Introduction
Colorectal cancer (CRC) is a common life threatening
disease worldwide and is also one of the most common causes of
cancer-associated mortality in developed countries (1,2). In
addition, 700,000 people succumb to this disease annually, making
it the fourth most deadly cancer among males and females (3). The development of CRC is a
heterogeneous process involving numerous genetic and epigenetic
variations, which are affected by environmental conditions, diet,
inflammatory responses and microbial adhesion; however, the
etiology of CRC remains unknown (4).
Previously, gut microbial dysbiosis has been reported to serve a
vital role in homeostasis and the development of CRC and has
received increasing attention in this research field (5,6).
Therefore, improved understanding of the differential combination
of microorganisms that comprise the gut microbiota may provide
novel insight for developments in the treatment of CRC.
Further investigation into the interactions between
host microbes is required to understand how the gut microbiota
promotes disease progression. Based on differences in embryology,
morphology and anatomy, Bufill (7)
proposed the existence of distinct types of CRC according to the
location of the tumor relative to the proximal (right) or distal
(left) segments of the splenic flexor muscle in the colon.
Epidemiological studies revealed differences between these proximal
and distal segments (8,9). The association between the gut
microbiota and the initiation and progression of CRC is well
reported (10). Alterations in gut
microecology, which is linked to disease progression, may be
applied for the diagnosis of gastrointestinal diseases. Of note,
Escherichia-Shigella has been reported to be linked to the
development of CRC and variations in conditional pathogens, which
may be the main cause of gut microbial dysbiosis in patients with
CRC (11,12). Prevotella has been associated
with the expression of Th17 response-related genes and correlated
with the reduced survival of CRC patients (13). Additionally, studies into the fecal
microbiota may improve understanding of the general composition of
the gut microbiota and its imbalance under different conditions
(14,15).
Understanding the factors that affect the
composition of the microbial community in the gut is essential for
developing CRC treatments. In the present study, the V3-V4 region
of 16S rRNA was sequenced using an Illumina sequencing platform to
analyze the composition of the gut microbiota of fecal samples from
67 patients with CRC and 30 healthy controls. The present study
also aimed to investigate differences in the gut microbiota among
various stages of CRC and between distal and proximal segments of
the colon in CRC.
Materials and methods
Patients and samples
In total, 67 patients with CRC (aged 33–86 years, 42
males and 25 females) and 30 healthy controls (aged 27–79 years, 15
males and 15 females) were recruited between October 2016 and
August 2017 from The First Affiliated Hospital of Zhejiang
University (Hangzhou, China). All patients with CRC were classified
according to their postoperative clinical data, using the
tumor-node-metastasis (TNM) staging system for malignant tumors
(16). The CRC patients were
selected according to the following criteria: No complications
(such as chronic bowel disease, diabetes, other signs of infections
or hypertension); no family history of CRC or recurrence of CRC, no
radiotherapy and chemotherapy prior to surgical resection; no use
of antibiotics, non-steroidal anti-inflammatory drugs, statins or
probiotics within the past 3 months prior to the collection of
stool samples; no food allergies and no potential immunodeficiency.
The 30 healthy individuals were selected as controls and were
matched according to sex and age during a routine physical
examination; healthy controls corresponded to stage O of the
staging system applied for classifying CRC patients. In addition,
the healthy controls did not have gastrointestinal tract disorders
or other complications and were not administered antibiotics or
probiotics during the 3 months prior to sample collection. The
present study was approved by the Research Ethics Committee of The
First Affiliated Hospital, College of Medicine, Zhejiang University
(approval no. 2016-436). All patients indicated that they had
obtained written informed consent in the present study. On the
morning of the day prior to surgical resection, one fecal sample
was collected in a sterile container from each healthy control and
patient with CRC. The data recorded included the general
information (age and sex) and the clinical data of patients (tumor
stages, tumor sites and pathological data). Fecal samples were
frozen immediately after collection and stored at −80°C until DNA
was extracted.
DNA extraction
Genomic DNA was extracted from each fecal sample
(~200 mg) using the QIANamp DNA Stool Mini kit (Tiangen
Biotechnology Co., Ltd.) according to the manufacturer's protocols
(15,17). The concentration and quality of the
extracted DNA were determined using a NanoDrop spectrophotometer;
the integrity of the DNA samples was analyzed via 2% (w/v) agarose
gel electrophoresis. All DNA samples were stored at −80°C until use
for microbial characterization.
Polymerase chain reaction (PCR)
amplification and sequencing
The extracted DNA samples were sent to an external
company (Jingbai Biotechnology Co., Ltd.) for library construction
and sequencing using the paired-end protocol on the MiSeq Illumina
platform. The V3-V4 region of 16S rRNA was amplified by PCR. The
sequences for the universal primers (V3-V4) were: Forward, (515F)
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′ and
reverse, (806R)
5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′.
Briefly, PCR was performed using a reaction volume of 50 µl
containing 10 ng DNA template, 25 µl 2X Phanta Max Master Mix
buffer solution (Vazyme Biotech Co., Ltd), 2 µl each primer (10 M)
and ddH2O (to 50 µl). Amplifications were performed
under the following conditions: Initial denaturation at 95°C for 3
min, followed by 25 cycles of denaturation at 95°C for 30 sec,
primer annealing at 55°C for 30 sec and extension at 72°C for 45
sec, followed by final elongation at 72°C for 5 min. The PCR
products were purified using the MiniE-lute PCR purification kit
(Axygen; Thermo Fisher Scientific, Inc.) and quantified using a
detection system (Light Cycler® 96 Flex Real-time PCR
System; Roche Diagnostics). Then, samples were pooled at equal
concentrations. The thermocycling conditions were as follows: 95°C
for 3 min, followed by 8 cycles of denaturation at 95°C for 30 sec;
primer annealing at 55°C for 30 sec and extension at 72°C for 45
sec and final elongation at 72°C for 5 min. Library construction
and sequencing with specific tags were performed on a MiSeq
Benchtop Sequencer (Illumina, Inc.).
Bioinformatics analyses
Based on the overlap of paired-end reads, the
microbial data obtained via the Illumina platform were optimized
and the paired reads were combined into sequences. The quality of
reads and the merged results were filtered as follows: The maximum
mismatch ratio of the overlap portion was 0.15; the minimum overlap
of merging paired reads was 10 bp and an average of <20 bases at
the end of reads and sequences other than 300–480 bp in length were
filtered. In order to obtain high-quality and more accurate
biological information, sequences containing some point mutations
and macromolecular homopolymers were used (Qiime, version 1.17
http://qiime.org/) (18). Then, the selected sequences were
clustered into optional taxonomic units (OTUs) using Usearch
(version 7.1, http://drive5.com/uparse/) with a standard similarity
of ≥97%. The chimera sequences generated by PCR amplification were
detected and excluded using Uchime (version 4.2.40 http://drive5.com/usearch/manual/uchime_algo.html)
(19). The representative OTUs were
compared with the optimized sequences to obtain the abundance of
OTUs within each sample for subsequent analysis.
Richness estimators (Ace, Chao) and α-diversity
estimators (Shannon and Simpson indexes) were calculated using the
mothur software package (version 1.35.1) (15). Unweighted Unifrac distance metrics
analysis of relative abundance at different levels in each sample
was conducted using OTUs and principal coordinate analysis (PCoA)
was performed to demonstrate the clustering of different samples
according to the distance matrix (20). This method takes into account the
divergence between different sequences. In addition, R package
(version 2.15.3; R Foundation for Statistical Computing) was also
employed for other statistical analyses.
Based on the Kyoto Encyclopedia of Genes and Genomes
(KEGG) database, the functional classifications in the 16 sRNA
genes of fecal gut microbiota were predicted by the software
Phylogenetic Investigation of Communities by Reconstruction of
Unobserved States (PICRUSt). PICRUSt was used online in the Galaxy
workflow framework.
Statistical analyses
As the majority of the datasets did not meet the
assumptions of normal distribution, non-parametric Dunn's tests
with Kruskal-Wallis tests or Mann-Whitney U test where applicable
were used, and analysis of variance were performed to analyze
differences in relative expression between patients with CRC and
healthy controls using GraphPad Prism 6.00 (GraphPad Software,
Inc.). Unless otherwise indicated, data were expressed as the mean
± standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinicopathological status of
patients
A total of 67 patients with CRC were enrolled in the
present study, including 42 males and 25 females. The diagnosis of
CRC was assessed by two experienced pathologists. The average age
of males was 63.69±12.85 years and that of females was 61.32±10.37
years. All patients with CRC were divided into stages I–IV:18 as
stage I, 17 as stage II, 27 as stage III and 5 as stage IV. The 30
healthy controls were considered as stage O, in accordance with the
staging system employed. In addition, the number of proximal
(right) and the distal (left) segments collected were 23 and 44,
respectively. The clinical and pathological characteristics of
patients with CRC are presented in Table
I.
 | Table I.Clinicopathological characteristics
of patients with CRC and healthy controls enrolled in the present
study. |
Table I.
Clinicopathological characteristics
of patients with CRC and healthy controls enrolled in the present
study.
| Characteristic | Male | Female | Total |
|---|
| CRC, n | 42 | 25 | 67 |
| Age, years | 63.69±12.85 | 61.32±10.37 | 62.81±11.96 |
| Tumor stage |
|
|
|
| I | 12 | 6 | 18 |
| II | 7 | 10 | 17 |
|
III | 19 | 8 | 27 |
| IV | 4 | 1 | 5 |
| Tumor site |
|
|
|
| Distal
(left) segment | 32 | 12 | 44 |
|
Proximal (right) segment | 10 | 13 | 23 |
| Healthy controls,
n | 15 | 15 | 30 |
| Age, years | 51.79±14.14 | 50.14±16.28 | 50.96±14.99 |
Richness and diversity analysis
In total, 32,992 and 32,704 high-quality and usable
reads were obtained from fecal samples of 67 patients with CRC and
30 healthy controls from sequencing the 16S rRNA gene, with an
average length of 416 and 418 bp, respectively. At the level of 3%
difference, there were 531,164 OTUs in all samples, with an average
of 149,755 OTUs (n=97) per sample. The Good's coverage value of
each group was >93%, indicating that the 16S rRNA sequences
identified in each group represented the majority of bacteria
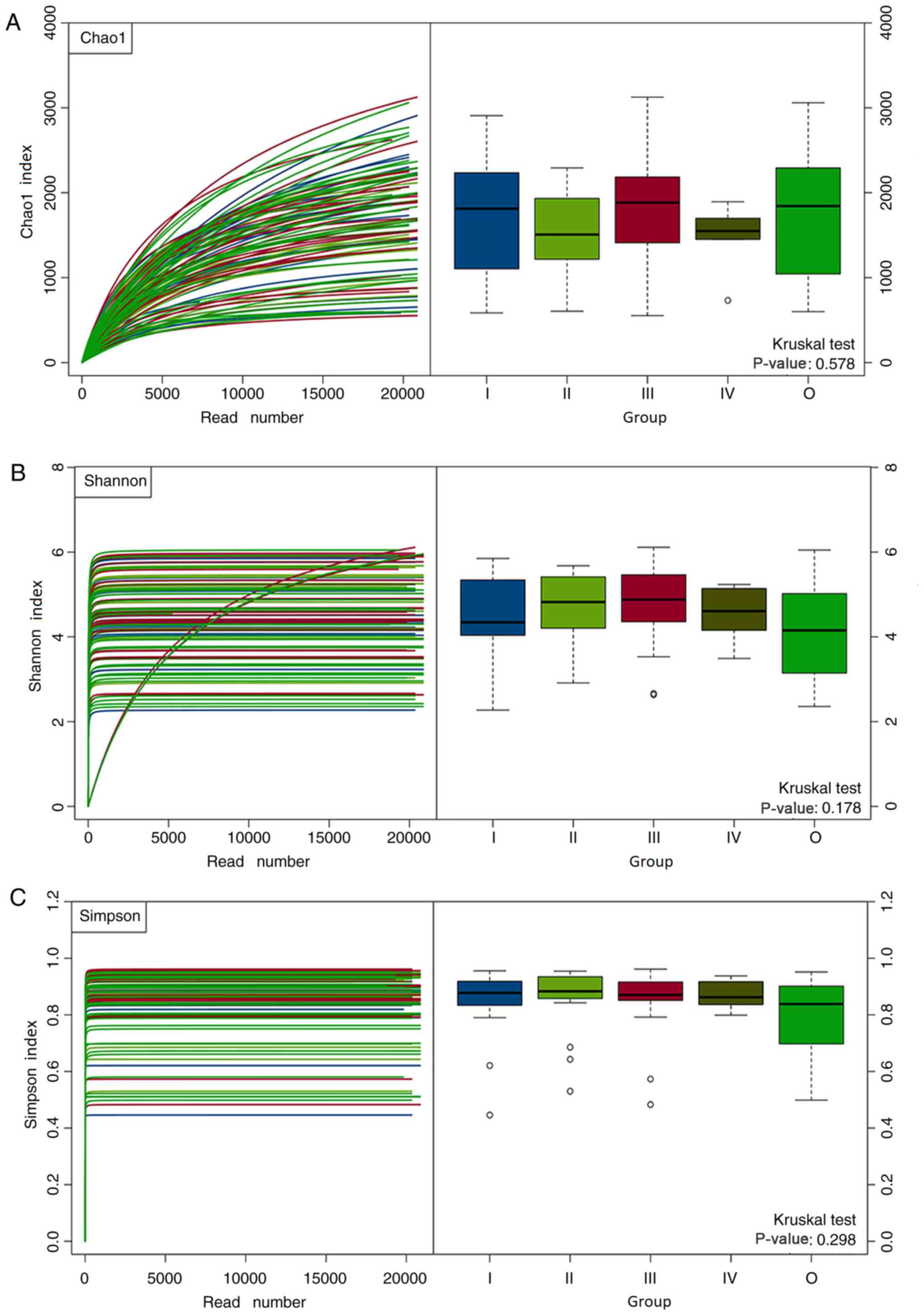
present in the samples analyzed. The Chao1 index was used to
evaluate microbial richness; the Shannon and Simpson diversity
indexes were applied to assess the diversity of fecal gut
microbiota between patients with CRC and healthy controls. The
results revealed that the Shannon and Simpson diversity indexes
(Shannon, 4.63±0.92 vs. 4.12±1.09; P=0.015; Simpson, 0.86±0.11 vs.
0.79±0.15; P=0.011) of CRC patients were significantly increased
compared with the healthy controls. This indicated that the
microbiota diversity of patients with CRC patients was
significantly increased compared with the healthy controls; no
significant difference in the Chao1 index (1,659±600 vs. 1,763±722;
P=0.45) between CRC patients and healthy controls was observed.
Furthermore, there were no significant differences in the Chao1,
Shannon diversity and Simpson diversity indexes among the gut
microbiota of patients with CRC stages I–IV and healthy controls
(P>0.05; Fig. 1).
Bacteria comprising the fecal
microbial community of healthy controls and patients with CRC
The relative abundance of dominant bacterial phyla
for each fecal sample was presented in Fig. S1. At the phylum level, dominant
bacterial phyla were analyzed and presented according to relative
abundance. The present study reported that all bacterial phyla were
detected from the interpretable sequences among patients with CRC
and the healthy controls. The first five dominant bacterial phyla
were Firmicutes, Bacteroidetes, Proteobacteria,
Verrucomicrobia and Actinobacteria in the CRC patients
and healthy controls. Among these top 20 dominant bacterial phyla,
no significant differences in the fecal gut microbiota were
observed within patients with CRC of stages I–IV and healthy
controls (Fig. S1).
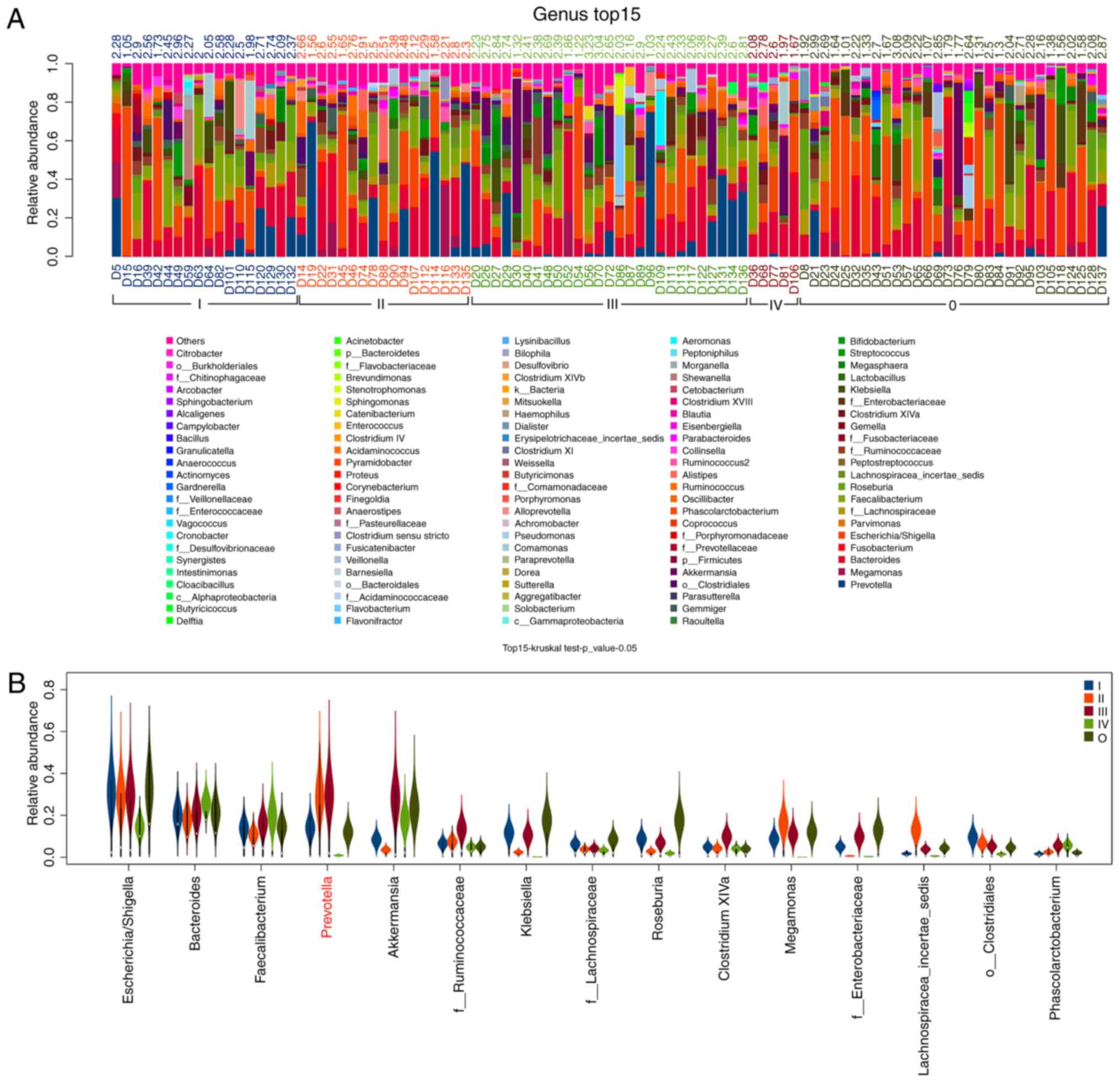
The relative abundance of the dominant bacterial
genera within each fecal sample was presented in Fig. 2. At the genus level, the dominant
bacterial genera were analyzed and presented according to relative
abundance. The results revealed that all bacterial genera were
detected from the interpretable sequences among patients with CRC
and healthy controls; the first five dominant bacterial were
Escherichia-Shigella, Bacteroides, Faecalibacterium,
Prevotella and Akkermansia in the CRC patients and
healthy controls. Among these dominant bacterial genera, according
to the analysis of the top 15 genera, the abundance of
Prevotella differed within the fecal gut microbiota of
patients with CRC of stages I–IV and healthy controls (Fig. 2).
Differences in fecal microbial
communities between healthy controls and CRC patients based on
classification comparisons
Principal component analysis (PCA) and PCoA were
performed according to the relative abundance of OTUs in each
group, and the composition of the gut microbiota of all samples was
compared. Distance-based PCA revealed the relative abundances of
the fecal microbial community among patients with CRC of stages
I–IV and healthy controls, which were separated by the first two
principal component scores of PC1 and PC2, accounting for 12.7 and
9.5% of the total variations, respectively (Fig. 3A). PCoA according to weighted UniFrac
metrics confirmed the aforementioned results, indicating
differences between patients with CRC of stages I–IV and healthy
controls; however, the results of stage I–IV CRC patients
overlapped with healthy controls and could not be separated well
from the PC1 and PC2 values (9.99 and 8.05% of the interpreted
variance, respectively; Fig.
3B).
Gene functional classifications using
the KEGG database
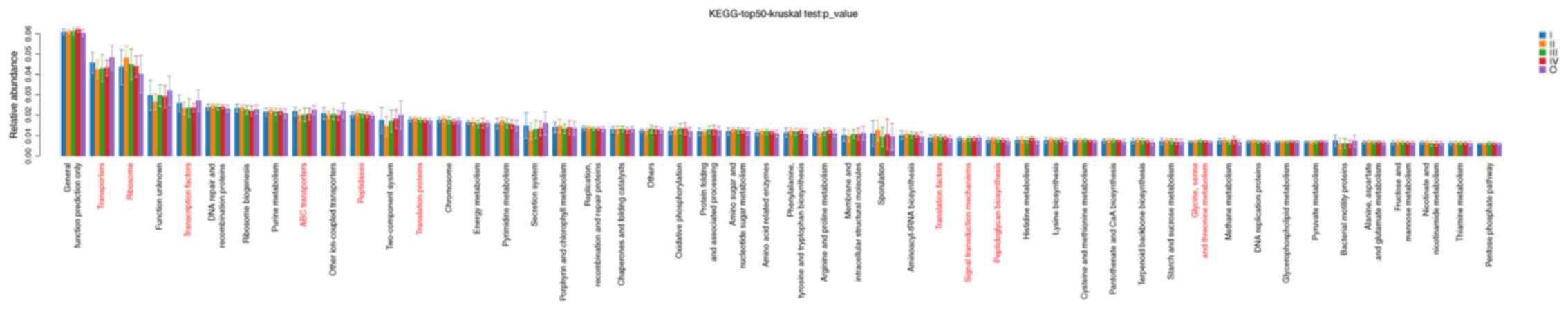
Based on KEGG database analysis, gene functional
classifications of the top 50 assembled unigenes are presented in
Fig. 4. Significant differences in
the function of the unigenes were reported between healthy controls
and patients with CRC of stages I–IV (P<0.05), including
‘transporters’, ‘ribosome’, ‘transcription factors’, ‘ABC
transporters’, ‘peptidases’, ‘translation proteins’, ‘translation
factors’, ‘signal transduction mechanisms’, ‘peptidoglycan
biosynthesis’ and ‘glycine, serine and threonine metabolism’.
The bacterial communities in fecal samples of
patients with CRC and healthy controls were investigated; all fecal
samples comprised 28 phyla, 61 classes, 99 orders, 191 families and
442 genera. The bacterial phyla of all gut microbiota were
analyzed. The differences at the phylum level in the fecal gut
microbiota were determined between healthy controls and patients
with CRC of stages I–IV. The results revealed that, compared with
the healthy controls (stage O), the relative abundance of
Fusobacteria (7.351±21.960 vs. 16.251±24.253%; P=0.028) was
increased in patients with stage I CRC. In addition, the relative
abundance of Lentisphaerae was greater in patients with
stage II CRC than those of stage I (0.169±0.504 vs. 0.370±1.155%;
P=0.031). Compared with stage II CRC patients, the abundance of
Fusobacteria (16.731±31.397 vs. 7.103±15.111%; P=0.043) was
significantly decreased in patients with stage III CRC (Fig. S2); that of the phylum
Verrucomicrobia (10.141±19.264 vs. 90.380±158.505%; P=0.035)
was significantly increased in patients with stage III CRC
(Fig. S2). On the contrary, no
differences between patients with CRC of stages III and IV were
reported.
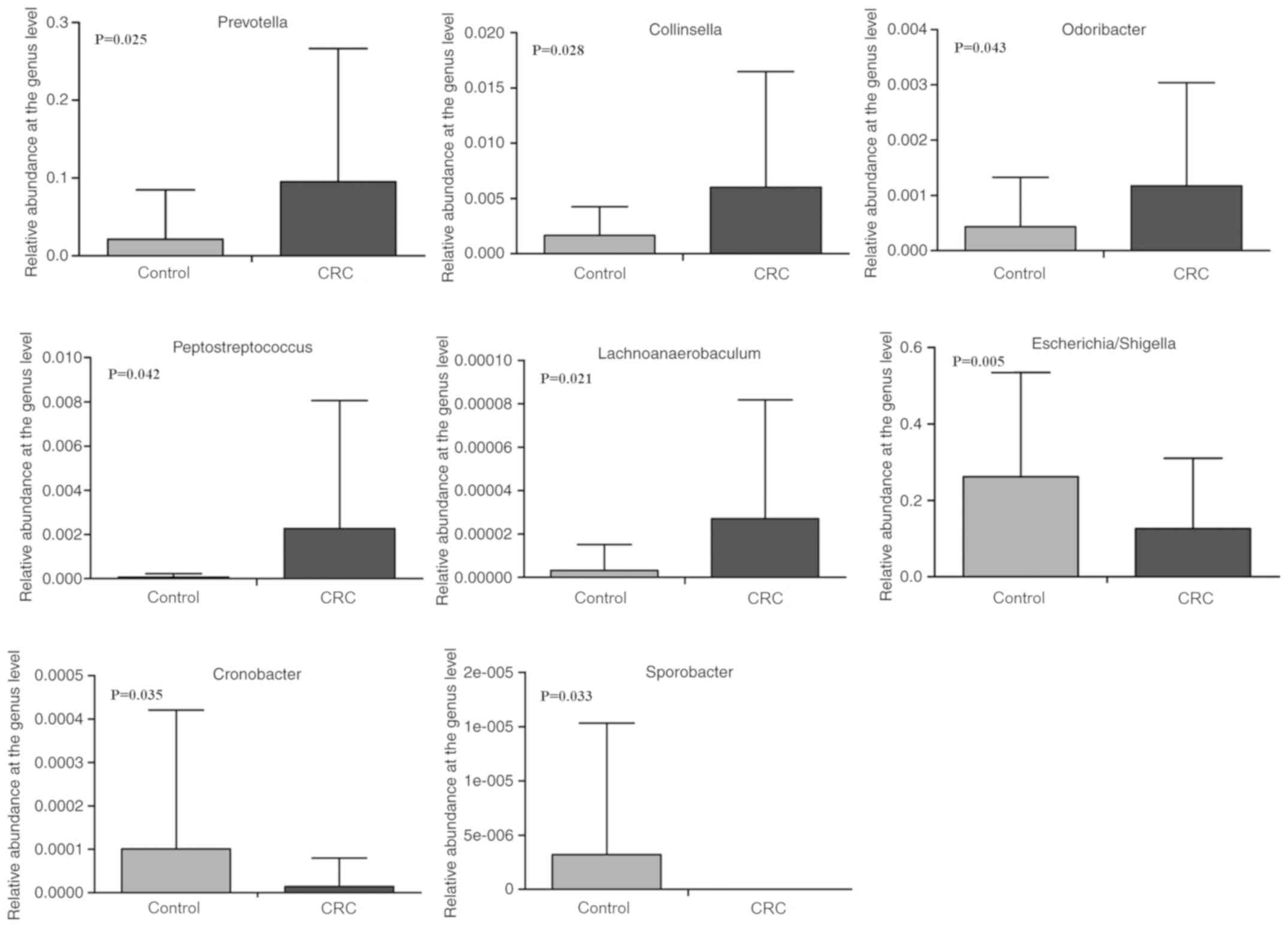
At the genus level, the bacterial genera of the gut
microbiota were analyzed. The results indicated significant
differences in the microbial composition of dominant genera between
CRC patients and healthy controls (Fig.
5). Compared with healthy controls, the genera
Prevotella (0.021±0.063 vs. 0.095±0.172; P=0.025),
Collinsella (1.671±2.595 vs. 6.002±10.466%; P=0.028),
Odoribacter (0.436±0.894 vs. 1.173±1.866%; P=0.043),
Peptostreptococcus (0.070±0.149 vs. 2.262±5.794%; P=0.042)
and Lachnoanaerobaculum (0.003±0.012 vs. 0.027±0.055%;
P=0.021) were significantly enriched in patients with CRC. However,
the abundances of Escherichia-Shigella (0.262±0.273 vs.
0.126±0.183; P=0.005), Cronobacter (0.101±0.320 vs.
0.014±0.066%; P=0.035) and Sporobacter (0.003±0.012 vs. 0‰;
P=0.033) were significantly reduced in patients with CRC.
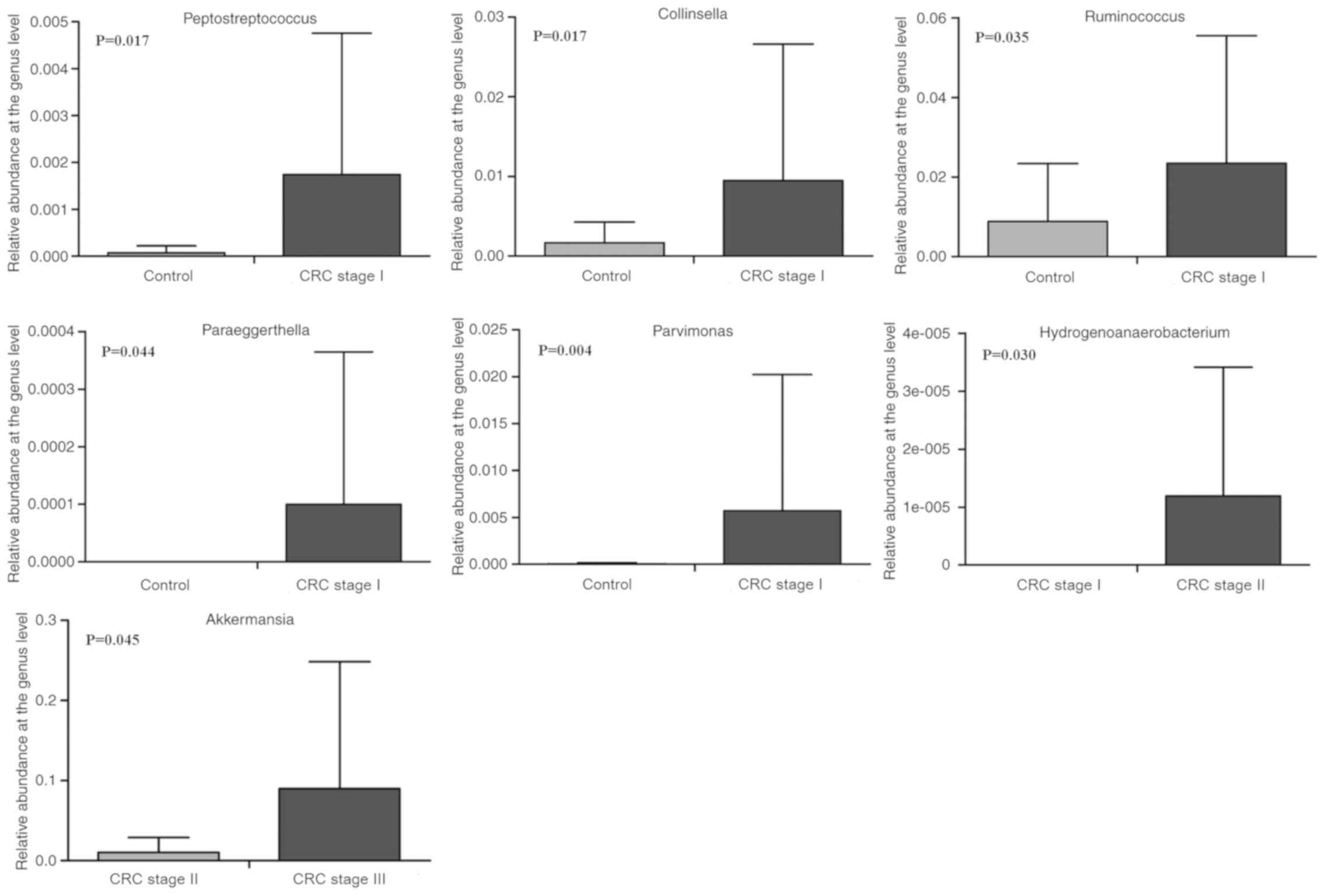
In addition, the effects of different tumor stages
on the composition of the gut microbiota were investigated in
healthy controls and patients with CRC of stages I–IV. The results
revealed that, compared with the healthy controls (stage O), the
relative abundances of Ruminococcus (8.803±14.593 vs.
23.469±31.995%; P=0.035), Collinsella (1.671±2.595 vs.
9.496±17.125%; P=0.017), Parvimonas (0.047±0.149 vs.
1.737±3.013%; P=0.004) and Peptostreptococcus (0.070±0.012
vs. 23.469±31.995%; P=0.017) were significantly enriched in stage I
CRC patients (Fig. 6). Furthermore,
Paraeggerthella (0 vs. 0.100±0.265%; P=0.044) was only
detected in patients with stage I CRC. The relative abundance of
Hydrogenoanaerobacterium was determined and detected in
patients with stage II CRC, but not stage I patients (0 vs.
0.012±0.022%; P=0.030). On the contrary, compared with patients
with stage II CRC, the abundance of Akkermansia was
significantly increased in those with stage III CRC (Fig. 6). Nevertheless, compared with
patients with stage III CRC, the genera Phascolarctobacterium,
Parasutterella, Comamonas, Cloacibacillus and Olsenella
were relatively enriched in those with stage IV CRC; the abundances
of Alistipes, Blautia, Eisenbergiella, Intestinimonas,
Eggerthella and Anaeroglobus were significantly
decreased in patients with stage IV CRC (Table II). Furthermore,
Methylophilus (0.003±0.012 vs. 0%; P=0.017) and
Synergistes (0.003±0.012 vs. 0%; P=0.017) were detected only
in stage III CRC patients (data not shown).
| Table II.Relative abundance of genera in fecal
samples and statistical differences in the gut microbiota at the
genus level between patients with stage III and IV CRC. |
Table II.
Relative abundance of genera in fecal
samples and statistical differences in the gut microbiota at the
genus level between patients with stage III and IV CRC.
| Genus | CRC stage III,
% | CRC stage IV,
% | P-value |
|---|
|
Phascolarctobacterium | 13.563±14.576 | 13.756±24.568 | 0.033 |
|
Parasutterella | 0.785±1.789 | 1.378±2.233 | 0.010 |
|
Comamonas | 0.781±2.250 | 1.870±8.948 | 0.026 |
|
Cloacibacillus | 0.090±0.257 | 0.280±1.069 | 0.025 |
|
Olsenella | 0.003±0.012 | 0.042±0.127 | 0.021 |
|
Alistipes | 24.969±60.888 | 8.852±13.432 | 0.016 |
| Blautia | 7.467±8.095 | 5.138±4.640 | 0.047 |
|
Eisenbergiella | 2.267±4.444 | 0.809±1.387 | 0.036 |
|
Intestinimonas | 0.305±1.134 | 0.239±0.613 | 0.027 |
|
Eggerthella | 0.234±0.523 | 0.153±0.255 | 0.026 |
|
Anaeroglobus | 0.070±0.243 | 0.002±0.009 | 0.009 |
Additionally, the key microorganisms of the gut
microbiota in the proximal (right) and distal (left) segments of
patients with CRC were analyzed. At the genus level, the abundance
of 442 genera in the distal (left) and proximal (right) segments
were inversely associated with splenic flexure syndrome. The
present study reported that these genera Veillonella,
Granulicatella and Coprobacter were more abundant in the
proximal (right) segment than in the distal (left) segment and the
differences were statistically significant (P<0.05; Table III). By contrast, the genera
Sneathia (0 vs. 0.024±0.065%; P=0.014),
Acetanaerobacterium (0 vs. 0.005±0.014%; P=0.036),
Phocaeicola (0 vs. 0.007±0.023%; P=0.048) and
Anaerofustis (0 vs. 0.007±0.023%; P=0.047) were detected
only in the proximal (right) segments (data not shown).
| Table III.Relative abundance of genera in the
fecal samples and statistical differences in the gut microbiota at
the genus level between the distal (left) and proximal (right)
segments of patients with CRC. |
Table III.
Relative abundance of genera in the
fecal samples and statistical differences in the gut microbiota at
the genus level between the distal (left) and proximal (right)
segments of patients with CRC.
| Genus | Distal (left)
segment, % | Proximal (right)
segment, % | P-value |
|---|
|
Veillonella | 1.548±3.792 | 16.747±33.805 | 0.004 |
|
Granulicatella | 0.191±0.300 | 0.468±0.630 | 0.018 |
|
Coprobacter | 0.058±0.161 | 0.716±2.135 | 0.042 |
Discussion
CRC is a major public health problem worldwide; the
cause of CRC is largely due to a combination of genetic
susceptibility and lifestyle and environmental factors (21). In recent years, differences in the
composition of the gut microbiota were reported to potentially
affect the initiation and progression of CRC; the dysbiosis of the
symbiotic microbiota may also be associated with systemic
inflammatory disorders and various types of cancer (22,23). At
present, tumor staging is the most important prognostic indicator
for patients with CRC and various strategies have been developed
for the TNM staging of certain types of cancer (24). In addition, the 16S rRNA gene
sequencing method has been widely used as an effective tool to
analyze the global microbial community (25). In the present study, the 16S rRNA
sequencing approach was applied to examine differences in the gut
microbiota between patients with CRC of stages I–IV and healthy
controls, and the proximal (right) and distal (left) segments
relative to the splenic flexor muscle.
The richness and diversity of the gut microbiota of
patients with CRC and healthy controls was compared. The richness
was represented by the Chao1 index and diversity was expressed by
the Shannon and Simpson indexes. No significant differences in the
Chao1, Shannon and Simpson indexes were observed between CRC
patients of stages I–IV and healthy controls. The possible cause
for these findings may be that the microbiota of fecal samples in
CRC patients of stages I–IV and healthy controls were examined at
relatively close stages, and the sample sizes could be insufficient
or unequal. The results also demonstrated no significant
differences in the Chao1 index between patients with CRC and
healthy controls but were reported for the Shannon and Simpson
indexes. This indicated the increased diversity of the gut
microbiota of patients with CRC compared with the healthy
controls.
Furthermore, the present study demonstrated that,
compared with healthy controls, there were differences in the
composition of the gut microbiota of CRC patients, regardless of
disease stage. The relative abundance of the top 5 dominant genera
Escherichia-Shigella, Bacteroides, Faecalibacterium,
Prevotella and Akkermansia differed. At the genus level,
compared with healthy controls, the relative abundances of
Prevotella, Collinsella and Peptostreptococcus were
increased in CRC patients. Previous studies have reported that
Prevotella, Peptostreptococcus and other opportunistic
bacteria are relatively abundant at tumor sites (17,26),
which was consistent with the gut microbiota of fecal samples
analyzed in the present study. The genus Peptostreptococcus,
formerly known as Peptococcus, has recently been determined
to be involved in CRC and was identified to promote the
proliferation of CRC cells by inducing the biosynthesis of
intracellular cholesterol (27,28).
Additionally, patients with symptomatic atherosclerosis, classified
as atherosclerotic plaques associated with carotid stenosis that
lead to cerebrovascular events, exhibited a high abundance of
Collinsella (29). The
present study identified that Escherichia-Shigella was
significantly reduced in CRC patients. Of note, the number of
Escherichia-Shigella in the tumor tissue of patients with
CRC was determined to be reduced (11); however, the increased abundance of
Escherichia-Shigella suggested its association with the
pathogenesis of inflammatory bowel disease (IBD) (30). These results indicate that
Escherichia-Shigella leads to different manifestations in
patients with CRC and IBD, yet further investigation is required to
determine whether these strains are pathogenic in CRC. In addition,
CRC-associated Prevotella has also been reported to be
linked with variations in mucosal gene-expression profiles, which
could be used as a tool for screening CRC in high-risk populations
(5,31). These findings and the results of the
present study suggested that Prevotella, Collinsella,
Peptostreptococcus and Escherichia-Shigella may
contribute to variations in the gut microbiota of patients with
CRC. In addition, gene and species markers may indicate alterations
in the microbiota during early stages of neoplastic growth, which
suggests the potential of sensitive microbial markers of advanced
adenomas, such as CRC (16).
Nevertheless, the present study reported that the relative
abundance of these bacteria was associated with CRC patients;
however, the direct link between bacterial imbalance and CRC
requires further investigation.
At the genus level, differences were also identified
in the gut microbiota among patients with CRC stages I–IV and
healthy controls. The results revealed that the relative abundances
of Collinsella and Peptostreptococcus were
substantially increased in patients with stage I CRC compared with
healthy controls (stage O), which was consistent with differences
in the gut microbiota between healthy controls and all CRC
patients. The relative abundance of Ruminococcus was also
increased in stage I CRC patients. A previous study demonstrated
that, compared with healthy controls and advanced adenomas,
Alistipes were enriched in patients with CRC (32). In addition, the abundance of
OTU-related Alistipes fnegoldii gradually increased during
the development of CRC, P53, K-RAS and BRAF,
demonstrating that biological evolutionary transformation of gut
microbiota, characterized by the increase in DNA damage-causing
bacteria, is associated with tumorigenesis in CRC models (33). On the contrary, the present study
identified that, compared with stage III CRC, the relative
abundance of Alistipes was decreased in patients with stage
IV CRC. This was inconsistent with previous results and could be
due to the small sample size of the present study. Therefore,
studies involving more samples should be conducted to determine
potential differential characteristics of the gut microbiota at
different stages of CRC.
Differences in the gut microbiota of the proximal
(right) and distal (left) colon segments of patients with CRC were
determined. At the genus level, compared with the distal (left)
segment, the genera Veillonella, Granulicatella and
Coprobacter were highly enriched in the proximal (right)
segment. Veillonella dispar was associated with patients
with adenocarcinoma (34); however,
Sneathia, Acetanaerobacterium, Phocaeicola and
Anaerofustis were detected only in the proximal (right)
segments, suggesting that these gut microbiota could be related to
these regions within patients with CRC.
The present study demonstrated differences in the
gut microbiota of stool samples at the genus level between stage
I–IV CRC patients and healthy controls, and the proximal (right)
and distal (left) segments relative to the splenic flexor muscle.
Of note, the sample size of patients within the different groups
employed in the present study was relatively small, which may pose
certain limitations. For instance, other intestinal bacterial
species may not be identified, such as Fusobacterium
nucleatum (Fn), which is a gram-negative anaerobe that
is enriched in the oral cavity, but is rarely detected in other
organs under physiological conditions (35). Fn serves an important role in
the organization of biofilms in the oral cavity and may be a
dominant microbe that creates physical and metabolic scaffolds that
support the microbial shift of numerous microbes in developing
tumors over time (36). However,
this bacterium is not a predominant species in stool samples and
has been detected in cancer biopsies of patients with CRC by two
independent research groups using whole-genome sequencing
techniques (37). Regarding
Fn, its abundance was lower in the present study; bacteria
of higher abundance at the genus level were selected for further
analysis. Fn has been recently identified as a pathogenic
bacteria in CRC (38). Additionally,
the present study reported that the levels of Fn were
increased in tumor tissues compared with in adjacent normal tissues
of the same patients, supporting the authors' previous study
(39), which revealed its role in
promoting the initiation and development of CRC. Future
investigations with a larger number of patients are required to
determine the effects of Fn.
In conclusion, despite the low number of samples
included in the present study, dysbiosis and increased diversity of
the gut microbiota were reported in CRC. In addition,
Prevotella, Collinsella, Peptostreptococcus and
Escherichia-Shigella were proposed as potential fecal
markers for the early detection of CRC. The results of the present
study also suggest that bacterial communities and CRC might be
further investigated for their possible correlations in order to
assess the opportunity of detecting colon cancer through the
analysis of specific fecal bacterial markers (40). In addition, differences were also
reported in the bacterial populations present in the gut microbiota
of different sites and at various stages of disease. Therefore, the
constituents of the gut microbiota may be associated with the
biological characteristics of CRC; however, whether variations in
biological disorders are related to the pathogenesis of CRC, or
simply the results of competitive bacteria utilization in the tumor
microenvironment, remains to be confirmed in larger clinical and
experimental studies. Of note, to improve understanding of the
occurrence and development of CRC, other candidate pathogens should
be investigated in the future using tumor samples of patients with
CRC, oral microbiota and related animal models. Furthermore,
additional metagenomic data can enable the in-depth analysis of
cancer-associated differences in gene function, genomic content and
variation. This could serve as a basis for determining the
potential mechanisms underlying the roles of the microbiota in the
development and progression of cancer (16,41).
Future investigations may aid the monitoring of the microbiota in
the early detection of CRC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Natural Science
Foundation of Zhejiang Province (grant no. LY18H160018) and the
Medical and Health Science and Technology Project of Zhejiang
Province (grant no. 2017KY360).
Availability of data and materials
The data used to support the finding of this study
are available from the corresponding author upon request.
Authors' contributions
QS, HD and JL analyzed and interpreted the patient
data regarding the patients with colorectal cancer; QS wrote the
manuscript; HD, XFC and XBC participated in the experimental study
and data analysis; YT, LP and QW participated data collecting and
statistical analysis; JL conceived the idea and designed the study
as the corresponding authors of this paper; All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of The First Affiliated Hospital, College of
Medicine, Zhejiang University (approval no. 2016-436). All patients
indicated that they had obtained written informed consent in the
present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Russo E, Taddei A, Ringressi MN, Ricci F
and Amedei A: The interplay between the microbiome and the adaptive
immune response in cancer development. Therap Adv Gastroenterol.
9:594–605. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Flemer B, Lynch DB, Brown JM, Jeffery IB,
Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F and O'Toole PW:
Tumour-associated and non-tumour-associated microbiota in
colorectal cancer. Gut. 66:633–643. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakatsu G, Li X, Zhou H, Sheng J, Wong SH,
Wu WK, Ng SC, Tsoi H, Dong Y, Zhang N, et al: Gut mucosal
microbiome across stages of colorectal carcinogenesis. Nat Commun.
6:8727–8736. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bufill JA: Colorectal cancer: Evidence for
distinct genetic categories based on proximal or distal tumor
location. Ann Intern Med. 113:779–788. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tamas K, Walenkamp AM, de Vries EG, van
Vugt MA, Beets-Tan RG, van Etten B, de Groot DJ and Hospers GA:
Rectal and colon cancer: Not just a different anatomic site. Cancer
Treat Rev. 41:671–679. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Missiaglia E, Jacobs B, D'Ario G, Di Narzo
AF, Soneson C, Budinska E, Popovici V, Vecchione L, Gerster S, Yan
P, et al: Distal and proximal colon cancers differ in terms of
molecular, pathological, and clinical features. Ann Oncol.
25:1995–2001. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shan L, Konstantinov SR, Smits R and
Peppelenbosch MP: Bacterial biofilms in colorectal cancer
initiation and progression. Trends Mol Med. 23:18–30. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao Z, Guo B, Gao R, Zhu Q and Qin H:
Microbiota disbiosis is associated with colorectal cancer. Front
Microbiol. 6:202015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang
X, Jia W, Cai S and Zhao L: Structural segregation of gut
microbiota between colorectal cancer patients and healthy
volunteers. ISME J. 6:320–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Flemer B, Herlihy M, O'Riordain M,
Shanahan F and O'Toole PW: Tumour-associated and
non-tumour-associated microbiota: Addendum. Gut Microbes. 8:1–5.
2018. View Article : Google Scholar
|
|
14
|
Sabino J, Vieira-Silva S, Machiels K,
Joossens M, Falony G, Ballet V, Ferrante M, Van Assche G, Van der
Merwe S, Vermeire S and Raes J: Primary sclerosing cholangitis is
characterised by intestinal dysbiosis independent from IBD. Gut.
65:1681–1689. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y,
Chen Y, Yang F, Lu N, Wang Z, et al: Dysbiosis signature of fecal
microbiota in colorectal cancer patients. Microb Ecol. 66:462–470.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Russo E, Bacci G, Chiellini C, Fagorzi C,
Niccolai E, Taddei A, Ricci F, Ringressi MN, Borrelli R, Melli F,
et al: Preliminary comparison of oral and intestinal human
microbiota in patients with colorectal cancer: A pilot study. Front
Microbiol. 8:2699–2712. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao R, Kong C, Huang L, Li H, Qu X, Liu Z,
Lan P, Wang J and Qin H: Mucosa-associated microbiota signature in
colorectal cancer. Eur J Clin Microbiol Infect Dis. 36:2073–2083.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamady M, Walker JJ, Harris JK, Gold NJ
and Knight R: Error-correcting barcoded primers allow hundreds of
samples to be pyrosequenced in multiplex. Nat Methods. 5:235–237.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edgar RC: UPARSE: Highly accurate OTU
sequences from microbial amplicon reads. Nat Methods. 10:996–998.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lozupone C, Lladser ME, Dan K, Stombaugh J
and Knight R: UniFrac: An effective distance metric for microbial
community comparison. Isme J. 5:169–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rodriguez-Castaño GP, Caro-Quintero A,
Reyes A and Lizcano F: Advances in gut microbiome research, opening
new strategies to cope with a western lifestyle. Front Genet.
7:2242017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
García-Castillo V, Sanhueza E, Mcnerney E,
Onate SA and García A: Microbiota dysbiosis: A new piece in the
understanding of carcinogenesis puzzle. J Med Microbiol.
65:1347–1362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rajagopala SV, Vashee S, Oldfield LM,
Suzuki Y, Venter JC, Telenti A and Nelson KE: The human microbiome
and cancer. Cancer Prev Res (Phila). 10:226–234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei Z, Cao S, Liu S, Yao Z, Sun T, Li Y,
Li J, Zhang D and Zhou Y: Could gut microbiota serve as prognostic
biomarker associated with colorectal cancer patients' survival? A
pilot study on relevant mechanism. Oncotarget. 7:46158–46172. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caporaso JG, Lauber CL, Walters WA,
Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N and Knight R:
Global patterns of 16S rRNA diversity at a depth of millions of
sequences per sample. Proc Natl Acad Sci USA. 108:4516–4522. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen W, Liu F, Ling Z, Tong X and Xiang C:
Human intestinal lumen and mucosa-associated microbiota in patients
with colorectal cancer. PLoS One. 7:e397432012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu J, Feng Q, Wong SH, Zhang D, Liang QY,
Qin Y, Tang L, Zhao H, Stenvang J, Li Y, et al: Metagenomic
analysis of faecal microbiome as a tool towards targeted
non-invasive biomarkers for colorectal cancer. Gut. 66:70–78. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu
G, Ng SC, Chan AWH, Chan FKL, Sung JJY and Yu J: Peptostreptococcus
anaerobius induces intracellular cholesterol biosynthesis in colon
cells to induce proliferation and causes dysplasia in mice.
Gastroenterology. 152:1419–1433. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karlsson FH, Fåk F, Nookaew I, Tremaroli
V, Fagerberg B, Petranovic D, Bäckhed F and Nielsen J: Symptomatic
atherosclerosis is associated with an altered gut metagenome. Nat
Commun. 3:12452012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Wang W, Zhou R, Ng SC, Li J, Huang
M, Zhou F, Wang X, Shen B, A Kamm M, et al: Characteristics of
fecal and mucosa-associated microbiota in Chinese patients with
inflammatory bowel disease. Medicine (Baltimore). 93:e512014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Purcell RV, Visnovska M, Biggs PJ,
Schmeier S and Frizelle FA: Distinct gut microbiome patterns
associate with consensus molecular subtypes of colorectal cancer.
Sci Rep. 7:115902017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng Q, Liang S, Jia H, Stadlmayr A, Tang
L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al: Gut microbiome
development along the colorectal adenoma-carcinoma sequence. Nat
Commun. 6:65282015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun T, Liu S, Zhou Y, Yao Z, Zhang D, Cao
S, Wei Z, Tan B, Li Y, Lian Z and Wang S: Evolutionary biologic
changes of gut microbiota in an ‘adenoma-carcinoma sequence’ mouse
colorectal cancer model induced by 1,2-Dimethylhydrazine.
Oncotarget. 8:444–457. 2017.PubMed/NCBI
|
|
34
|
Kasai C, Sugimoto K, Moritani I, Tanaka J,
Oya Y, Inoue H, Tameda M, Shiraki K, Ito M, Takei Y, et al:
Comparison of human gut microbiota in control subjects and patients
with colorectal carcinoma in adenoma: Terminal restriction fragment
length polymorphism and next-generation sequencing analyses. Oncol
Rep. 35:325–333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han YW: Fusobacterium nucleatum: A
commensal-turned pathogen. Curr Opin Microbiol. 23:141–147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brennan CA and Garrett WS: Gut microbiota,
inflammation, and colorectal cancer. Annu Rev Microbiol.
70:395–411. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kostic AD, Gevers D, Pedamallu CS, Michaud
M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et
al: Genomic analysis identifies association of Fusobacterium with
colorectal carcinoma. Genome Res. 22:292–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yan X, Liu L, Li H, Qin H and Sun Z:
Clinical significance of Fusobacterium nucleatum,
epithelial-mesenchymal transition, and cancer stem cell markers in
stage III/IV colorectal cancer patients. Onco Targets Ther.
10:5031–5046. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sheng QS, He KG, Li JJ, Zhong ZF, Wang FX,
Pan LL and Lin JJ: Comparison of gut microbiome in colorectal
cancer in paired tumor and adjacent normal tissues. OncoTargets and
Therapy. 2019.(In Press).
|
|
40
|
Schloissnig S, Arumugam M, Sunagawa S,
Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J,
et al: Genomic variation landscape of the human gut microbiome.
Nature. 493:45–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zeller G, Tap J, Voigt AY, Sunagawa S,
Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et
al: Potential of fecal microbiota for early-stage detection of
colorectal cancer. Mol Syst Biol. 10:7662014. View Article : Google Scholar : PubMed/NCBI
|