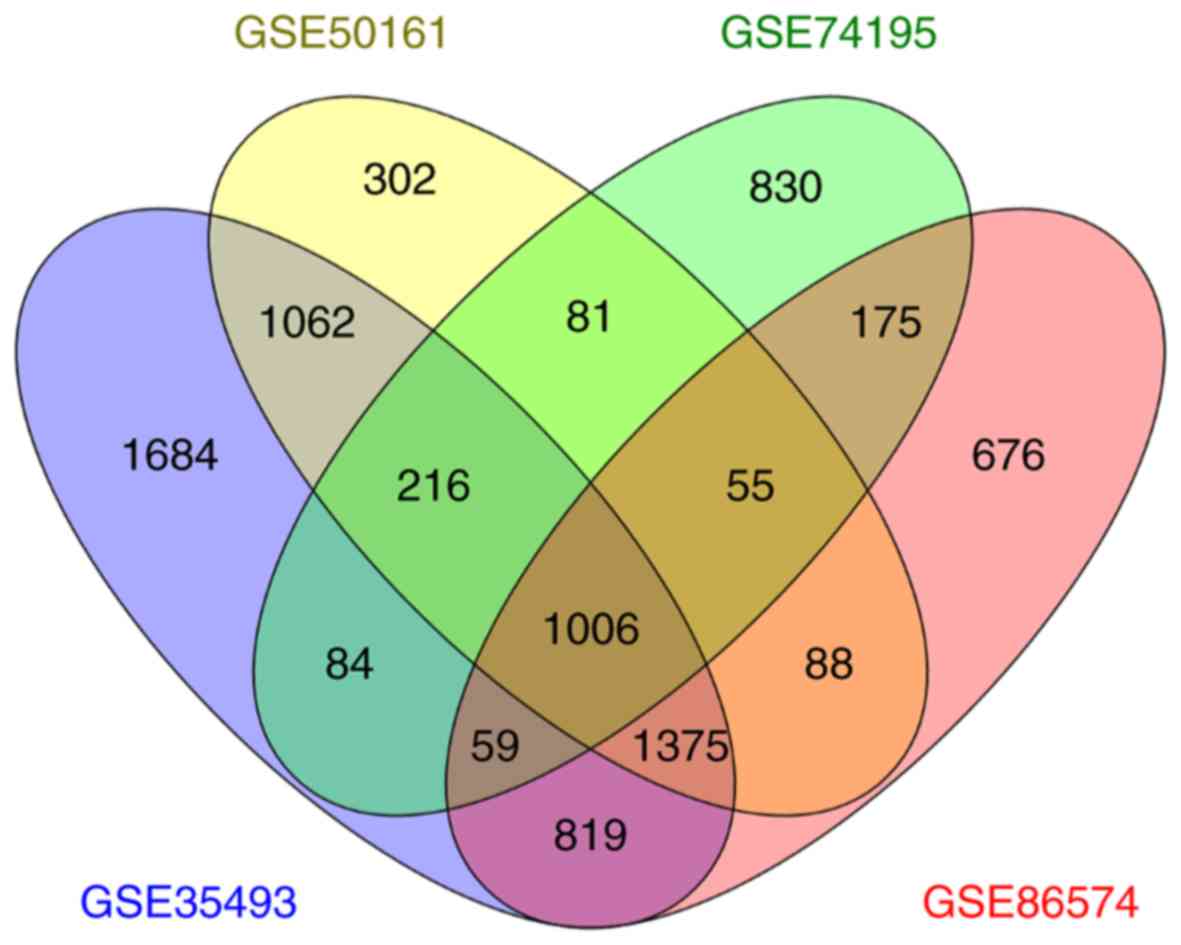
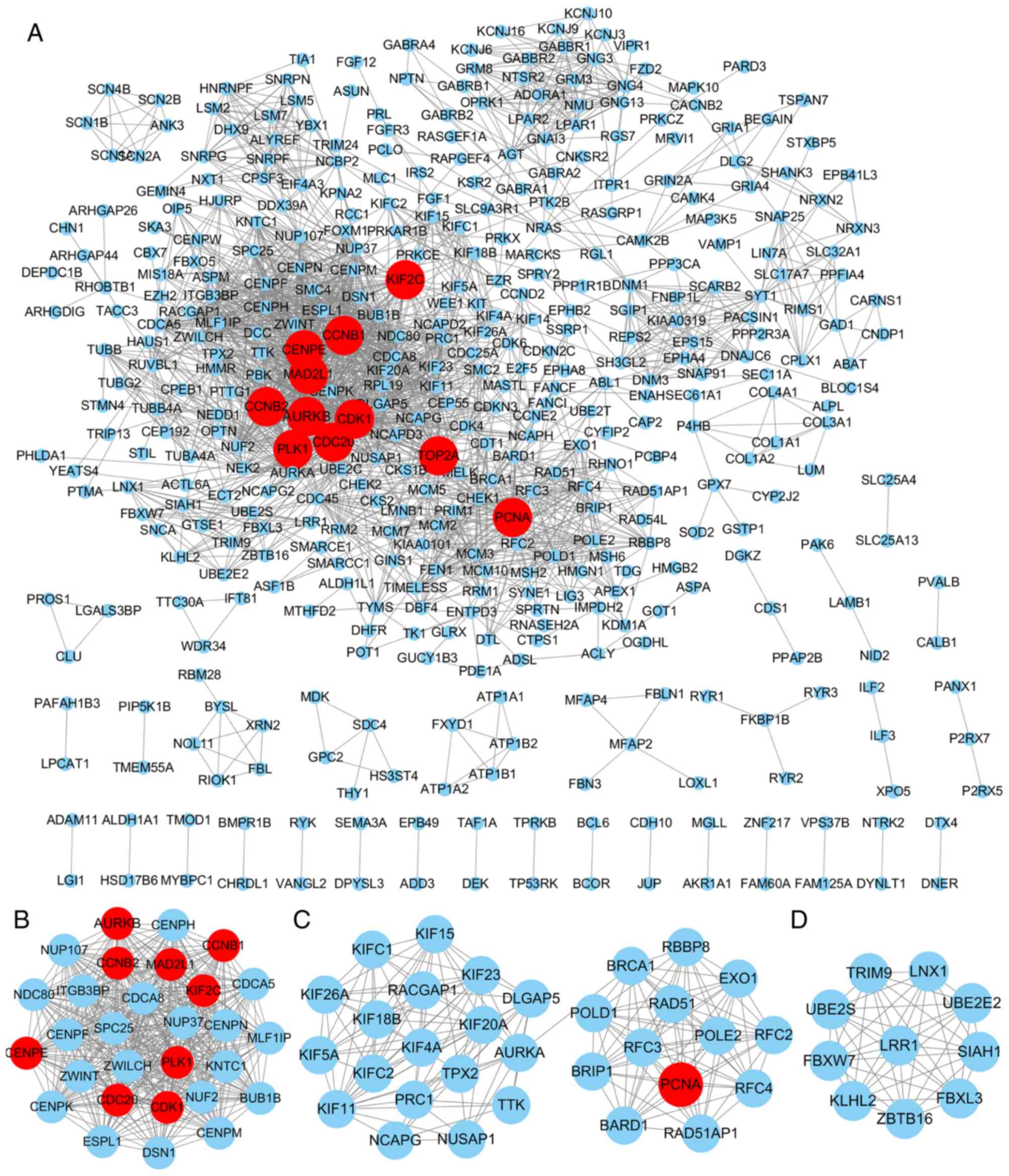
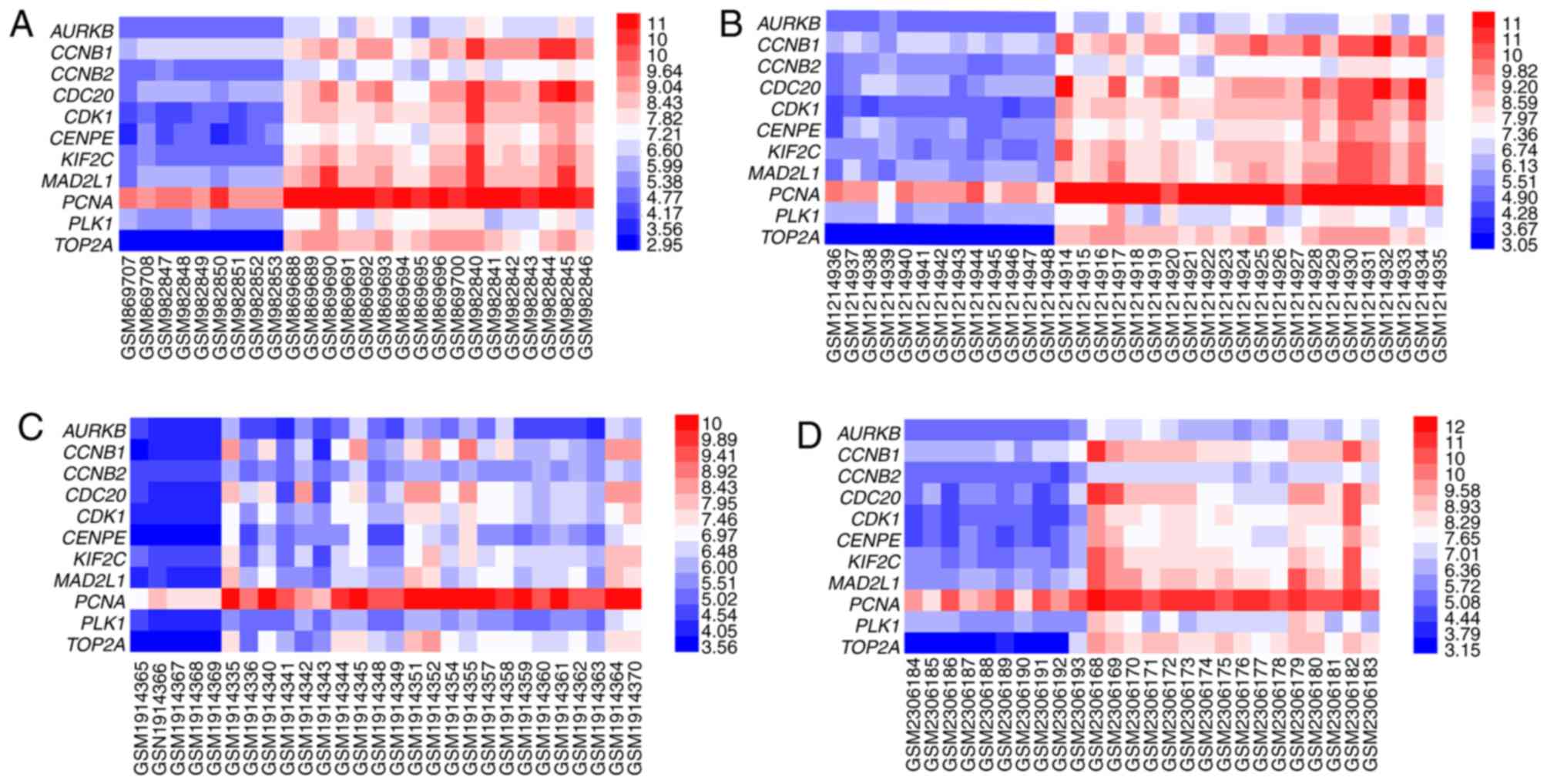
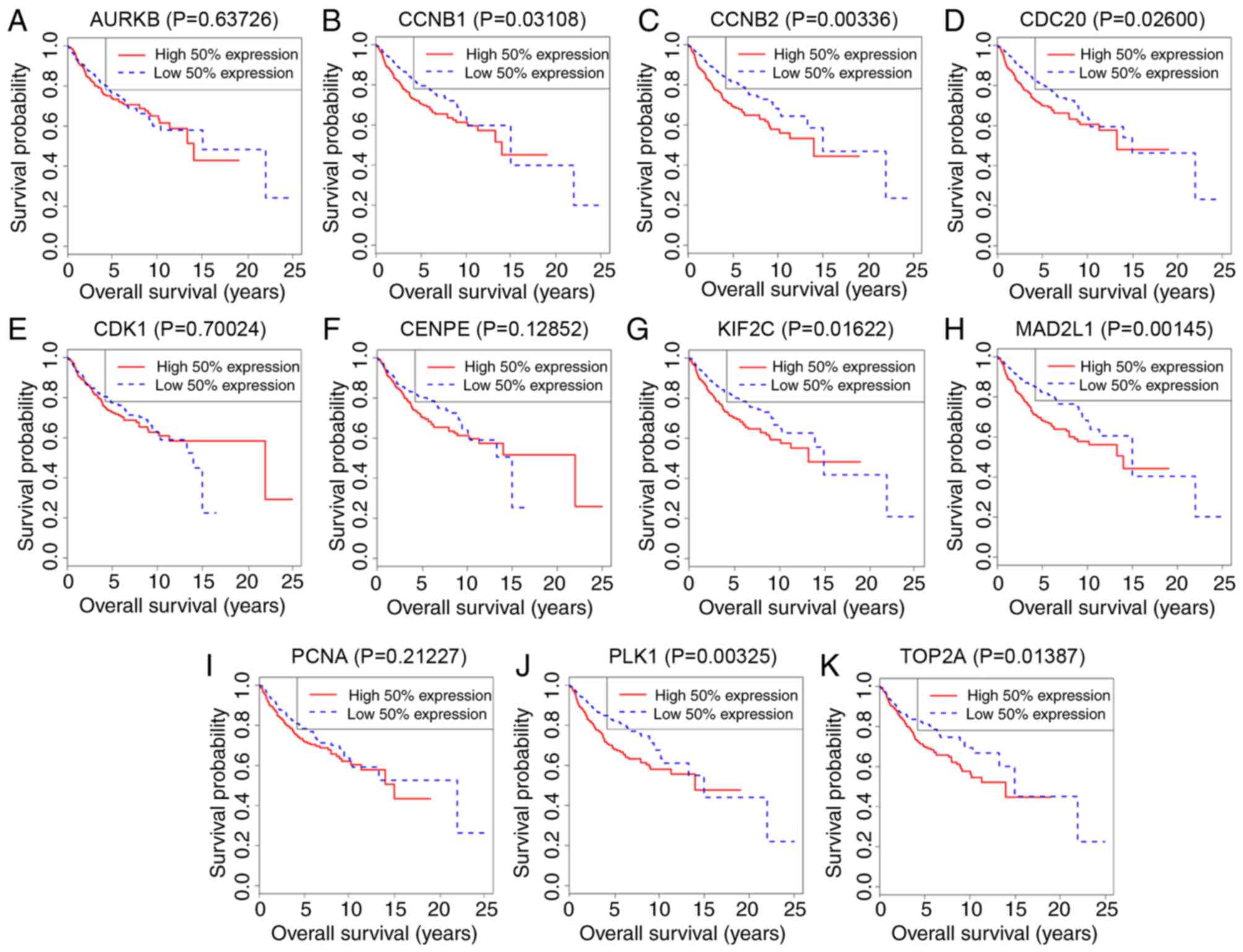
|
1
|
Gilbertson RJ: Medulloblastoma: Signalling
a change in treatment. Lancet Oncol. 5:209–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartlett F, Kortmann R and Saran F:
Medulloblastoma. Clin Oncol (R Coll Radiol). 25:36–45. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gerber NU, Mynarek M, von Hoff K,
Friedrich C, Resch A and Rutkowski S: Recent developments and
current concepts in medulloblastoma. Cancer Treat Rev. 40:356–365.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quinlan A and Rizzolo D: Understanding
medulloblastoma. JAAPA. 30:30–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sengupta S, Pomeranz Krummel D and Pomeroy
S: The evolution of medulloblastoma therapy to personalized
medicine. F1000 Res. 6:4902017. View Article : Google Scholar
|
|
7
|
Archer TC, Mahoney EL and Pomeroy SL:
Medulloblastoma: Molecular classification-based personal
therapeutics. Neurotherapeutics. 14:265–273. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shou Y, Robinson DM, Amakye DD, Rose KL,
Cho YJ, Ligon KL, Sharp T, Haider AS, Bandaru R, Ando Y, et al: A
five-gene hedgehog signature developed as a patient preselection
tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer
Res. 21:585–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khatua S and Zaky W: The biologic era of
childhood medulloblastoma and clues to novel therapies. Future
Oncol. 10:637–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harris PS, Venkataraman S, Alimova I,
Birks DK, Balakrishnan I, Cristiano B, Donson AM, Dubuc AM, Taylor
MD, Foreman NK, et al: Integrated genomic analysis identifies the
mitotic checkpoint kinase WEE1 as a novel therapeutic target in
medulloblastoma. Mol Cancer. 13:722014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Birks DK, Donson AM, Patel PR, Sufit A,
Algar EM, Dunham C, Kleinschmidt-DeMasters BK, Handler MH, Vibhakar
R and Foreman NK: Pediatric rhabdoid tumors of kidney and brain
show many differences in gene expression but share dysregulation of
cell cycle and epigenetic effector genes. Pediatr Blood Cancer.
60:1095–1102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Griesinger AM, Birks DK, Donson AM, Amani
V, Hoffman LM, Waziri A, Wang M, Handler MH and Foreman NK:
Characterization of distinct immunophenotypes across pediatric
brain tumor types. J Immunol. 191:4880–4888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Bont JM, Kros JM, Passier MM,
Reddingius RE, Sillevis Smitt PA, Luider TM, den Boer ML and
Pieters R: Differential expression and prognostic significance of
SOX genes in pediatric medulloblastoma and ependymoma identified by
microarray analysis. Neuro Oncol. 10:648–660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jensen LJ, Kuhn M, Stark M, Chaffron S,
Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et
al: STRING 8-a global view on proteins and their functional
interactions in 630 organisms. Nucleic Acids Res. 37:D412–416.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 4 (Suppl 8):S112014. View Article : Google Scholar
|
|
20
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brandes AA, Bartolotti M, Marucci G,
Ghimenton C, Agati R, Fioravanti A, Mascarin M, Volpin L, Ammannati
F, Masotto B, et al: New perspectives in the treatment of adult
medulloblastoma in the era of molecular oncology. Crit Rev Oncol
Hematol. 94:348–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diaz RJ, Golbourn B, Faria C, Picard D,
Shih D, Raynaud D, Leadly M, MacKenzie D, Bryant M, Bebenek M, et
al: Mechanism of action and therapeutic efficacy of Aurora kinase B
inhibition in MYC overexpressing medulloblastoma. Oncotarget.
6:3359–3374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alimova I, Ng J, Harris P, Birks D, Donson
A, Taylor MD, Foreman NK, Venkataraman S and Vibhakar R: MPS1
kinase as a potential therapeutic target in medulloblastoma. Oncol
Rep. 36:2633–2640. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ringer L, Sirajuddin P, Heckler M, Ghosh
A, Suprynowicz F, Yenugonda VM, Brown ML, Toretsky JA, Uren A, Lee
Y, et al: VMY-1-103 is a novel CDK inhibitor that disrupts
chromosome organization and delays metaphase progression in
medulloblastoma cells. Cancer Biol Ther. 12:818–826. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bish R and Vogel C: RNA binding
protein-mediated post-transcriptional gene regulation in
medulloblastoma. Mol Cells. 37:357–364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Milla LA, Arros A, Espinoza N, Remke M,
Kool M, Taylor MD, Pfister SM, Wainwright BJ and Palma V: Neogenin1
is a sonic hedgehog target in medulloblastoma and is necessary for
cell cycle progression. Int J Cancer. 134:21–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ellert-Miklaszewska A, Grajkowska W,
Gabrusiewicz K, Kaminska B and Konarska L: Distinctive pattern of
cannabinoid receptor type II (CB2) expression in adult and
pediatric brain tumors. Brain Res. 1137:161–169. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sepsova V, Krusek J, Zdarova Karasova J,
Zemek F, Musilek K, Kuca K and Soukup O: The interaction of
quaternary reversible acetylcholinesterase inhibitors with the
nicotinic receptor. Physiol Res. 63:771–777. 2014.PubMed/NCBI
|
|
29
|
Sardi I, la Marca G, Giovannini MG,
Malvagia S, Guerrini R, Genitori L, Massimino M and Arico M:
Detection of doxorubicin hydrochloride accumulation in the rat
brain after morphine treatment by mass spectrometry. Cancer
Chemother Pharmacol. 67:1333–1340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
de Haas T, Hasselt N, Troost D, Caron H,
Popovic M, Zadravec-Zaletel L, Grajkowska W, Perek M, Osterheld MC,
Ellison D, et al: Molecular risk stratification of medulloblastoma
patients based on immunohistochemical analysis of MYC, LDHB, and
CCNB1 expression. Clin Cancer Res. 14:4154–4160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pezuk JA, Brassesco MS, Ramos PMM,
Scrideli CA and Tone LG: Polo-like kinase 1 pharmacological
inhibition as monotherapy or in combination: Comparative effects of
polo-like kinase 1 inhibition in medulloblastoma cells. Anticancer
Agents Med Chem. 17:1278–1291. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Triscott J, Lee C, Foster C, Manoranjan B,
Pambid MR, Berns R, Fotovati A, Venugopal C, O'Halloran K,
Narendran A, et al: Personalizing the treatment of pediatric
medulloblastoma: Polo-like kinase 1 as a molecular target in
high-risk children. Cancer Res. 73:6734–6744. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uesaka T, Shono T, Kuga D, Suzuki SO,
Niiro H, Miyamoto K, Matsumoto K, Mizoguchi M, Ohta M, Iwaki T and
Sasaki T: Enhanced expression of DNA topoisomerase II genes in
human medulloblastoma and its possible association with etoposide
sensitivity. J Neurooncol. 84:119–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kayaselcuk F, Zorludemir S, Gumurduhu D,
Zeren H and Erman T: PCNA and Ki-67 in central nervous system
tumors: Correlation with the histological type and grade. J
Neurooncol. 57:115–121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie Q, Wu Q, Mack SC, Yang K, Kim L,
Hubert CG, Flavahan WA, Chu C, Bao S and Rich JN: CDC20 maintains
tumor initiating cells. Oncotarget. 6:13241–13254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang L, Zhang J, Wan L, Zhou X, Wang Z and
Wei W: Targeting Cdc20 as a novel cancer therapeutic strategy.
Pharmacol Ther. 151:141–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji P, Zhou X, Liu Q, Fuller GN, Phillips
LM and Zhang W: Driver or passenger effects of augmented c-Myc and
Cdc20 in gliomagenesis. Oncotarget. 7:23521–23529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bie L, Zhao G, Cheng P, Rondeau G,
Porwollik S, Ju Y, Xia XQ and McClelland M: The accuracy of
survival time prediction for patients with glioma is improved by
measuring mitotic spindle checkpoint gene expression. PLoS One.
6:e256312011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ko YH, Roh JH, Son YI, Chung MK, Jang JY,
Byun H, Baek CH and Jeong HS: Expression of mitotic checkpoint
proteins BUB1B and MAD2L1 in salivary duct carcinomas. J Oral
Pathol Med. 39:349–355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Krapf G, Kaindl U, Kilbey A, Fuka G,
Inthal A, Joas R, Mann G, Neil JC, Haas OA and Panzer-Grumayer ER:
ETV6/RUNX1 abrogates mitotic checkpoint function and targets its
key player MAD2L1. Oncogene. 29:3307–3312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Z, Katsaros D, Shen Y, Fu Y, Canuto
EM, Benedetto C, Lu L, Chu WM, Risch HA and Yu H: Biological and
clinical significance of MAD2L1 and BUB1, genes frequently
appearing in expression signatures for breast cancer prognosis.
PLoS One. 10:e01362462015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Manni I, Mazzaro G, Gurtner A, Mantovani
R, Haugwitz U, Krause K, Engeland K, Sacchi A, Soddu S and Piaggio
G: NF-Y mediates the transcriptional inhibition of the cyclin B1,
cyclin B2, and cdc25C promoters upon induced G2 arrest. J Biol
Chem. 276:5570–5576. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Halvorson KG, Barton KL, Schroeder K,
Misuraca KL, Hoeman C, Chung A, Crabtree DM, Cordero FJ, Singh R,
Spasojevic I, et al: A high-throughput in vitro drug screen in a
genetically engineered mouse model of diffuse intrinsic pontine
glioma identifies BMS-754807 as a promising therapeutic agent. PLoS
One. 10:e01189262015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu Z, Sun Y, She X, Wang Z, Chen S, Deng
Z, Zhang Y, Liu Q, Liu Q, Zhao C, et al: SIX3, a tumor suppressor,
inhibits astrocytoma tumorigenesis by transcriptional repression of
AURKA/B. J Hematol Oncol. 10:1152017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liang ML, Hsieh TH, Ng KH, Tsai YN, Tsai
CF, Chao ME, Liu DJ, Chu SS, Chen W, Liu YR, et al: Downregulation
of miR-137 and miR-6500-3p promotes cell proliferation in pediatric
high-grade gliomas. Oncotarget. 7:19723–19737. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bie L, Zhao G, Wang YP and Zhang B:
Kinesin family member 2C (KIF2C/MCAK) is a novel marker for
prognosis in human gliomas. Clin Neurol Neurosurg. 114:356–360.
2012. View Article : Google Scholar : PubMed/NCBI
|