Introduction
Bisphosphonates (BPs) are C-substituted
pyrophosphate analogs used to treat and prevent osteoporosis,
cancer and bone metastasis (1,2). BPs are
divided into two classes based on differences in the structure of
the R2 side chain, this includes non-nitrogen-containing BPs
(non-N-BPs), including etidronate and clodronate, and
nitrogen-containing BPs (N-BPs), including alendronate, risedronate
and pamidronate (3). Non-N-BPs are
less potent antiresorptive agents compared with N-BPs; however,
they exhibit other properties, including anticancer activities
(4). These anticancer activities
include inhibition of proliferation, induction of apoptosis,
reduction of migration and invasion, and inhibition of angiogenesis
(1,5). BPs have demonstrated in vitro
anticancer activity against various cancer cell lines, including
breast (6), prostate (7), lung (8),
endometrium (9) and colon cancer
(10). However, further studies are
required to investigate the specificity of different BPs for
various cancer cell types and to elucidate the precise mechanisms
of the anticancer activity.
It is important to investigate direct anticancer
effects of BPs on cancer cell death, apoptosis and migration. In
the past decade, BPs have been identified to be potent inhibitors
of important enzymes in the mevalonate pathway, including farnesyl
pyrophosphate synthase (FPPS) (11).
FPPS is a key enzyme that catalyzes the reaction of isopentenyl
pyrophosphate (IPP) and dimethylallyl pyrophosphate to generate
farnesyl pyrophosphate (FPP) (5).
This results in an increase in geranylgeranyl pyrophosphate (GGPP),
which plays an important role in the production of small GTPases,
including Ras, Rac, Rho and cell division control protein 42
homolog (CDC42) (11), and can
subsequently control cancer cell proliferation. BPs strongly
inhibit FPPS, which reduces the levels of FPP and GGPP and
expression of small GTPases (5).
Furthermore, BPs cause an accumulation of IPP that is converted to
the cytotoxic adenosine 5′-triphoshpate analogue ApppI, which
induces cancer cell death (12).
It has been suggested that BPs stimulate cancer cell
death and apoptosis by inhibiting the mevalonate pathway and
reducing the number of small GTPases (13,14).
This inhibits integrin-mediated cancer cell adhesion to the bone
(15), increase Rap1 unprenylation,
reduce the growth of mesothelioma cells (16) and deactivate Rho protein, which leads
to inhibition of cancer cell migration (17). Small GTPases affect cancer cell cycle
progression and/or growth by modulating the transcription of
certain genes, including cyclin D, which stimulates the G1 to S
transition and cancer cell proliferation (18,19). The
transcription of cyclin D1 is controlled by a number of
transcription factors, including activator protein-1 and nuclear
factor-κB, the activity of which are regulated by small GTPases
(18,19). Accordingly, direct inhibition of
small GTPase activity induces cell cycle arrest and apoptosis in
cancer cells by leading to decreased cell function and eventually
programmed cell death (20).
Zoledronic acid exhibits pronounced antiproliferative and
proapoptotic effects in breast cancer MDA-MB-231 cells by
increasing tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) production and enhancing the TRAIL/osteoprotegerin (OPG)
ratio, which affects cell integrity and survival (21).
The present study investigated the anticancer
properties of three second-generation BPs, alendronate, risedronate
and pamidronate, in MCF-7 human breast cancer cells using
sulforhodamine B (SRB), colony formation and flow cytometry assays.
The mechanism of BP-induced apoptosis was also explored by
analyzing expression levels of apoptosis-associated proteins,
reactive oxygen species (ROS) production, caspase-3 activity and
mitochondrial function. Finally, effects of BPs on cancer cell
migration were determined using a wound healing assay, gelatin
zymography and by analyzing expression levels of genes associated
with migration. The current study provides a valuable overview of
the cytotoxic, apoptotic and antimigratory effects of different BPs
on MCF-7 breast cancer cells.
Materials and methods
Chemicals and reagents
Alendronate, risedronate, pamidronate, protease
inhibitor cocktail, dihydroethidium (DHE), radioimmunoprecipitation
assay (RIPA) lysis buffer, Caspase-3 Fluorometric assay kit, GGPP,
doxorubicin and SRB were obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). Primary antibodies against cyclin D1 (cat.
no. 2992), p21 (cat. no. 2947), cytochrome c (cat. no. 4272),
caspase-3 (cat. no. 9662) and the internal control β-actin (cat.
no. 4967), and the anti-rabbit IgG horseradish peroxidase
(HRP)-conjugated antibody (cat. no. 7074) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). All cell culture
reagents were purchased from Gibco; Thermo Fisher Scientific, Inc.
(Waltham, MA, USA).
Cell lines, culture condition
The human breast cancer cell line MCF-7 was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
maintained according to the manufacturer's protocol. The MCF-7 cell
line was cultured in complete Dulbecco's modified Eagle's medium
(DMEM) consisting of 10% fetal bovine serum, 100 U/ml penicillin G
and 100 mg/ml streptomycin, and maintained under an atmosphere of
5% CO2 at 37°C. The cells were subcultured every 3 days
or after cells reached 70–80% confluence using 0.25% trypsin-EDTA.
Cells were plated in new complete DMEM until required for future
experiments.
Cell viability assay
A SRB assay was used to measure the effect of the
BPs alendronate, risedronate and pamidronate on the viability of
MCF-7 cells. The assay was performed as previously described
(22). In brief, MCF-7 cells were
seeded into 96-well culture plates at 1×104 cells/well
and allowed to attach for 24 h. Subsequently, cultured cells were
treated with various concentrations of BPs (0–1,000 µM alendronate
and risedronate, 0–250 µM pamidronate) for 24 and 48 h at 37°C.
Cells were then washed, fixed with 10% ice-cold trichloroacetic
acid for 30 min at 4°C, stained with 0.4% SRB for 30 min at room
temperature, washed with 1% acetic acid to remove unbound dye and
the remaining protein-bound dye was then solubilized with 10 mM
unbuffered Tris. The absorbance was measured at 540 nm using a
microplate reader. Cell viability was expressed in terms of
percentage of untreated control absorbance following subtraction of
mean background absorbance. The IC50 concentrations were
calculated from the dose-response curves. Assays were performed in
triplicate.
The effects of GGPP and doxorubicin with or without
BPs on the MCF-7 cell line were also determined by SRB assay. This
involved seeding MCF-7 cells into 96-well culture plates at
1×104 cells/well, allowing cells to attach for 24 h and
then treating the cells with 5 µM GGPP or 1 µM doxorubicin with BPs
(250 µM alendronate and risedronate, and 100 µM pamidronate) or
without BPs [0.25% dimethyl sulfoxide (DMSO)] at 37°C for 24 h.
Cell viability was measured using the aforementioned protocol for
SRB assay.
Clonogenic assay
The clonogenic assay was performed as previously
described (22). MCF-7 cell lines
were seeded into six-well culture plates at 500 cells/well for 24 h
and following cell attachment fresh DMEM was added with BPs (100 µM
alendronate and risedronate, and 50 µM pamidronate) or without BPs
(0.25% DMSO) at 37°C for 24 h. The medium containing the BPs was
subsequently removed from the culture plates, cells were washed
with PBS, fresh DMEM was added and medium was renewed every 2 days.
Following 2 weeks of treatment, the cell culture medium was removed
for analysis and cells were washed with PBS. Cells were fixed in
100% methanol for 30 min at −20°C. Colonies were stained at room
temperature for 1 h with 0.5% crystal violet in 25% (v/v) methanol
and excess dye was removed by washing several times with water.
Following washing and drying, the colonies were captured using a
Nikon camera and counted to contain >50 cells/colony. Colony
formation was expressed as a percentage relative to untreated
cells. The clonogenic assay was performed in triplicate.
Wound healing assay
A wound healing assay was performed as previously
described (22). MCF-7 cells were
seeded into 24-well culture plates at 2.5×105 cells/well
and allowed to attach for 24 h. Subsequently, monolayers of
confluent cultures were scratched with a 0.2 ml pipette tip, cell
debris was washed away with PBS, cells were treated with BPs (0–50
µM alendronate, 0–100 µM risedronate, 0–25 µM pamidronate) and
without BPs (0.25% DMSO) and images of the scratched wound were
obtained at 0 and 72 h using phase contrast microscopy
(magnification, ×40). The wound distance was determined as the
width of the scratch and treated and untreated groups were
compared. The wound healing assay was performed in triplicate.
ROS formation assay
A ROS formation assay was performed by oxidation of
DHE to the ethidium cation, which binds to intracellular ROS, as
previously described (22). Briefly,
MCF-7 cells were seeded into white 96-well culture plates at
1×104 cells/well and allowed to attach for 24 h.
Subsequently, cells were incubated with BPs (0–1,000 µM alendronate
and risedronate, and 0–250 µM pamidronate) or without BPs (0.25%
DMSO) and 25 µM DHE at 37°C for 90 min in a 5% CO2
incubator in the dark. ROS production was assessed by measuring the
fluorescence intensity with 518 nm excitation and 605 nm emission
wavelengths. Data are expressed as the percentage of ROS relative
to the control groups. The ROS formation assay was performed in
triplicate.
Caspase-3 activity assay
Caspase-3 activity was measured using a caspase-3
fluorometric assay kit, according to the manufacturer's
instructions. Briefly, MCF-7 cells were seeded into 6-well culture
plates at 2.5×105 cells/well and allowed to attach for
24 h. Cells were treated with BPs (0–250 µM alendronate and
pamidronate, and 0–1,000 µM risedronate) or without BPs (0.25%
DMSO) for 24 h, cells were then lysed with lysis buffer and the
protein concentration was measured using Bradford reagent.
Caspase-3 activity reactions comprised of cell lysates and buffer
containing the caspase-3 substrate Ac-DEVD-AMC with the final
volume being ~200 µl. The reaction mixture was incubated at 37°C in
the dark for 90 min and the fluorescence intensity was measured
with 360 nm excitation and 460 nm emission wavelengths. Caspase-3
activity was calculated using fluorescent 7-amino-4-methylcoumarin
(AMC) as a standard. The caspase-3 activity assay was performed in
triplicate.
Mitochondrial transmembrane potential
assay
To measure alterations in the mitochondrial
transmembrane potential, the fluorescent dye JC-1 (JC-1
mitochondrial membrane potential assay kit; cat no. 10009172;
Cayman Chemical Company, Ann Arbor MI, USA) was used. In brief,
MCF-7 cells were seeded into black 96-well culture plates at
1×104 cells/well and allowed to attach for 24 h. Cells
were then treated with BPs (0–1,000 µM alendronate and risedronate,
and 0–250 µM pamidronate) or without BPs (0.25% DMSO) for 24 h and
cells were incubated with JC-1 dye for 30 min at 37°C in the dark.
Subsequently, cells were rinsed and incubated in 200 µl JC-1 assay
buffer. Fluorescence intensity was measured with 485 nm excitation
and 535 nm emission wavelengths. Assays were performed in
triplicate.
Acridine orange/ethidium bromide
(AO/EB) staining
The AO/EB assay was performed as previously
described (22). Briefly, MCF-7
cells were seeded into 96-well culture plates at 1×104
cells/well and allowed to attach for 24 h. Subsequently, cells were
incubated with or without BPs (0–1,000 µM alendronate and
risedronate, and 0–250 µM pamidronate) or without BPs (0.25% DMSO)
for an additional 24 h. Cells were then washed with PBS and stained
with 1 µg/ml AO and EB in PBS for 10–15 min at room temperature in
the dark. Images were obtained using an inverted fluorescent
microscope with excitation and long-pass emission filters of 480
and 535 nm (magnification, ×200). Subsequently, images were
examined and the number of viable, apoptotic and necrotic cells in
the same area were counted. Data are expressed as percentage values
relative to the control. Assays were performed in triplicate.
Cell apoptosis by flow cytometry
MCF-7 cell apoptosis was measured by flow cytometry.
MCF-7 cells were seeded in six-well culture plates at
2.5×105 cells/well and allowed to attach for 24 h. Cells
were then exposed to BPs (0–250 µM alendronate and risedronate, and
0–50 µM pamidronate) or without BPs (0.25% DMSO) for 24 h prior to
collection by trypsinization and washing with cold PBS. Cells were
resuspended in 200 µl binding buffer. Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) staining was
performed according to the manufacturer's protocol (FITC Annexin V
apoptosis detection kit I; cat. no. 556547; BD Biosciences, San
Jose, CA, USA). In brief, 5 µl Annexin V-FITC (1 mg/ml) and 5 µl PI
(2.5 mg/ml) were added to 100 µl of suspended cells and incubated
at room temperature in the dark for 15 min prior to the addition of
400 µl assay buffer. Data are expressed as the percentage of cells
in each quadrant, which represent different stages of apoptosis
following BPs treatment. Analysis was performed using BD Accuri C6
software (BD Biosciences). Assays were performed in triplicate.
Cell cycle analysis by flow
cytometry
The cell cycle phase of MCF-7 cells was determined
by flow cytometry. MCF-7 cells were seeded in 6-well culture plates
at 2.5×105 cells/well and allowed to attach for 24 h.
Cells were then exposed to BPs (250 µM alendronate and risedronate,
and 100 µM pamidronate) or without BPs (0.25% DMSO) for 24 h prior
to collection and overnight fixation with 70% cold ethanol at
−20°C. Following washing three times with PBS, cells were
resuspended in 200 µl Guava Cell Cycle Reagent (cat. no. 4500-0220;
Guava Technologies, Hayward, CA, USA) at room temperature in the
dark for 30 min. The DNA content of the cells was analyzed using a
flow cytometer and the percentage of cells in different phases of
the cell cycle was calculated using the Guava EasyCyte system with
Cytosoft software v2.0 (Guava Technologies). Assays were performed
in triplicate.
Matrix metallopeptidase (MMP)-9
expression by gelatin zymography
The effects of BPs on MMP-9 protein levels were
assessed by gelatin zymography analysis. In brief, MCF-7 cells were
seeded in 24-well culture plates at 2.5×105 cells/well
and allowed to attach for 24 h. DMEM containing various
concentrations of BPs (250 µM alendronate and risedronate, and 100
µM pamidronate) or without BPs (0.25% DMSO) was then added for 24 h
at 37°C. Subsequently, the medium was collected and concentrated by
centrifugation at 12,000 × g for 5 min using centrifugal filter
units. The protein concentration was subsequently measured using
Bradford's reagent. The proteins were mixed with 2X loading sample
buffer [1.5 ml 0.5 M Tris-HCl (pH 6.8), 2.5 ml glycerol, 4 ml 10%
(w/v) SDS, 0.1 ml 1% bromophenol blue and 2.15 ml deionized water]
for 20 min at room temperature in the dark. The 20 µg protein
samples were loaded onto 10% SDS-PAGE gel containing 0.01% (w/v)
gelatin and subjected to electrophoresis at 120 V for 90 min. The
gel was then washed three times with 2.5% Triton X-100 and
incubated with developing buffer [50 mM Tris-HCl buffer (pH 7.45)
and 10 mM CaCl2] overnight at 37°C. Subsequently, the
gels were stained with 0.5% Coomassie Brilliant Blue R-250 for 1 h
at room temperature with shaking and gels were washed with
destaining buffer [methanol (25 ml), acetic acid (37.5 ml) and
deionized water (437.5 mm)] until clear bands were observed against
an intensely stained background. The band intensity was calculated
using ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Assays were performed in triplicate.
Gene expression by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was used to determine the effect of BPs on
Ras-related C3 botulinum toxin substrate 1 (Rac1), Ras homolog gene
family member A (RhoA), cell division control protein 42 homolog
(CDC42), MMP-2, MMP-9, intracellular adhesion molecule 1 (ICAM1)
and vascular endothelial growth factor A (VEGFA) gene expression.
In brief, MCF-7 cells were seeded in six-well culture plates at
2.5×105 cells/well and allowed to attach for 24 h at
37°C. MCF-7 cells were then treated with BPs (250 µM alendronate
and risedronate, and 100 µM pamidronate) or without BPs (0.25%
DMSO) at 37°C for 24 h prior to the addition of 0.5 ml
TRIzol® reagent (cat. no. T9424; Sigma-Aldrich; Merck
KGaA). Subsequently, RNA was isolated and complementary DNA was
prepared using the iScript™ cDNA synthesis kit (cat. no.
1708898; Bio-Rad Laboratories, Inc.) at 42°C for 60 min. PCR
amplification was performed using primers for the target genes with
β-actin used as an internal control. PCR primer sequences are
presented in Table I and final
reaction volume of 20 µl was prepared containing SsoFast™ EvaGreen
Supermix with low Rox (Bio-Rad Laboratories, Inc.), and primers for
the target gene and the internal control β-actin. The PCR
conditions were as follows: Denaturation at 95°C for 3 min and
amplification by cycling 40 times at 95°C for 15 sec and 60°C for
30 sec. Expression was detected using a CFX96 Touch™ real-time PCR
detection system (Bio-Rad Laboratories, Inc.). Differences in gene
expression levels were calculated using the 2−ΔΔCq
method (23) for relative
quantification and expressed as the fold change relative to the
untreated control. Assays assessing target gene expression were
performed in triplicate.
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer
sequence (5′-3′) | Reverse primer
sequence (5′-3′) |
|---|
| Rac1 |
ATGTCCGTGCAAAGTGGTATC |
CTCGGATCGCTTCGTCAAACA |
| RhoA |
GGAAAGCAGGTAGAGTTGGCT |
GGCTGTCGATGGAAAAACACAT |
| CDC42 |
CCATCGGAATATGTACCGACTG |
CTCAGCGGTCGTAATCTGTCA |
| MMP-2 |
GATACCCCTTTGACGGTAAGGA |
CCTTCTCCCAAGGTCCATAGC |
| MMP-9 |
GGGACGCAGACATCGTCATC |
TCGTCATCGTCGAAATGGGC |
| ICAM1 |
GTATGAACTGAGCAATGTGCAAG |
GTTCCACCCGTTCTGGAGTC |
| VEGFA |
AGGGCAGAATCATCACGAAGT |
AGGGTCTCGATTGGA-TGGCA |
| β-actin |
GTGACGTTGACATCCGTAAAGA |
GCCGGACTCATCGTACTCC |
Protein extraction and western blot
analysis
Protein expression was assessed as described
previously (22). In brief, MCF-7
cells were seeded in 75 mm flasks, allowed to attach for 24 h at
37°C and then treated with BPs (250 µM alendronate and risedronate,
and 100 µM pamidronate) or without BPs (0.25% DMSO) at 37°C for 24
h. Subsequently, pellets were collected and lysed with RIPA lysis
buffer to extract the total protein, which was quantified using
Bradford's reagent. Equal samples of 20 µg protein were separated
using 12% SDS-PAGE gels and transferred to polyvinylidene
difluoride membranes. The membranes were blocked in 5% non-fat
powdered milk/TBS/Tween-20 for 1 h at room temperature with shaking
and incubated overnight at 4°C with the relevant primary antibodies
against p21 (1:1,000), cyclin D1 (1:1,000), cytochrome c (1:1,000),
caspase-3 (1:1,000) and β-actin (1:2,500). This was followed by a 2
h treatment with HRP-conjugated secondary antibody (1:5,000) at
room temperature with shaking. Blots were washed and visualized
using Clarity™ Western Enhanced Chemiluminescent
Substrate (cat. no. 1705060; Bio-Rad Laboratories, Inc.) and
calculated using ChemiDoc™ Touch Imaging System (Bio-Rad,
Laboratories, Inc.). Assays were performed in triplicate.
Statistical analysis
Control and treatment groups were statistically
compared using Student's t-test or one-way analysis of variance
followed by Tukey's post-hoc test using SigmaStat software version
3.5 (Systat Software Inc., San Jose, CA, USA) and values are
expressed as the mean ± standard error of the mean of three
experiments. P<0.05 was considered to indicate a statistically
significant result.
Results
Effects of BPs on cell death and
colony formation efficacy
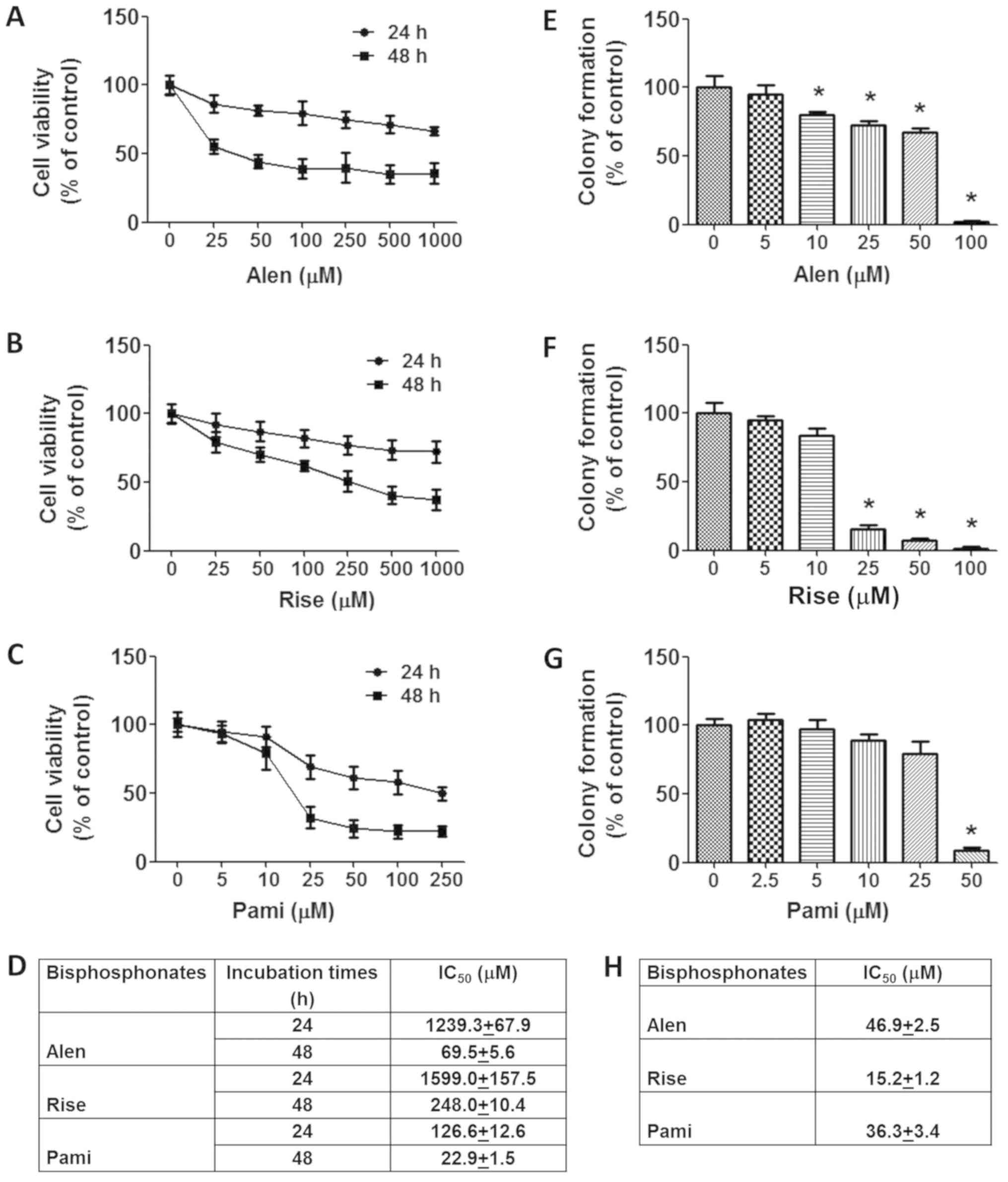
The cytotoxic effect of the BPs alendronate,
risedronate and pamidronate in MCF-7 human breast cancer cells and
their ability to disrupt MCF-7 cell replication were assessed by
SRB and colony formation assay. The results demonstrated that the
BPs exerted a cytotoxic effect on MCF-7 cells in a concentration-
and time-dependent manner, with IC50 values for
alendronate, risedronate and pamidronate at 48 h calculated as
69.5±5.6, 248.0±10.4 and 22.9±1.5 µM, respectively (Fig. 1A-D). Accordingly, pamidronate
exhibited greater cytotoxicity compared with alendronate and
risedronate against human breast cancer cells.
The effects of BPs on the longer term viability and
replicative ability of MCF-7 cells was determined using a colony
formation assay. It was identified that cells treated with BPs
underwent a concentration-dependent decrease in colony forming
ability (Fig. 1E-H). A significant
inhibitory effect was observed at doses >10, >25 and 50 µM
for alendronate, risedronate and pamidronate, respectively. The
greatest inhibition of replicative ability was by risedronate
followed by alendronate and pamidronate, with IC50
values of 15.2±1.2, 46.9±2.5 and 36.3±3.4 µM, respectively. In
summary, these results indicate that BPs inhibit MCF-7 cell growth
and induce MCF-7 cell death.
To determine if the effects of BPs on cell viability
were inhibited by the mevalonate product GGPP, or if the BPs
exerted any synergistic effects with the anticancer drug
doxorubicin, their combined effects were examined using a SRB
assay. The results demonstrated that pretreatment of MCF-7 cells
with 5 µM GGPP in the absence of BPs slightly stimulates their
growth but not significantly. The addition of GGPP in the presence
of the BPs risedronate and pamidronate significantly reversed the
effects of the BPs on cell viability (Fig. 2A). To determine if doxorubicin was
potentiated by the BPs, MCF-7 cell viability was assessed in the
presence of the two types of drugs. By adding 250 µM of
alendronate, 250 µM of risedronate, or 100 µM of pamidronate to the
culture medium containing 1 µM doxorubicin, the antiproliferative
effects of doxorubicin were improved in all treatment groups
(Fig. 2B).
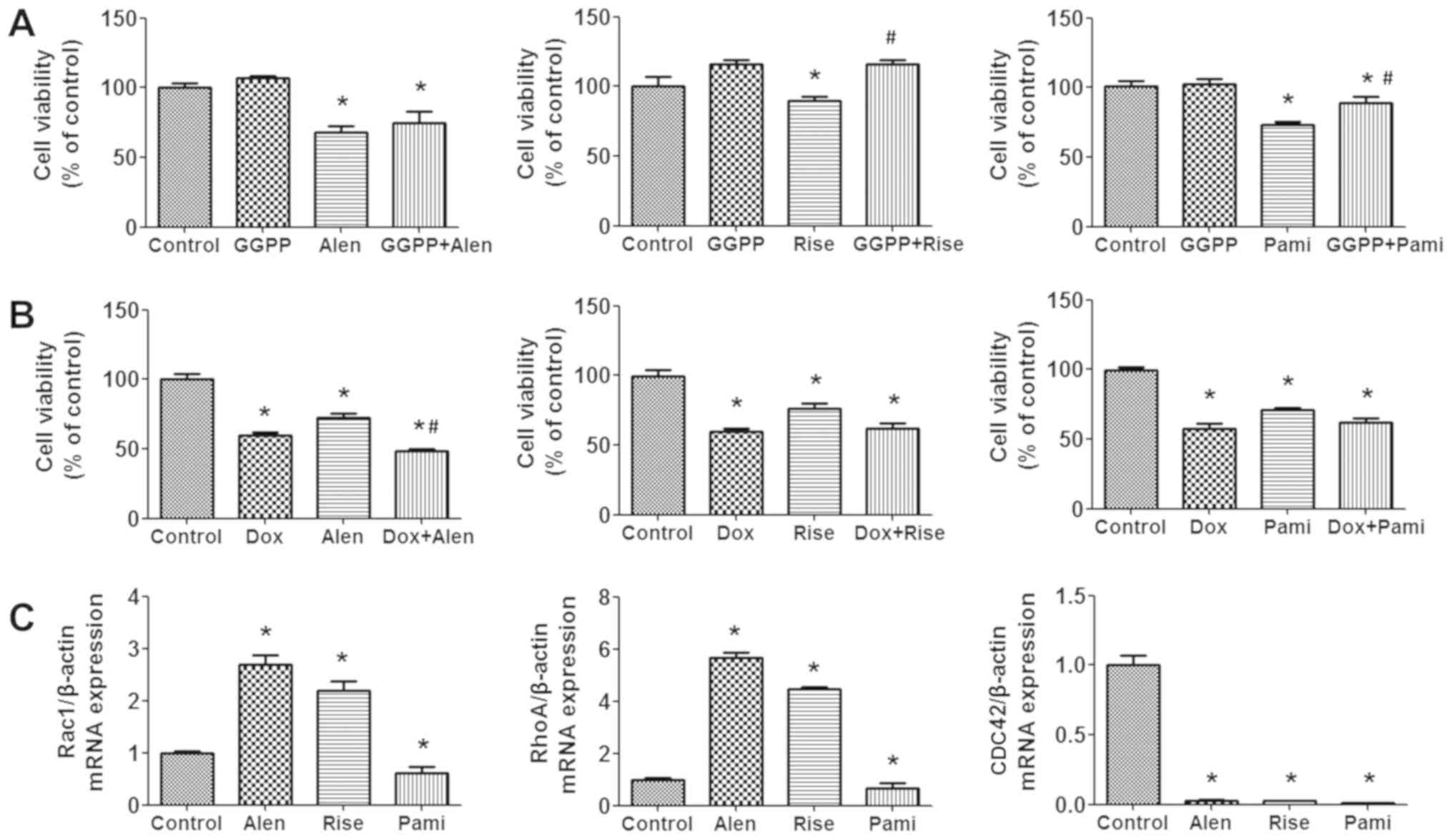 | Figure 2.Effect of GGPP and doxorubicin in
combination with BPs on cell viability and Rho GTPase gene
expression. MCF-7 cells were treated with 250 µM Alen, 250 µM Rise
and 100 µM Pami with or without (A) 5 µM GGPP or (B) 1 µM Dox for
24 h and cell viability was measured by sulforhodamine B assay. (C)
Cells were treated with 250 µM Alen, 250 µM Rise and 100 µM Pami
for 24 h and Rac1, RhoA and CDC42 mRNA expression levels were
determined by reverse transcription-quantitative polymerase chain
reaction. Data are presented as the mean ± standard error of the
mean. *P<0.05 vs. control group. #P<0.05 vs. BP-treated
group. Alen, alendronate; Rise, risedronate; Pami, pamidronate;
GGPP, geranylgeranyl pyrophosphate; Dox, doxorubicin; Rac,
Ras-related C3 botulinum toxin substrate; Rho, Ras homolog gene
family; CDC42, cell division control protein 42 homolog; BP,
bisphosphonate. |
Effects of BPs on Rac1, RhoA and CDC42
gene expression
Members of the Rho GTPase family, including RhoA,
Rac1 and CDC42, are involved in regulating a variety of normal and
cancer cellular processes (24), and
Rho GTPase genes are dysregulated in numerous types of cancer cells
(25). Results from the present
study revealed that treatment of MCF-7 cells with BPs for 24 h
resulted in altered expression levels of the Rac1, RhoA and CDC42
genes (Fig. 2C). Rac1 and RhoA
levels were significantly upregulated in the alendronate and
risedronate treatment groups, but significantly reduced in the
pamidronate treatment group. Furthermore, all BPs significantly
reduced CDC42 gene expression. In conclusion, the current results
demonstrated that BPs may modulate Rho GTPase gene expression in
the mevalonate pathway. Therefore, it was suggested that BPs
interrupted an important step of cholesterol synthesis, which may
induce breast cancer cell death, however, additional studies are
required.
Effects of BPs on MCF-7 cell cycle
progression and expression of proteins associated with cell
cycle
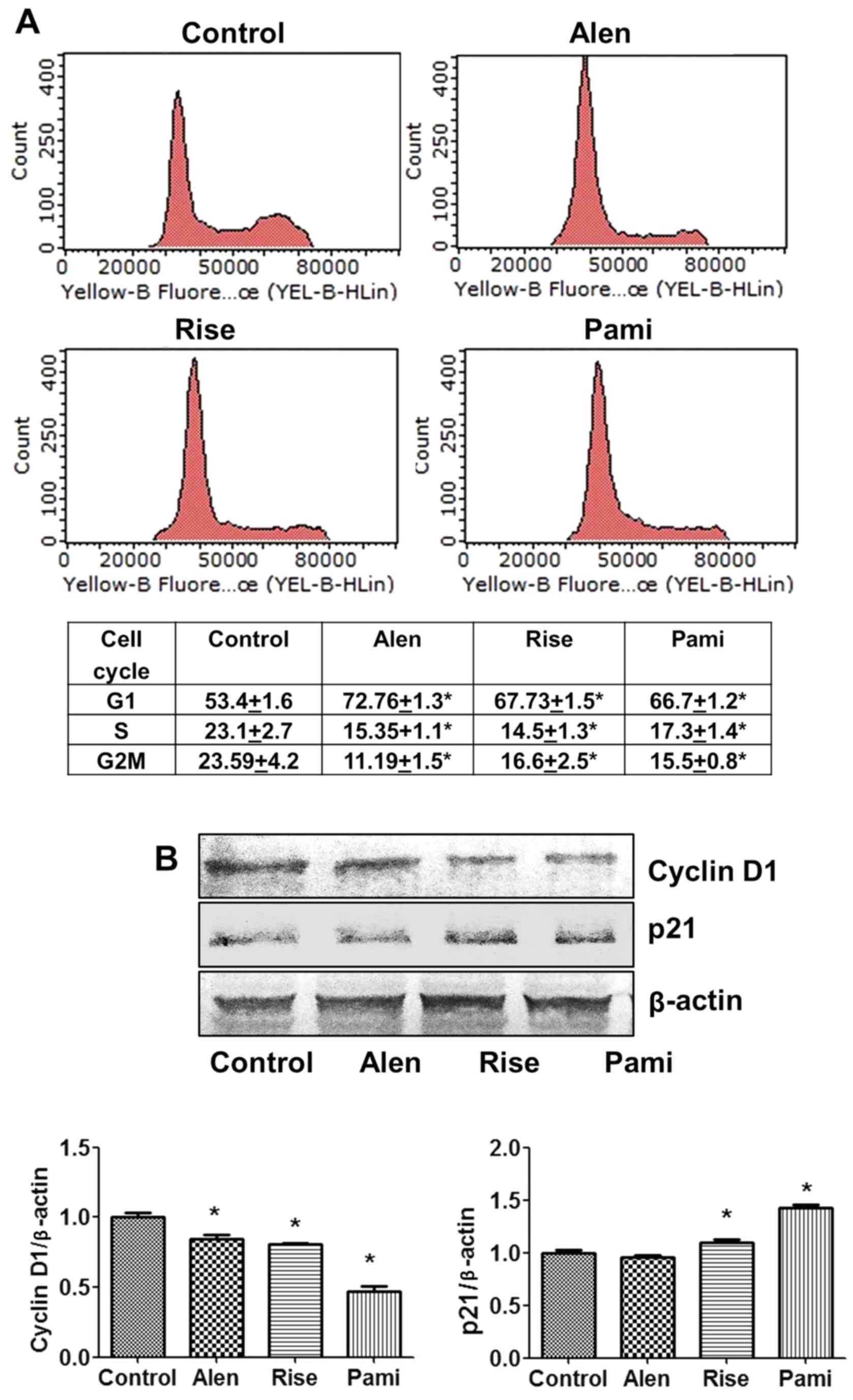
To investigate whether BP-induced inhibition of
MCF-7 cell growth was mediated via the alteration of the cell cycle
progression, flow cytometry was used to determine the cell cycle
phase distribution in MCF-7 cells. The results demonstrated that
BPs caused an increase in the percentage of cells in the G1 phase
with significant effects occurring at doses of 250 µM alendronate,
250 µM risedronate and 100 µM pamidronate (Fig. 3A). Furthermore, BPs reduced the
percentage of cells in the S and G2/M phases, which is consistent
with G1 phase arrest. These data demonstrated that alendronate
induced cell cycle arrest, particularly G1 phase arrest, in the
MCF-7 cells to a greater extent compared with the other BPs.
To further investigate which molecules were affected
by BPs, the expression levels of cell cycle-associated proteins
were assessed by western blot analysis. The results demonstrated
that treatment with 250 µM alendronate, 250 µM risedronate and 100
µM pamidronate was associated with a significant decrease in the
levels of cyclin D1 (Fig. 3B), with
pamidronate inducing the greatest change. Furthermore, the BPs
risedronate and pamidronate inhibited cancer cell proliferation by
significantly inducing the cyclin Dependent kinase inhibitor p21
(Fig. 3B). In conclusion, the
results indicated that BPs inhibited cell proliferation by inducing
MCF-7 cell arrest in the G1 phase, inhibiting cell cycle protein
cyclin D1 and inducing cell cycle inhibitor p21 protein.
Effect of BPs on ROS formation,
caspase-3 activity and mitochondrial membrane potential
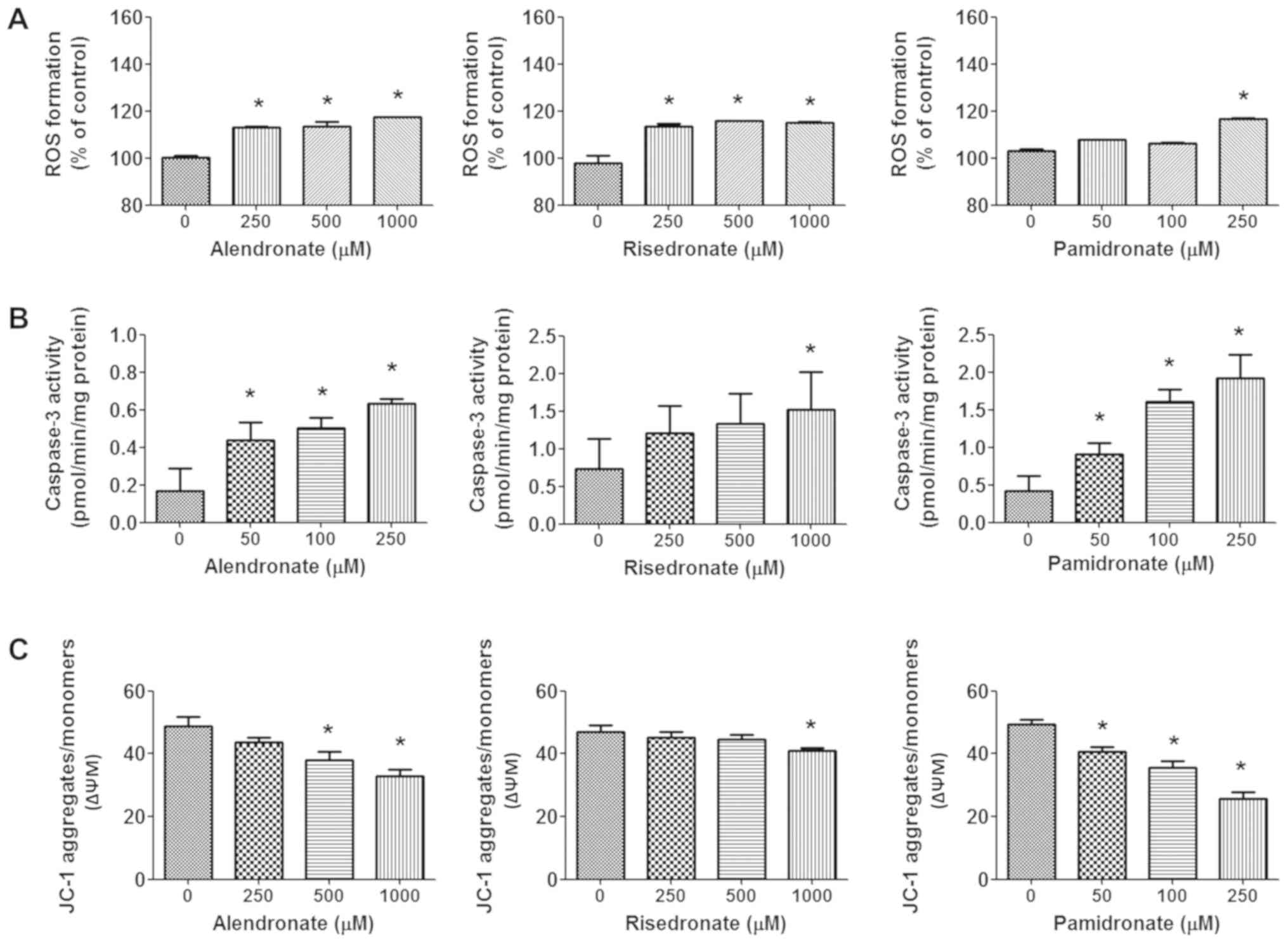
To investigate the mechanism by which BPs induced
MCF-7 apoptotic cell death and ROS formation, caspase-3 activity
and mitochondrial transmembrane potential were assessed. A
significant increase in ROS formation was induced by all BPs at a
concentration of 250 µM (Fig. 4A). A
high concentration of alendronate and risedronate (1,000 µM)
further induced a significant increase in ROS production compared
with the control group. In addition, all BPs increased caspase-3
activity in a dose-dependent manner, with significant inductive
effects observed with >50 µM alendronate, >50 µM pamidronate
and 1,000 µM risedronate (Fig. 4B).
It was also identified that BPs significantly decreased the
mitochondrial transmembrane potential of MCF-7 cells compared with
the control, with pamidronate exhibiting the most significant
reduction in mitochondrial activity (Fig. 4C).
Effect of BPs on induction of
apoptotic cell death
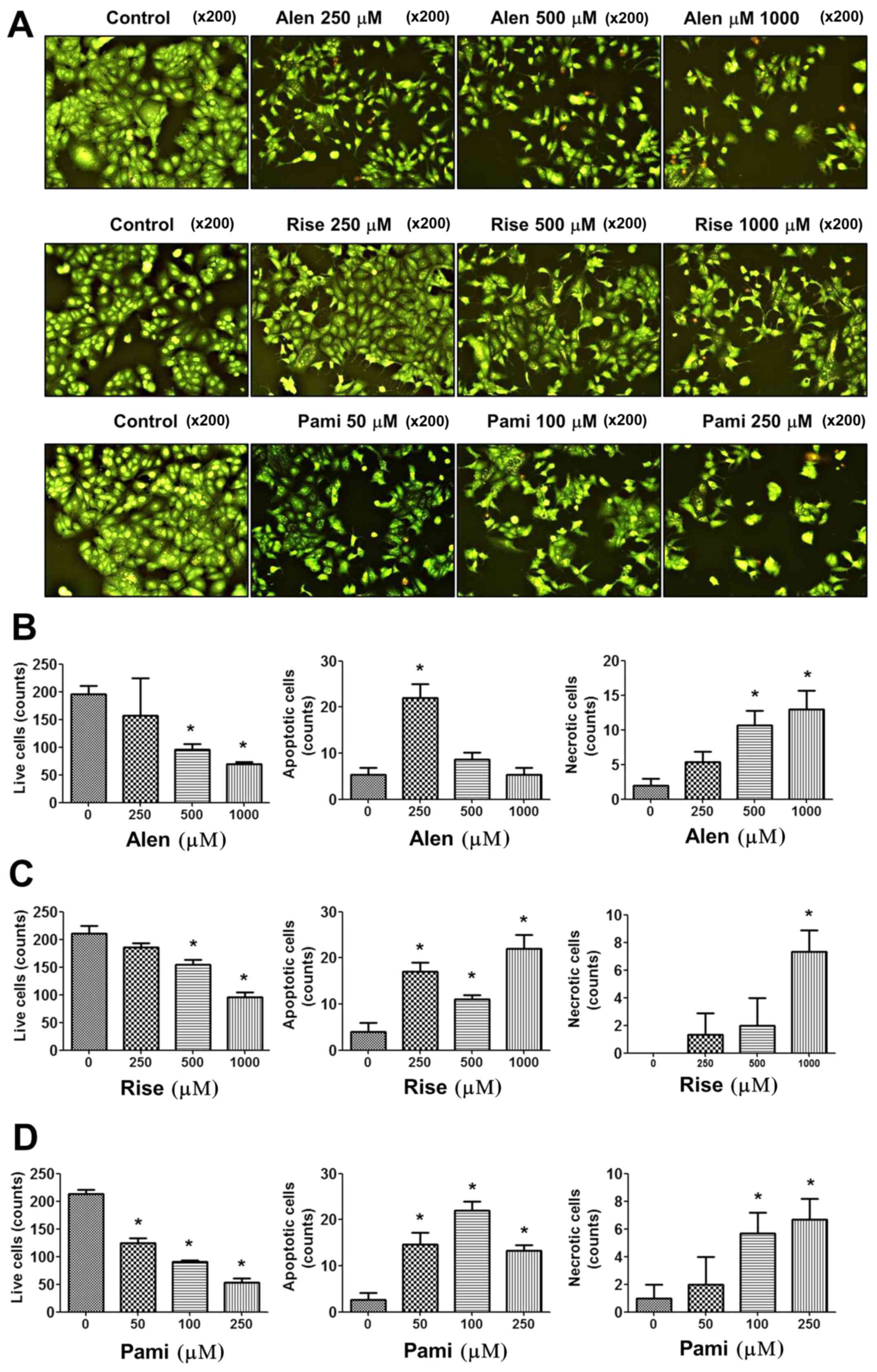
To determine the effect of BPs on the apoptotic cell
death of MCF-7 cells, AO/EB staining, flow cytometry and western
blot analysis were performed. To investigate the mode of cell
killing by BPs, BP-treated MCF-7 cells were stained with the
fluorescent dye AO/EB. The results revealed that cells underwent
cell death, apoptosis and necrosis following incubation with the
BPs for 24 h. The number of viable cells decreased in a
dose-dependent manner following treatment with BPs, with necrotic
cells increasing in number. Notably, significant induction of
apoptotic cell death was observed following treatment with 250 µM
alendronate, 250–1,000 µM risedronate and 50–250 µM pamidronate
(Fig. 5). Furthermore, high
concentrations of the BPs also induced dose-dependent necrotic cell
death following treatment with 500–1,000 µM alendronate, 1,000 µM
risedronate and 100–250 µM pamidronate. These results indicate that
BPs exhibit significant effects on breast cancer cell apoptosis and
cell death.
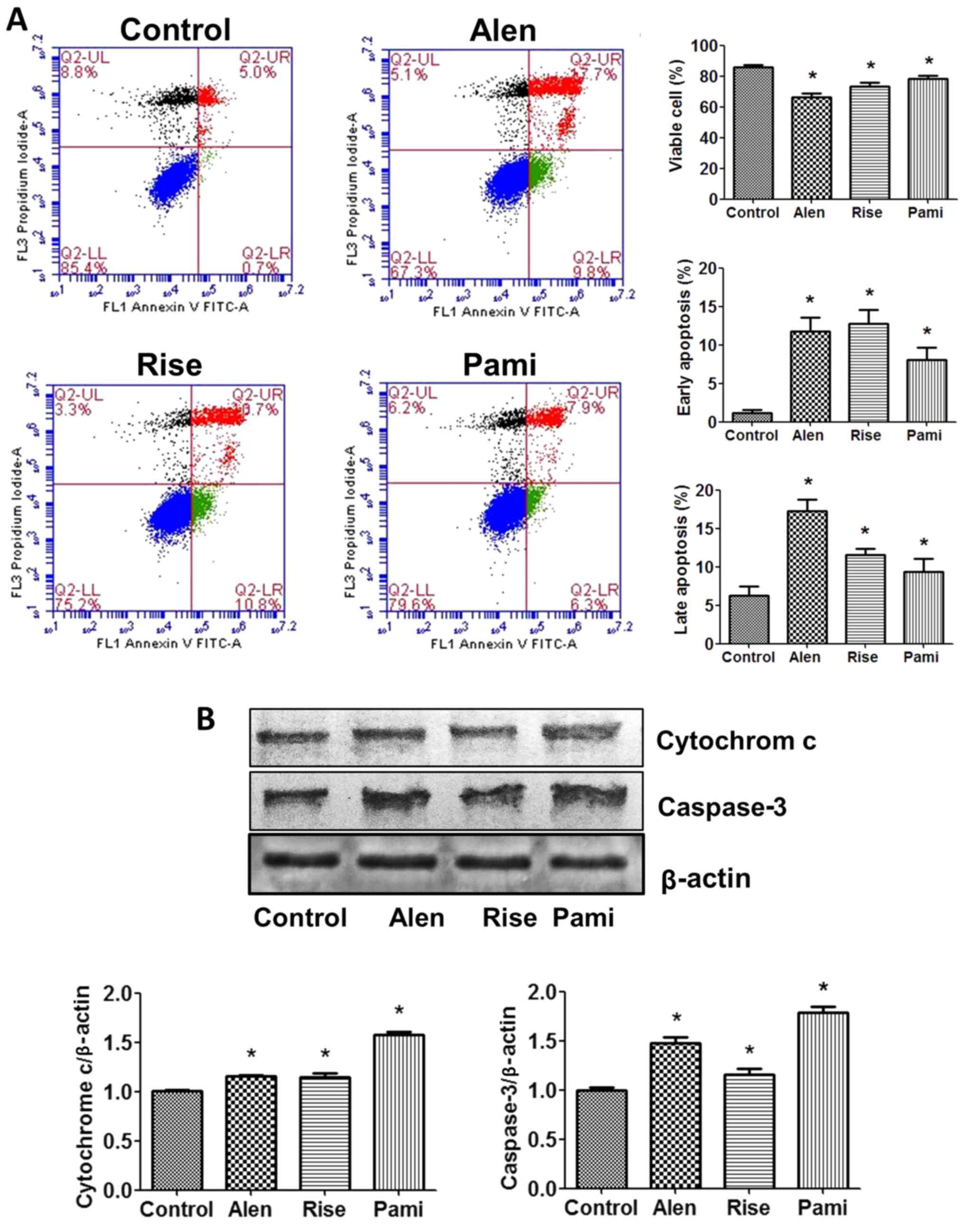
To investigate whether apoptosis was involved in the
action of BPs on MCF-7 cells, the proportion of apoptotic cells
were determined using a flow cytometer by staining samples with PI
and Annexin V-FITC. Significant BP-induced apoptosis of MCF-7 cells
was identified, as demonstrated in Fig.
6A. Compared with the control group, the proportion of viable
cells decreased and the proportion of early and late apoptotic
cells significantly increased following 24 h incubation with the
BPs.
To gain insight into the molecular mechanism by
which BPs induced MCF-7 cell death, western blot analysis was
performed. All BPs demonstrated a similar pattern of changes in the
expression levels of apoptosis-associated proteins in MCF-7 cells.
The BPs significantly increased cytochrome c and caspase-3 protein
expression levels compared with the untreated control group
(Fig. 6B).
Effect of BPs on cell migration
To determine the effect of BPs on MCF-7 cell
migration, a wound healing assay, gelatin zymography and RT-qPCR
were performed. BPs were identified to significantly inhibit MCF-7
cell migration in a dose-dependent manner (Fig. 7A). The inhibitory effects of
risedronate and pamidronate on MCF-7 cell migration were revealed
to be markedly higher compared with alendronate. Risedronate and
pamidronate also significantly inhibited MMP-9 protein expression
levels in the culture medium to a greater extent compared with
alendronate (Fig. 7B). In addition,
treatment with BPs was associated with increased MMP-2, MMP-9,
VEGFA and ICAM1 gene expression levels compared with the untreated
control groups (Fig. 7C).
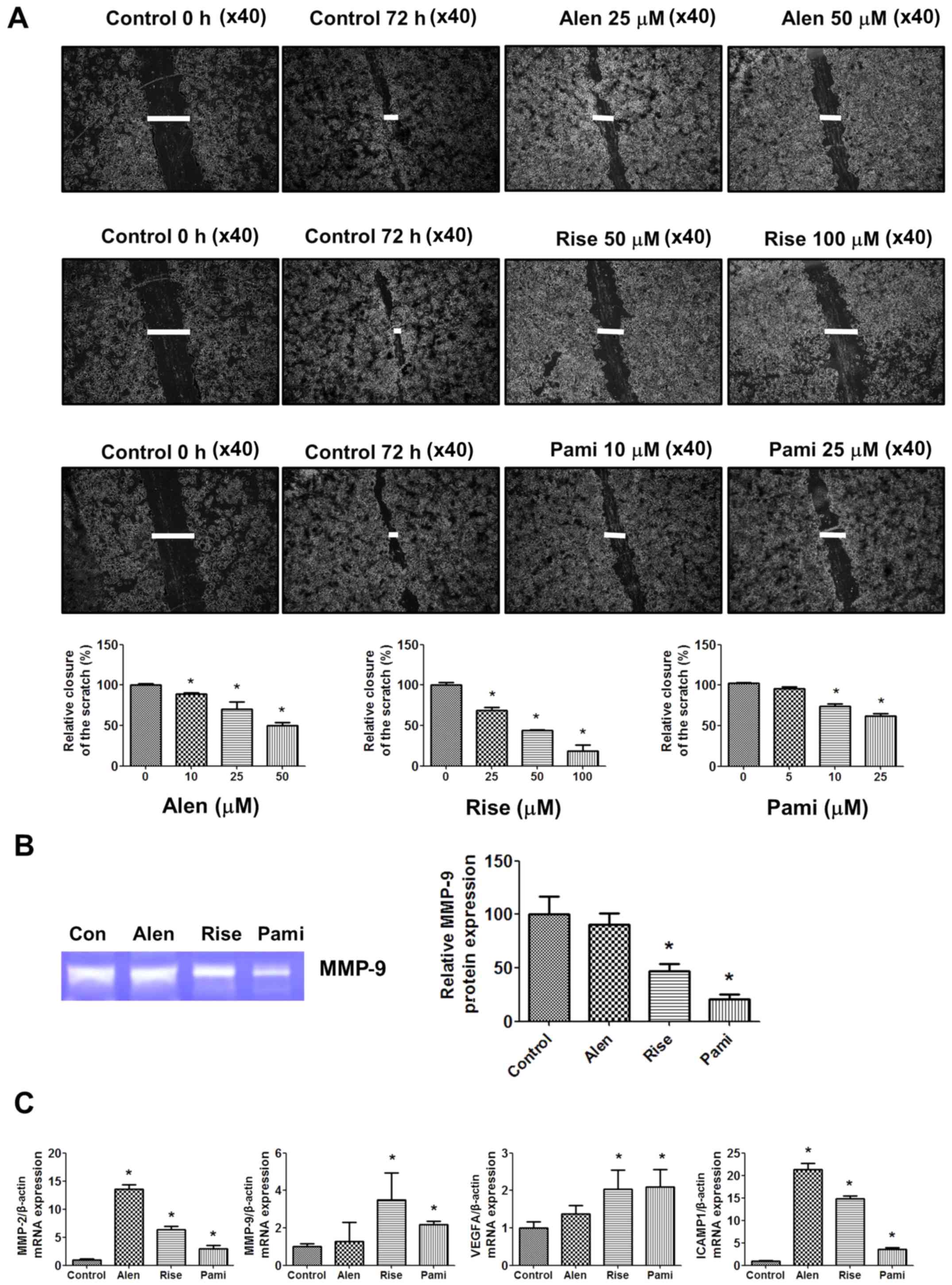 | Figure 7.Effects of BPs on MCF-7 cell
migration. (A) MCF-7 cells were scratched using 0.2 ml pipette tips
and treated with various concentration of BPs for 72 h. Cell
migration was monitored using phase contrast microscopy
(magnification, ×40). Quantified cell migration is presented as the
percentage of wound closure at 72 h. (B) Effects of BPs on MMP-9
expression levels in conditioned media analyzed by gelatin
zymography. (C) Cells were treated with 250 µM alendronate, 250 µM
risedronate and 100 µM pamidronate for 24 h and MMP-2, MMP-9, VEGFA
and ICAM1 mRNA expression levels were determined by reverse
transcription-quantitative polymerase chain reaction. Data are
presented at the mean ± standard error of the mean from three
independent experiments with a duplicate in each experiment.
*P<0.05 vs. control group. Alen, alendronate; Rise, risedronate;
Pami, pamidronate; MMP, matrix metallopeptidase; VEGFA, vascular
endothelial growth factor; ICAM1, intracellular adhesion molecule
1; BP, bisphosphonate. |
Discussion
BPs influence cell proliferation and programmed cell
death in different cancer cell lines, possibly by inhibiting the
production of mevalonate products (3). However, not all BPs exhibit identical
effects on breast cancer MCF-7 cells in vitro. The present
study examined the BPs alendronate, risedronate and pamidronate for
their capacity to inhibit MCF-7 breast cancer cell proliferation
and suppress the metastatic potential of MCF-7 cells. It was
identified that pamidronate exhibited the most potent effects. In
addition, the mevalonate product GGPP was revealed to reverse
BP-induced cancer cell death. When the BPs were combined with the
anticancer drug doxorubicin, an enhanced antiproliferative effect
was observed compared with doxorubicin alone. The BPs were also
identified to modulate the expression of the Rho GTPase family
genes Rac1, RhoA and CDC42. Regarding the mechanism underlying the
aforementioned effects on MCF-7 cells, the BPs were revealed to
inhibit cell proliferation by inducing cell cycle arrest in the G1
phase, which was associated with a downregulation of cyclin D1 and
an upregulation of p21. The BPs induced MCF-7 cell death by
increasing ROS formation, increasing caspase-3 activity and
reducing mitochondrial function. Furthermore, an alternative
mechanism of BP-induced cell death was revealed to be associated
with an increase in cytochrome c and caspase-3 protein expression
levels. Finally, the BPs inhibited cancer cell migration by
reducing MMP-9 protein expression levels. Therefore, BPs may
potentially be developed as drugs for the treatment or prevention
of breast cancer, or for the potentiation of anticancer drugs that
are currently in clinical use.
Results from the present study demonstrated that BPs
reduce both cancer cell proliferation and cell growth. This was
hypothesized to occur via interactions between BPs and cell
cycle-associated proteins, which may lead to cell cycle arrest.
Investigation of the effect of BPs on MCF-7 cells indicated that
BPs induce cell death and inhibit cell growth in a dose-dependent
manner. A previous study has demonstrated that BPs inhibit FFPS in
the mevalonate pathway, causing a reduction of GGPP and FPP, and
reducing the expression levels of Rho GTPase proteins, including
Rac1, RhoA and CDC42 (26). Rho
GTPases are important regulators of cell cycle progression and
proliferation; they have been identified to be overexpressed in
human tumors and their overexpression levels have been associated
with cancer progression (27). The
present study revealed that BP-induced cancer cell death was
reversed by GGPP treatment. It was also identified that BPs
potentiated the effect of anticancer drug doxorubicin and that BPs
modulated Rho GTPase gene expression. Notably, pamidronate was
demonstrated to be the most potent BP, as pamidronate significantly
reduced the expression levels of small GTPases Rac1, RhoA and
CDC42. However, Rac1 and RhoA were significantly upregulated in the
alendronate and risedronate treatment groups, which may explain why
pamidronate exhibited the highest cytotoxic effects on MCF-7
cells.
Furthermore, to determine the effects of BPs on the
cell cycle and cell cycle-associated proteins, flow cytometry
analysis and western blot analysis were performed. The results
revealed that BPs significantly increased the percentage of cells
in the G1 phase and decreased the percentage of cells in the S and
G2 phases. Alendronate exhibited the most potent activity. The
effect of BPs on MCF-7 cells was associated with inhibition of
cyclin D1 expression, induction of p21 expression and cell cycle
arrest. p21 is a cell cycle dependent kinase inhibitor that
regulates the cell cycle (28). In
addition, p21 serves an essential role in growth arrest following
DNA damage and overexpression of p21 leads to G1 and G2 or S-phase
arrest (29,30). Cyclin D1 overexpression has also been
demonstrated to induce cell proliferation and has been be
associated with genomic instability in breast cancer, which may
contribute to oncogenesis (31,32).
Therefore, sustained G1 arrest, which is initially triggered by
downregulation of cyclin D1, depends on p21 expression (33). These results indicate that BPs
inhibit MCF-7 cell growth via cell cycle arrest.
Apoptosis may be induced via several mechanisms,
including alteration of the intracellular mitochondrial pathway
(34). In the present study, BPs
were identified to induce ROS formation, increase caspase-3
activity and decrease mitochondrial function. Furthermore,
apoptotic cell death, caspase-3 expression and cytochrome c
expression were substantially increased following BP treatment. In
summary, the results suggest that BP-induced antiproliferative
effects and induction of apoptosis in MCF-7 cells may be caused by
ROS formation, and associated DNA damage and alteration of
mitochondrial function. Senaratne et al (35) have previously demonstrated that other
BPs significantly increase cell apoptosis in breast cancer cell
lines. In this previous study, zoledronate was identified as the
most potent BP, increasing the proportion of cells with
morphological features characteristic of apoptosis, including
fragmented chromosomal DNA, and it is associated with a
downregulation of B-cell lymphoma 2 protein and proteolytic
cleavage of poly (ADP-ribose) polymerase (35). Furthermore, Rachner et al
(21) demonstrated that zoledronate
induces breast cancer cell apoptosis by inducing TRAIL expression
and enhancing the TRAIL/OPG ratio in TRAIL-sensitive MDA-MB-231
cells. Ebert et al (36)
identified that ApppI expression increases following treatment with
zoledronate to a greater extent compared with risedronate and
alendronate, and this increase may lead to cancer cell apoptosis in
MDA-MB-231 cells; however, BPs were revealed to suppress cell
viability with minor effects on apoptosis. In summary, these
studies indicate that BPs exert direct anticancer effects by
inducing cancer cell apoptosis via several mechanisms; however, the
precise mechanisms of BPs on cancer cell apoptosis require further
investigation in future studies.
Cancer cell adhesion, invasion and metastasis are
essential steps in the metastatic process, which enable cancer
cells to establish persistence at the site of metastasis (37). Inhibition of the mevalonate pathway
by statins and BPs is thought to impair the adhesive abilities of
circulating cancer cells and thereby impact their metastatic
potential (38). Treatment with
statins has been demonstrated to cause a concentration-dependent
decrease in cholangiocarcinoma KKU-100 cell migration (22). As cancer cell migration also requires
digestion of the basement membrane by protease enzymes, this step
in the metastatic cascade represents a potential target for
reduction and/or inhibition (37).
BPs inhibit the activity of several MMPs with IC50
values in the range of 50–150 µM (39,40). The
present study identified that the three tested BPs decreased cancer
cell migration in a dose-dependent manner. Similar findings have
been reported by Stearns and Wang (41), who demonstrated that alendronate
reduces cellular secretion of MMP-2 and MMP-9 in PC3 prostate
cancer cells. Rac1 and CDC42 are both required by migrating cells,
and they act downstream of the mevalonate pathway (42). Statin- and BP-induced inhibition of
the mevalonate pathway therefore inhibits cancer cell
migration.
All three BPs tested in the present study exhibited
cytotoxic activity against human breast cancer MCF-7 cells. This
activity was partly attributed to cell cycle arrest and the
induction of apoptosis. The BPs also inhibited breast cancer
progression by inhibiting cyclin D1 expression and inducing p21,
caspase-3 and cytochrome c expression. Finally, the BPs were
identified to induce breast cancer cell apoptosis by stimulating
ROS formation and caspase-3 activity, and by decreasing
mitochondrial function. Additionally, BPs inhibited cancer cell
migration by suppressing MMP-9 expression in the culture medium.
The aforementioned findings indicate that BPs possess promising
activities against MCF-7 breast cancer cells and may represent a
new approach for breast cancer therapy. Caution regarding the
concentration of BPs used is required as permanent exposure to high
doses of BPs induces apoptosis in both tumor cells and osteogenic
precursors (43). However, high
concentrations are required as the cellular uptake of BPs is poor
in cells other than macrophages and osteoclasts, which has been
described for zoledronate in ovarian cancer cells (36,44).
In conclusion, alendronate, risedronate and
pamidronate demonstrated cytotoxic and antimigratory activity
against human breast cancer cells by inducing cell cycle arrest and
increasing cancer cell apoptosis. All BPs suppressed breast cancer
MCF-7 cell progression by inhibiting cyclin D1 and inducing p21,
caspase-3 and cytochrome c expression. Furthermore, BPs were
identified to induce ROS formation and caspase-3 activity, and
decrease mitochondrial function. BPs decreased cancer cell
migration via a reduction of MMP-9. In summary, these results
indicated that BPs may potentially serve as candidates for breast
cancer therapy and deserve to be further investigated in future
studies.
Acknowledgements
Not applicable.
Funding
This study was financially supported by the Office
of the Higher Education Commission (grant no. 2559A10962004), the
Thailand Research Fund (grant no. MRG6080071) and the National
Research Council of Thailand (grant no. 2559A10902073).
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
BB and SB designed the study. BB performed the
research and wrote the manuscript. All authors analyzed the data
and were involved in writing the manuscript. BB and SB read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Gnant M and Clézardin P: Direct and
indirect anticancer activity of bisphosphonates: A brief review of
published literature. Cancer Treat Rev. 38:407–415. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Labropoulou VT, Theocharis AD, Symeonidis
A, Skandalis SS, Karamanos NK and Kalofonos HP: Pathophysiology and
pharmacological targeting of tumor-induced bone disease: Current
status and emerging therapeutic interventions. Curr Med Chem.
18:1584–1598. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fleisch H: Development of bisphosphonates.
Breast Cancer Res. 4:30–34. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rogers MJ, Gordon S, Benford HL, Coxon FP,
Luckman SP, Monkkonen J and Frith JC: Cellular and molecular
mechanisms of action of bisphosphonates. Cancer. 88:2961–2978.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clezardin P: Potential anticancer
properties of bisphosphonates: Insights from preclinical studies.
Anticancer Agents Med Chem. 12:102–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jagdev SP, Coleman RE, Shipman CM, Rostami
HA and Croucher PI: The bisphosphonate, zoledronic acid, induces
apoptosis of breast cancer cells: Evidence for synergy with
paclitaxel. Br J Cancer. 84:1126–1134. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goffinet M, Thoulouzan M, Pradines A,
Lajoie-Mazenc I, Weinbaum C, Faye JC and Séronie-Vivien S:
Zoledronic acid treatment impairs protein geranyl-geranylation for
biological effects in prostatic cells. BMC Cancer. 6:602006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koshimune R, Aoe M, Toyooka S, Hara F,
Ouchida M, Tokumo M, Sano Y, Date H and Shimizu N: Anti-tumor
effect of bisphosphonate (YM529) on non-small cell lung cancer cell
lines. BMC Cancer. 7:82007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ou YJ, Chiu HF, Wong YH and Yang YH:
Bisphosphonate use and the risk of endometrial cancer: A
meta-analysis of observational studies. Pharmacoepidemiol Drug Saf.
25:1107–1115. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Passarelli MN, Newcomb PA, LaCroix AZ,
Lane DS, Ho GY and Chlebowski RT: Oral bisphosphonate use and
colorectal cancer incidence in the Women's Health Initiative. J
Bone Miner Res. 28:2043–2048. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo RT, Cao R, Liang PH, Ko TP, Chang TH,
Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, et al:
Bisphosphonates target multiple sites in both cis- and
trans-prenyltransferases. Proc Natl Acad Sci USA. 104:10022–10027.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mönkkönen H, Auriola S, Lehenkari P,
Kellinsalmi M, Hassinen IE, Vepsäläinen J and Mönkkönen J: A new
endogenous ATP analog (ApppI) inhibits the mitochondrial adenine
nucleotide translocase (ANT) and is responsible for the apoptosis
induced by nitrogen-containing bisphosphonates. Br J Pharmacol.
147:437–445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luckman SP, Hughes DE, Coxon FP, Graham R,
Russell G and Rogers MJ: Nitrogen-containing bisphosphonates
inhibit the mevalonate pathway and prevent post-translational
prenylation of GTP-binding proteins, including Ras. J Bone Miner
Res. 13:581–589. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wada A, Fukui K, Sawai Y, Imanaka K, Kiso
S, Tamura S, Shimomura I and Hayashi N: Pamidronate induced
anti-proliferative, apoptotic, and anti-migratory effects in
hepatocellular carcinoma. J Hepatol. 44:142–150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Coxon JP, Oades GM, Kirby RS and Colston
KW: Zoledronic acid induces apoptosis and inhibits adhesion to
mineralized matrix in prostate cancer cells via inhibition of
protein prenylation. BJU Int. 94:164–170. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wakchoure S, Merrell MA, Aldrich W,
Millender-Swain T, Harris KW, Triozzi P and Selander KS:
Bisphosphonates inhibit the growth of mesothelioma cells in vitro
and in vivo. Clin Cancer Res. 12:2862–2868. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Denoyelle C, Hong L, Vannier JP, Soria J
and Soria C: New insights into the actions of bisphosphonate
zoledronic acid in breast cancer cells by dual RhoA-dependent and
-independent effects. Br J Cancer. 88:1631–1640. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qie S and Diehl JA: Cyclin D1, cancer
progression, and opportunities in cancer treatment. J Mol Med
(Berl). 94:1313–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshida T, Zhang Y, Rivera Rosado LA, Chen
J, Khan T, Moon SY and Zhang B: Blockade of Rac1 activity induces
G1 cell cycle arrest or apoptosis in breast cancer cells through
downregulation of cyclin D1, survivin, and X-linked inhibitor of
apoptosis protein. Mol Cancer Ther. 9:1657–1668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rachner TD, Singh SK, Schoppet M, Benad P,
Bornhäuser M, Ellenrieder V, Ebert R, Jakob F and Hofbauer LC:
Zoledronic acid induces apoptosis and changes the TRAIL/OPG ratio
in breast cancer cells. Cancer Lett. 287:109–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buranrat B, Senggunprai L, Prawan A and
Kukongviriyapan V: Simvastatin and atorvastatin as inhibitors of
proliferation and inducers of apoptosis in human cholangiocarcinoma
cells. Life Sci. 153:41–49. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Porter AP, Papaioannou A and Malliri A:
Deregulation of Rho GTPases in cancer. Small GTPases. 7:123–138.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orgaz JL, Herraiz C and Sanz-Moreno V: Rho
GTPases modulate malignant transformation of tumor cells. Small
GTPases. 5:e290192014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mahtani R and Jahanzeb M: Bisphosphonates
as anticancer therapy for early breast cancer. Clin Breast Cancer.
10:359–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sahai E and Marshall CJ: RHO-GTPases and
cancer. Nat Rev Cancer. 2:133–142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cayrol C, Knibiehler M and Ducommun B: p21
binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient
cells. Oncogene. 16:311–320. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brugarolas J, Chandrasekaran C, Gordon JI,
Beach D, Jacks T and Hannon GJ: Radiation-induced cell cycle arrest
compromised by p21 deficiency. Nature. 377:552–557. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Niculescu AB III, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: pRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lung JC, Chu JS, Yu JC, Yue CT, Lo YL,
Shen CY and Wu CW: Aberrant expression of cell-cycle regulator
cyclin D1 in breast cancer is related to chromosomal genomic
instability. Genes Chromosomes Cancer. 34:276–284. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nelsen CJ, Kuriyama R, Hirsch B, Negron
VC, Lingle WL, Goggin MM, Stanley MW and Albrecht JH: Short term
cyclin D1 overexpression induces centrosome amplification, mitotic
spindle abnormalities, and aneuploidy. J Biol Chem. 280:768–776.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sandor V, Senderowicz A, Mertins S,
Sackett D, Sausville E, Blagosklonny MV and Bates SE: P21-dependent
g(1)arrest with downregulation of cyclin D1 and upregulation of
cyclin E by the histone deacetylase inhibitor FR901228. Br J
Cancer. 83:817–825. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ and
Los M: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Senaratne SG, Pirianov G, Mansi JL, Arnett
TR and Colston KW: Bisphosphonates induce apoptosis in human breast
cancer cell lines. Br J Cancer. 82:1459–1468. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ebert R, Meissner-Weigl J, Zeck S, Määttä
J, Auriola S, Coimbra de Sousa S, Mentrup B, Graser S, Rachner TD,
Hofbauer LC and Jakob F: Probenecid as a sensitizer of
bisphosphonate-mediated effects in breast cancer cells. Mol Cancer.
13:2652014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morrissey MA, Hagedorn EJ and Sherwood DR:
Cell invasion through basement membrane: The netrin receptor DCC
guides the way. Worm. 2:e261692013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Green JR: Antitumor effects of
bisphosphonates. Cancer. 97:840–847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boissier S, Ferreras M, Peyruchaud O,
Magnetto S, Ebetino FH, Colombel M, Delmas P, Delaissé JM and
Clézardin P: Bisphosphonates inhibit breast and prostate carcinoma
cell invasion, an early event in the formation of bone metastases.
Cancer Res. 60:2949–2954. 2000.PubMed/NCBI
|
|
40
|
Teronen O, Heikkilä P, Konttinen YT,
Laitinen M, Salo T, Hanemaaijer R, Teronen A, Maisi P and Sorsa T:
MMP inhibition and downregulation by bisphosphonates. Ann N Y Acad
Sci. 878:453–465. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stearns ME and Wang M: Alendronate blocks
metalloproteinase secretion and bone collagen I release by PC-3 ML
cells in SCID mice. Clin Exp Metastasis. 16:693–702. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Raftopoulou M and Hall A: Cell migration:
Rho GTPases lead the way. Dev Biol. 265:23–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Idris AI, Rojas J, Greig IR, Van't Hof RJ
and Ralston SH: Aminobisphosphonates cause osteoblast apoptosis and
inhibit bone nodule formation in vitro. Calcif Tissue Int.
82:191–201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shmeeda H, Amitay Y, Gorin J, Tzemach D,
Mak L, Ogorka J, Kumar S, Zhang JA and Gabizon A: Delivery of
zoledronic acid encapsulated in folate-targeted liposome results in
potent in vitro cytotoxic activity on tumor cells. J Control
Release. 146:76–83. 2010. View Article : Google Scholar : PubMed/NCBI
|