Introduction
Lung cancer, one of the most common malignant
tumors, is the leading cause of cancer-associated morbidity in the
population worldwide; it is the most common cancer among males and
the fourth most common tumor in women (1). Lung cancer is divided into different
pathological subtypes, including adenocarcinoma, squamous cell
carcinoma and small cell lung cancer (SCLC) (1). The occurrence, development and
metastasis of lung cancer include a number of orchestrated steps,
including DNA mutations and injury (2). Despite an increased understanding of
the underlying molecular mechanisms of the disease and the
implementation of novel therapeutic strategies, the 5-year survival
rate remains low. The study of the molecular mechanism of cancer
guides the classification and treatment of lung cancer, and
promotes the rapid progress of targeted therapy and immunotherapy.
The large-scale research and clinical trials of these new therapies
provide prospects for the individualized treatment of lung
cancer.
Much progress has been made with lung cancer
biomarkers over the last decade, and biomarkers have been widely
applied in the diagnosis, treatment and prognosis evaluation of
lung cancer, with further biomarkers now being studied. For
example, anaplastic lymphoma kinase (ALK) was initially identified
to be abnormally downregulated in lung cancer and a fusion of
echinoderm microtubule-associated protein-like 4 (EML4) and ALK
genes was found in 3.7–7% of non-SCLC (NSCLC) (3). Due to ALK fusion, 57–74% of patients
with lung adenocarcinoma respond well to ALK inhibitors such as
crizotinib (3). The study revealed
that the median progression-free survival (PFS) and response rates
of patients who received crizotinib were significantly improved
compared with those of patients treated with chemotherapy (4). The epidermal growth factor receptor
(EGFR), a tyrosine kinase receptor, was overexpressed in 62% of
patients with NSCLC (5). Tyrosine
kinase inhibitors have been the standard treatment of patients with
EGFR mutations due to their high response rate (55–78%) and PFS
rate (1). Therefore, the discovery
of new diagnostic and therapeutic targets is of great significance
for the early diagnosis, drug development and targeted therapy of
lung cancer.
Bioinformatics analysis has been commonly applied in
cancer research to identify genetic changes associated with cancer.
Previous studies have performed bioinformatics analysis to identify
differentially expressed genes (DEGs) in various types of cancer,
as well as to determine their roles in biological processes,
molecular functions and different pathways (6,7).
Accordingly, the present study analyzed data generated by
microarray technology to explore the potential pathogenesis of lung
cancer. Specifically, given the high number false-positives
associated with the analysis of a single microarray, four public
mRNA datasets were screened in the present study to identify DEGs
between lung cancer and adjacent non-cancerous tissue samples.
Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes
and Genomes (KEGG) analyses were performed, and a protein-protein
interaction (PPI) network analysis was used to assist in
demonstrating the molecular pathogenesis underlying the
carcinogenesis and development of lung cancer. A total of 552 DEGs
and 16 hub genes were identified and they may serve as candidate
biomarkers in lung cancer.
Materials and methods
Public mRNA datasets
Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) is an open platform to store
genetic data (8). Four gene
expression profiles (GSE102287, GSE89047, GSE67061 and GSE74706)
were acquired from the GEO The GSE102287 dataset contained 32
cancer samples and 34 normal samples (9). The GSE89047 dataset consisted of 8
cancer samples and 8 normal samples. The GSE67061 contained 56
cancer samples and 17 normal samples. The GSE74706 contained 18
cancer samples and 18 normal samples (10). The datasets consisted of a number of
pathological subtypes of lung cancer, including NSCLC and lung
squamous cell carcinoma. In the current study, in order to be more
representative, a specific pathological type was not specified when
selecting datasets.
Identification of DEGs
GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r) is an interactive
online tool to identify DEGs from GEO series (11). GEO2R was applied to distinguish DEGs
between normal and lung cancer tissue samples. Duplicate and absent
probe sets were removed. The cut-off criteria for the
identification of DEGs were |log2 fold-change|>1 and
adjusted P<0.05.
Functional annotation for DEGs with
KEGG and GO analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; www.david.abcc.ncifcrf.gov) provides typical batch
annotation and GO (www.geneontology.org) analysis to highlight the most
relevant GO terms associated with a given gene list (12). GO covers three aspects of biology,
including biological process, molecular function and cellular
component. KEGG (version 90.0; www.kegg.jp), is
one of the most commonly used biological information databases in
the world (13). Following KEGG and
GO analysis in DAVID, functional annotation for DEGs was performed.
P<0.05 was considered to indicate a statistically significant
difference.
Construction of the PPI network and
identification of a significant module
The Search Tool for the Retrieval of Interacting
Genes (version 11.0; string.embl.de) (14),
an online open tool, was applied to construct a PPI network, and
Cytoscape (version 3.7.1) (15) was
used to present the network. Using a confidence cutoff of >0.4,
a node score cutoff of 0.2, a degree cutoff of 10, a maximum depth
of 100 and a k-core of 2, the significant modules in the
aforementioned PPI network were identified using the Molecular
Complex Detection tool (version 1.5.1) (16). Subsequently, functional annotation
for the genes in this module were performed using KEGG and GO
analysis in DAVID.
Analysis and identification of hub
genes
Hub genes with ≥43 degrees were selected. cBioPortal
(www.cbioportal.org) integrates The
Cancer Genome Atlas (TCGA; portal.gdc.cancer.gov), the International Cancer
Genome Consortium (icgc.org) and other cancer genome
database data to provide online visualization tools. Based on the
hub genes screened, a gene co-expression network was constructed
and cBioportal was used to search for genes with a similar
expression pattern to the hub genes in lung cancer and to
investigate the interaction between genes (17). Furthermore, hub genes were analyzed
with the biological process analysis, and were visualized using the
BiNGO tool in Cytoscape (version 3.7.1) (18). The Kaplan-Meier plotter (kmplot.com/analysis) and the log rank test were used
to plot and compared survival curves, respectively. The
Kaplan-Meier plotter is an online tool that integrates gene
expression data and clinical data from TCGA, GEO and the European
Genome-Phenome Archive databases (www.ebi.ac.uk/ega/home). According to the different
quantile expression levels of the proposed biomarkers, patients
were divided into two groups to analyze the prognostic value of
specific genes (19).
Results
Screening of DEGs in lung cancer
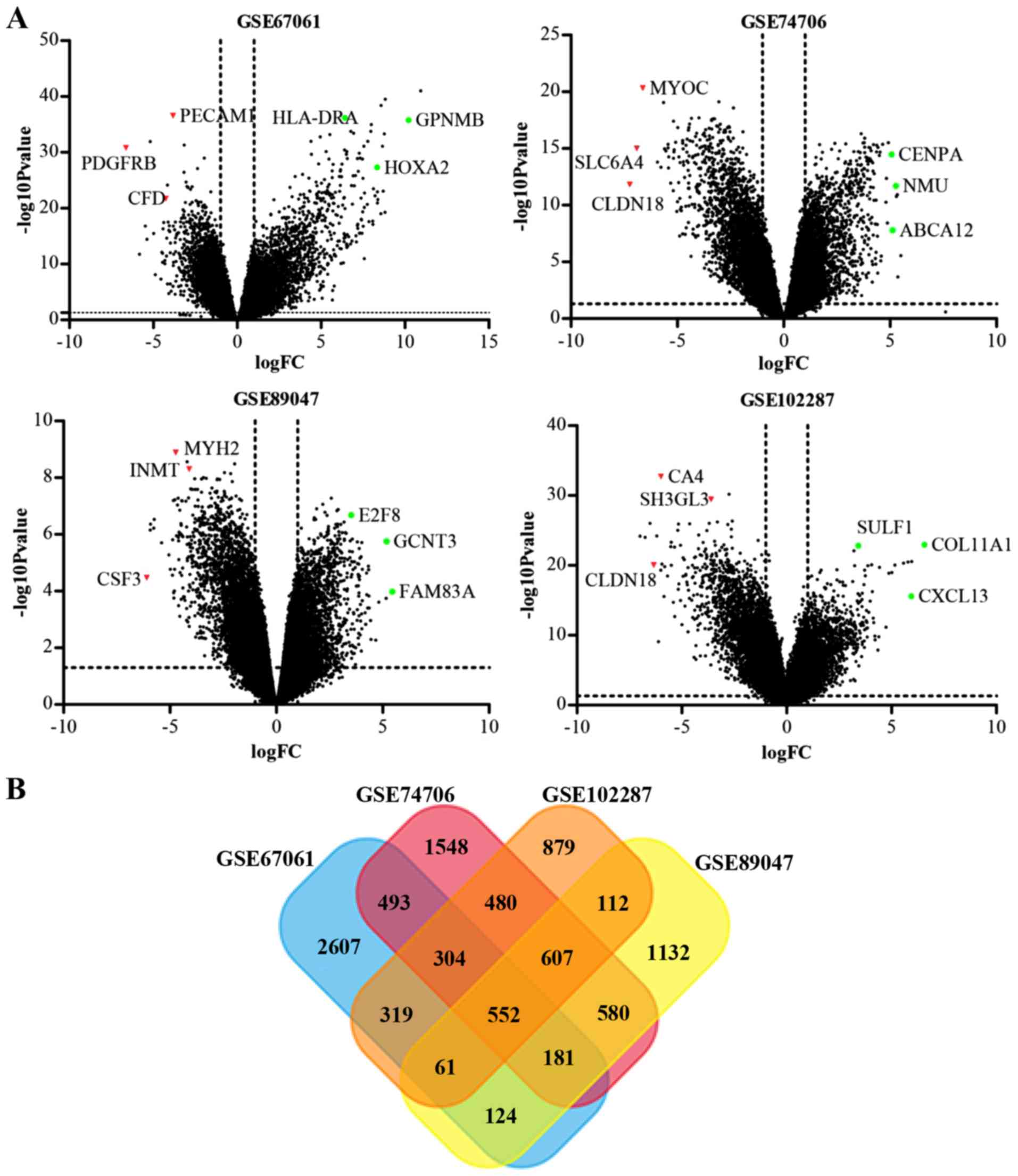
The analysis of the GSE67061, GSE74706, GSE89047 and
GSE102287 datasets revealed 5,553, 5,562, 4,028 and 4,703 DEGs,
respectively (Fig. 1A). Venn diagram
analysis revealed that 552 DEGs (389 downregulated and 163
upregulated genes) were present in the four datasets (Fig. 1B; Table
SI).
Functional annotation for DEGs using
KEGG and GO analysis
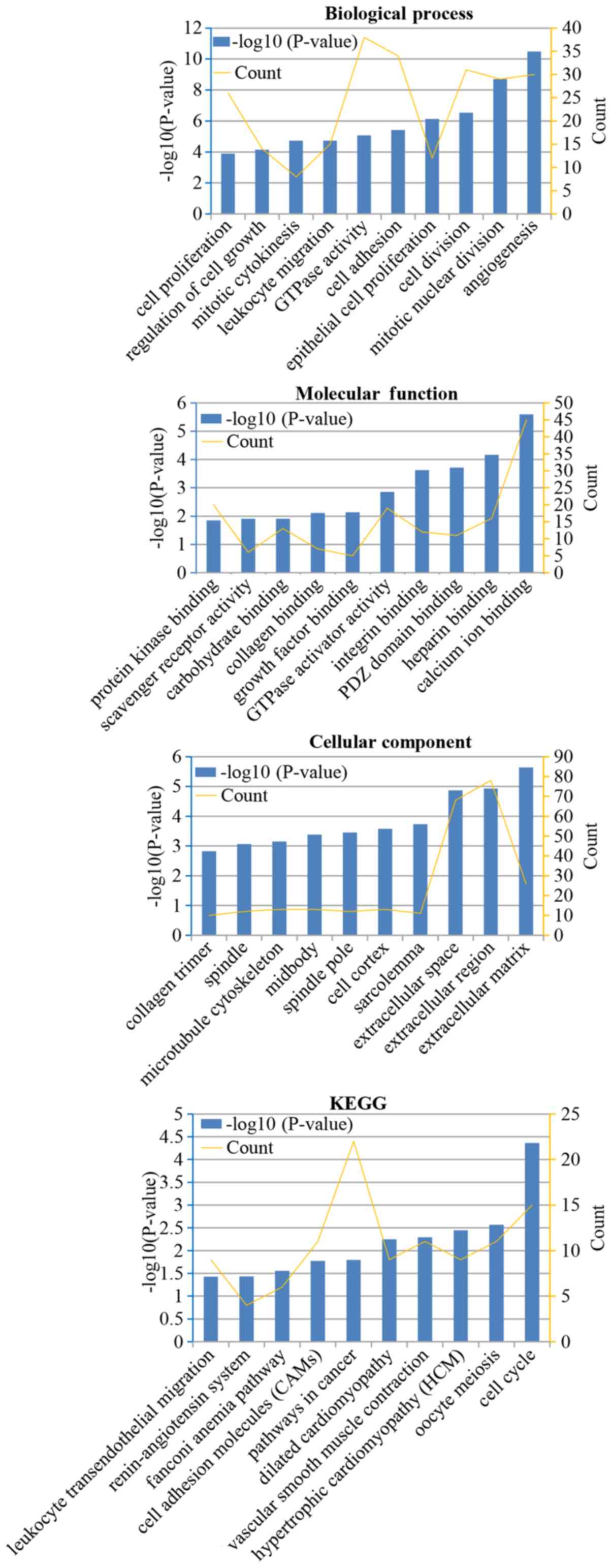
The results of GO analysis revealed that the
biological processes were primarily enriched in ‘cell
proliferation’, ‘cell growth’, ‘cell division’, ‘cell adhesion’,
‘angiogenesis’, ‘mitotic nuclear division’, ‘mitotic cytokinesis’,
‘leukocyte migration’, ‘GTPase activity’ and ‘epithelial cell
proliferation’. Variations in molecular function were enriched in
‘calcium ion binding’, ‘heparin binding’, ‘PDZ domain binding’,
‘integrin binding’, ‘GTPase activator activity’, ‘growth factor
binding’, ‘collagen binding’, ‘carbohydrate binding’, ‘scavenger
receptor activity’ and ‘protein kinase binding’. Changes in
cellular component were mainly enriched in ‘extracellular matrix’,
‘extracellular region’, ‘extracellular space’, ‘sarcolemma’, ‘cell
cortex’, ‘spindle pole’, ‘midbody’, ‘microtubule cytoskeleton’,
‘spindle’ and ‘collagen trimer’. KEGG pathway analysis revealed
that DEGs were mainly enriched in ‘cell cycle’, ‘oocyte meiosis’,
‘hypertrophic cardiomyopathy’, ‘vascular smooth muscle
contraction’, ‘dilated cardiomyopathy’, ‘pathways in cancer’, ‘cell
adhesion molecules’, ‘fanconi anemia pathway’, ‘renin-angiotensin
system’ and ‘leukocyte transendothelial migration’ (Fig. 2).
Construction of the PPI network and
identification of a significant module
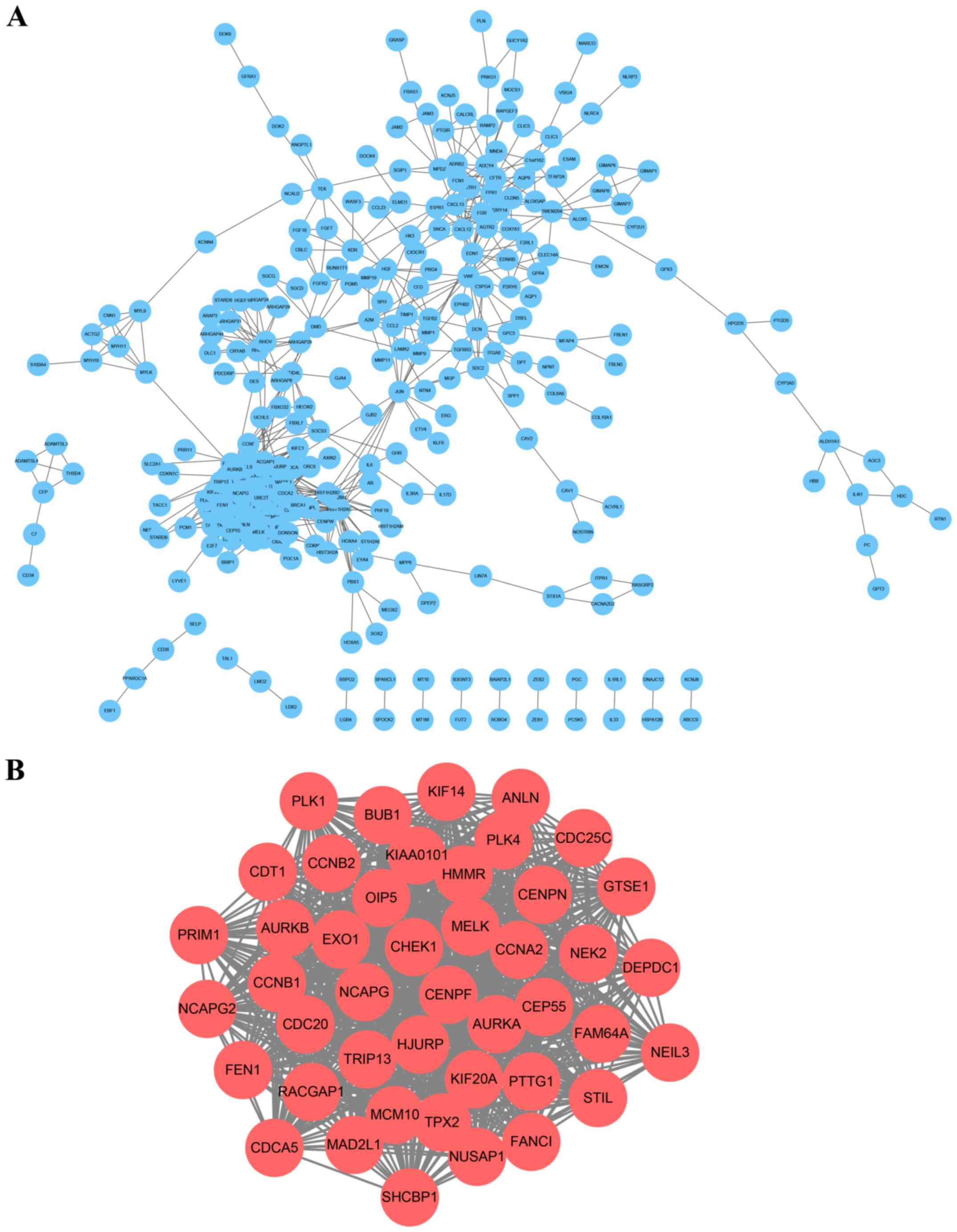
A PPI network was constructed and a significant
module with 44 nodes and 886 edges was identified (Fig. 3; Table
SII). KEGG pathway and GO analysis of the DEGs involved in this
module were analyzed using DAVID. Results revealed that genes in
this module were significantly enriched in ‘cell division’, ‘cell
cycle’ and ‘mitotic nuclear division’ (Table I).
 | Table I.GO and KEGG pathway enrichment
analysis of the differentially expressed genes in the most
significant module. |
Table I.
GO and KEGG pathway enrichment
analysis of the differentially expressed genes in the most
significant module.
| Category | Term | Count in gene
set | P-value |
|---|
| GOTERM_BP | Mitotic nuclear
division | 19 | <0.001 |
| GOTERM_BP | Cell division | 18 | <0.001 |
| GOTERM_BP | G2/M
transition of mitotic cell cycle | 11 | <0.001 |
| GOTERM_BP | Mitotic
cytokinesis | 5 | <0.001 |
| GOTERM_MF | Protein
binding | 39 | <0.001 |
| GOTERM_MF | Protein
serine/threonine kinase activity | 8 | <0.001 |
| GOTERM_MF | Protein kinase
binding | 8 | <0.001 |
| GOTERM_CC | Nucleoplasm | 29 | <0.001 |
| GOTERM_CC | Spindle | 9 | <0.001 |
| GOTERM_CC | Midbody | 9 | <0.001 |
| KEGG_PATHWAY | Cell cycle | 10 | <0.001 |
| KEGG_PATHWAY | p53 signaling
pathway | 4 | <0.001 |
| KEGG_PATHWAY | FoxO signaling
pathway | 4 | 0.006 |
Hub gene selection and analysis
Hub genes with ≥43 degrees were selected and a total
of 16 genes were identified as previously described (20): Opa interacting protein 5 (OIP5),
exonuclease 1 (EXO1), PCNA clamp-associated factor (KIAA0101),
checkpoint kinase 1 (CHEK1), hyaluronan-mediated motility receptor
(HMMR), maternal embryonic leucine zipper kinase (MELK), non-SMC
condensin I complex subunit G (NCAPG), centromere protein F
(CENPF), BUB1 mitotic checkpoint serine/threonine kinase (BUB1),
cyclin A2 (CCNA2), thyroid hormone receptor interactor 13 (TRIP13),
TPX2 microtubule nucleation factor (TPX2), nucleolar and spindle
associated protein 1 (NUSAP1), kinesin family member 20A (KIF20A),
aurora kinase A (AURKA) and centrosomal protein 55 (CEP55; Table II). A co-expression network of these
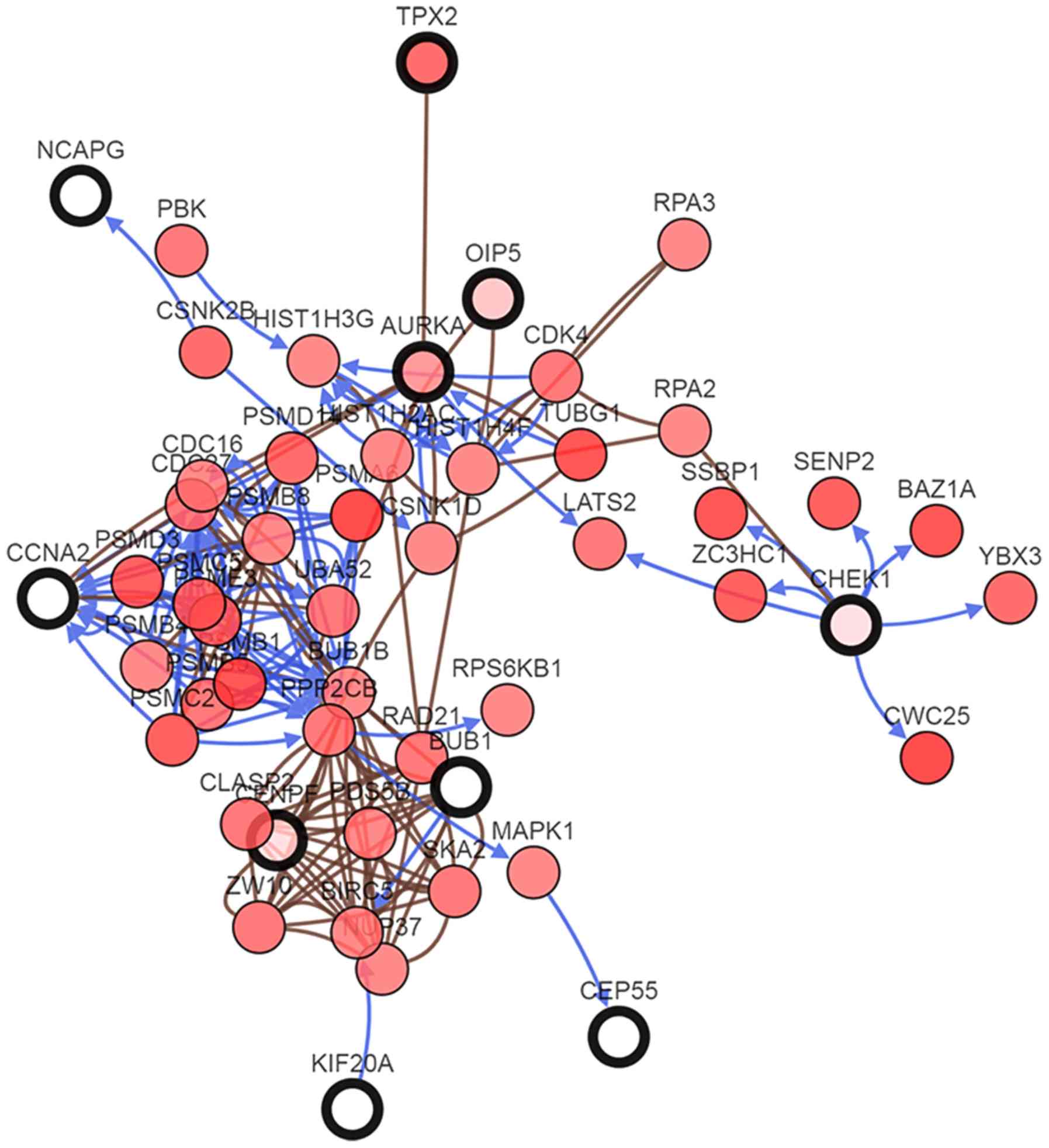
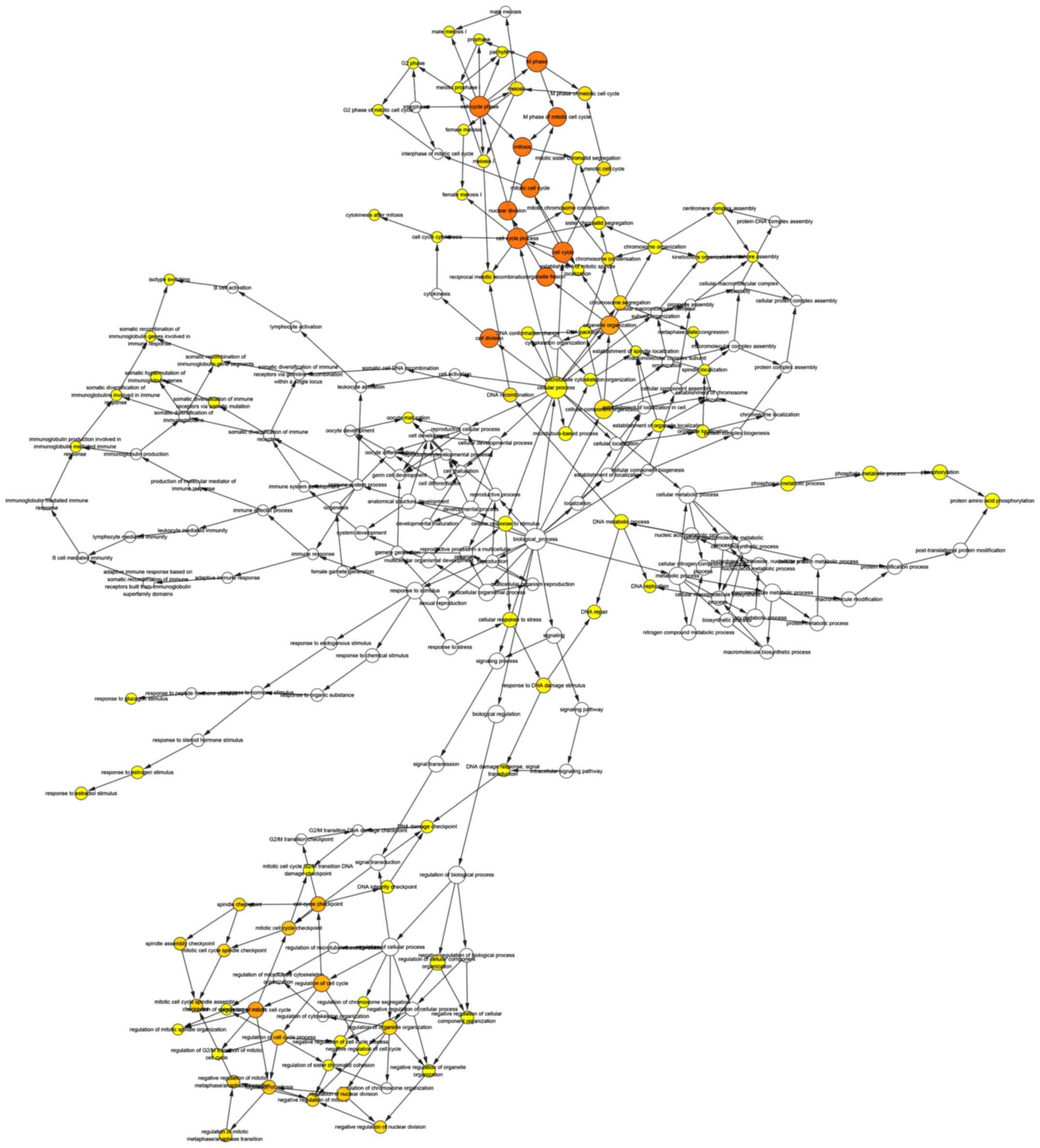
genes was obtained using cBioPortal (Fig. 4). The biological process analysis for
these genes is presented in Fig. 5.
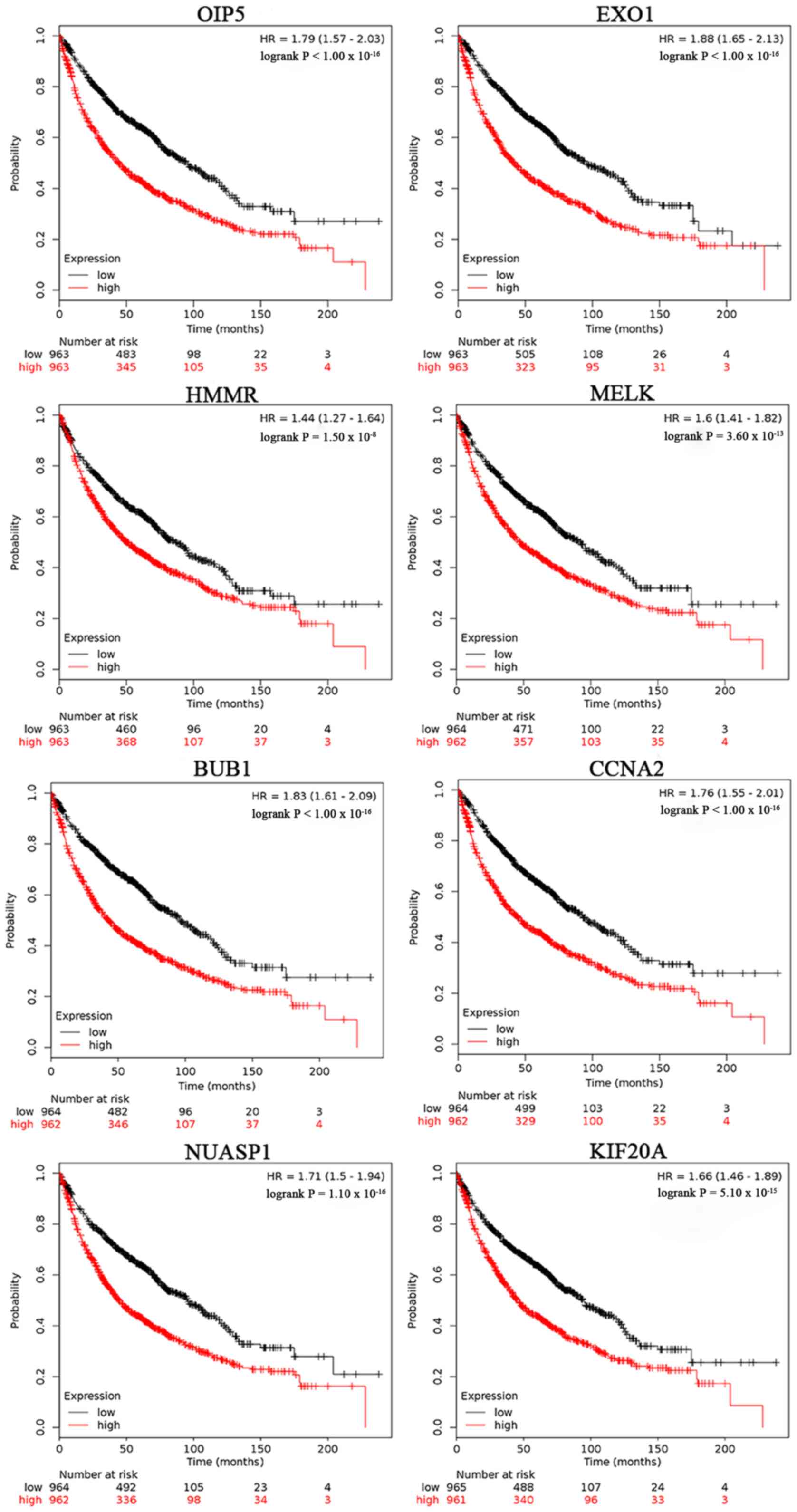
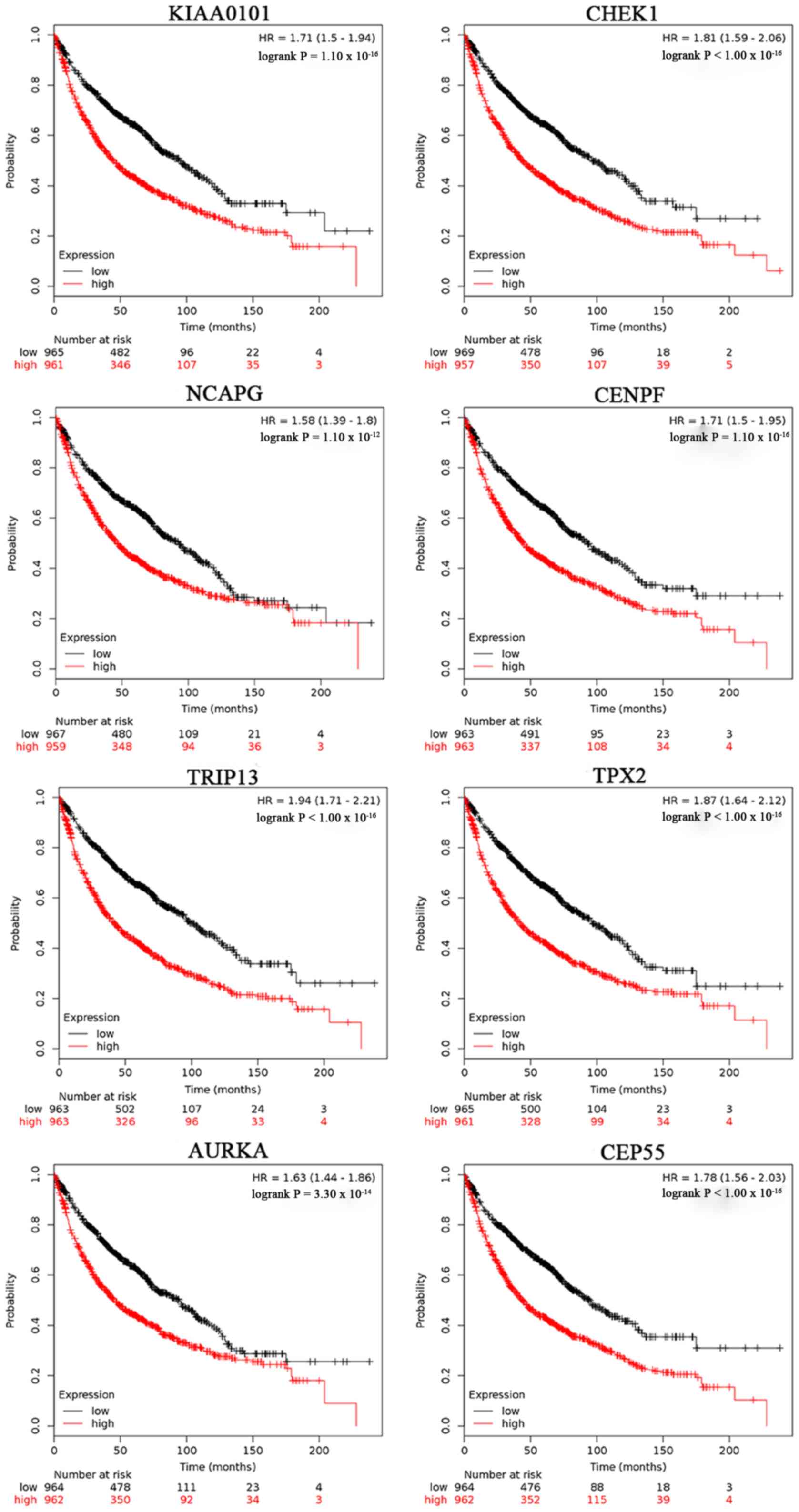
Kaplan-Meier survival curves were used to perform the overall
survival analysis. Patients with lung cancer with a high expression
level of OIP5, EXO1, KIAA0101, CHEK1, HMMR, MELK, NCAPG, CENPF,
BUB1, CCNA2, TRIP13, TPX2, NUSAP1, KIF20A, AURKA and CEP55
exhibited a worse 5-year overall survival time compared with
patients with low expression (Figs.
6 and 7).
| Table II.Functional roles of 16 hub genes with
≥43 degrees of interaction. |
Table II.
Functional roles of 16 hub genes with
≥43 degrees of interaction.
| Gene symbol | Gene name | Function |
|---|
| OIP5 | Opa interacting
protein 5 | Required for
recruitment of centromere protein A to centromeres and normal
chromosome segregation during mitosis. Expression of this gene is
upregulated in several types of cancer, making it a putative
therapeutic target. |
| EXO1 | Exonuclease 1 | Encodes a protein
with 5′ to 3′ exonuclease activity, as well as an RNase H
activity. |
| KIAA0101 | PCNA
clamp-associated factor | PCNA-binding
protein that acts as a regulator of DNA repair during DNA
replication. Also acts as a regulator of centrosome number. |
| CHEK1 | Checkpoint kinase
1 | Required for
checkpoint-mediated cell cycle arrest in response to DNA damage or
the presence of unreplicated DNA. |
| HMMR | Hyaluronan-mediated
motility receptor | Encodes a protein
involved in cell motility. |
| MELK | Maternal embryonic
leucine zipper kinase |
Serine/threonine-protein kinase involved
in various processes, such as cell cycle regulation, self-renewal
of stem cells, apoptosis and splicing regulation. |
| NCAPG | Non-SMC condensin I
complex subunit G | Encodes a subunit
of the condensin complex, which is responsible for the condensation
and stabilization of chromosomes during mitosis and meiosis. |
| CENPF | Centromere protein
F | Encodes a protein
that associates with the centromere-kinetochore complex. |
| BUB1 | BUB1 mitotic
checkpoint | Encodes a
serine/threonine-protein kinase that serves a central role in |
|
| serine/threonine
kinase | mitosis. Mutations
in this gene have been associated with aneuploidy and several forms
of cancer. |
| CCNA2 | Cyclin A2 | Encodes a protein
that binds and activates cyclin-dependent kinase 2 and promotes
transition through G1/S and G2/M. |
| TRIP13 | Thyroid hormone
receptor interactor 13 | Encodes a protein
that interacts with thyroid hormone receptors, which may serve a
role in early-stage non-small cell lung cancer. |
| TPX2 | Targeting protein
for Xklp2 | Spindle assembly
factor required for normal assembly of mitotic spindles. |
| NUSAP1 | Nucleolar and
spindle-associated protein 1 |
Nucleolar-spindle-associated protein that
serves a role in spindle microtubule organization. |
| KIF20A | Kinesin family
member 20A | Mitotic kinesin
required for chromosome passenger complex-mediated
cytokinesis. |
| AURKA | Aurora kinase
A | Encodes a cell
cycle-regulated kinase involved in microtubule formation and/or
stabilization at the spindle pole during chromosome
segregation. |
| CEP55 | Centrosomal protein
55 | Serves a role in
mitotic exit and cytokinesis. |
Discussion
Lung cancer is one of the most common malignancies
worldwide, both in terms of incidence and mortality (21,22).
Despite significant advances in diagnostic and treatment
strategies, the prognosis of patients with lung cancer remains
unsatisfactory. Therefore, there is a requirement for the
identification of lung cancer biomarkers to serve as novel
diagnostic and therapeutic targets. Bioinformatics analysis has
been widely applied to investigate genetic alterations in the
progression of diseases, and may enable the identification of novel
therapeutic targets.
Previous studies have screened biomarkers associated
with the different pathological subtypes of lung cancer (23–26).
Similarly, the present study screened potential biomarkers of lung
cancer. However, the present study differs from the previous
literature in a number of ways. In the current study, research data
was derived from different datasets, which allows diversification
of data results. Four datasets were selected to reduce the errors
associated with a single dataset and differences of sequencing
platforms, so as to improve the credibility of the results. The aim
of the present study was to screen common biomarkers and drug
targets in various pathological types of lung cancer using
bioinformatics analysis. Finally, different results were achieved
due to the different data sources and statistical methods used.
However, certain biomarkers identified in the current study are
consistent with previously published studies (27–31).
In the present study, 552 common DEGs were
identified in the four microarray datasets. GO enrichment analysis
revealed that changes in the most significant module were mainly
enriched in ‘cell division’, ‘mitotic nuclear division’ and
‘G2/M transition of mitotic cell cycle’, while changes
in KEGG analysis were mainly enriched in the ‘cell cycle’ and ‘p53
signaling pathway’. Previous studies demonstrated that
dysregulation of the cell cycle is associated with carcinogenesis
and the progression of tumors (32,33). In
the current study, a PPI network consisting of 44 nodes and 886
edges was constructed. The 16 genes with the highest degrees in the
PPI network included OIP5, EXO1, KIAA0101, CHEK1, HMMR, MELK,
NCAPG, CENPF, BUB1, CCNA2, TRIP13, TPX2, NUSAP1, KIF20A, AURKA and
CEP55. Subsequently, survival analysis of these genes revealed that
they were significantly associated with a worse 5-year overall
survival time of patients with lung cancer.
The mechanism of lung cancer is driven by specific
genetic and epigenetic changes (34). In certain types of cancer, such as
gastric colorectal cancer, the expression of OIP5 is upregulated
and may be associated with the occurrence of cancer (35,36).
However, its function in lung cancer remains unknown. EXO1 is a
nuclease that modulates DNA recombination, maintains genomic
stability and mediates cell cycle arrest. Several reports have
indicated that functional polymorphisms of EXO1 may be associated
with the occurrence of lung cancer, and it may serve as a novel
biomarker for the diagnosis and treatment of lung cancer (37,38).
KIAA0101 is involved in cell cycle regulation and DNA repair and is
expressed at high levels in several types of cancer, including
gastric and lung cancer (27,39,40).
Previous studies reported that high expression levels of KIAA0101
and CHEK1 in lung cancer are associated with a poor prognosis
(27,41).
Man et al (28) revealed that HMMR, the receptor for
hyaluronic acid, was upregulated in lung adenocarcinoma samples
compared with healthy adjacent non-cancerous tissues. MELK is
expressed in several types of human cancer (42,43),
including SCLC. Inoue et al (42) reported that inhibition of MELK may be
a therapeutic strategy for SCLC. Zhang et al (44) reported that NCAPG may be implicated
in hepatocellular carcinoma cell proliferation and migration, and
may provide a promising novel therapeutic target for the treatment
of advanced hepatocellular carcinoma. However, the clinical
significance of NCAPG in lung cancer remains unknown.
Previous studies reported that CENPF serves a role
in the tumorigenesis of hepatocellular carcinoma and prostate
cancer (45,46); however, its role in lung cancer
requires further investigation. A number of studies demonstrated
that BUB1 serves important roles in breast and endometrial cancer
(47–49). However, Haruki et al (47–49)
reported that the BUB gene family members, including BUB1, are not
commonly associated with mitotic checkpoint defects in lung cancer.
The potential association between BUB1 and lung cancer requires
further investigation. Kim et al (29) reported that a functional single
nucleotide polymorphism in the promoter region of CCNA2 was
associated with an increased risk of lung cancer. TRIP13 is an
ATPase that serves a key role in mitotic checkpoint complex
inactivation and is associated with the progression of lung
adenocarcinoma (30). Li et
al (30) demonstrated that
increased TRIP13 expression promoted lung adenocarcinoma
progression and may serve as a potential therapeutic target or
biomarker for the disease.
Yang et al (50,51)
revealed that TPX2 was associated with lung squamous carcinoma cell
radioresistance and may serve as a therapeutic target to enhance
cell radiosensitivity in lung squamous carcinoma. Furthermore,
Schneider et al (50,51) demonstrated that the expression of the
TPX2, mitosis-associated gene, was associated with the prognosis of
patients with NSCLC. Previous studies reported that overexpression
of NUSAP1 was associated with a poor prognosis in prostate cancer,
hepatocellular and oral squamous cell carcinoma (52,53);
however; little is known about the association of NUSAP1 with lung
cancer. Zhao et al (54)
demonstrated that KIF20A may confer a malignant phenotype in lung
adenocarcinoma by regulating cell proliferation and apoptosis.
AURKA, an oncogene, encodes a serine-threonine kinase that
regulates mitotic processes in mammalian cells and serves as a
potential therapeutic target of NSCLC (55,56). Lo
et al (55,56) reported that AURKA upregulation is
restricted to specific subtypes and poorly differentiated tumors in
NSCLC. Ma et al (31)
revealed that CEP55 was upregulated in lung cancer cells and was
associated with poor clinical outcomes in patients with lung
cancer, and that it may serve as a prognostic biomarker for the
disease.
The current study is only a preliminary report, and
heterogeneous results due to the limitations of the source and
quantity of samples may have occurred. Furthermore, statistical
differences may not translate to the expected clinical
significance. In order to be more representative, a specific
pathological type of lung cancer was not selected in the current
study. However, this may lead to poor specificity in lung cancer
subtypes. The 16 hub genes identified revealed clinical
significance in the validation of survival analysis. However,
further validation in the subsequent basic and clinical trial
studies is required. In addition to DEGs, further studies
investigating differentially expressed microRNAs and their
association with genes, particularly DEGs, are required.
In summary, the current study identified DEGs that
may be involved in the carcinogenesis or progression of lung
cancer. A total of 552 DEGs and 16 hub genes were identified, and
these may serve as potential diagnostic biomarkers or therapeutic
targets for lung cancer. The results suggested that data mining and
integration may be a promising tool for the identification of
biomarkers in malignant tumors. As tumor biomarkers only have
meaning if they are integrated with clinical data, further
experiments should be conducted to verify the results obtained in
the current study.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the GEO repository (www.ncbi.nlm.nih.gov/geo).
Authors' contributions
ZHL and BES designed the overall research. ZL, JL
and FZ collected the data. ZHL, MXS, ZQT, ZL and BES contributed to
data analysis and visualization. JL and FZ drafted and revised the
manuscript. All authors approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DEGs
|
differentially expressed genes
|
|
PPI
|
protein-protein interaction
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
GEO
|
Gene Expression Omnibus
|
|
DAVID
|
Database for Annotation, Visualization
and Integrated Discovery
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nana-Sinkam SP and Powell CA: Molecular
biology of lung cancer: Diagnosis and management of lung cancer,
3rd ed: American College of Chest Physicians evidence-based
clinical practice guidelines. Chest. 143:e30S–e39S. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa
K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al:
First-line crizotinib versus chemotherapy in ALK-positive lung
cancer. N Engl J Med. 371:2167–2177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mok TS: Personalized medicine in lung
cancer: What we need to know. Nat Rev Clin Oncol. 8:661–668. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shao Y, Liang B, Long F and Jiang SJ:
Diagnostic microRNA biomarker discovery for non-small-cell lung
cancer adenocarcinoma by integrative bioinformatics analysis.
Biomed Res Int. 2017:25630852017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang LQ, Zhao LH and Qiao YZ:
Identification of potential therapeutic targets for lung cancer by
bioinformatics analysis. Mol Med Rep. 13:1975–1982. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Edgar R, Domrachev M and Lash AE: Gene
Expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mitchell KA, Zingone A, Toulabi L,
Boeckelman J and Ryan BM: Comparative transcriptome profiling
reveals coding and noncoding RNA differences in NSCLC from African
Americans and European Americans. Clin Cancer Res. 23:7412–7425.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marwitz S, Depner S, Dvornikov D, Merkle
R, Szczygieł M, Müller-Decker K, Lucarelli P, Wäsch M, Mairbäurl H,
Rabe KF, et al: Downregulation of the TGFβ pseudoreceptor BAMBI in
non-small cell lung cancer enhances TGFβ signaling and invasion.
Cancer Res. 76:3785–3801. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID Gene Functional Classification Tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–103. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nagy Á, Lánczky A, Menyhárt O and Győrffy
B: Validation of miRNA prognostic power in hepatocellular carcinoma
using expression data of independent datasets. Sci Rep. 8:92272018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li T, Gao X, Han L, Yu J and Li H:
Identification of hub genes with prognostic values in gastric
cancer by bioinformatics analysis. World J Surg Oncol. 16:1142018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bunn PA Jr: Karnofsky award 2016: A lung
cancer journey, 1973 to 2016. J Clin Oncol. 35:243–252. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mendell JT: MicroRNAs: Critical regulators
of development, cellular physiology and malignancy. Cell Cycle.
4:1179–1184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao Y, Feng M, Ran H, Han X and Li X:
Identification of key differentially expressed genes associated
with non-small cell lung cancer by bioinformatics analyses. Mol Med
Rep. 17:6379–6386. 2018.PubMed/NCBI
|
|
25
|
Wen P, Chidanguro T, Shi Z, Gu H, Wang N,
Wang T, Li Y and Gao J: Identification of candidate biomarkers and
pathways associated with SCLC by bioinformatics analysis. Mol Med
Rep. 18:1538–1550. 2018.PubMed/NCBI
|
|
26
|
Tang Q, Zhang H, Kong M, Mao X and Cao X:
Hub genes and key pathways of non-small lung cancer identified
using bioinformatics. Oncol Lett. 16:2344–2354. 2018.PubMed/NCBI
|
|
27
|
Kato T, Daigo Y, Aragaki M, Ishikawa K,
Sato M and Kaji M: Overexpression of KIAA0101 predicts poor
prognosis in primary lung cancer patients. Lung Cancer. 75:110–118.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Man Y, Cao J, Jin S, Xu G, Pan B, Shang L,
Che D, Yu Q and Yu Y: Newly identified biomarkers for detecting
circulating tumor cells in lung adenocarcinoma. Tohoku J Exp Med.
234:29–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim DH, Park SE, Kim M, Ji YI, Kang MY,
Jung EH, Ko E, Kim Y, Kim S, Shim YM and Park J: A functional
single nucleotide polymorphism at the promoter region of cyclin A2
is associated with increased risk of colon, liver, and lung
cancers. Cancer. 117:4080–4091. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Zhang G, Li X, Wang X, Li Q, Hong L,
Shen Y, Zhao C, Gong X, Chen Y and Zhou J: Thyroid hormone receptor
interactor 13 (TRIP13) overexpression associated with tumor
progression and poor prognosis in lung adenocarcinoma. Biochem
Biophys Res Commun. 499:416–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma XP, Zhang W, Wu BQ and Qin J:
Correlations between mRNA levels of centrosomal protein 55 (CEP55)
and clinical features of patients with lung cancer. Med Sci Monit.
24:3093–3097. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiang IT, Wang WS, Liu HC, Yang ST, Tang
NY and Chung JG: Curcumin alters gene expression-associated DNA
damage, cell cycle, cell survival and cell migration and invasion
in NCI-H460 human lung cancer cells in vitro. Oncol Rep.
34:1853–1874. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wan J, Zou S, Hu M, Zhu R, Xu J, Jiao Y
and Fan S: Thoc1 inhibits cell growth via induction of cell cycle
arrest and apoptosis in lung cancer cells. Mol Med Rep.
9:2321–2327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wen J, Fu J, Zhang W and Guo M: Genetic
and epigenetic changes in lung carcinoma and their clinical
implications. Mod Pathol. 24:932–943. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chun HK, Chung KS, Kim HC, Kang JE, Kang
MA, Kim JT, Choi EH, Jung KE, Kim MH, Song EY, et al: OIP5 is a
highly expressed potential therapeutic target for colorectal and
gastric cancers. BMB Rep. 43:349–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gong M, Xu Y, Dong W, Guo G, Ni W, Wang Y,
Wang Y and An R: Expression of Opa interacting protein 5 (OIP5) is
associated with tumor stage and prognosis of clear cell renal cell
carcinoma. Acta Histochem. 115:810–815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tang J, Tang S, Liu J, Wu Q, Wan L and Xu
Q: Genetic risk of lung cancer associated with a single nucleotide
polymorphism from EXO1: A meta analysis. Int J Clin Exp Med.
8:11132–11138. 2015.PubMed/NCBI
|
|
38
|
Hsu NY, Wang HC, Wang CH, Chiu CF, Tseng
HC, Liang SY, Tsai CW, Lin CC and Bau DT: Lung cancer
susceptibility and genetic polymorphisms of Exo1 gene in Taiwan.
Anticancer Res. 29:725–730. 2009.PubMed/NCBI
|
|
39
|
Zhu K, Diao D, Dang C, Shi L, Wang J, Yan
R, Yuan D and Li K: Elevated KIAA0101 expression is a marker of
recurrence in human gastric cancer. Cancer Sci. 104:353–359. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fan S and Li X, Tie L, Pan Y and Li X:
KIAA0101 is associated with human renal cell carcinoma
proliferation and migration induced by erythropoietin. Oncotarget.
7:13520–13537. 2016.PubMed/NCBI
|
|
41
|
Liu B, Qu J, Xu F, Guo Y, Wang Y, Yu H and
Qian B: MiR-195 suppresses non-small cell lung cancer by targeting
CHEK1. Oncotarget. 6:9445–9456. 2015.PubMed/NCBI
|
|
42
|
Inoue H, Kato T, Olugbile S, Tamura K,
Chung S, Miyamoto T, Matsuo Y, Salgia R, Nakamura Y and Park JH:
Effective growth-suppressive activity of maternal embryonic
leucine-zipper kinase (MELK) inhibitor against small cell lung
cancer. Oncotarget. 7:13621–13633. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pitner MK, Taliaferro JM, Dalby KN and
Bartholomeusz C: MELK: A potential novel therapeutic target for
TNBC and other aggressive malignancies. Expert Opin Ther Targets.
21:849–859. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Q, Su R, Shan C, Gao C and Wu P:
Non-SMC condensin I complex, subunit G (NCAPG) is a novel mitotic
gene required for hepatocellular cancer cell proliferation and
migration. Oncol Res. 26:269–276. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim HE, Kim DG, Lee KJ, Son JG, Song MY,
Park YM, Kim JJ, Cho SW, Chi SG, Cheong HS, et al: Frequent
amplification of CENPF, GMNN and CDK13 genes in hepatocellular
carcinomas. PLoS One. 7:e432232012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Aytes A, Mitrofanova A, Lefebvre C,
Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A,
Pienta KJ, Shen MM, et al: Cross-species regulatory network
analysis identifies a synergistic interaction between FOXM1 and
CENPF that drives prostate cancer malignancy. Cancer Cell.
25:638–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Takagi K, Miki Y, Shibahara Y, Nakamura Y,
Ebata A, Watanabe M, Ishida T, Sasano H and Suzuki T: BUB1
immunolocalization in breast carcinoma: Its nuclear localization as
a potent prognostic factor of the patients. Horm Cancer. 4:92–102.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li L, Xu DB, Zhao XL and Hao TY:
Combination analysis of Bub1 and Mad2 expression in endometrial
cancer: Act as a prognostic factor in endometrial cancer. Arch
Gynecol Obstet. 288:155–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Haruki N, Saito H, Harano T, Nomoto S and
Takahashi T, Osada H, Fujii Y and Takahashi T: Molecular analysis
of the mitotic checkpoint genes BUB1, BUBR1 and BUB3 in human lung
cancers. Cancer Lett. 162:201–205. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang J, Gao F, Xu X, Wang Y and Zhu S:
Targeting protein for Xenopus kinesin-like protein 2 knockdown
enhances radiation sensitivity of human lung squamous carcinoma
cell. Clin Exp Pharmacol Physiol. 44:1060–1068. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schneider MA, Christopoulos P, Muley T,
Warth A, Klingmueller U, Thomas M, Herth FJ, Dienemann H, Mueller
NS, Theis F and Meister M: AURKA, DLGAP5, TPX2, KIF11 and CKAP5:
Five specific mitosis-associated genes correlate with poor
prognosis for non-small cell lung cancer patients. Int J Oncol.
50:365–372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang M, Yang D, Liu X, Liu Y, Liang J, He
H, Zhong K, Lin L, Tao G, Zhang C and Zhou J: Expression of Nusap1
in the surgical margins of hepatocellular carcinoma and its
association with early recurrence. Nan Fang Yi Ke Da Xue Xue Bao.
33:937–938, inside back cover. 2013.(In Chinese). PubMed/NCBI
|
|
53
|
Gordon CA, Gulzar ZG and Brooks JD: NUSAP1
expression is upregulated by loss of RB1 in prostate cancer cells.
Prostate. 75:517–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao X, Zhou LL, Li X, Ni J, Chen P, Ma R,
Wu J and Feng J: Overexpression of KIF20A confers malignant
phenotype of lung adenocarcinoma by promoting cell proliferation
and inhibiting apoptosis. Cancer Med. 7:4678–4689. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL,
Zhao BT, Jin Y, Li YL and Jin YX: Tanshinones suppress AURKA
through up-regulation of miR-32 expression in non-small cell lung
cancer. Oncotarget. 6:20111–20120. 2015.PubMed/NCBI
|
|
56
|
Lo Iacono M, Monica V, Saviozzi S, Ceppi
P, Bracco E, Papotti M and Scagliotti GV: Aurora Kinase A
expression is associated with lung cancer histological-subtypes and
with tumor de-differentiation. J Transl Med. 9:1002011. View Article : Google Scholar : PubMed/NCBI
|