Introduction
Tumors are the second leading cause of mortality
worldwide, behind cardiovascular disease (1,2). In the
USA, the incidence and mortality rates of colorectal cancer (CRC)
are the third highest among all cancer types in both males and
females in 2018 (2). In China, CRC
ranks as the fifth most common malignancy in males and the fourth
most common malignancy in females, with 12,000 new cases being
diagnosed each year (3). Early
prevention, detection and treatment are crucial for cancer control
and management. Although significant progress has been made in
understanding the molecular mechanisms involved in CRC, and in CRC
diagnosis and treatment, the 5-year survival rate of patients with
CRC has not significantly improved (4–6).
Therefore, the early detection of CRC for prognostic management
with non-invasive methods is urgently necessary.
The nucleotide excision repair (NER) mechanism
serves an important role in maintaining the integrity of
chromosomes. Any factor that interferes with the NER mechanism can
affect cellular activity and lead to cell death. The normal
function of NER factors is particularly important for cell division
and differentiation (7). The SNF2
family is an important family of proteins that function as NER
factors and are essential for ribosome assembly, translation
initiation and cell growth (7).
Excision repair cross-complementation group 6 (ERCC6) is a member
of the SNF2 family and serves a key role in transcription-coupled
DNA repair, allowing access of the DNA-repair apparatus to DNA
(7). ERCC6 is upregulated in CRC
tissues, and increased expression levels of ERCC6 have been
associated with a poor response to chemotherapy and worse survival
for patients with CRC (8). A newly
identified ERCC6-like gene in mice, ERCC6L, also known as polo-like
kinase 1 (PLK1)-interacting checkpoint helicase, has been
demonstrated to be a development-associated member of the SNF2
family (9,10). Nielsen et al (9) identified that ERCC6L cooperates with
topoisomerase II in mitosis to promote sister chromatid
disjunction. Furthermore, ERCC6L has been revealed to function as a
DNA-dependent ATPase that interacts with PLK to form a complex that
maintains the chromosome architecture during prometaphase (10). These studies suggest that ERCC6L is
not a typical NER factor but rather serves a role in the
segregation of sister chromatids during mitosis.
The ERCC6L protein is assembled in the cytoplasm and
then enters the cell nucleus to exert its function (10). It is strongly expressed in the mouse
embryonic stage, particularly in embryonic brain, heart, kidney,
liver and lung. However, ERCC6L expression is significantly
downregulated following birth, with no expression detected in the
majority of adult organs (7,11). Previous studies have demonstrated
that ERCC6L is highly expressed in numerous types of human solid
tumor; therefore, it is considered a potential target for cancer
therapy (12,13). Pu et al (13) demonstrated that the mRNA expression
level of ERCC6L increases during the progression of breast and
kidney cancers, and increased ERCC6L expression is associated with
poor overall survival. In addition, ERCC6L silencing has been
revealed to inhibit the proliferation of cancer cells, which
suggests that ERCC6L may be an effective biomarker for cancer
progression (13). However, to the
best of our knowledge, the role of ERCC6L in CRC remains to be
investigated.
The present study first investigated the expression
of ERCC6L in CRC tissues using immunohistochemistry (IHC), reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis with the aim of determining the role of
ERCC6L in CRC. Subsequently, the associations between ERCC6L
expression level and the clinicopathological characteristics of
patients with CRC were evaluated. Finally, the functional role of
ERCC6L in CRC cell proliferation, cycle, apoptosis and invasion was
explored by in vitro experiments.
Materials and methods
Tissue specimens
The present study included 30 patients with primary
CRC from the Department of Colorectal Surgery at the Zhongnan
Hospital of Wuhan University (Wuhan, China) from June to Setember
2017. Written informed consent was obtained from all patients prior
to enrolment in the study. This prospective study was approved by
the institutional Ethics Committee of Zhongnan Hospital of Wuhan
University (Wuhan, China) and conducted in accordance with the
ethical guidelines of the Declaration of Helsinki. All patients
underwent surgical resection without preoperative chemotherapy and
radiotherapy. Detailed patient clinical information is listed in
Table I. The cohort consisted of 11
females and 19 males, with a median age of 51 years (range, 22–80
years). The diagnosis of all patients was confirmed to be CRC by
histopathology. The tissue size as 1 cm3 and was stored
in a liquid nitrogen until subsequent use.
 | Table I.Association of ERCC6L expression with
clinicopathological parameters. |
Table I.
Association of ERCC6L expression with
clinicopathological parameters.
|
|
| ERCC6L
expression |
|
|---|
|
|
|
|
|
|---|
|
Characteristics | Cases, n | Low (n=9) | High (n=21) | P-value |
|---|
| Age, years |
|
|
| 0.589 |
|
<51 | 8 | 3 | 5 |
|
|
≥51 | 22 | 6 | 16 |
|
| Sex |
|
|
| 0.563 |
|
Male | 19 | 5 | 14 |
|
|
Female | 11 | 4 | 7 |
|
| Median tumor size,
cm |
|
|
| 0.011 |
|
<5 | 10 | 6 | 4 |
|
| ≥5 | 20 | 3 | 17 |
|
| Tumor
differentiation |
|
|
| 0.109 |
|
Well/moderately | 25 | 6 | 19 |
|
|
Poor | 5 | 3 | 2 |
|
| TNM stage |
|
|
| 0.523 |
|
I–II | 16 | 4 | 12 |
|
|
III–IV | 14 | 5 | 9 |
|
Cell cultures
Human CRC (HCT116, SW480 and HT29) and normal
colonic mucosal (NCM460) cell lines were obtained from The Cell
Bank of the Chinese Academy of Sciences (Shanghai, China). All
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10%
fetal bovine serum (FBS; Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd., Hangzhou, China) and 1%
penicillin/streptomycin. Cells were incubated in a humidified
chamber with 5% CO2 at 37°C.
RNA interference and transfection
Small interfering RNA (siRNA) sequences targeting
human ERCC6L (siRNA-ERCC6L) and a negative control (si-NC) were
purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The
sequences of the siRNAs were as follows: si-ERCC6L-101 forward,
5′-GCAGGCTGCTCATTACCTA-3′ and reverse, 5′-TAGGTAATGAGCAGCCTGC-3′;
si-ERCC6L-102 forward, 5′-GTAGGTGGTGTCGGTTTAA-3′ and reverse,
5′-TTAAACCGACACCACCTAC-3′; si-ERCC6L-103 forward,
5′-GCTGGTTAATGACGTCTAA-3′ and reverse, 5′-TTAGACGTCATTAACCAGC-3′;
and NC forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. SW480 and HT29 cells were seeded in a
6-well plate at a density of 50,000 cells/ml and transfection was
initiated when the cell confluence reached 50–60%. The amount of
siRNA transfected was 5.0 pmoles. Cells were transfected with the
siRNA using GenMute™ siRNA Transfection Reagent (SignaGen
Laboratories, Rockville, MD, USA), according to the manufacturer's
protocol. Following ~24 h, cells were harvested for further
experiments.
RNA isolation and RT-qPCR
CRC tissue (0.5 cm3) was placed in an
Eppendorf tube containing 1 ml TRIzol reagent (Thermo Fisher
Scientific, Inc.) and then ground with a homogenizer. Total RNA
extraction was performed according to the manufacturer's protocol.
The resulting RNA was dissolved in RNase-free water and immediately
stored at −80°C. The RNA concentration was measured using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
Complementary DNA was then synthesized using a ReverTra Ace qPCR RT
kit (Toyobo Life Science, Osaka, Japan). qPCR was then performed
using SYBR-green PCR master mix (CWBIO, Beijing, China) in a
Bio-Rad 7500 real-time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The reverse transcription reaction was
carried out at 37°C for 15 min, then the enzyme inactivation at
98°C for 5 min, and finally stored at 4°C. The primers for ERCC6L
and GAPDH were purchased from Qingke Biotechnology Co., Ltd.
(Wuhan, China). The specific primers used were as follows: ERCC6L
forward, 5′-ATCGGTGCCTCAGCGTTCGG-3′ and reverse,
5′-CTGTCCTCGCCGTCACACCG-3′; and GAPDH forward,
5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse,
5′-GCAGGAGGCATTGCTGATGAT-3′. GAPDH was used as a control. The
reaction conditions were as follows: Pre-denaturation at 95°C for
10 min; denaturation at 95°C for 10 sec; annealing at 60°C for 20
sec; extension at 72°C for 10 sec; full extension at 72°C for 3
min; the number of gene cycles was 39. All reactions were run in
triplicate, and the results were analyzed and expressed relative to
threshold cycle (Cq) values and then converted to fold change
values (2−ΔΔCq) (14).
Western blot analysis
CRC tissues and cells were lysed with
radioimmunoprecipitation assay protein extraction reagent (Beyotime
Institute of Biotechnology, Haimen, China) supplemented with 1%
phenylmethanesulfonyl fluoride (Seebio Science & Technology
Co., Ltd., Shanghai, China). Protein concentrations were measured
using an enhanced BCA protein assay kit (Beyotime Institute of
Biotechnology). Each lane was loaded with an equal amount of
protein (30 µg), and proteins were then separated using 8% SDS-PAGE
and transferred to a polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
milk for 2 h at room temperature. The membranes were then placed in
a TBS and Tween-20 (TBST) solution containing anti-ERCC6L antibody
(1:1,000; cat. no. BC008808; Wuhan Sanying Biotechnology, Wuhan,
China) and were allowed to react at 4°C overnight for ~12 h. The
following day, following washing with TBST, the blots were
incubated with a horseradish peroxidase-labeled anti-rabbit
secondary antibody (1:5,000; cat. no. GB233303-1; Servicebio,
Woburn, MA, USA) for 2 h at room temperature. The blots were
visualized using a Super ECL detection reagent (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China). Each set of
analyses was repeated a minimum of three times.
Wound healing assay
CRC cells were seeded in a 6-well plate (50,000
cells/ml). At ~24 h post-transfection, the cells had reached 70–80%
confluency. A straight scratch was created using a sterile pipette
tip. PBS was added to the six-well plate to remove floating cells.
The original culture medium was replaced with 0.5% FBS for a
further 24 h. All tests were repeated three times. Cell migration
was observed and imaged at 0 and 24 h with a light microscope (×40
magnification; OLYMPUS U-RFL-T; Olympus Corporation, Tokyo,
Japan).
Cell proliferation assay
At 24 h post-transfection, HT29 and SW480 cells were
trypsinized to provide suspensions that were then seeded in 96-well
plates at densities of 3,000 and 2,000 cells/well, respectively.
The cell proliferation rates were calculated using the MTT method
at 0, 24, 48, 72 and 96 h. Briefly, 10 µl MTT was added to each
well and the cells were incubated for 4 h. After 4 h, the medium
was replaced with 200 µl dimethyl sulfoxide to dissolve the
formazan. The absorbance value of each well at 490 nm was recorded.
Each experiment was repeated a minimum of three times.
Colony formation assay
The colony formation ability of SW480 and HT29 cells
transfected with siRNA was measured as follows. Cells were plated
in 6-well plates at 1,000 cells/well and maintained in DMEM
containing 10% FBS. The cells were incubated in a humidified
chamber with 5% CO2 at 37°C. Following 12–14 days, the
cells were washed twice with PBS, fixed with 4% paraformaldehyde
for 25 min and then stained with 0.1% crystal violet for 20 min at
room temperature. All experiments were performed in triplicate.
Cell invasion assay
A 24-multiwell insert plate with a small chamber (BD
Biosciences, San Jose, CA, USA) containing an 8.0-µm pore size
Matrigel-coated membrane was used for the cell invasion assays.
Briefly, 2×105 cells in serum-free medium were seeded
into the upper chambers, which were coated in Matrigel. The lower
chambers of the 24-well plates were filled with 600 µl DMEM
containing 20% FBS as a chemo-attractant. Following incubation of
the plates at 37°C for 24 h, the non-invasive cells above the
chamber were gently wiped away with a wet cotton swab. The cells in
the lower chamber were fixed with 4% paraformaldehyde for 30 min,
stained with 0.1% crystal violet for 20 min at room temperature and
then counted under a light microscope (×200 magnification). Each
experiment was repeated a minimum of three times.
Analysis of cell apoptosis and the
cell cycle
The cells were seeded in 6-well plates at
5×105 cells/well. Following transfection with siRNA for
24 h, HT29 and SW480 cells were harvested, washed with PBS,
resuspended in binding buffer and incubated at room temperature for
15 min in the dark with propidium iodide (PI) and annexin
V/fluorescein isothiocyanate (Absin Bioscience Inc., Shanghai,
China). The apoptosis rate was immediately detected by flow
cytometry using a flow cytometer (Beckman CytoFLEX FCM, USA).
For analysis of the cell cycle, HT29 and SW480 cells
were transfected with siRNA for 24 h and then fixed in 70% ethanol
at −20°C for 24 h. The cells were washed twice with PBS and then
incubated with RNase A at 37°C for 30 min. PI (Nanjing KeyGen
Biotech Co., Ltd., Nanjing, China) was added to the cell suspension
(100,000/ml) and the cells were incubated for 30 min at room
temperature in the dark. The cell cycle distribution was then
analyzed using a flow cytometer. All experiments were performed in
triplicate.
IHC
CRC tissues were fixed in 10% neutral buffered
formalin solution for 24 h at room temperature and then embedded in
paraffin for IHC analysis. Briefly, 5-µm paraffin sections were
deparaffinized in xylene and gradually rehydrated in 100, 95 and
75% ethanol at 60°C. The 5-µm paraffin sections were baked in a
60°C oven for 2 h, then deparaffinized in xylene and gradually
rehydrated in 100, 95 and 75% ethanol. The tissue sections were
blocked with 30% H2O2 for 20 min at room
temperature to quench the activity of endogenous peroxidases.
Following antigen retrieval in heated 10 mM citrate buffer for 10
min, the tissue sections were immunostained with primary antibody
(anti-ERCC6L; 1:1,000; cat. no. BC008808) overnight at 4°C. A mouse
horseradish peroxidase-conjugated secondary antibody
SignalStain® Boost IHC Detection Reagent (HRP, Rabbit)
(1:1,000; cat no. 8114; Cell Signaling Technology, Inc.) was then
added for 1 h at room temperature. Finally, images were observed
under a light microscope (x 200 mangification). The positive cells
were judged by the staining of nucleus and cytoplasm. The positive
rate was divided by the total number of stained positive cells in
different visual fields, and then the average value was taken for
statistical analysis.
Statistical analysis
Data are presented as the mean standard ± deviation.
The IHC staining results and patient data were evaluated using a
χ2 test. Other data presented in the text and figures
were analyzed using one-way analysis of variance with a Bonferroni
correction. All statistical analysis was performed using SPSS 22.0
(IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Expression of ERCC6L in CRC tissues
and cell lines
The expression level of ERCC6L was detected in 30
paired samples of CRC and adjacent histological noncancerous
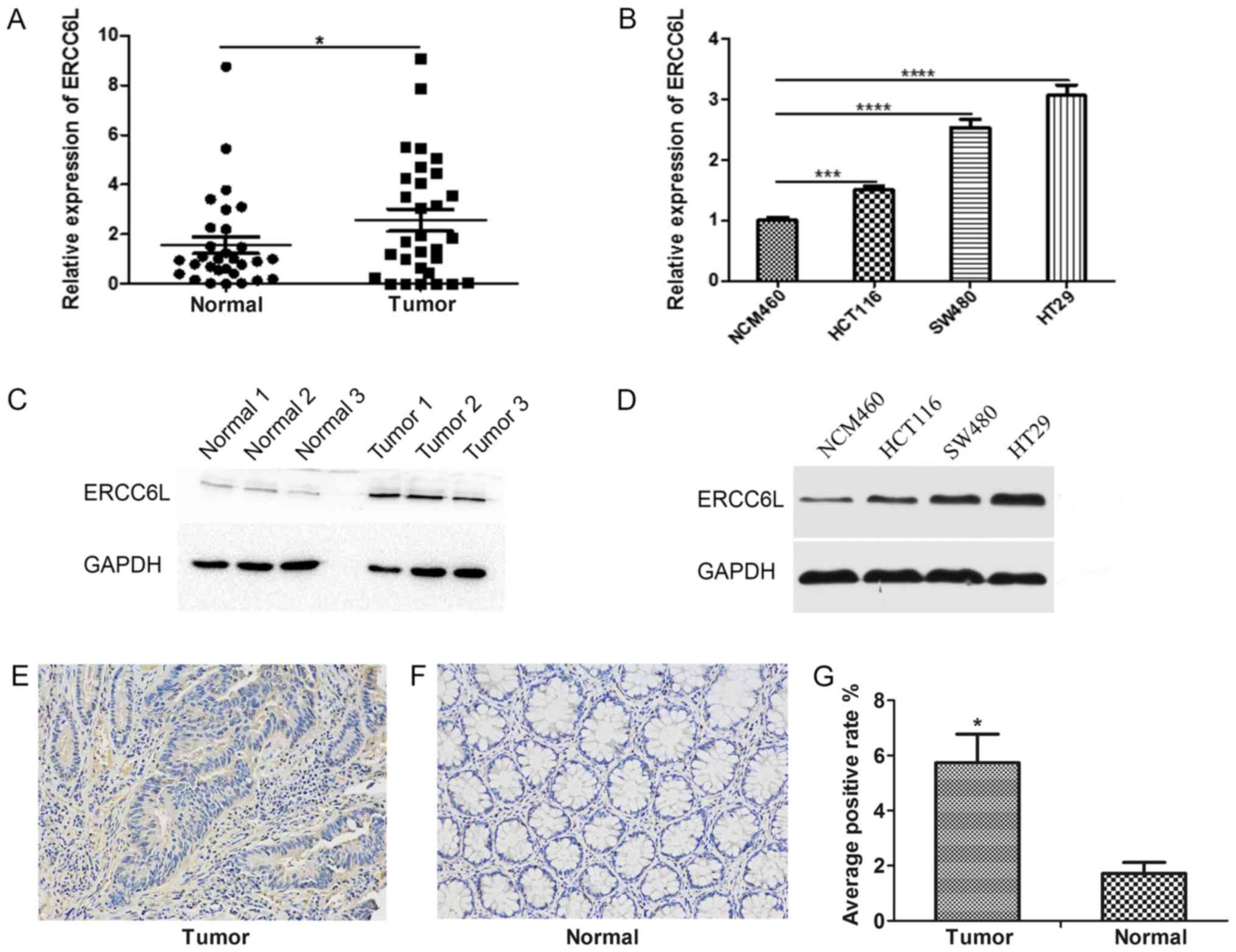
tissues by RT-qPCR. As demonstrated in Fig. 1A, the expression level of ERCC6L in
CRC tissues was significantly higher compared with the expression
level in paracancerous tissues (P<0.05). Similarly, the
expression levels of ERCC6L in a normal colonic mucosal (NCM460)
and three CRC (HT29, SW480, HCT116) cell lines were determined by
RT-qPCR. The expression level of ERCC6L in HT29, SW480 and HCT116
CRC cells was significantly higher compared with that in NCM460
cells (P<0.001; Fig. 1B). In
addition, western blot analysis revealed that the protein
expression level of ERCC6L was increased in CRC tissues and cancer
cell lines compared with that in normal tissues and cells (Fig. 1C and D). IHC demonstrated that ERCC6L
is expressed in the cytoplasm and nucleus of cells, and indicated
that its cytoplasmic expression is higher than that of the nucleus
(Fig. 1E and F). In addition, the
IHC results revealed that the expression level of ERCC6L was higher
in tumor tissues compared with the expression in matched non-tumor
tissues (P<0.05; Fig. 1G).
Association between ERCC6L expression
and the clinicopathological features of patients with CRC
To examine the association between ERCC6L expression
and the clinicopathological features of patients with CRC, the
patients were divided into low (n=9) and high (n=21) ERCC6L
expression groups based on comparison with the expression levels of
ERCC6L in adjacent noncancerous tissues. High ERCC6L expression
group, the expression of ERCC6L in tumor tissue was higher than
that in paracancerous tissue; and low ERCC6L expression group, the
expression of ERCC6L in tumor tissue was lower than that in
paracancerous tissue. As presented in Table I, the expression level of ERCC6L
exhibited a significant association with tumor size (P<0.05);
however, no associations were detected with other clinical
characteristics, including age, sex, tumor differentiation and
clinical stage.
Migration, invasion and proliferation
of CRC cells decreases following ERCC6L knockdown
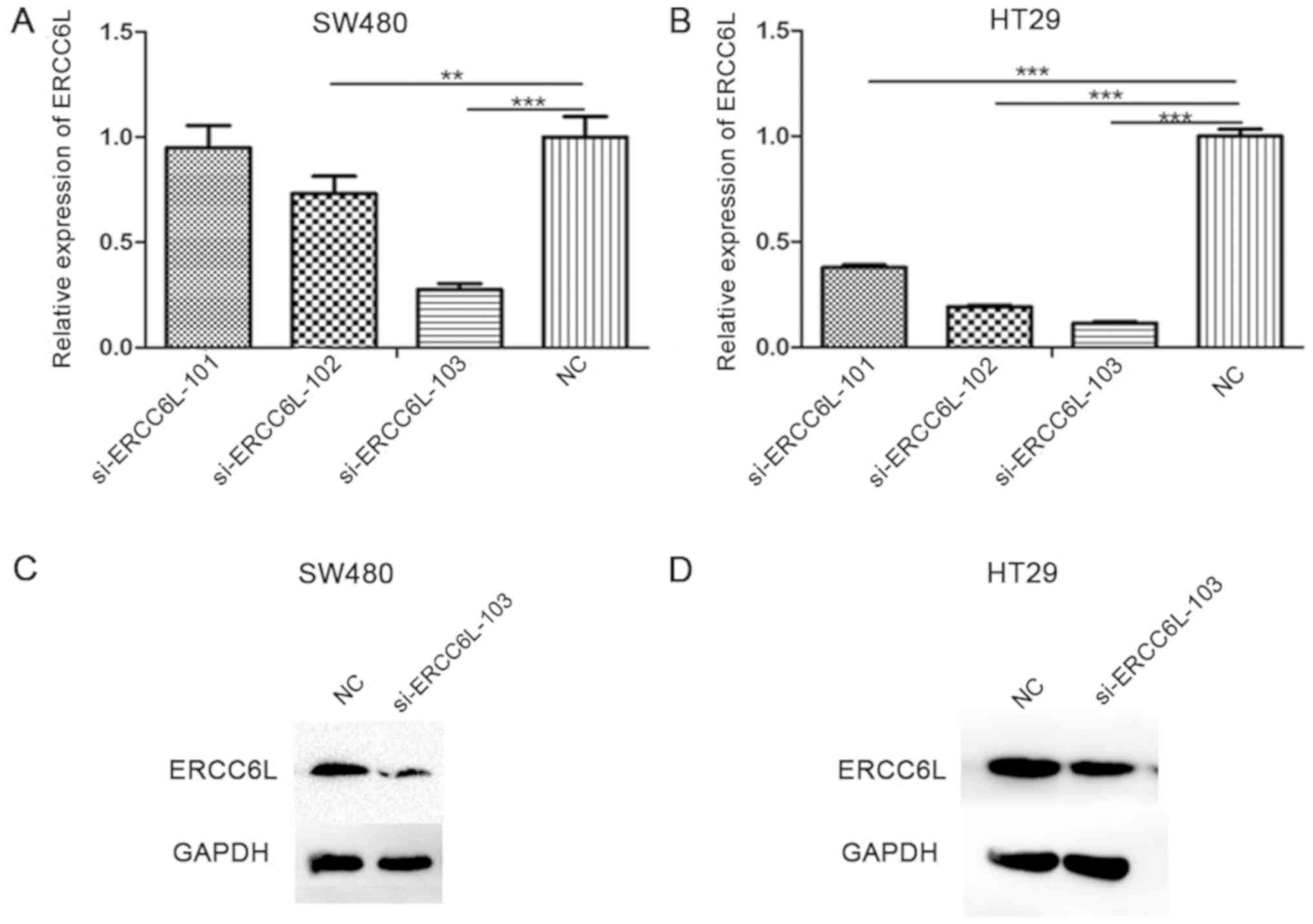
ERCC6L expression was knocked down using three
different siRNAs, and the silencing efficiency of these siRNAs was
detected by RT-qPCR (Fig. 2A and B).
The results demonstrated that si-ERCC6L-103 reduced the expression
level of ERCC6L to the greatest extent in the SW480 and HT29 cell
lines (P<0.001); therefore, this siRNA was selected for
subsequent experiments. Similarly, at the protein level (Fig. 2C and D), si-ERCC6L-103 could
significantly decrease the expression of ERCC6L in SW480 and HT29
cells.
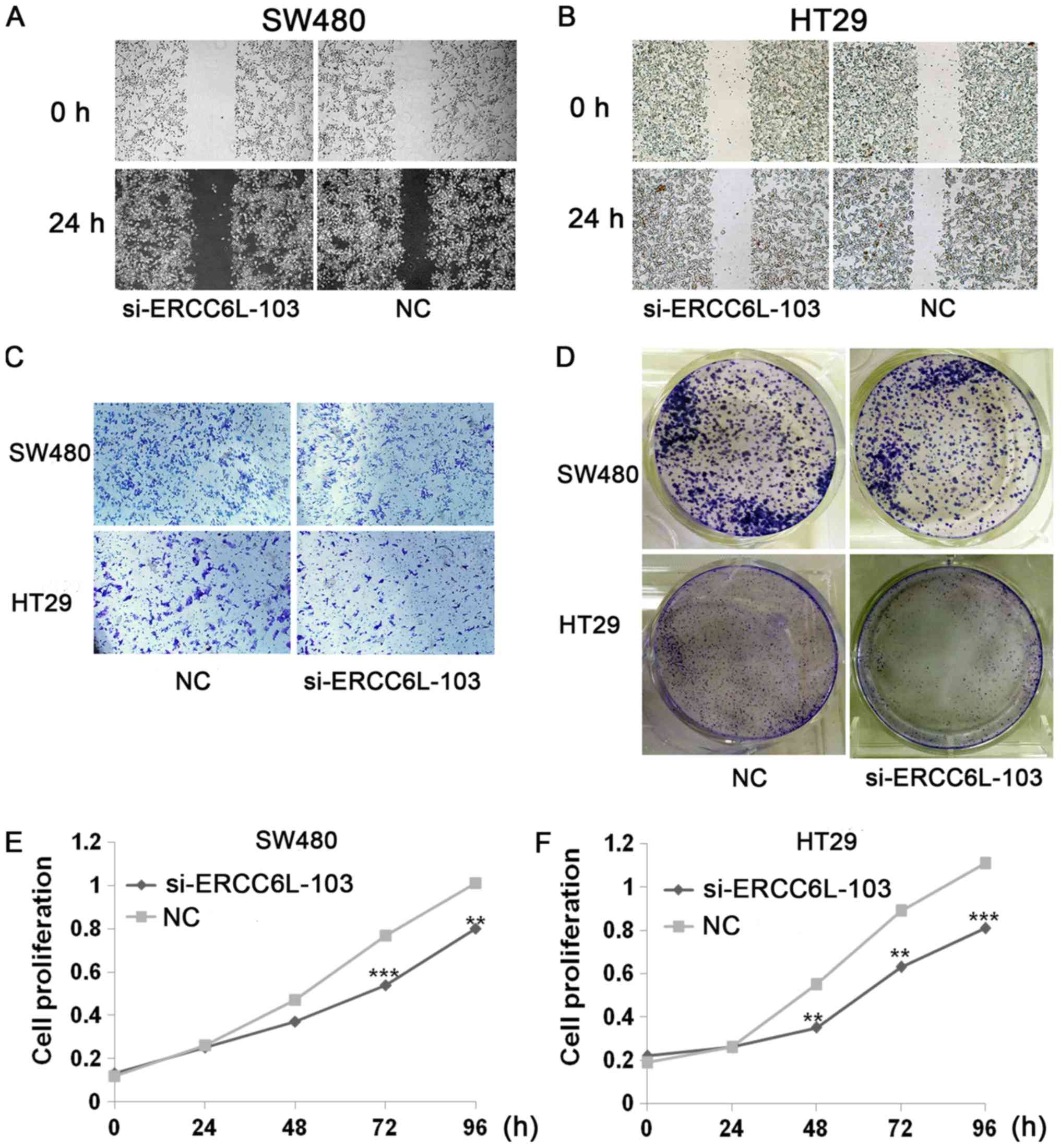
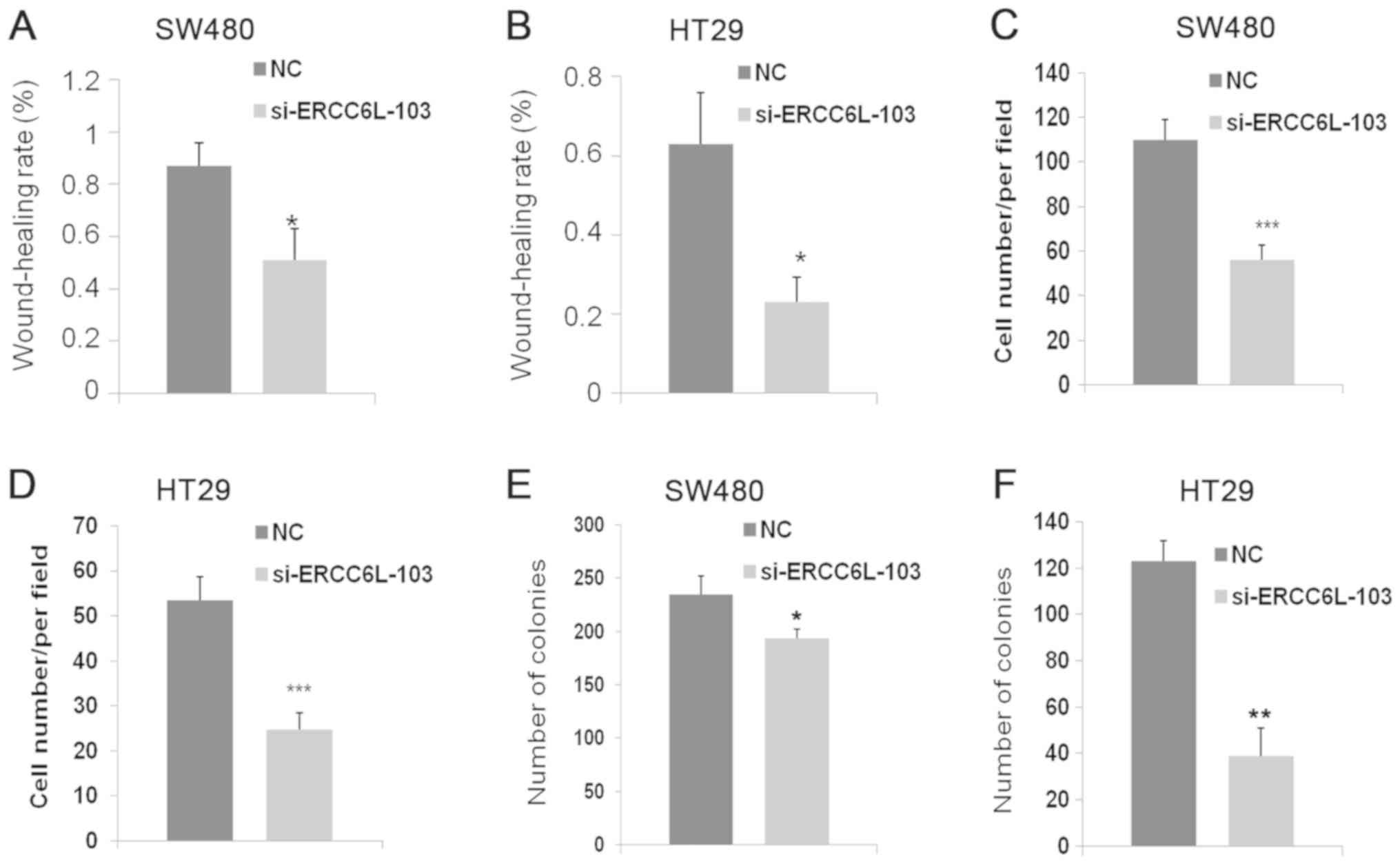
Wound healing, Transwell, colony formation and
proliferation assays were conducted to evaluate the effects of
ERCC6L knockdown (Figs. 3 and
4). Wound healing assays revealed
that knockdown of ERCC6L inhibited the migration of SW480 and HT29
cells compared with that in the respective control group
(P<0.05; Figs. 3A and B, 4A and B). Transwell assays were performed
to evaluate the effect of ERCC6L on the invasion capability of CRC
cells. Knockdown of ERCC6L in SW480 and HT29 CRC cell lines
significantly decreased the number of invading cells compared with
the respective cells treated with NC (P<0.001; Figs. 3C, 4C and
D). Clonogenic survival assays revealed a significant reduction
in the colony formation efficiency of cells transfected with
ERCC6L-siRNA compared with the control group (P<0.01; Figs. 3D, 4E and
F). To detect the effect of ERCC6L knockdown on CRC cell
proliferation, SW480 and HT29 cells were treated with ERCC6L-siRNA
or a control siRNA for 72 h, and cell viability was determined via
an MTT assay. It was demonstrated that ERCC6L knockdown
significantly inhibits CRC cell proliferation (Fig. 3E and F). These results indicate that
ERCC6L promotes the migration, invasion and proliferation
capability of CRC cells.
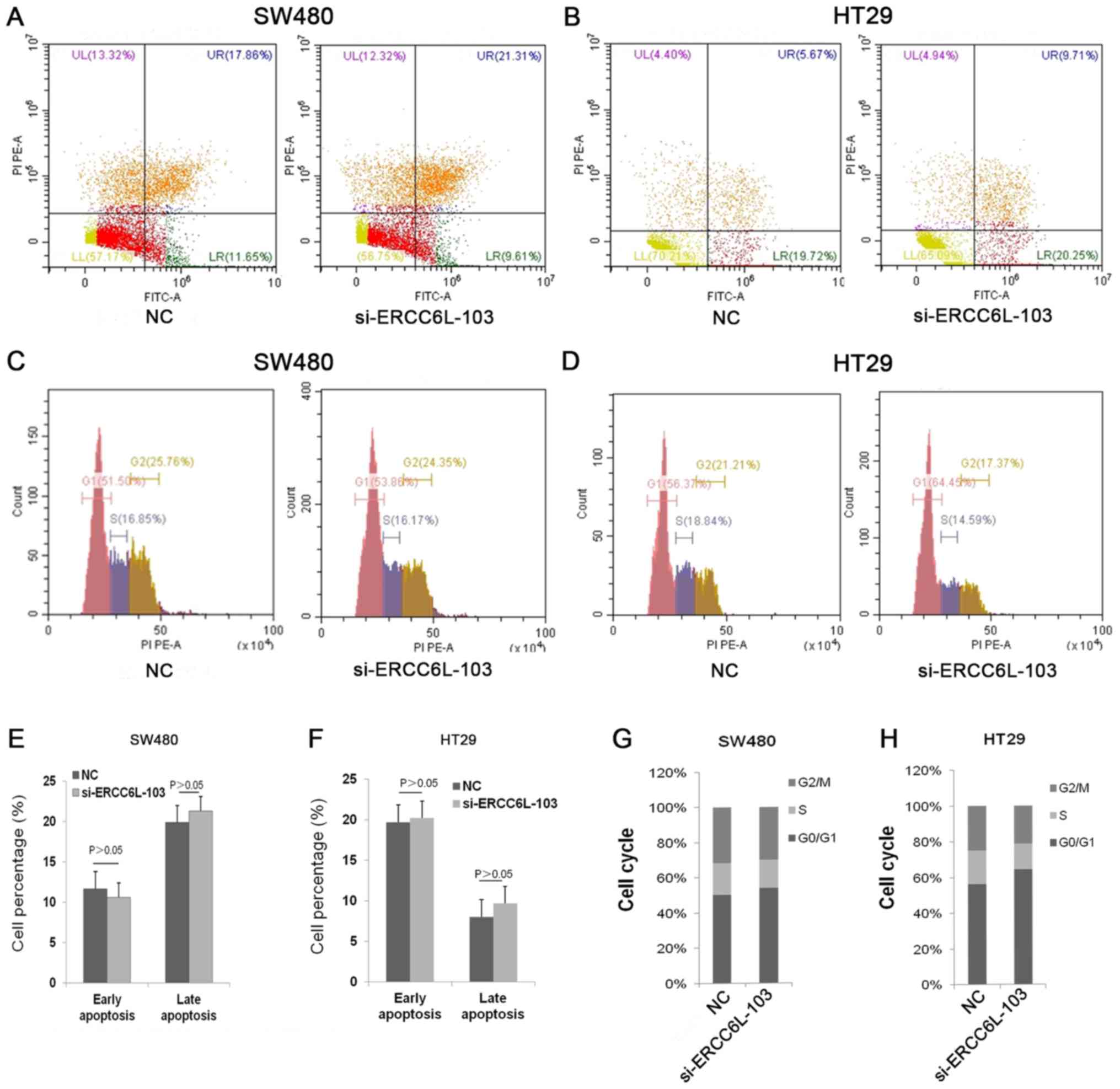
ERCC6L silencing induces cell cycle
arrest at the G0/G1 phase in CRC cells
To evaluate the effect of ERCC6L on cell apoptosis,
ERCC6L was knocked down in SW480 and HT29 CRC cells, and apoptosis
was then examined via flow cytometry (Fig. 5). The results revealed that cells
treated with si-ERCC6L-103 and a siRNA control exhibited similar
rates of apoptosis (Fig. 5A, B, E and
F). Next, the effect of ERCC6L inhibition on the cell cycle in
SW480 and HT29 cells was examined. The results demonstrated that
ERCC6L silencing induced cell cycle arrest at the G0/G1 phase
compared with the control group (Fig.
5C, D, G and H). In summary, these results indicate that ERCC6L
silencing inhibits cell proliferation by blocking cells at the
G0/G1 phase.
Discussion
CRC is a complex, multi-step disease with a variety
of genetic alterations (15,16). A number of proteins have been
described as tumor biomarkers that aid the identification of CRC
tumors, including A-kinase anchoring protein 4 (17), X-box binding protein 1 (18) and solute carrier family 38 member 1
(19). However, several different
CRC subtypes have been identified, including subtypes based on DNA
mismatch repair status, oncogene genotypes, or the consensus
molecular subtypes derived from transcriptional profiling;
therefore, one biomarker is unlikely to identify all types of
colorectal tumor (20). Thus, it is
essential to identify therapeutic targets and reliable prognostic
biomarkers to improve the diagnosis and treatment of CRC.
ERCC6, a member of the SWI/SNF-associated ATPase
family, has been associated with numerous diseases (21,22). For
example, it has been reported that Cockayne syndrome and
cerebro-oculofacio-skeletal syndrome are caused by ERCC6 gene
mutations (23–25). In addition, ERCC6 genetic
polymorphism may provide early diagnosis, precise therapy, and
improved clinical prognosis and quality of life in patients with
bladder cancer (26). ERCC6L,
another development-associated member of the SNF2 family, has a
different function and role compared with ERCC6. A number of
studies have examined the effects of disrupting ERCC6L function on
chromosome structure and stability. For example, ERCC6L knockdown
in human cancer cells was demonstrated to result in the loss of
PLK1 from the chromosome arm and an increase in the occurrence of
chromosomal abnormalities, including chromatin bridges and
micronuclei (27–30). Furthermore, studies have revealed
that PLK1 serves an important role in regulating biological
processes, including apoptosis, the cell cycle and cell
proliferation (28,31–33).
These findings indicate potential mechanisms by which ERCC6L
regulates cellular biological functions.
The current study first used RT-qPCR and western
blot analysis to detect the expression levels of ERCC6L mRNA and
protein in CRC tissues and cell lines. In addition, IHC was used to
detect the expression of ERCC6L protein in CRC tissues. The results
revealed that the expression of ERCC6L was significantly increased
in CRC compared with normal cells and tissues. Clinicopathological
data confirmed that a high ERCC6L expression level was associated
with increased tumor size. These findings suggest that ERCC6L
overexpression may serve an important role in the development of
CRC.
To investigate the role of ERCC6L in the development
and progression of CRC, the expression of ERCC6L was inhibited
using siRNA. It was identified that knockdown of ERCC6L led to the
attenuated cell growth, cell cycle arrest, and decreased migration
and invasion capability of CRC cells. However, the ability to
migrate, invade and clone was higher in SW480 cells compared with
HT29 cells, and a significant cell cycle arrest at G0/G1 was only
observed in HT29 cells. These findings may result from differences
in the KRAS and BRAF genotypes of these cell lines (34,35).
KRAS and BRAF have been demonstrated to be signal transduction
molecules that serve a role in the mitogen-activated protein kinase
pathway (34,35). KRAS and BRAF gene mutations are
understood to stimulate tumor growth and are present in various
organs, including the colon (36),
pancreas (37) and lungs (38). It may be beneficial to investigate
the potential association between KRAS and BRAF gene mutations and
ERCC6L in different patients with CRC and cell lines, as this may
also help to elucidate the underlying mechanism of ERCC6L in
tumors.
In conclusion, the present study identified that
ERCC6L is upregulated in CRC tissues and cells, and that ERCC6L
overexpression is closely associated with tumor size. It also
demonstrated that ERCC6L promotes CRC cell growth and proliferation
in vitro, possibly via acceleration of the cell cycle. In
summary, to the best of our knowledge, the current study provided
the first evidence that ERCC6L serves a role in the development of
CRC, which indicates that ERCC6L may serve as a novel biomarker and
therapeutic target for CRC. Future studies should evaluate the
mechanisms of ERCC6L at different stages of the cell cycle and its
regulatory signaling pathways.
Acknowledgements
This abstract was presented at the 8th Shanghai
International Conference of Gastroenterology in Shanghai in August
2018, and was published as Abstract ID: 1 in J Dig Dis 19, 696–718,
2018. The abstract was also presented at the 26th United European
Gastroenterology (UEG) Week, October 20–24, 2018 in Vienna,
Austria.
Funding
The present study was supported by the Applied Basic
Research Programs of the Wuhan Science and Technology Department
(grant no. 2015061701011642) and the Wuhan University Graduate
Student Exchange Program.
Availability of data and materials
The datasets used and/or analysed during the present
study are available fro. the corresponding author on reasonable
request
Authors' contributions
All authors were responsible for the conception and
design of the present study. FW and ML were responsible for the
provision of the study materials. YX and JY were responsible for
the collection and assembly of the data. YX, JY, FW, ML, XQ, YL and
JQ performed the data analysis and interpretation. YX, JY, XQ, and
JQ contributed in writing the manuscript. YX, JY, FW, ML, XQ, YL
and JQ read and gave the final approval of the manuscript.
Ethical approval and consent to
participate
The study was approved by the Ethics Committee of
Zhongnan Hospital of Wuhan University. The patient included in the
case provided consent for his or her data to be used in this
publication.
Patient consent for publication
All patients included in this study at the time of
data collection provided consent for their data to be used in this
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Miller K and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maurel J and Postigo A: Prognostic and
predictive biomarkers in colorectal cancer. From the preclinical
setting to clinical practice. Curr Cancer Drug Targets. 15:703–715.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schreuders EH, Ruco A, Rabeneck L, Schoen
RE, Sung JJ, Young GP and Kuipers EJ: Colorectal cancer screening:
A global overview of existing programmes. Gut. 64:1637–1649. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dickinson BT, Kisiel J, Ahlquist DA and
Grady WM: Molecular markers for colorectal cancer screening. Gut.
64:1485–1494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu Y, Chen X and Li Y: Ercc6l, a gene of
SNF2 family, may play a role in the teratogenic action of alcohol.
Toxicol Lett. 157:233–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Z, Zhang G and Li W: Elevated
expression of ERCC6 confers resistance to 5-fluorouracil and is
associated with poor patient survival in colorectal cancer. DNA
Cell Biol. 36:781–786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nielsen CF, Huttner D, Bizard AH, Hirano
S, Li TN, Palmai-Pallag T, Bjerregaard VA, Liu Y, Nigg EA, Wang LH
and Hickson ID: PICH promotes sister chromatid disjunction and
co-operates with topoisomerase II in mitosis. Nat Commun.
6:89622015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baumann C, Körner R, Hofmann K and Nigg
EA: PICH, a centromere-associated SNF2 family ATPase, is regulated
by Plk1 and required for the spindle checkpoint. Cell. 128:101–114.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen XG, Li Y, Zang MX, Pei XR, Xu YJ and
Gao LF: cDNA cloning and expression analysis of mouse gene encoding
the protein Ercc61 which is a novel member of SNF2 family. Prog
Biochem Biophys. 31:443–448. 2004.
|
|
12
|
Santamaria A, Neef R, Eberspächer U, Eis
K, Husemann M, Mumberg D, Prechtl S, Schulze V, Siemeister G,
Wortmann L, et al: Use of the novel Plk1 inhibitor
ZK-thiazolidinone to elucidate functions of Plk1 in early and late
stages of mitosis. Mol Biol Cell. 18:4024–4036. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pu SY, Yu Q, Wu H, Jiang JJ, Chen XQ, He
YH and Kong QP: ERCC6L, a DNA helicase, is involved in cell
proliferation and associated with survival and progress in breast
and kidney cancers. Oncotarget. 8:42116–42124. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jagadish N, Parashar D, Gupta N, Agarwal
S, Purohit S, Kumar V, Sharma A, Fatima R, Topno AP, Shaha C and
Suri A: A-kinase anchor protein 4 (AKAP4) a promising therapeutic
target of colorectal cancer. J Exp Clin Cancer Res. 34:1422015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mhaidat NM, Alzoubi KH and Abushbak A:
X-box binding protein 1 (XBP-1) enhances colorectal cancer cell
invasion. J Chemother. 27:167–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou FF, Xie W, Chen SQ, Wang XK, Liu Q,
Pan XK, Su F and Feng MH: SLC38A1 promotes proliferation and
migration of human colorectal cancer cells. J Huazhong Univ Sci
Technolog Med Sci. 37:30–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gil-Raga M, Jantus-Lewintre E, Gallach S,
Giner-Bosch V, Frangi-Caregnato A, Safont-Aguilera MJ,
Garde-Noguera J, Zorraquino-Pina E, García-Martínez M and
Camps-Herrero C: Molecular subtypes in early colorectal cancer
associated with clinical features and patient prognosis. Clin
Transl Oncol. 20:1422–1429. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Deng N, Xu Q, Sun L, Tu H, Wang Z,
Xing C and Yuan Y: Polymorphisms of multiple genes involved in NER
pathway predict prognosis of gastric cancer. Oncotarget.
7:48130–48142. 2016.PubMed/NCBI
|
|
22
|
Xu Q, Liu JW, He CY, Sun LP, Gong YH, Jing
JJ, Xing CZ and Yuan Y: The interaction effects of pri-let-7a-1
rs10739971 with PGC and ERCC6 gene polymorphisms in gastric cancer
and atrophic gastritis. PLoS One. 9:e892032014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taghdiri M, Dastsooz H, Fardaei M,
Mohammadi S, Farazi Fard MA and Faghihi MA: A novel mutation in
ERCC8 gene causing cockayne syndrome. Front Pediatr. 5:1692017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie H, Li X, Peng J, Chen Q, Gao Z, Song
X, Li W, Xiao J, Li C, Zhang T, et al: A complex intragenic
rearrangement of ERCC8 in Chinese siblings with Cockayne syndrome.
Sci Rep. 7:442712017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He C, Sun M, Wang G, Yang Y, Yao L and Wu
Y: Two novel mutations in ERCC6 cause Cockayne syndrome B in a
Chinese family. Mol Med Rep. 15:3957–3962. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Truta A, Popon TA, Saraci G, Ghervan L and
Pop IV: Novel non invasive diagnostic strategies in bladder cancer.
Clujul Med. 89:187–192. 2016.PubMed/NCBI
|
|
27
|
Kurasawa Y and Yu-Lee LY: PICH and
cotargeted Plk1 coordinately maintain prometaphase chromosome arm
architecture. Mol Biol Cell. 21:1188–1199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leng M, Besusso D, Jung SY, Wang Y and Qin
J: Targeting Plk1 to chromosome arms and regulating chromosome
compaction by the PICH ATPase. Cell Cycle. 7:1480–1489. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ke Y, Huh JW, Warrington R, Li B, Wu N,
Leng M, Zhang J, Ball HL, Li B and Yu H: PICH and BLM limit histone
association with anaphase centromeric DNA threads and promote their
resolution. EMBO J. 30:3309–3321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kaulich M, Cubizolles F and Nigg EA: On
the regulation, function, and localization of the DNA-dependent
ATPase PICH. Chromosoma. 121:395–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma X, Wang L, Huang, Li Y, Yang D, Li T,
Li F, Sun L, Wei H, He K, et al: Polo-like kinase 1 coordinates
biosynthesis during cell cycle progression by directly activating
pentose phosphate pathway. Nat Commun. 8:15062017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Helmke C, Becker S and Strebhardt K: The
role of Plk3 in oncogenesis. Oncogene. 35:135–147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yen TJ: Polo delivers a PICH to the
kinetochore. Cell. 128:20–21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baines AT, Xu D and Der CJ: Inhibition of
Ras for cancer treatment: The search continues. Future Med Chem.
3:1787–1808. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sclafani F, Gullo G, Sheahan K and Crown
J: BRAF mutations in melanoma and colorectal cancer: A single
oncogenic mutation with different tumour phenotypes and clinical
implications. Crit Rev Oncol Hematol. 87:55–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang J, Zheng J, Yang Y, Lu J, Gao J, Lu
T, Sun J, Jiang H, Zhu Y, Zheng Y, et al: Molecular spectrum of
KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer
patients: analysis of 1,110 cases. Sci Rep. 5:186782015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McAllister F, Bailey JM, Alsina J, Nirschl
CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H,
et al: Oncogenic Kras activates a hematopoietic-to-epithelial IL-17
signaling axis in preinvasive pancreatic neoplasia. Cancer Cell.
25:621–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nguyen-Ngoc T, Bouchaab H, Adjei AA and
Peters S: BRAF alterations as therapeutic targets in non-small-cell
lung cancer. J Thorac Oncol. 10:1396–1403. 2015. View Article : Google Scholar : PubMed/NCBI
|