blood. 128:42962016.
|
|
18
|
Yang H, Bueso-Ramos C, DiNardo C, Estecio
MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, et al:
Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic
syndromes is enhanced by treatment with hypomethylating agents.
Leukemia. 28:1280–1288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Azuma T, Yao S, Zhu G, Flies AS, Flies SJ
and Chen L: B7-H1 is a ubiquitous antiapoptotic receptor on cancer
cells. Blood. 111:3635–3643. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ball B, Zeidan A, Gore SD and Prebet T:
Hypomethylating agent combination strategies in myelodysplastic
syndromes: Hopes and shortcomings. Leuk Lymphoma. 58:1022–1036.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clark CA, Gupta HB, Sareddy G, Pandeswara
S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K,
et al: Tumor-intrinsic PD-L1 signals regulate cell growth,
pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer
Res. 76:6964–6974. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lainey E, Wolfromm A, Marie N, Enot D,
Scoazec M, Bouteloup C, Leroy C, Micol JB, De Botton S, Galluzzi L,
et al: Azacytidine and erlotinib exert synergistic effects against
acute myeloid leukemia. Oncogene. 32:4331–4342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maiso P, Colado E, Enrique MO, Garayoa M,
Atadja P, Pandiella A and San Miguel JF: Panobinostat (LBH589) a
promising new partner for combination with doxorubicin in acute
myeloid leukemia. Blood. 112:16382008.PubMed/NCBI
|
|
24
|
Chiappinelli KB, Strissel PL, Desrichard
A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et
al: Inhibiting DNA methylation causes an interferon response in
cancer via dsRNA including endogenous retroviruses. Cell.
162:974–986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roulois D, Loo Yau H, Singhania R, Wang Y,
Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al:
DNA-Demethylating agents target colorectal cancer cells by inducing
viral mimicry by endogenous transcripts. Cell. 162:961–973. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dear AE: Epigenetic modulators and the new
immunotherapies. N Engl J Med. 374:684–686. 2016. View Article : Google Scholar : PubMed/NCBI
|