Introduction
Liver cancer is among the most prevalent
malignancies in the world (1); it
has a high degree of malignancy and a poor prognosis. However,
relatively limited treatment approaches are available in the
clinic. Therefore, there is an urgent need to identify novel
therapeutic targets and to develop effective strategies for liver
cancer therapy. Emerging evidence has demonstrated a key role for
epigenetics in tumorigenesis (2–4). The
occurrence and development of liver cancer is also dependent on
epigenetics-related disorders of various signal transduction
pathways (5). Epigenetic regulation
is defined as genomic modifications of heritable factors that do
not involve alterations in the DNA sequence and affect the
transcriptional activity of genes instead (6). Several mechanisms of epigenetic
regulation, including histone modification, noncoding RNAs and DNA
methylation, have been extensively studied (7). Among these mechanisms, histone
modification is one of the important modes of epigenetic regulation
in liver cancer (8).
Histones are well-conserved and highly alkaline
proteins, which appear as an octameric core and help to package and
order DNA into nucleosomes. The N-terminal tails of histones extend
beyond the nucleosome and undergo a variety of post-translational
modifications, such as acetylation, methylation, ubiquitination and
phosphorylation (9). Acetylation of
histones is mediated through histone acetylases (HATs) and histone
deacetylases (HDACs) (10–12). Some studies have unveiled the
importance of the balance of HATs and HDACs for the expression of
several key genes, including p21, Fas, Bcl-2 and Bax (13). Once this balance is broken, an
imbalance of gene transcription occurs, which may lead to
tumorigenesis or abnormal cell proliferation. Moreover, the high
expression of HDAC family members and the downregulation of histone
acetylation in liver cancer tissues have been confirmed in certain
studies (14,15). Therefore, the inhibition of HDAC
activity by an HDAC inhibitor (HDACi) is expected to be an
effective approach for the treatment of liver cancer.
Suberoylanilide hydroxamic acid (SAHA), the first
HDACi approved for clinical use by the US Food and Drug
Administration, has been widely acknowledged for its antitumor
activity (16). It has been reported
that SAHA decreases proliferation, induces differentiation and
elicits apoptosis in many tumor cells during in vitro
culturing (17–19). Additionally, SAHA has been
demonstrated to be a potential inducer of endoplasmic reticulum
(ER) stress that led to induction of apoptosis in lung cancer
cells, mediated by the activation of the ER stress-mediated
apoptotic signaling pathway (20).
However, the effects of SAHA in liver cancer and the mechanism of
SAHA in regulating ER stress are largely unknown.
In the present study, the effects of SAHA on
apoptosis in the HepG2 liver cancer cells and the potential
mechanisms involved in histone acetylation and ER stress were
explored. Following treatment with various doses of SAHA, HepG2
cells were subjected to an apoptosis assay. Furthermore, the levels
of the ER-stress related molecules 78 kDa glucose-regulated protein
(GRP78), activating transcription factor 4 (ATF4) and
C/EBP-homologous protein (CHOP), as well as the acetylation levels
of the histone proteins H4 (acH4), H4-lysine 5 (H4K5) and H4-lysine
12 (H4K12) were quantitated. The effects of SAHA on acetylation of
H4 in the promoter regions of GRP78, ATF4 and CHOP were evaluated
by chromatin immunoprecipitation (ChIP) experiments.
Materials and methods
Cell culture
The HepG2 liver cancer cell line (cat. no. KCB
200507YJ) was purchased from the cell bank of the Type Culture
Collection of the Chinese Academy of Sciences. The HepG2 cells were
authenticated by short tandem repeat DNA profiling analysis and
tested to be free of mycoplasma and other contaminants by the
vendor. The cells were maintained in Dulbecco's modified Eagle's
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal bovine serum (ScienCell Research Laboratories, Inc.), 100
U/ml penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) and grown in a humidified atmosphere with 5%
CO2 at 37°C. The cells were treated with SAHA (Abcam) or
mock-treated with DMSO (Sigma-Aldrich; Merck KGaA), as
indicated.
Real-time cellular analysis
Cell proliferation was monitored using an
xCELLigence Real-Time Cell Analysis instrument (xCELLigence DP
System; ACEA Biosciences, Inc.), which can continuously monitor
live cell proliferation, morphology and viability with a label-free
assay. The experimental procedures used were as previously
described (21,22). Briefly, HepG2 cells were seeded at a
density of 1×104 cells/well in a 16-well µl plate
(E-plate 16), cultured in complete medium supplemented with various
doses of SAHA (0, 0.1, 1, 6, 12, 25, 50 and 100 µM), and placed at
37°C in a humidified incubator containing 5% CO2. The
effects of the various doses of SAHA on the proliferation of HepG2
cells were continuously monitored for 90 h. The cell index is a
dimensionless parameter that is translated from the electrical
impedance measured to denote cell proliferation.
Apoptosis assay
HepG2 cells (1×106) were seeded onto
10-cm diameter dishes and treated with various doses of SAHA (0, 1,
6 and 12 µM) for 48 h. The cells were harvested and subjected to an
apoptosis assay using an annexin V/propidium iodide (PI) cell
apoptosis detection kit (Nanjing KeyGen Biotech Co., Ltd.),
according to the manufacturer's instructions. Briefly, the cells
were washed with cold phosphate-buffered saline (PBS). After
centrifugation at 110 g for 5 min, the cell pellet was resuspended
in 1 ml PBS (final concentration ~1–5×105 cells/ml).
After washing with 1X annexin V binding solution, the cells were
stained with 5 µl of fluorescein isothiocyanate (FITC)-conjugated
annexin V and 10 µl PI staining solution at room temperature for 20
min. After washing once with the 1X annexin V binding solution, the
labeled cells (1×104 cells) were detected immediately by
a flow cytometer (FACSCalibur™; BD Biosciences). The data were
analyzed by BD CellQuest Pro software (version 1.0; BD
Biosciences). The apoptotic rate was defined as the percentage of
FITC-annexin V single positive cells (early apoptotic cells) among
total cells.
Western blot analysis
HepG2 cells (1×106) were seeded onto
10-cm diameter dishes and treated at various doses of SAHA (0, 1, 6
and 12 µM) for 48 h. The cells were then lysed with 0.2 ml of
radioimmunoprecipitation assay buffer (Thermo Fisher Scientific,
Inc.). Total protein samples (60 µg) were separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (10% gels),
transferred to a polyvinylidene difluoride membrane and blocked
with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween
20 (TBST) for 60 min at room temperature. The membrane was
incubated overnight at 4°C with the following primary antibodies:
acH4 (1:1,000), acH4K5 (1:5,000), acH4K12 (1:1,000), GRP78
(1:1,500), PERK (1:1,500), phosphorylated (p)-PERK (1:1,500), ATF4
(1:1,500) and CHOP (1:1,500). After washing with TBST buffer, the
membrane was incubated with the peroxidase-conjugated anti-rabbit
secondary antibody (1:4,000; Abcam; cat. no. ab205718) for 90 min
at room temperature. Following treatment with enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Inc.), the
Image Lab software (version 4.1; Bio-Rad Laboratories, Inc.) was
used to quantitate the band intensities. The antibodies against
β-actin (cat. no. ab8227), GRP78 (cat. no. ab21685), ATF4 (cat. no.
ab184909), CHOP (cat. no. ab10444), PERK (cat. no. ab65142), acH4K5
(cat. no. ab51997) and acH4K12 (cat. no. ab46983) were purchased
from Abcam. The antibody against acH4 (cat. no. 39925) was
purchased from Active Motif, Inc. the antibody against p-PERK (cat.
no. AF4499) was purchased from Affinity Biosciences.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was isolated from HepG2 cells using a
TRIzol RNA isolation kit (Thermo Fisher Scientific, Inc.) and the
purity and concentration of the RNA samples were determined by a
DN2000 ultramicro nucleic acid analyzer (Thermo Fisher Scientific,
Inc.). The total RNA was reverse transcribed into cDNA using a
First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc),
according to the manufacturer's instructions. RT-qPCR reactions
were performed as follows: 25°C for 10 min, 48°C for 60 min and
95°C for 5 min. The qPCR was performed using SYBR green mix from
Thermo Fisher Scientific, Inc. The reaction conditions included an
initial pre-denaturation step at 95°C for 30 sec, followed by 40
cycles of thermal steps consisting of 95°C for 5 sec and 60°C for
30 sec. β-Actin was used as an internal control. The fold-change
was calculated by the 2−∆∆Cq method (23). The primer sequences used for each
gene are shown in Table I.
 | Table I.DNA sequences of primers used for RT-
and ChIP-quantitative PCR. |
Table I.
DNA sequences of primers used for RT-
and ChIP-quantitative PCR.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| RT-qPCR |
|
|
|
GRP78 |
GCCTGTATTTCTAGACCTGCC |
TTCATCTTGCCAGCCAGTTG |
|
PERK |
CTCACAGGCAAAGGAAGGAG |
AACAACTCCAAAGCCACCAC |
|
ATF4 |
GACCGAAATGAGCTTCCTGA |
ACCCATGAGGTTTGAAGTGC |
|
CHOP |
CTGCTTCTCTGGCTTGGCTG |
GCTCTGGGAGGTGCTTGTGA |
|
β-actin |
GCACCCAGCACAATGAAGAT |
ACTCCTGCTTGCTGATCCAC |
| ChIP-qPCR |
|
|
|
GRP78 |
GGGATGGAGGAAGGGAGAAC |
GAGGCATTTCCGCTGGTAAC |
|
ATF4 |
GGTGGGTTCCATGGTCAAAT |
AACACATCCACCACTGC |
|
CHOP |
CACGACCTCAGCCTGTCAAG |
ACTGGAGTGGTGTGGCAATG |
ChIP-qPCR
HepG2 cells (1×106) were seeded onto
10-cm diameter dishes and were mock-treated with DMSO (0 µM SAHA)
or treated with 6 µM SAHA for 36 h. Following treatment, the cells
were subjected to a ChIP assay using a ChIP kit (Sigma-Aldrich;
Merck KGaA), according to the manufacturer's instructions. The
HepG2 cells were cross-linked with 1% formaldehyde and lysed with
600 µl of radioimmunoprecipitation assay lysis buffer (Thermo
Fisher Scientific, Inc.), and the lysates were sonicated to
fragment the DNA into lengths between 200–1,000 base pairs.
Immunoprecipitation was performed with the following antibodies:
Anti-acH4 (1:50; Active Motif, Inc.; cat. no. 39925), rabbit IgG
(1:50; Abcam; cat. no. ab205718) and anti-RNA Polymerase II (1:50;
Abcam; cat. no. ab51462) at 4°C overnight. Protein G-sepharose
beads were used for immunoprecipitation, and the immunoprecipitates
were washed and eluted, according to the manufacturer's protocol.
The immunoprecipitated DNA was recovered by reversing the
cross-linking, and were purified and dissolved in distilled water.
A corresponding sample handled without the addition of any antibody
served as an input control. The ChIP DNA and input DNA were
analyzed by qPCR. The abundance of the immunoprecipitated target
DNA was expressed as a percentage of the input chromatin DNA. The
primers used for detecting the promoter regions of the GRP78, ATF4
and CHOP genes are listed in Table
I.
Statistical analysis
SPSS version 20 statistical software (IBM Corp.) was
used for statistical analysis. Data were expressed as the mean ±
SD. The one-way analysis of variance method was used for the
multivariate comparison, and the least significant difference
method was used as a post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
SAHA inhibits the proliferation of
HepG2 cells in a dose- and time-dependent manner
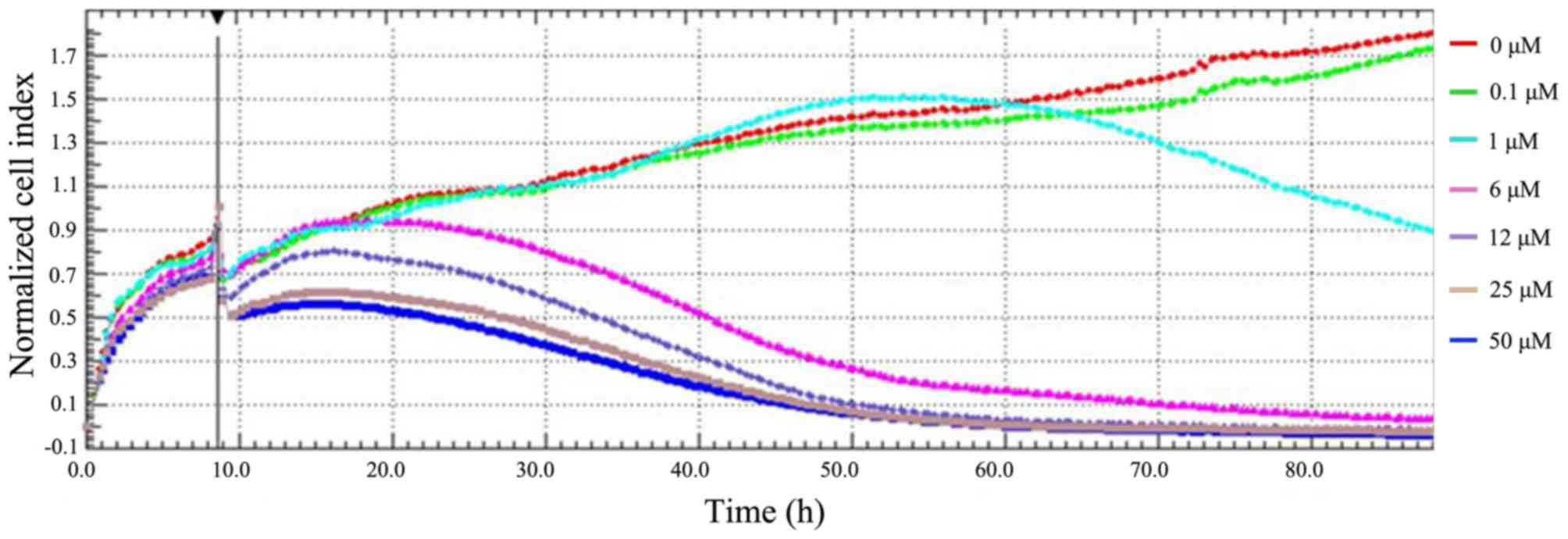
Firstly, the effect of SAHA on the proliferation of
HepG2 cells was determined by real-time cellular analysis. This
involved live monitoring of cell proliferation in a label-free
manner. The electrical impedance of adherent cells was detected by
the electrode at the bottom of an E-plate 16, and the cell index is
a dimensionless parameter that was translated from the measured
electrical impedance to denote cell proliferation. After HepG2
cells were incubated for 10 h to allow cell adhesion, SAHA was
added at various doses in the culture medium to treat cells. This
resulted in a rapid decrease in HepG2 cell proliferation at 10 h
(Fig. 1), which was probably due to
the transient effect of SAHA vehicle DMSO on cell proliferation.
Compared with the mock-treated group (0 µM SAHA), treatment with
SAHA at concentrations >1 µM resulted in a notable inhibitory
effect on the proliferation of HepG2 cells (Fig. 1). Furthermore, SAHA at 6 µM resulted
in significantly (P<0.01) decreased normalized cell index, by
>70%, at 50 h after treatment. A significant (P<0.01)
suppression of proliferation was observed in HepG2 cells treated
with higher doses of SAHA (12, 25 and 50 µM) for longer periods of
time. Thus, the antiproliferative effect of SAHA on HepG2 cells
occurs in a dose- and time-dependent manner.
SAHA induces apoptosis in HepG2 cells
in a dose-dependent manner
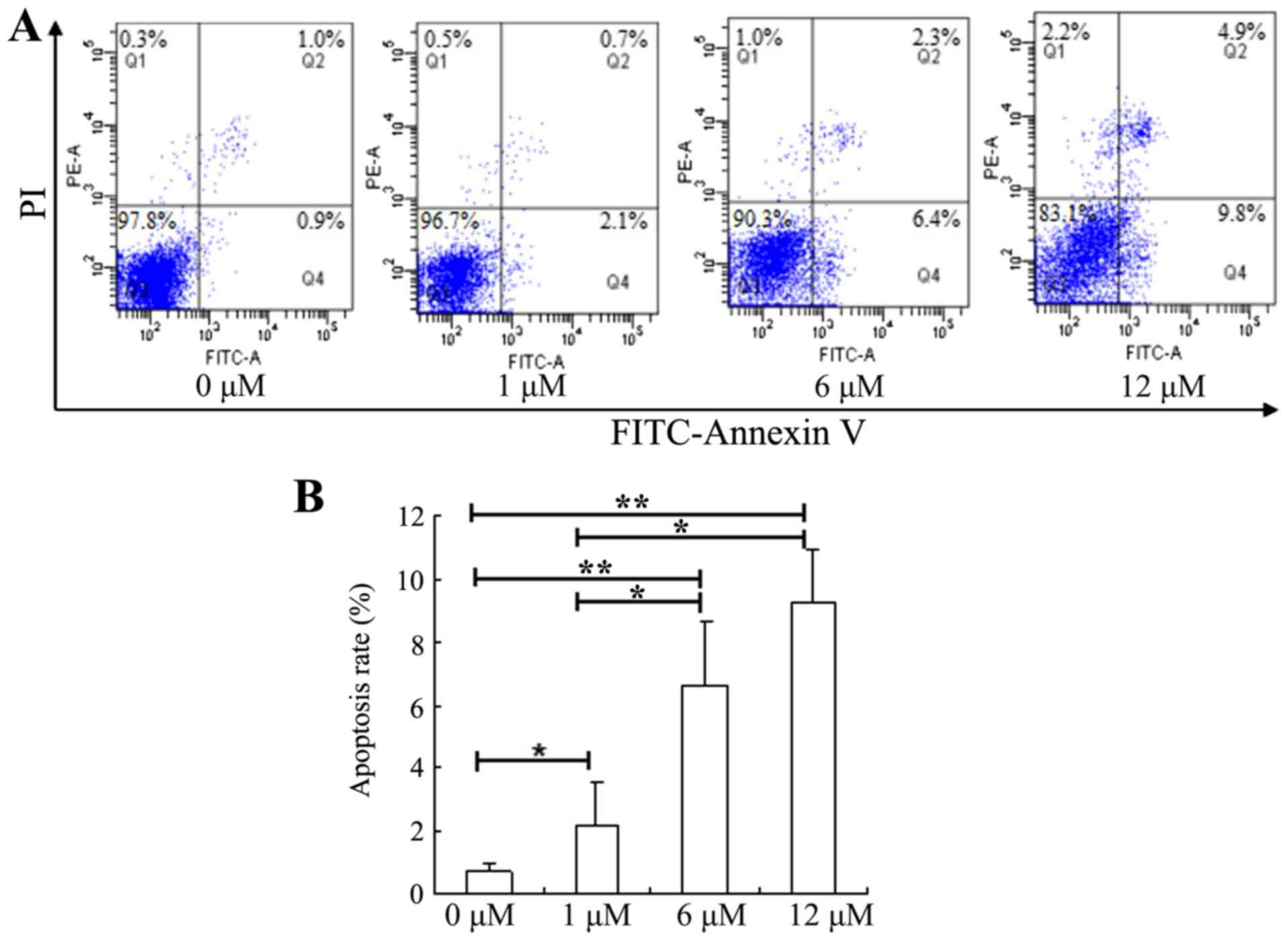
The effect of SAHA on apoptosis in HepG2 cells was
investigated by annexin V-FITC and PI staining. Following treatment
with SAHA for 48 h, the apoptotic rates were significantly elevated
in HepG2 cells treated with 1, 6 or 12 µM SAHA, compared with the
untreated cells (Fig. 2). While the
control group (0 µM SAHA) had an average apoptotic rate of ~1%, 12
µM SAHA led to an increased apoptotic cell population to ~10%.
These results indicate that SAHA could significantly promote
apoptosis in HepG2 cells in a dose-dependent manner.
SAHA induces ER stress-mediated
apoptotic signaling pathway in HepG2 cells
Since SAHA was revealed to be a potential inducer of
ER stress, the effect of SAHA on ER stress in HepG2 cells was
determined by measuring the levels of ER stress-associated
molecules, including GRP78, PERK, p-PERK, ATF4 and CHOP. No
significant changes were observed in the expression of GRP78 at
either the mRNA (Fig. 3A) or protein
level (Fig. 3B and C) in cells
treated with 1 µM SAHA, whereas cells treated with 6 and 12 µM SAHA
had significantly increased levels of GRP78 mRNA and protein
compared with control cells (0 µM SAHA). No significant changes in
the level of PERK mRNA were observed following treatment with any
of the tested doses of SAHA (Fig.
3A). However, the expression of PERK protein was significantly
decreased in the HepG2 cells treated with different concentrations
of SAHA, while the level of p-PERK protein was significantly
increased (Fig. 3B and C), which
resulted in an increased ratio of p-PERK/PERK following SAHA
treatments (Fig. 3D). Notably,
following treatment with SAHA, the mRNA and protein expression
levels of ATF4 and CHOP were significantly elevated in a
dose-dependent manner (Fig. 3A and
C). Moreover, the protein levels of ATF4 and CHOP in the HepG2
cells were significantly upregulated by over 3-fold following
treatment with 12 µM SAHA.
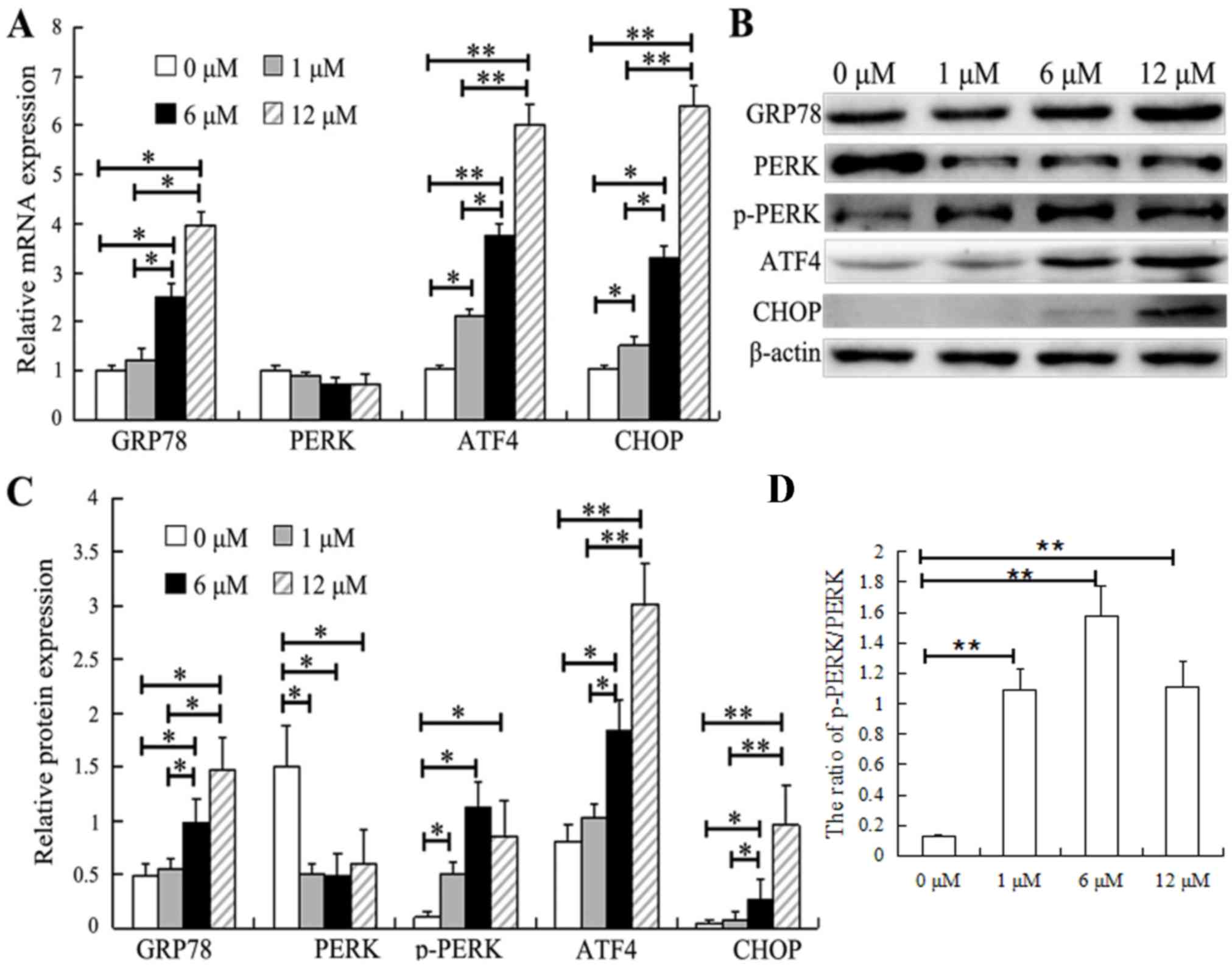 | Figure 3.SAHA induces ER stress in HepG2
cells. (A) HepG2 cells were treated with SAHA at the indicated
concentrations for 48 h, and the mRNA levels of endoplasmic
reticulum stress-associated genes (GRP78, PERK, ATF4 and CHOP) were
analyzed by reverse transcription-quantitative PCR. (B and C)
Representative images (one of three experiments) showing the (B)
western blot analysis of GRP78, PERK, p-PERK, ATF4 and CHOP protein
expression in HepG2 cells exposed to SAHA at the indicated
concentrations and (C) their relative protein expression levels.
(D) Relative ratio of p-PERK/PERK. N=3. *P<0.05, **P<0.01.
SAHA, suberoylanilide hydroxamic acid; GRP78, 78 kDa
glucose-regulated protein; ATF4, activating transcription factor 4;
CHOP, C/EBP-homologous protein; PERK, PRKR-like endoplasmic
reticulum kinase; p, phosphorylated. |
SAHA significantly upregulates the
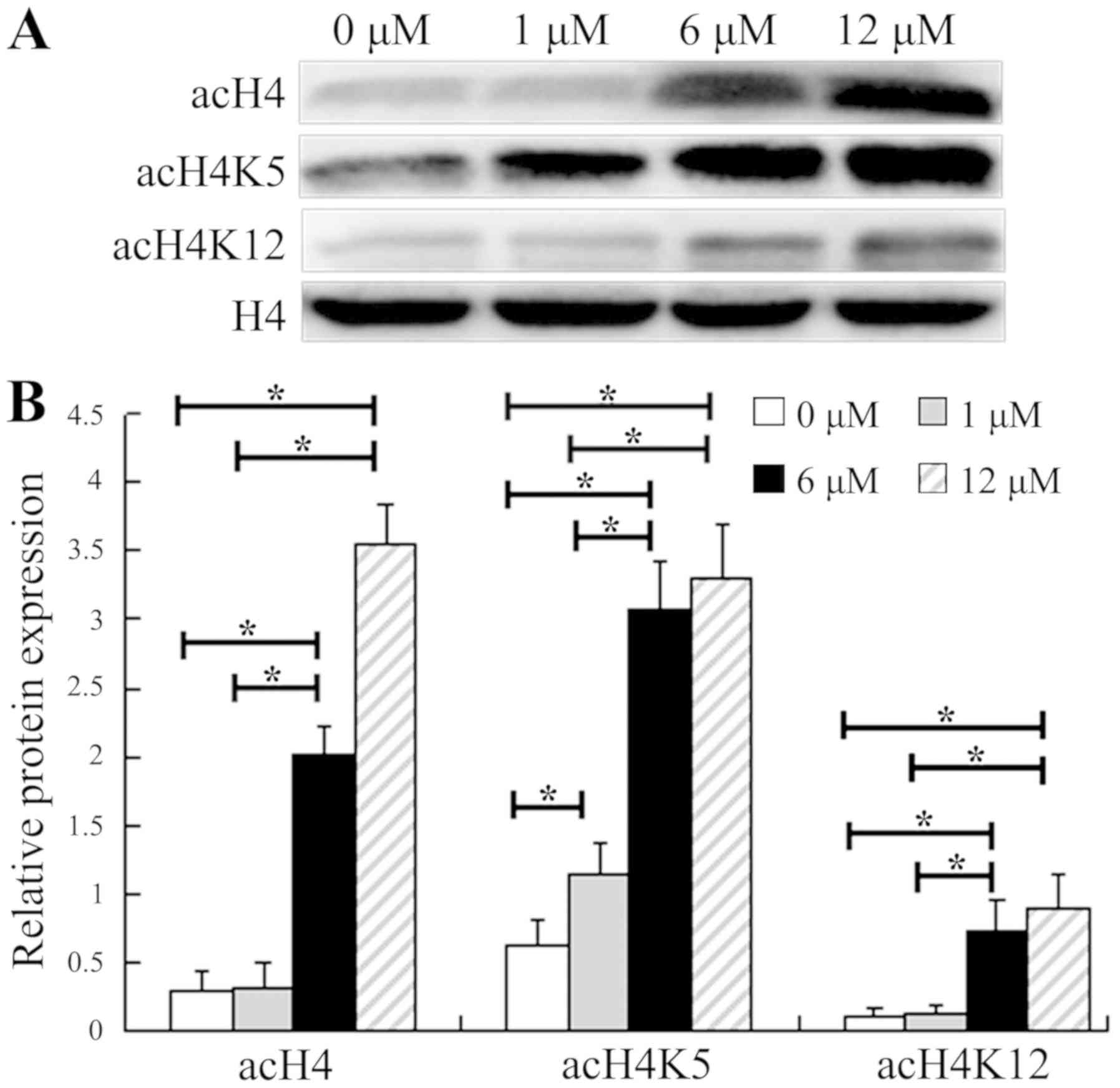
level of acH4 in HepG2 cells
The role of SAHA, as an HDACi, in the acetylation of
histone H4 was determined by western blot analysis. The protein
levels of total acH4, acH4K5 and acH4K12 were detected in HepG2
cells treated at various doses of SAHA for 48 h (Fig. 4). Compared with the control group (0
µM SAHA), the levels of acH4 and acH4K12 were not significantly
increased in the HepG2 cells treated with 1 µM SAHA, whereas the
level of acH4K5 was significantly increased; these findings were
likely due to the varied sensitivities of histone deacetylases to
inhibition by SAHA. In cells treated with 6 and 12 µM SAHA, the
levels of total acH4, acH4K5 and acH4K12 were all significantly
higher than in those treated with 0 and 1 µM SAHA. These results
suggest that 6 µM SAHA was sufficient to markedly elevate the
acetylation of histone H4.
SAHA treatment enhances the
acetylation level of histone H4 in the promoter regions of the
GRP78, ATF4 and CHOP genes in HepG2 cells
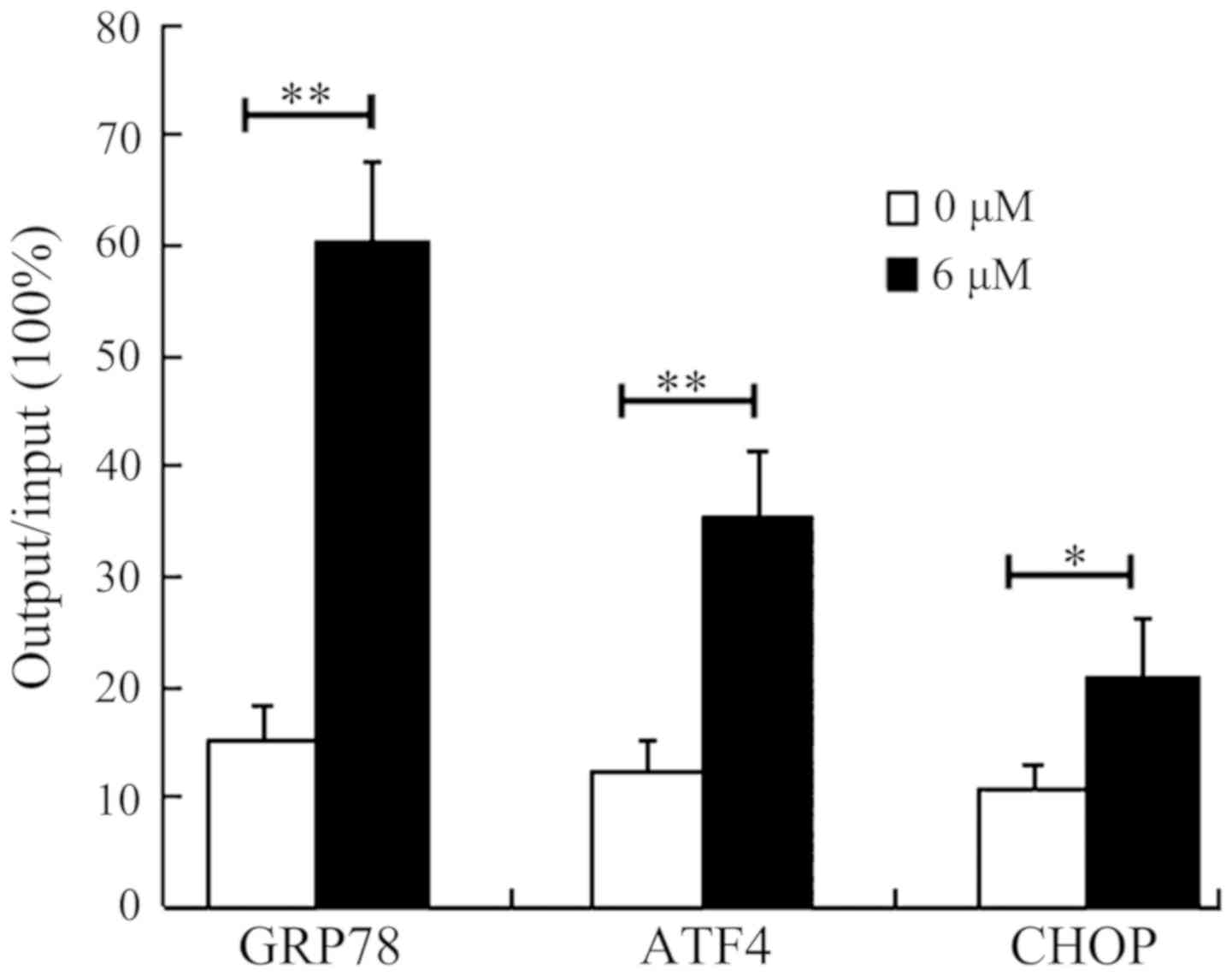
In order to confirm whether the regulation of
transcription of GRP78, ATF4 and CHOP genes by SAHA is mediated by
the upregulation of acH4, ChIP assays were conducted. This involved
determination and quantitation of the acH4-associated promoter
regions of these genes in HepG2 cells (Fig. 5). Following treatment with DMSO (0 µM
SAHA) or 6 µM SAHA for 36 h, the HepG2 cells were lysed and
immunoprecipitated with specific anti-acH4 antibody. qPCR results
demonstrated significant increase in the acH4-associated promoter
regions of the GRP78, ATF4 and CHOP genes in HepG2 cells treated
with 6 µM SAHA. The promoter regions of ATF4 and CHOP were
increased by 2-fold and 3-fold, respectively, the promoter region
of GRP78 was enriched by ~4-fold (the output to input ratio
increased from ~15 to ~60%). These results confirmed that SAHA
induces the expression of ER stress-associated molecules by
increasing their transcription, at least partially through
enhancing the acetylation of histone H4 in the promoter regions of
these genes.
Discussion
Epigenetic therapy using an HDACi, alone or in
combination with other treatments, has shown therapeutic potential
in clinical trials for the treatment of several types of cancer,
including non-small cell lung cancer and breast cancer (24,25). As
an HDACi, SAHA specifically induces tumor cells to undergo
apoptosis by regulating the expression of key genes involved in
apoptotic signaling pathways (26).
The present study revealed that SAHA induced apoptosis in HepG2
liver cancer cells by activating the ER stress-mediated apoptotic
signaling pathway, at least partially through enhancing the
acetylation of histone H4 on the promoter regions of ER-stress
associated genes, including GRP78, ATF4 and CHOP. This suggests
that ER stress-mediated apoptotic signaling may play a central role
in HDACi-induced apoptosis of HepG2 cells and that SAHA alone or in
combination with inducers of ER stress can potentially be applied
in patients with liver cancer.
SAHA-induced apoptosis has been demonstrated to be
associated with activation of the intrinsic apoptotic pathways
(27). It can be concluded that in
tumor cells exposed to SAHA, proapoptotic genes (such as Bax and
Bim) are upregulated, whereas the expression levels of
antiapoptotic genes (such as Bcl-2 and Bcl-XL) are suppressed
(28). SAHA has also been shown to
influence the expression of death receptors (such as Fas and
TNF-related apoptosis-inducing ligand receptor) and death receptor
ligands in leukemia, which are responsible for the extrinsic
apoptotic pathways (29). SAHA was
also demonstrated to activate the ER stress-associated apoptotic
signaling pathway. Kim et al (30) demonstrated increased expression
levels of p-PERK, ATF4 and CHOP in papillary thyroid cancer and
anaplastic thyroid cancer cells treated with SAHA, and increased
apoptotic rates of these cells. However, the mechanism of SAHA on
regulating the expression of the ER stress signaling
pathway-related molecules remains unclear.
Certain studies have shown the inhibitory effect of
SAHA on the proliferation of liver cancer cells and the induction
of apoptosis (31,32). Furthermore, another study
demonstrated significant inhibition of proliferation of MHCC97L
hepatocellular carcinoma cells in vitro by SAHA (Cai et
al, unpublished data). In the present study, SAHA was
demonstrated to suppress cell proliferation and induce apoptosis in
a dose-dependent manner in HepG2 cells. Notably, the expression of
GRP78 at both the mRNA and protein levels in HepG2 cells was
significantly increased following treatment with 6 or 12 µM SAHA.
Since GRP78 is a protein marker of ER stress (33), its increased expression suggests that
SAHA may induce ER stress in HepG2 cells. In addition, it was found
that the mRNA level of PERK was unaffected by SAHA treatment,
indicating that SAHA does not regulate the expression of PERK at
the transcriptional level. Nevertheless, treatment with SAHA
resulted in significantly decreased expression of PERK and
increased expression of p-PERK in HepG2 cells at protein level.
This may be because SAHA-induced HepG2 cells undergo ER stress,
upon which GRP78 changes its binding preference towards
unfolded/misfolded proteins accumulated in the ER, resulting in the
release of PERK (34). Following the
release from GRP78, PERK may undergo self-phosphorylation and
dimerization (35–37), which may lead to a decreased level of
PERK protein and an increased level of p-PERK protein. Studies have
demonstrated that p-PERK can upregulate the expression levels of
ATF4 and CHOP in cells and activate the ER stress-induced apoptotic
signaling pathway (38). In the
present study, the increased expression of ATF4 and CHOP, at both
the mRNA and protein levels, and augmented apoptosis were observed
in HepG2 cells. These results suggest that induction of apoptosis
in HepG2 cells by SAHA can be partially attributed to the
activation of the PERK-ATF4-CHOP signaling pathway. However, it is
worth noting that 12 µM SAHA resulted in significantly decreased
proliferation of HepG2 cells (Fig.
1), however apoptosis was only induced in 10% of HepG2 cells
treated for 48 h (Fig. 2). This
suggests that SAHA may also induce cell death in other ways to
inhibit tumor cell proliferation. For example, another study
demonstrated that SAHA induced autophagic death of HepG2 cells
in vitro (Cai et al, unpublished data).
The present study also unveiled an unappreciated
mechanism of SAHA-mediated activation of the PERK-ATF4-CHOP
signaling pathway in HepG2 cells. It was inferred that this may be
associated with the essential role of SAHA, as an HDACi, in the
upregulation of histone acetylation. Changes in histone acetylation
influence chromatin condensation, and these alterations affect gene
transcription (39). In previous
studies, it was found that SAHA significantly upregulated the
acetylation levels of histones H3K9 and H3K27 in HepG2 cells
(40). In the present study, SAHA
was identified to be able to upregulate the acetylation levels of
histones H4, H4K5 and H4K12, at concentrations of 6 and 12 µM. In
addition, it was reported that the acetylation levels of histone H3
in the promoter region of the GRP78 gene was significantly
increased, following treatment with an HDACi and thereby promoting
the genetic transcription of GRP78 (41). However, it was not clear whether the
increased acetylation level of H4 regulates the transcription of
genes associated with the ER stress-mediated apoptotic signaling
pathway. The ChIP-qPCR results in the present study suggest that
the acetylation levels of histone H4 in the gene promoter regions
of GRP78, ATF4 and CHOP were all significantly increased, which was
associated with significantly increased mRNA levels of these
genes.
In conclusion, the HDACi SAHA induces apoptosis in
HepG2 cells by activating the ER stress-mediated apoptotic pathway,
at least partially through upregulation of the acH4 level on the
promoter regions of the ER stress-associated molecules GRP78, ATF4
and CHOP. Further studies will be conducted to investigate the
effect of acetylation modification at different sites of H4 on the
expression of molecules associated with the ER stress-mediated
apoptotic pathway. In addition, a study that aims to demonstrate
whether SAHA has the same inhibitory effect in a mouse xenograft
model in vivo will also be conducted. Overall, the present
study underscores the critical roles of ER stress in mediating
apoptosis in HepG2 cells and also suggests the potential
application of SAHA and other inducers of ER stress for the
treatment of patients with liver cancer.
Acknowledgements
The authors would like to thank Mrs Xue Shen
(Department of Central Laboratory, The Affiliated Hospital of
Guizhou Medical University) for technical help and advice on the
flow cytometry experiments, the Basic Medical Science Research
Center of Guizhou Medical University for providing the xCELLigence
Real-Time Cell Analysis instrument, and Dr Tengxiang Chen
(Department of Physiology, College of Basic Medical Sciences,
Guizhou Medical University) for critical reading of the manuscript
and suggestions.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81560105), the
Foundation of the Department of Science and Technology [grant no.
LH (2014) 7074] and the Natural Science Foundation of the
Department of Education [grant no. KY (2014) 269].
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LY, TT, LZ and LT performed the majority of the
experiments. BH, SC, ZM and TY provided analytical tools and
performed the statistical analysis. RX and QY designed the study.
RX and BH provided financial support for this work and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Buurman R, Sandbothe M, Schlegelberger B
and Skawran B: HDAC inhibition activates the apoptosome via Apaf1
upregulation in hepatocellular carcinoma. Eur J Med Res. 21:262016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen QW, Zhu XY, Li YY and Meng ZQ:
Epigenetic regulation and cancer (review). Oncol Rep. 31:523–532.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muhammad JS, Khan MR and Ghias K: DNA
methylation as an epigenetic regulator of gallbladder cancer: An
overview. Int J Surg. 53:178–183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shanmugam MK, Arfuso F, Arumugam S,
Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS,
Sethi G and Lakshmanan M: Role of novel histone modifications in
cancer. Oncotarget. 9:11414–11426. 2017.PubMed/NCBI
|
|
5
|
Khan FS, Ali I, Afridi UK, Ishtiaq M and
Mehmood R: Epigenetic mechanisms regulating the development of
hepatocellular carcinoma and their promise for therapeutics.
Hepatol Int. 11:45–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Yan L, Zhang Z, Prado E, Fu L, Xu
X and Du L: Epigenetic regulation and its therapeutic potential in
pulmonary hypertension. Front Pharmacol. 9:2412018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng L and Zhong X: Epigenetic regulation
of drug metabolism and transport. Acta Pharm Sin B. 5:106–112.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu KY, Wang LT and Hsu SH: Modification
of epigenetic histone acetylation in hepatocellular Carcinoma.
Cancers (Basel). 10(pii): E82018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reddy D, Khade B, Pandya R and Gupta S: A
novel method for isolation of histones from serum and its
implications in therapeutics and prognosis of solid tumours. Clin
Epigenetics. 9:302017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vahid F, Zand H, Nosrat-Mirshekarlou E,
Najafi R and Hekmatdoost A: The role dietary of bioactive compounds
on the regulation of histone acetylases and deacetylases: A review.
Gene. 562:8–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schneider A, Chatterjee S, Bousiges O,
Selvi BR, Swaminathan A, Cassel R, Blanc F, Kundu TK and Boutillier
AL: Acetyltransferases (HATs) as targets for neurological
therapeutics. Neurotherapeutics. 10:568–588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou Y, Peng J and Jiang S: Role of
histone acetyltransferases and histone deacetylases in adipocyte
differentiation and adipogenesis. Eur J Cell Biol. 93:170–177.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chrun ES, Modolo F and Daniel FI: Histone
modifications: A review about the presence of this epigenetic
phenomenon in carcinogenesis. Pathol Res Pract. 213:1329–1339.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanno K, Kanno S, Nitta H, Uesugi N, Sugai
T, Masuda T, Wakabayashi G and Maesawa C: Overexpression of histone
deacetylase 6 contributes to accelerated migration and invasion
activity of hepatocellular carcinoma cells. Oncol Rep. 28:867–873.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu W, Xiao J, Lan J, et al: The effects
of histone acetylation on the migration and invasion of
hepatocellular carcinoma cells. J Guizhou Med Univ. 42:1365–1369.
2017.(In Chinese).
|
|
16
|
Mrakovcic M, Kleinheinz J and Fröhlich LF:
Histone deacetylase inhibitor-induced autophagy in tumor cells:
Implications for p53. Int J Mol Sci. 18(pii): E18832017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Z, Jing S, Li Y, Gao Y, Yu S, Li Z,
Zhao Y, Piao J, Ma S and Chen X: The effects of SAHA on
radiosensitivity in pancreatic cancer cells by inducing apoptosis
and targeting RAD51. Biomed Pharmacother. 89:705–710. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu H, Yang XF, Tian XQ, Tang SL, Li LQ,
Zhao S and Zheng HC: The in vitro and vivo anti-tumor effects and
molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) and
MG132 on the aggressive phenotypes of gastric cancer cells.
Oncotarget. 7:56508–56525. 2016.PubMed/NCBI
|
|
19
|
Xue K, Gu JJ, Zhang Q, Mavis C,
Hernandez-Ilizaliturri FJ, Czuczman MS and Guo Y: Vorinostat, a
histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest
and re-sensitizes rituximab- and chemo-resistant lymphoma cells to
chemotherapy agents. J Cancer Res Clin Oncol. 142:379–387. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hanke NT, Garland LL and Baker AF:
Carfilzomib combined with suberanilohydroxamic acid (SAHA)
synergistically promotes endoplasmic reticulum stress in non-small
cell lung cancer cell lines. J Cancer Res Clin Oncol. 142:549–560.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Teng Z, Kuang X, Wang J and Zhang X:
Real-time cell analysis-a new method for dynamic, quantitative
measurement of infectious viruses and antiserum neutralizing
activity. J Virol Methods. 193:364–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zandi K: A real-time cell analyzing assay
for identification of novel antiviral compounds against chikungunya
virus. Methods Mol Biol 1426. 255–262. 2016. View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lakshmaiah KC, Jacob LA, Aparna S,
Lokanatha D and Saldanha SC: Epigenetic therapy of cancer with
histone deacetylase inhibitors. J Cancer Res Ther. 10:469–478.
2014.PubMed/NCBI
|
|
25
|
Ahuja N, Sharma AR and Baylin SB:
Epigenetic therapeutics: A new weapon in the war against cancer.
Annu Rev Med. 67:73–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hurwitz JL, Stasik I, Kerr EM, Holohan C,
Redmond KM, McLaughlin KM, Busacca S, Barbone D, Broaddus VC, Gray
SG, et al: Vorinostat/SAHA-induced apoptosis in malignant
mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur
J Cancer. 48:1096–1107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamamoto S, Tanaka K, Sakimura R, Okada T,
Nakamura T, Li Y, Takasaki M, Nakabeppu Y and Iwamoto Y:
Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or
autophagy-associated cell death in chondrosarcoma cell lines.
Anticancer Res. 28:1585–1591. 2008.PubMed/NCBI
|
|
28
|
Hrabeta J, Stiborova M, Adam V, Kizek R
and Eckschlager T: Histone deacetylase inhibitors in cancer
therapy. A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech
Repub. 158:161–169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arhoma A, Chantry AD, Haywood-Small SL and
Cross NA: SAHA-induced TRAIL-sensitisation of multiple myeloma
cells is enhanced in 3D cell culture. Exp Cell Res. 360:226–235.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim SM, Park KC, Jeon JY, Kim BW, Kim HK,
Chang HJ, Choi SH, Park CS and Chang HS: Potential anti-cancer
effect of N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA), a novel
histone deacetylase inhibitor, for the treatment of thyroid cancer.
BMC Cancer. 15:10032015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li YL, Zhang NY, Hu X, Chen JL, Rao MJ, Wu
LW, Li QY, Zhang B, Yan W and Zhang C: Evodiamine induces apoptosis
and promotes hepatocellular carcinoma cell death induced by
vorinostat via downregulating HIF-1α under hypoxia. Biochem Biophys
Res Commun. 498:481–486. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kunnimalaiyaan S, Sokolowski K, Gamblin TC
and Kunnimalaiyaan M: Suberoylanilide hydroxamic Acid, a histone
deacetylase inhibitor, alters multiple signaling pathways in
hepatocellular carcinoma cell lines. Am J Surg. 213:645–651. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuo K, Gray MJ, Yang DY, Srivastava SA,
Tripathi PB, Sonoda LA, Yoo EJ, Dubeau L, Lee AS and Lin YG: The
endoplasmic reticulum stress marker, glucose-regulated protein-78
(GRP78) in visceral adipocytes predicts endometrial cancer
progression and patient survival. Gynecol Oncol. 128:552–559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shi-Chen Ou D, Lee SB, Chu CS, Chang LH,
Chung BC and Juan LJ: Transcriptional activation of endoplasmic
reticulum chaperone GRP78 by HCMV IE1-72 protein. Cell Res.
21:642–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang M, Law ME, Castellano RK and Law BK:
The unfolded protein response as a target for anticancer
therapeutics. Crit Rev Oncol Hematol. 127:66–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoo YS, Han HG and Jeon YJ: Unfolded
protein response of the endoplasmic reticulum in tumor progression
and immunogenicity. Oxid Med Cell Longev 2017. 29692712017.
|
|
37
|
Nakka VP, Prakash-Babu P and Vemuganti R:
Crosstalk between endoplasmic reticulum stress, oxidative stress,
and autophagy: Potential therapeutic targets for acute CNS
injuries. Mol Neurobiol. 53:532–544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen Y, Gui D, Chen J, He D, Luo Y and
Wang N: Down-regulation of PERK-ATF4-CHOP pathway by Astragaloside
IV is associated with the inhibition of endoplasmic reticulum
stress-induced podocyte apoptosis in diabetic rats. Cell Physiol
Biochem. 33:1975–1987. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ketchum CC, Larsen CD, McNeil A,
Meyer-Ficca ML and Meyer RG: Early histone H4 acetylation during
chromatin remodeling in equine spermatogenesis. Biol Reprod.
98:115–129. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lei YU, Han B, Tian T, Zheng L, Yang T,
Liu X, Tang L, Luo X, Yang Q and Xie JR: Suberoylanilide hydroxamic
acid induces apoptosis of HepG2 cells by endoplasmic reticulum
stress apoptotic pathway. Chin J Pathophysiol. 33:2151–2156.
2017.
|
|
41
|
Chen M, Liu Q, Chen L, Zhang L and Gu E:
Remifentanil postconditioning ameliorates histone H3 acetylation
modification in H9c2 cardiomyoblasts after hypoxia/reoxygenation
via attenuating endoplasmic reticulum stress. Apoptosis.
22:662–671. 2017. View Article : Google Scholar : PubMed/NCBI
|