Introduction
Cholangiocarcinoma (CCA) is a primary adenocarcinoma
and malignancy arising from the epithelium of the bile duct. CCA
can be classified by its origin into intrahepatic, perihilar, and
distal CCA. The pathogenesis of CCA is poorly understood and needs
further investigation. Furthermore, the incidence and mortality
rate of CCA has increased worldwide over past decades and now
accounts for 3% of all gastrointestinal malignancies (1). It is also highly prevalent in Southeast
Asia particularly in Thailand due to the endemic parasitic biliary
tract infestation (2). Lack of
effective biomarkers makes early diagnosis of CCA difficult
(3). The occurrence of symptoms may
not be apparent until the cancer reaches an advanced stage
resulting in severe outcomes (4).
Previous studies have reported the association of chronic liver
diseases, hepatolithiasis, chronic biliary inflammation and
cholestasis with the development of CCA (3). Under liver inflammation, accumulation
of cholesterol was observed and caused extreme damage to the cells
(5). This indicates that cholesterol
plays a significant role in liver diseases.
Bile consisting of cholesterol, phospholipids,
bilirubin conjugates, bile salts and toxic substances is secreted
from hepatocytes and passes along bile duct through gallbladder or
small intestine. Excess cholesterols are excreted via bile and
eliminated into feces (6). Some of
cholesterols and unconjugated bile acids can passively diffuse into
cholangiocytes (7). Cholangiocytes
have important roles in modifying and delivering the bile to its
destination by secreting bicarbonate and water thus, preventing
bile acid diffusion and maintaining osmolality of the cell
(7). At the same time,
cholangiocytes maintain their cholesterol homeostasis and form
junction preventing the hepatic interstitial tissue from these
secreted toxic substances and bile (6). When the cholestasis occurred by the
bile duct obstruction, this led to overexposure of cholangiocytes
to bile lipid contents and toxic substances (8). Oxysterols are found to be increased in
bile acids of patients with biliary tract inflammation. They are
cholesterol oxidation derivatives in human bile and activators of
the hedgehog signaling pathway which associates in cell
proliferation, migration, and invasion of CCA (9,10).
Cholesterol transport is a crucial cellular
homeostatic mechanism. ATP-binding cassette (ABC) A1 and ABCG1 are
well characterized as cholesterol transporters in various cell
types (11–14). ABCA1 transports cholesterol and
phospholipids to apolipoprotein A-1 (ApoA-1) while ABCG1 transports
cholesterol to mature high density lipoprotein (HDL). In this
context, ABCA1 malfunction is associated with atherosclerosis.
Cholesterol accumulation causes a decrease in ABCA1 level enhancing
intracellular cholesterol excess in macrophages. This leads to
inflammation and cell apoptosis which eventually resulting in
atherosclerosis (15). Furthermore,
ABCA1 function is disrupted by epigenetic alteration of promoter
hypermethylation in prostate cancer. Decrease in ABCA1 export
ability enhances accumulation of intracellular cholesterol.
Extended cholesterol pools are consistent with prostate cancer
progression and aggressiveness (16). This underlines the importance of
ABCA1 and cholesterol in diseases and cancers.
Statins, 3-hydroxy-3-methyl glutaryl coenzyme A
(HMG-CoA) reductase inhibitors, are well-known for their LDL
cholesterol lowering effects. HMG-CoA reductase is involved in
cholesterol synthesis reducing HMG-CoA to mevalonate which is
eventually converted into cholesterol. Statins primarily inhibit
this process thus lowering cholesterol synthesis to balance
cholesterol homeostasis (17).
Multiple effects of statins in anti-proliferation and promotion of
apoptosis are found to be useful in reducing risk of CCA cancer
(18,19). For instance, statins diminished
insulin-like growth factor 1 (IGF-1) leading to the reduction of
CCA cell proliferation (20).
Statins also caused anti-proliferation and enhanced cell apoptosis
by cell cycle arrest at sub G1 fraction in CCA (19). Further studies also revealed that
simvastatin down-regulated serine/threonine kinase protein kinase B
(Akt). Signalling through Akt plays a prominent role in several
cancer types including prostate cancer (21), glioblastoma (22) and CCA (20). In addition, signaling through Akt
pathway regulates ABCA1 transporter in pancreatic beta cells
(23). Inhibition in Akt
subsequently decreased ABCA1 expression and enhanced cholesterol
efflux to Apo-A1 during autophagy which caused lipid raft imbalance
in hepatocytes and macrophages (24).
In this study, the role of statins in the expression
and function of major cholesterol transporters, ABCA1 and ABCG1 in
CCA cells was investigated. Also, the involvement of Akt pathway in
regulation of these transporters was examined. A specific condition
of pre-exposure of CCA cells to cholesterol was also set to
evaluate the effects of statins on ABCA1 and ABCG1-mediated efflux
and its contribution to CCA survival and progression.
Materials and methods
Cell cultures
CCA cell lines including KKU-100 (JCRB no.1568),
KKU-M213 (JCRB no. 1557) (25),
HuCCA-1 (JCRB no. 1657) (26) were
obtained from JCRB cell bank. RMCCA-1 cell line was cultured in our
laboratory (Pubmed 17072981) (27)
and HepG2 cells (ATCC no. 8065) were used to represent liver cancer
cells. They were maintained in HAM's F12 (Invitrogen; Thermo Fisher
Scientific, Inc.) and DMEM (Invitrogen) containing 10% fetal bovine
serum (FBS) (Sigma-Aldrich; Merck KGaA), 0.1 U/l penicillin and 0.1
g/l streptomycin (Invitrogen) at 37°C in a 5% CO2
incubator.
Preparation of statins and
cholesterol
Simvastatin and atorvastatin (both from Santa Cruz
Biotechnology, Inc.) were dissolved in DMSO (Sigma-Aldrich; Merck
KGaA) and dilutions were made for appropriate treatment conditions
at 1% final concentration of DMSO. Cholesterol (Santa Cruz
Biotechnology, Inc.) was dissolved in chloroform and a desired
concentration of cholesterol was left after being evaporated using
nitrogen gas. The cholesterol was then resuspended in
methyl-β-cyclodextrin (MCD; Sigma-Aldrich; Merck KGaA) at a ratio
of 1:4.
Clonogenic survival assay
KKU-100 cells were seeded at 700 cells per well in a
6-well plate and incubated for 24 h. Cells were subjected to
cholesterol loading for 1 h and statin treatment for 48 h. They
were allowed to grow into colonies for 14 days in a complete
medium. Cells were then fixed with 4% paraformaldehyde, stained
with crystal violet to examine their colony forming ability. Images
of colonies were taken. Crystal formation was dissolved with
glacial acetic acid before reading at 540 nm using an Infinite M200
PRO microplate reader (Tecan). The optimal statin and cholesterol
concentrations were determined from cell viability curve. In
subsequent assays, simvastatin at 25–50 µM and cholesterol at 10 µM
were used, 1% DMSO in medium and/or MCD was added in a non-treated
control cells.
Oil Red O staining
After statin and cholesterol treatment, KKU-100
cells were fixed with 4% paraformaldehyde, stained with 0.5% oil
red o (Santa Cruz Biotechnology, Inc.) and images were taken using
an inverted light microscope (Eclipse TS100; Nikon).
RNA isolation and gene expression
analysis by RT-PCR
Total RNA isolation was performed using RNeasy plus
mini kit (Qiagen) according to the manufacturer's instructions.
Total RNA concentration was determined by measuring a ratio of
260/280 nm absorbance using NanoVue (GE Healthcare Life Sciences).
First strand complementary DNA (cDNA) was created from 1 µg total
RNA template using RevertAidReverse Transcriptase (Thermo Fisher
Scientific, Inc.) according to manufacturer's instructions.
One-hundred ng of cDNA was amplified using specific primers and Taq
DNA (Vivantis). cDNA was used as a template in PCR analysis to
measure the relative expression of ABCA1, ABCG1, ABCG5, and
ABCG8 mRNA. The nucleotide sequences of the primers were as
follows: hABCA1 (NM_005502.3), hABCA1-F GACGCAAACACAAAAGTGGA,
hABCA1-R AACAAGCCATGTTCCCTCAG; hABCG1 (NM_207628.1),
hABCG1-FCAGGGACCTTTCCTATTCGG, hABCG1-RGGCCACCAACTCACCACTAT; hABCG5
(NM_022436.2), hABCG5-F GGCAGATCATGTGCATCCTA, hABCG5-R
ACATACACCTCCCCCAGGAA; hABCG8 (NM_022437.2)
hABCG8-FATTTCACAGCCATCGGCTAC, hABCG8-RCGAGTGACTGAGCCTTCTCC;
hβ-actin (NM_001101.4), hβ-actin-FGCACAGAGCCTCGCCTT,
hβ-actin-RCTTTGCACATGCCGGAG. PCR products were amplified (95°C, 1
min; followed by 40 cycles of 95°C, 45 sec, and 58°C, 45 sec; 72°C,
45 sec) and analyzed on a MC Nexus Gradient PCR cycler (Eppendorf
AG). PCR products were loaded onto 2% agarose gel and stained with
ethidium bromide. The sequences of the fragments amplified by PCR
were confirmed by DNA sequencing. Semi-quantitative mRNA expression
of ABCA1, ABCG1, ABCG5, and ABCG8 was normalized to
β-actin mRNA levels using ImageJ program (Version 1.48) to
calculate band intensity.
RNA isolation and gene expression
analysis by qPCR
Total RNA isolation was conducted and cDNA was
created from 1 µg total RNA template as above. A total of 100 ng of
cDNA was amplified using above specific primers and SYBR Green
real-time PCR master mix (Toyobo) according to manufacturer's
instructions to measure the relative expression of ABCA1 and
ABCG1 mRNA. PCR products were amplified (95°C, 1 min;
followed by 40 cycles of 95°C, 15 sec, and 60°C, 15 sec) and
analyzed on a CFX96 Touch real-time PCR cycler (Bio-Rad
Laboratories, Inc.). Fluorescence threshold cycles (CT) of each
sample were compared and normalized with the CT values of
housekeeping gene. The relative expression of ABCA1 and
ABCG1 were compared between controls and treatment.
Protein extraction and western blot
analysis
Cells were treated with or without simvastatin for
48 h and then lysed in a lysis buffer (10 mM Tris, 150 mM NaCl, 1mM
EDTA, 0.5% Triton X-100, and 1 mM PMSF protease inhibitor). Thirty
µg proteins were reduced by heating with loading buffer (225 mM
Tris, 25% (v/v) glycerol, 0.75 mM bromophenol blue and 10 mM DTT
with pH 6.8) at 95°C for 10 min and were separated by discontinuous
SDS polyacrylamide gel-electrophoresis (Bio-Rad Laboratories,
Inc.). Electrophoresis was performed using a Bio-Rad Mini
Trans-Blot Cell at 80 volts for 3 h and 8% gels were used for
ABCA1, ABCG1, Akt1 and pAkt (ser473). Proteins were transferred to
nitrocellulose membranes (HYBOND ECL; GE Healthcare Life Sciences)
using Bio-Rad Mini Trans-Blot Cell at 90 volts for 2 h before being
blocked with 5% skimmed milk in PBS 0.1% Tween-20 for 1.5 h. Mouse
monoclonal anti-actin (AC-15; Abcam), mouse monoclonal
anti-ABCA1(AB.H10; EMD Millipore), rabbit monoclonal anti-ABCG1
(EP1366Y; Abcam), mouse monoclonal anti-Akt1 (B-1; Santa Cruz
Biotechnology, Inc.) and rabbit monoclonal anti-pAkt (ser473)(D9E,
Cell Signaling Technology, Inc.) antibodies were added at 4°C for
16 h. Membranes were incubated with HRP-conjugated goat anti-mouse
(Invitrogen; Thermo Fisher Scientific, Inc.) and goat anti-rabbit
antibody (Invitrogen; Thermo Fisher Scientific, Inc.) diluted in
PBS at 25°C for 2 h. They were developed by SureBlue TMB membrane
substrate (KPL). Band intensity of TMB color was analyzed by ImageJ
program (version 1.48).
Cholesterol efflux assay
Cells were seeded at 2×104 cells per well
in black 96-well plates and incubated for 24 h. Cells were treated
with or without cholesterol for 1 h and replaced with serum-free
medium for 24 h before being treated with or without simvastatin.
Cells were then replaced with serum-free medium containing labeling
media for 1 h. Labeling medium consisted of bodipy cholesterol
(Avanti Polar Lipids), MCD, HEPES (Sigma-Aldrich; Merck KGaA), and
egg phosphatidylcholine (Avanti Polar Lipids) and prepared as
previously described (28). The
final concentrations of bodipy cholesterol, egg
phosphatidylcholine, and MCD in the labeling medium were 0.025 mM,
0.1 mM, and 10 mM, respectively. Cells were washed with HAM's
F12-HEPES and incubated with serum-free medium for 18 h. ApoA-1 and
HDL (both from Lee Biosolutions) were added for 6 h at 100 µg/ml
and 70 µg/ml, respectively. Cells were dissolved with 1% cholic
acid (Sigma-Aldrich; Merck KGaA) in 1N NaOH for 4 h with rocking
while the supernatant was collected and centrifuged at 10,000 ×g
for 5 min. The cholesterol fluorescence intensity value was
recorded using a microplate reader (excitation at 482 nm and
emission at 515 nm). The % cholesterol efflux = cholesterol efflux
intensity/(intracellular cholesterol + cholesterol efflux)
×100%.
Immunofluorescence of ABCA1 and ABCG1
transporter
Cells were seeded at 104 cells per well
in 96-well plates and incubated for 24 h. They were treated with or
without cholesterol for 1 h and replaced with serum-free medium for
24 h before being treated with or without simvastatin. Cells were
fixed with 4% paraformaldehyde in PBS for 20 min and permeabilized
with 0.1% saponin (Sigma-Aldrich; Merck KGaA) in PBS for 15 min.
They were then blocked with 2% FBS in PBS for 1 h at 25°C and
incubated with mouse monoclonal anti-ABCA1 (Millipore) and rabbit
polyclonal anti-ABCG1 (AAS52436C; Antibody Verify) antibodies at
1:50 dilution at 4°C for 16 h. Cells were washed with 0.1% saponin
in PBS and incubated with Alexa flour 488-conjugated goat
polyclonal anti-mouse (Invitrogen; Thermo Fisher Scientific, Inc.)
and Alexa flour 546-conjugated goat polyclonal anti-rabbit
(Invitrogen; Thermo Fisher Scientific, Inc.) antibodies for 2 h.
They were counterstained with Hoechst (33342; Molecular Probes) for
10 min. Fluorescence intensity was visualized and photographed
using an IX83 inverted fluorescence microscope with CellSens
software platform (Olympus Cooperation).
Statistical analysis
Quantitative data are presented as mean ± SEM.
Statistical significance between groups was analyzed using standard
t-tests or two-way ANOVA followed by the Bonferroni test.
Significant P-values are indicated within the figure panels. Error
bars indicate SEM.
Results
Statin inhibited cell viability and
decreased intracellular cholesterol level in KKU-100 cells
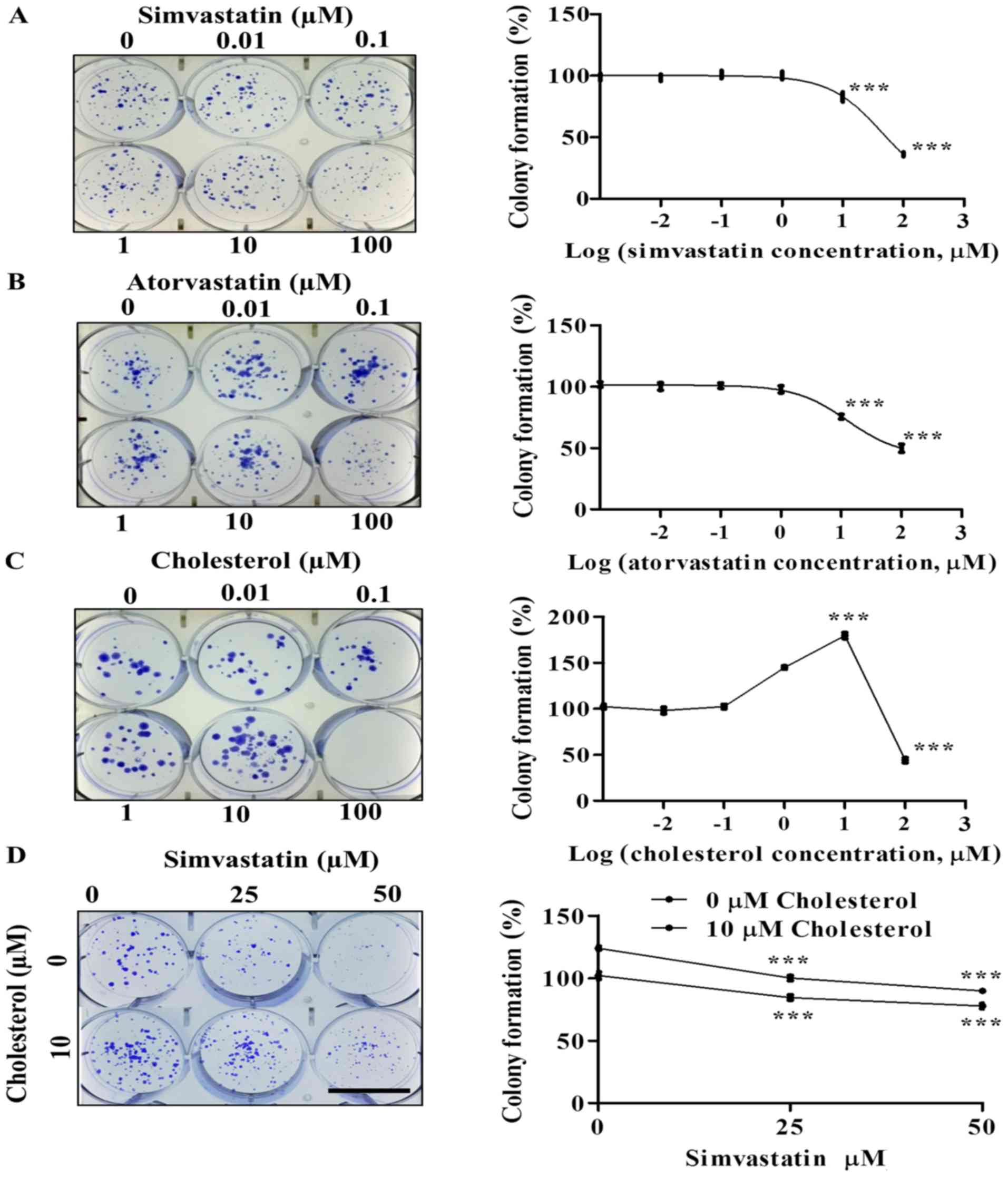
The effects of simvastatin, atorvastatin and
cholesterol on cell survival and growth of KKU-100 cells were
determined by clonogenic assay. These statins caused a
concentration-dependent decrease in the colony formation ability of
KKU-100 cells. At low concentrations (0.01–10 µM), simvastatin and
atorvastatin had minimal effects on colony formation of KKU-100
cells. However, they significantly inhibited KKU-100 cell growth at
100 µM, (P<0.001; Fig. 1A and B).
Using the MTT assay, cytotoxic activities of simvastatin and
atorvastatin were also observed. Both statins had different levels
of cytotoxicity to four CCA cell lines (Fig. S1). Cholesterol increased KKU-100
cell colony formation at 1–10 µM (P<0.001) but decreased it at
100 µM (P<0.001) compared to controls (Fig. 1C).
In order to examine the effect of simvastatin on
cell growth and survival in the presence of cholesterol, KKU-100
cells were loaded with 10 µM cholesterol prior to being incubated
with simvastatin (25–50 µM) for 48 h. In the absence of
cholesterol, KKU-100 cell viability decreased as the concentration
of simvastatin increased from 25–50 µM. In the presence of 10 µM
cholesterol, the reduction of cell viability was observed but at
lower degree than that without cholesterol (Fig. 1D).
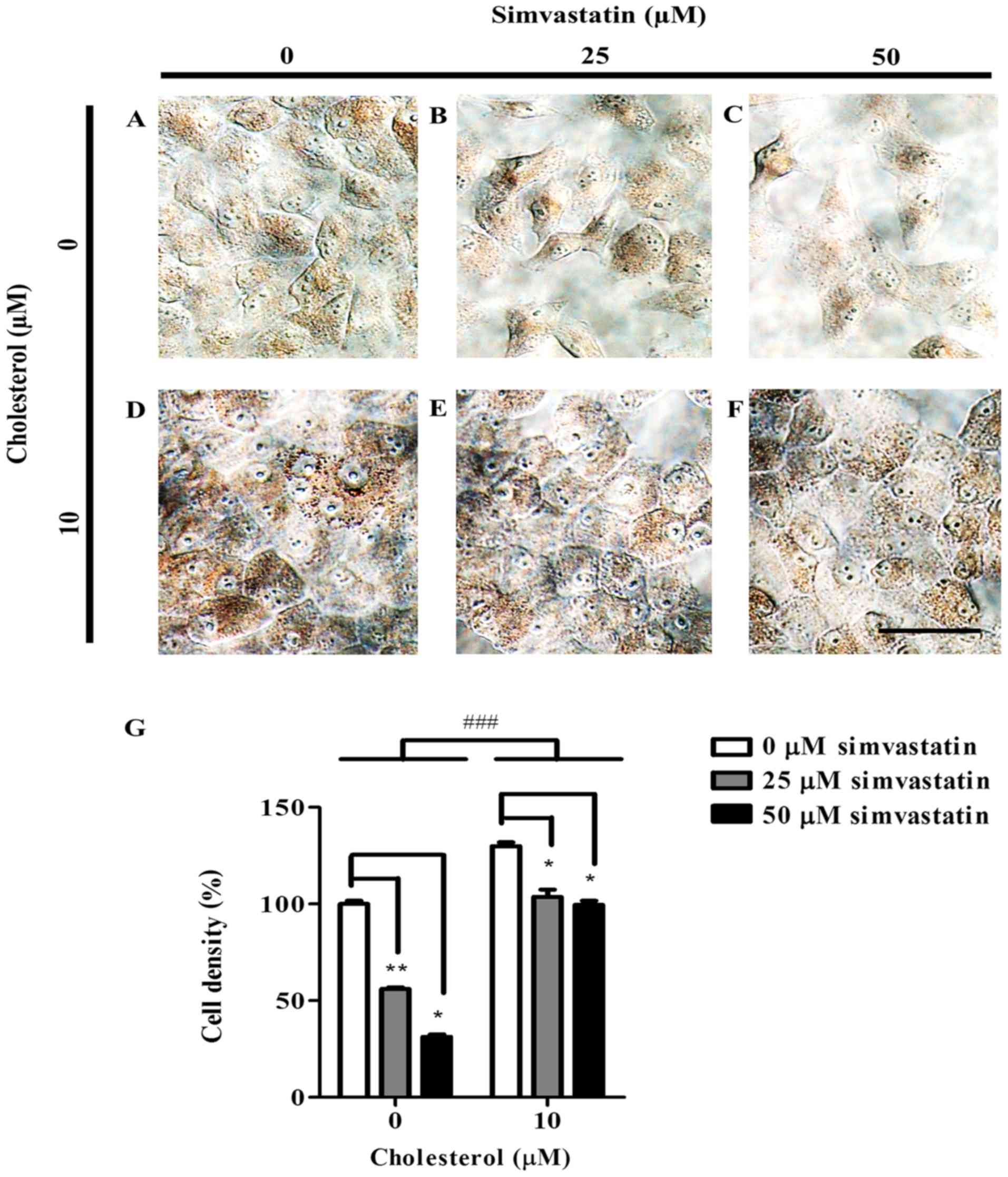
Oil red o staining revealed that without
cholesterol, the level of both intracellular lipids and neutral
triglycerides declined as KKU-100 cells were exposed to higher
concentrations of simvastatin (Fig. 2A
and C). Pre-cholesterol loaded KKU-100 cells showed greater
intensity of oil red o suggesting elevated levels of intracellular
lipids (Fig. 2D). However, with
higher concentration of simvastatin, oil red o staining intensity
reduced but at lesser extent than those without cholesterol
(Fig. 2D and F). In comparison with
0 µM, KKU-100 cell density at 25 and 50 µM simvastatin decreased by
44% (P<0.01) and by 69% (P<0.05), respectively (Fig. 2G). In the presence of 10 µM
cholesterol, KKU-100 cell density reduced by 26% (P<0.05) and by
30% (P<0.05), respectively when compared to a control group.
(Fig. 2G).
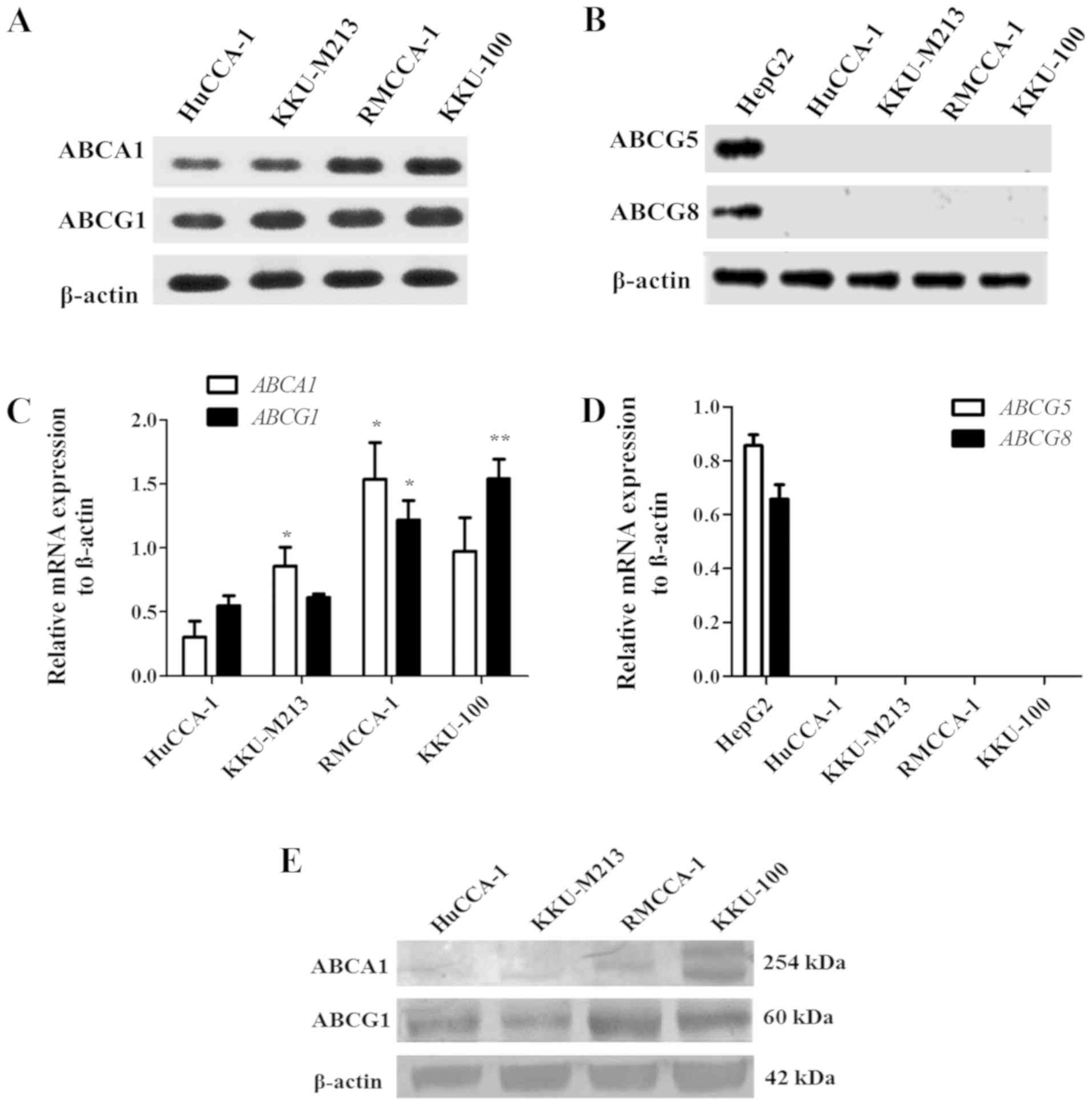
CCA cells expressed ABCA1 and ABCG1
transporters which mediated cholesterol efflux
CCA cell lines were examined for expression of
ABCA1, ABCG1, ABCG5 and ABCG8 by RT-PCR. RMCCA1 and
KKU-100 cells expressed relatively higher levels of ABCA1
and ABCG1 mRNA than those of HuCCA-1 and KKU-M213 cells
(Fig. 3A and C). Neither
ABCG5 nor ABCG8 mRNA was present in CCA cell lines.
We were able to detect ABCG5 and ABCG8 only in HepG2
cells which were used as a liver cancer cell line (Fig. 3B and D).
Western blot analysis revealed that KKU-100 cells
expressed the highest level of ABCA1. The expression level of ABCG1
was higher in HuCCA-1, RMCCA-1 and KKU-100 cells but slightly lower
in KKU-M213 cells (Fig. 3E). We
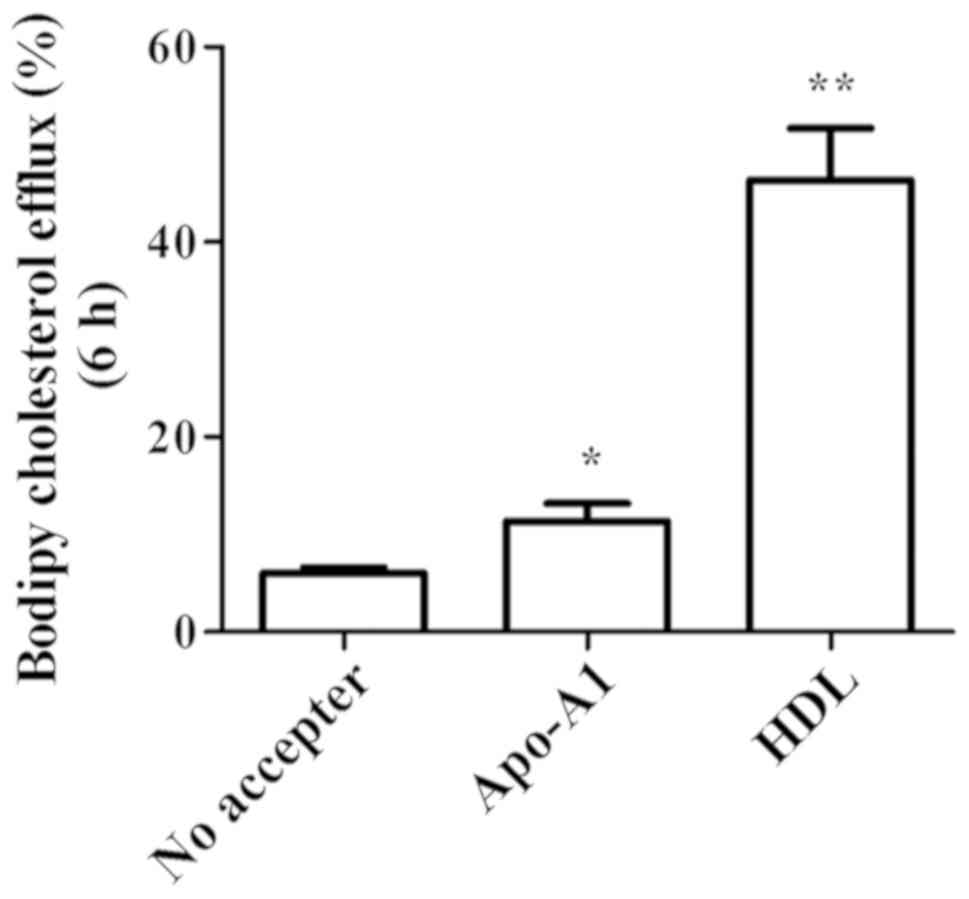
conducted cholesterol efflux assay using bodipy cholesterol in
KKU-100 cells which expressed highest level of ABCA1 and ABCG1.
Baseline condition was set in the absence of cholesterol acceptor
by which the constitutive efflux was at 6.00±0.58% (Fig. 4). By adding Apo-A1, cholesterol
translocation which was mediated specifically through ABCA1
increased to 11.33±1.86% (P<0.05; Fig. 4). HDL also triggered cholesterol
efflux via ABCG1. Export through this mechanism was obvious at
46.33±5.33% (P<0.01) (Fig.
4).
Simvastatin decreased ABCA1 and ABCG1
expression
The effect of simvastatin on ABCA1 and ABCG1
expression was evaluated by real-time PCR and western blot
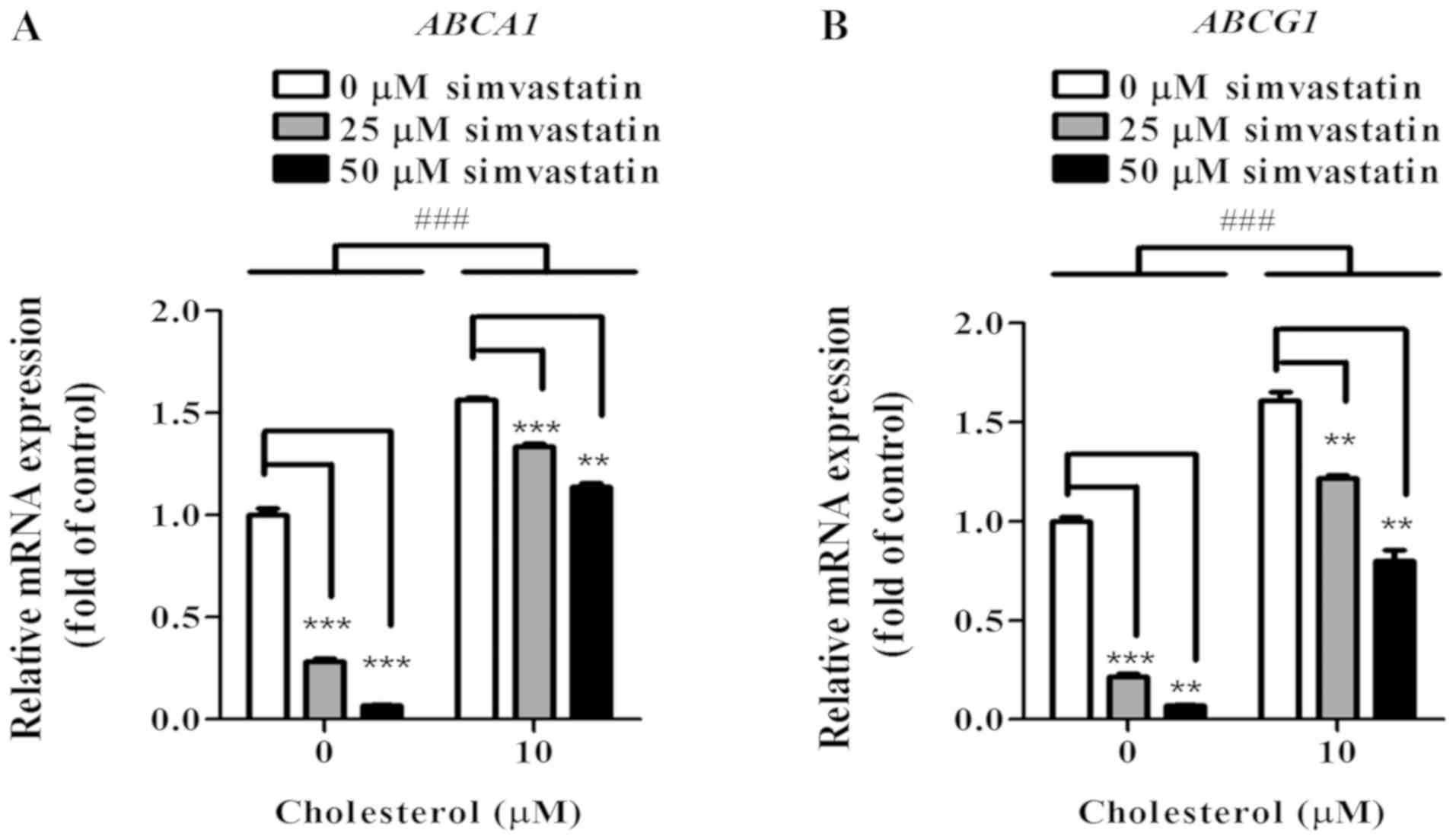
analysis. Compared to control, at 25 and 50 µM of simvastatin
treatment, the relative levels of ABCA1 mRNA expression
decreased significantly by 72% (P<0.001) and by 91%
(P<0.001), respectively (Fig.
5A). Compared to control, at 25 and 50 µM of simvastatin
treatment, the relative levels of ABCG1 mRNA expression
decreased significantly by 78% (P<0.001) and by 93%
(P<0.001), respectively (Fig.
5B). Following exposure of cholesterol at 10 µM, KKU-100 cells
showed significantly enhanced ABCA1 and ABCG1 expression at 50%
(P<0.001) and 60% (P<0.001), respectively comparing to no
cholesterol loaded cells (Fig. 5A and
B). However, with 25 and 50 µM simvastatin, expression of ABCA1
and ABCG1 decreased in a concentration-dependent manner (Fig. 5A and B).
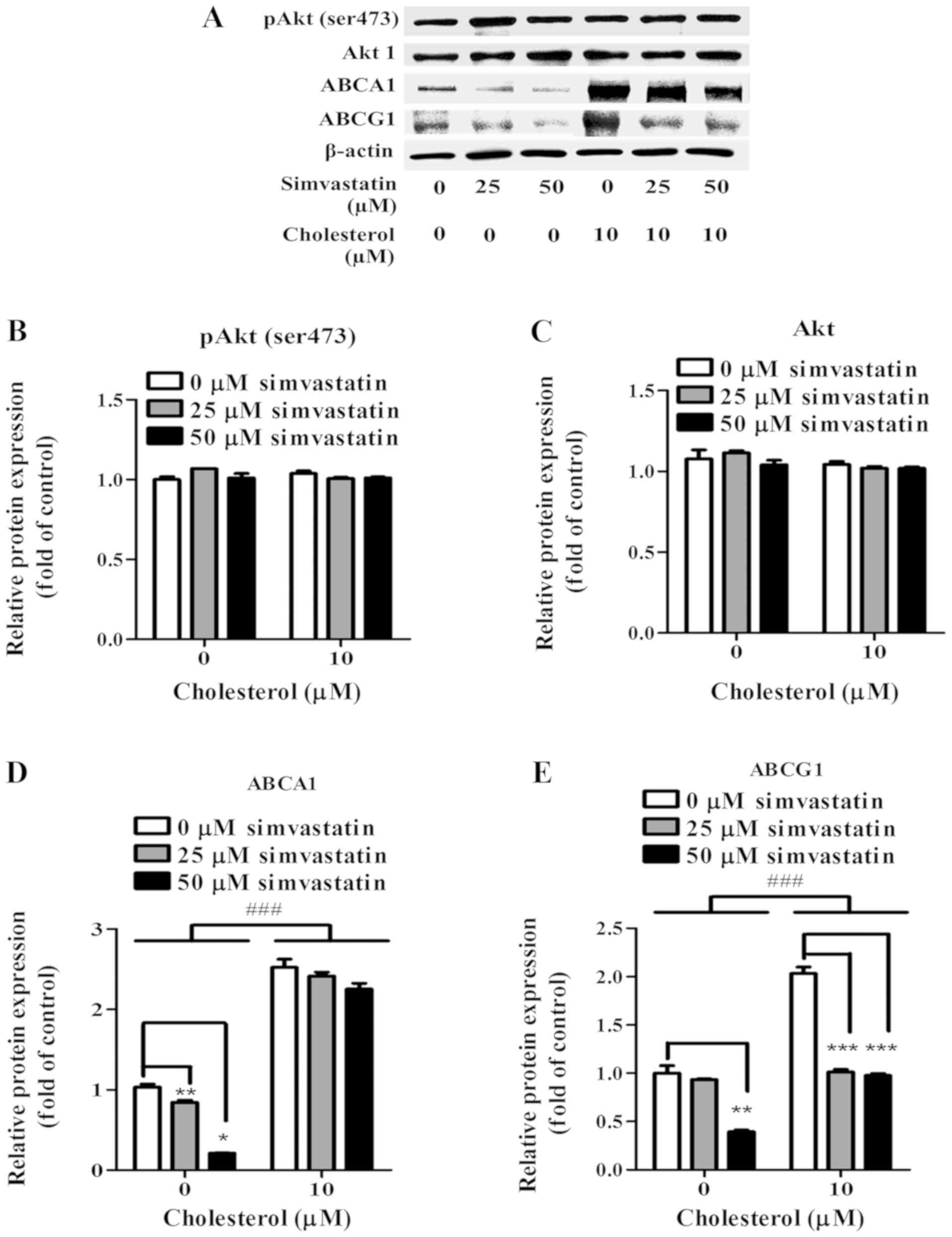
Simvastatin also caused a concentration-dependent
decrease in the levels of ABCA1 and ABCG1 protein expression in
KKU-100 cells. (Fig. 6A). At 25 µM
of simvastatin treatment, relative ABCA1 and ABCG1 proteins were
reduced by 15% (P<0.05) and by 8%, respectively (Fig. 6D and E). Expression of ABCA1 and
ABCG1 protein at 50 µM of simvastatin treatment were drastically
decreased by 79% (P<0.001) and by 61% (P<0.01), respectively
(Fig. 6D and E). Following exposure
of cholesterol at 10 µM, KKU-100 cells showed enhanced ABCA1 and
ABCG1 expression at 177% (P<0.001) and 100% (P<0.001),
respectively comparing to no cholesterol loaded cells (Fig. 6D and E). However, with 25 and 50 µM
simvastatin, expression of ABCA1 and ABCG1 decreased in a
concentration-dependent manner (Fig.
6A).
In order to investigate the role of Akt in ABCA1 and
ABCG1 expression in KKU-100 cells, Akt and pAkt (ser473 protein
expression were evaluated (Fig. 6A).
KKU-100 cells constitutively expressed Akt and pAkt (ser473)
(Fig. 6B and C). Nonetheless, in the
presence of simvastatin, KKU-100 cells retained the levels of those
proteins (Fig. 6B and C). Exposures
of cholesterol at 10 µM did not noticeably alter the level of Akt
and pAkt (ser473) expression (Fig. 6B
and C). Among pre-cholesterol loaded KKU-100 cells, levels of
those proteins were comparable between simvastatin-treated and
non-treated cells suggesting the Akt-independent manner of ABCA1
and ABCG1 expression in KKU-100 cells (Fig. 6B).
Intracellular localization of ABCA1
and ABCG1 in KKU-100 cells
We visualized the intracellular expression of ABCA1
and ABCG1 in KKU-100 cells by immunocytochemistry. The expression
of ABCA1 (green) was observed within cytoplasm. Intense staining of
ABCA1 was detected particularly in the proximity of the nucleus
(Fig. 7A). Similar to ABCA1, ABCG1
expression was recorded in the cytoplasmic region of KKU-100 cells.
However, the staining showed a more dispersed pattern of ABCG1
expression (red). This transporter could be detected throughout the
cytoplasm and in the submembrane region (Fig. 7B and C). Co-localization of ABCA1 and
ABCG1 staining showed overlapping expression of these two
transporters (Fig. 7C). Following
simvastatin treatment, at 25 µM, KKU-100 cells exhibited lower
levels of both ABCA1 and ABCG1 expression (Fig. 7D and F). Simvastatin seemed to act in
a concentration-dependent manner as lower expression of these two
transporters was observed at higher concentration (50 µM) of
simvastatin (Fig. 7G and I).
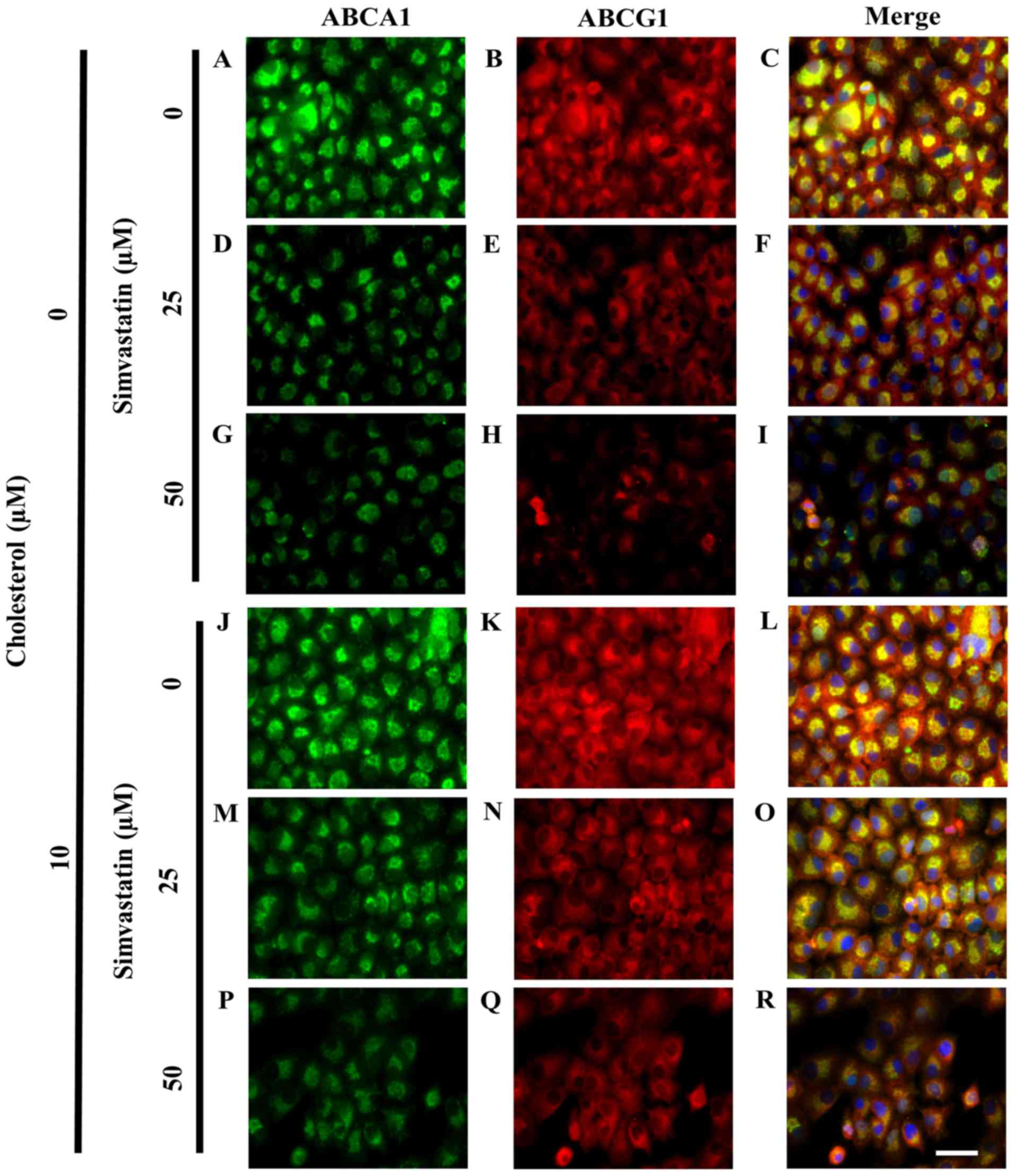 | Figure 7.Cholesterol loading prevented
simvastatin-induced reduction of ABCA1 and ABCG1 expression in
KKU-100 cells. Fluorescence micrographs show cells which were
loaded with cholesterol before being treated with simvastatin at
various concentrations for 48 h. They were fixed, permeabilized and
stained using specific antibodies against ABCA1 (green) and ABCG1
(red). Hoechst (blue) was counterstained for nucleus. At 0 µM
simvastatin, the localization of (A) ABCA1, (B) ABCG1 and the (C)
co-localization of ABCA1 and ABCG1 were determined. KKU-100 cells
were treated with 25 µM, the localization of (D) ABCA1, (E) ABCG1
and the (F) co-localization of ABCA1 and ABCG1 were examined. With
50 µM of simvastatin, the expression of (G) ABCA1, (H) ABCG1 and
(I) ABCA1 and ABCG1 were localized in KKU-100 cells. In
pre-cholesterol loading condition, (10 µM) (J-R), at 0 µM
simvastatin, the localization of (J) ABCA1, (K) ABCG1 and the (L)
co-localization of ABCA1 and ABCG1 were determined. At 25 µM
simvastatin, the localization of (M) ABCA1, (N) ABCG1 and the (O)
co-localization of ABCA1 and ABCG1 were examined with 50 µM of
simvastatin, the expression of (P) ABCA1, (Q) ABCG1 and (R) ABCA1
and ABCG1 were localized in KKU-100 cells. Scale bar is 50 µm for
A-R. Scale bar=50 µm for A-R. ABC, ATP-binding cassette. |
Pre-exposure to cholesterol minimized
simvastatin effect in CCA cells
Following exposure to cholesterol at 10 µM, KKU-100
cells showed enhanced ABCA1 and ABCG1 expression (Fig. 7J and L). With 25 µM simvastatin,
KKU-100 cells retained expression of ABCA1 and ABCG1 in
pre-cholesterol loaded cells compared with non-loaded cells
(Fig. 7M and O). This expression was
greatly decreased at greater simvastatin condition (50 µM). However
with cholesterol, expressions of both ABCA1 and ABCG1 was more
visible (Fig. 7P and R) than those
without the pre-exposure to cholesterol (Fig. 7G and I).
Different effect of simvastatin on
cholesterol transport in CCA cells
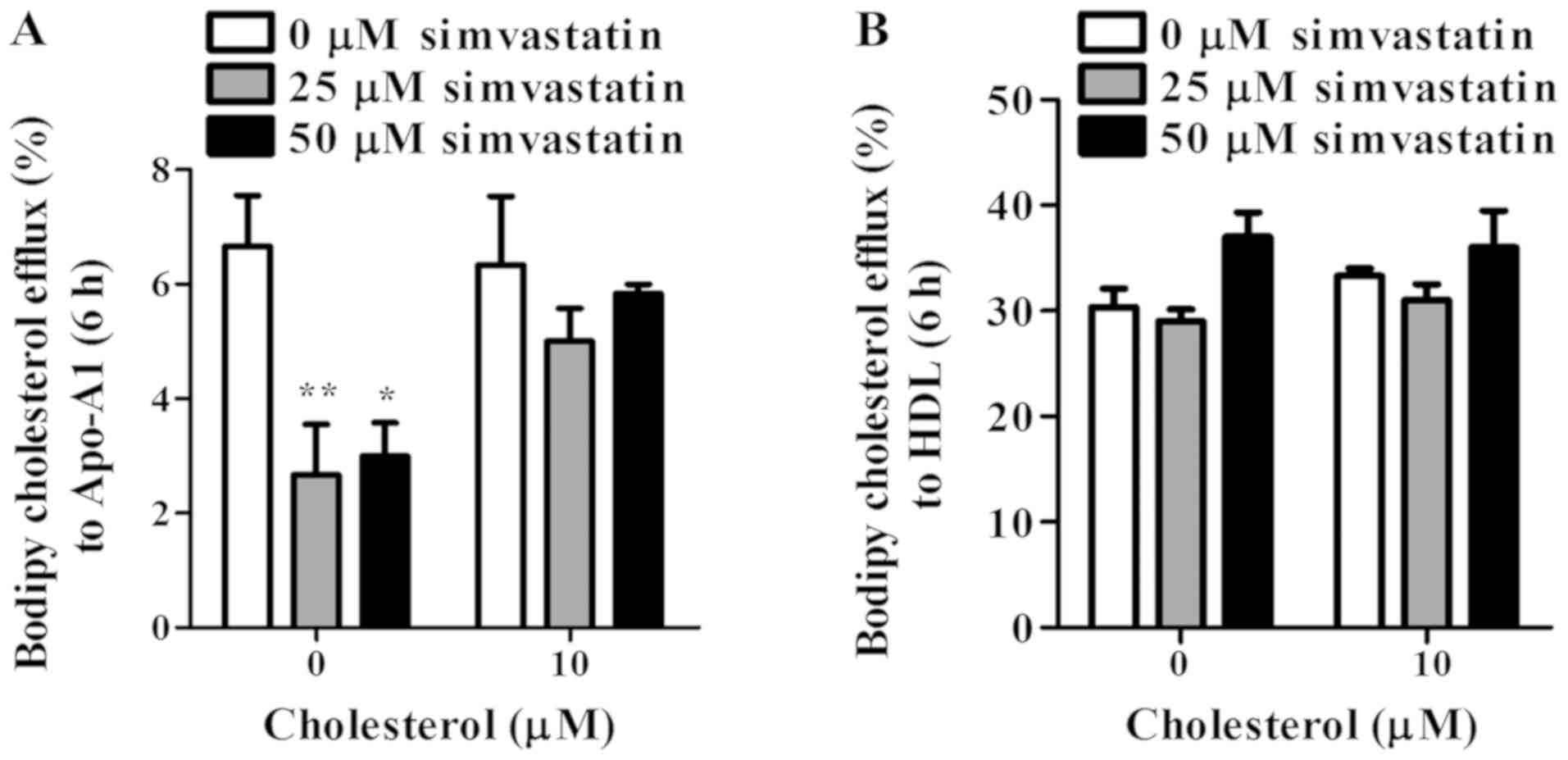
The effect of simvastatin on cholesterol efflux was
further examined. Cholesterol export through ABCA1 in
simvastatin-treated KKU-100 cells decreased by 60% (P<0.01) and
55% (P<0.05) at 25 and 50 µM simvastatin, respectively comparing
to non-treated controls (Fig. 8A).
Interestingly, pre-exposure of KKU-100 cells to cholesterol did not
change the ABCA1-mediated function, although these cells showed
up-regulated ABCA1 expression levels. In simvastatin treatment,
efflux to Apo-A1 of pre-cholesterol loaded KKU-100 cells decreased
but only at marginal levels (Fig.
8A). For ABCG1 function, simvastatin-treated cells showed
similar levels of cholesterol export to HDL to non-treated cells
(Fig. 8B). Under stimulation of
cholesterol, simvastatin had no considerable effect on this
cholesterol efflux compared to controls. ABCG1 knockdown experiment
was performed in HuCCA-1 cells using RNA interference. siABCG1
transfection successfully decreased ABCG1 expression level
(Fig. S2). However, comparable
cholesterol efflux to HDL was found between siABCG1 cells and
non-silenced (wild-type) cells (Fig.
S3).
Discussion
In our study, cholesterol is found to play an
important role in CCA cell viability. We demonstrated that
cholesterol (1–10 µM) significantly increased KKU-100 cell
viability. Growth promoting effect of cholesterol was also reported
in another CCA study (18). Similar
to our findings, cholesterol increased prostate cancer cell
proliferation (21). In addition,
in vivo experiments revealed that high fat diet-fed mice
showed enhanced breast tumor size and metastasis compared with
normal diet fed mice (29). By
increasing intracellular cholesterol viability, this favored cancer
cell proliferation because cholesterol is an important component
for cell membrane synthesis (30).
However, at a very high cholesterol concentration (100 µM), a
significant decrease in CCA cell proliferation was found. Studies
in gastric carcinoma and neuroblastoma cells also revealed this
phenomenon. High cholesterol concentrations caused cytotoxicity
which then decreased cell proliferation and induced cell apoptosis
(31,32). High accumulation of cholesterol also
caused foam cell formation in macrophages leading to their
malfunction in preventing the atherosclerosis (33). Exposure to high cholesterol enhanced
macrophage cell apoptosis via the mitochondrial Fas signaling
pathway (33). These studies
emphasize cholesterol as a cell growth driving substance and its
homeostasis is also important in cell survival, growth,
proliferation and apoptosis.
Statins have been well studied in term of their
cholesterol lowering ability. They have been widely used to treat
hyperlipidemia in humans. Natural cholesterol sources are from
plasma cholesterol which is obtained from cholesterol biosynthesis
and from intestinal absorption. Statins reduced serum lipids by
suppressing cholesterol biosynthesis which eventually caused
decrease in cell viability (18,19,34,35). In
our experiments, simvastatin reduced intracellular cholesterol
level of KKU-100 cells which is similar to other CCA studies that
simvastatin suppressed endogenous cholesterol synthesis (18) through the reduced HMG-CoA reductase
activity (36). Nonetheless, these
effects of simvastatin were prevented in the presence of
cholesterol. Cholesterol-loading CCA cells negated the decreased
intracellular cholesterol and decreased cell viability under
simvastatin (25 µM) exposure. Recent findings elsewhere also showed
that pre-cholesterol exposure of various cancer cells including
CCA, esophageal adenocarcinoma, macrophages, and T-cell leukemia
elevated unesterified cholesterol pools (18,37–41). It
is therefore possible that unesterified cholesterol supply resist
the effect of simvastatin (18,39,41,42).
In our paper, we confirm that in CCA, ABCA1 and
ABCG1 transporters mediate cholesterol efflux to Apo-A1 and HDL,
respectively. These transporters are well-known as predominant
cholesterol transporters in macrophages and hepatocarcinoma
assisting cholesterol export and maintaining cholesterol
homeostasis (11–14). ABCA1 is localized in proximity to the
nucleus and spread throughout the cytoplasm of KKU-100 cells. In
fibroblasts, ABCA1-mediated cellular cholesterol transport also
occurs in late endosome (43).
Apo-A1 is trafficked via ABCA1 from cell membrane into late
endosome in which lipidated Apo-A1 would subsequently become
nascent HDL (43). ABCG1 is
dominantly found perinuclear endosomes and plasma membrane. In
ABCG1-transfected HeLa cells, ABCG1 mediates cholesterol efflux by
either vesicular or non-vesicular pathway (44). ABCG1-mediated intracellular
cholesterol would be shuttled from late endocytic compartment to
the cell membrane in order to deliver cholesterol to extracellular
HDL In this study, we could not detect either ABCG5 or ABCG8
expression in our four CCA cell lines used. These transporters are
well studied, showing high expression in hepatocarcinoma cells
assisting cholesterol export as a part of bile composition
(45). Therefore, we speculated that
for CCA cells, ABCA1 and ABCG1, but not ABCG5 and ABCG8 potentially
play role in cholesterol transport. We demonstrated here in our
work that CCA cells constitutively exported cholesterol via ABCG1
to HDL to a greater extent than to Apo-A1 which was mediated
through ABCA1. This was similar to cholesterol export levels in
macrophages (28,46). This suggests a smaller range of
cholesterol translocation ability of ABCA1 compared to ABCG1.
Further examination was carried out to identify the
effects of simvastatin on ABCA1 and ABCG1 levels in CCA cell lines.
Our experiments are consistent with previous results that ABCA1 and
ABCG1 level decreased in simvastatin treatment. The influence of
simvastatin on cholesterol homeostasis genes has been shown in a
variety of cancer cell lines (37,47).
Statin treatment caused reduction in ABCA1 and ABCG1 expression in
epithelial colorectal adenocarcinoma cells (47) and macrophages (37). This supported the theory that
simvastatin decreased intracellular cholesterol synthesis, thereby
limiting mevalonate and sources of oxysterol production. Oxysterols
are important for the nuclear receptor, LXR, in maintaining
cholesterol homeostasis (48). In
macrophages, LXR controls ABCA1 and ABCG1 activation (49). Therefore, the condition which low
mevalonate together with oxysterols triggers down-regulation of
ABCA1 and ABCG1 expression caused by statins could occur in CCA.
Further experiments on the quantification of mevalonate and
oxysterols would clarify this point. In this paper, cholesterol
efflux to Apo-A1 decreased under simvastatin treatment suggesting a
possible role of ABCA1 in KKU-100 cells. Cholesterol efflux to
Apo-A1 was reduced in simvastatin treated macrophages (37,39). Our
results suggest that the inhibitory effect of simvastatin could be
specific to efflux to Apo-A1 but not to HDL, and more apparent in
non-cholesterol loaded cells. Cholesterol-loaded cells reversed the
effect of simvastatin in cholesterol translocation to Apo-A1.
Relevant pathways and ligands such as those in LXR signaling were
possibly restored under cholesterol-loaded conditions reversing the
effects of simvastatin at certain concentrations (37,39). In
contrast, other papers revealed a simvastatin-facilitated increment
in ABCA1 levels and function in hepatocarcinoma cells which
displayed an atheroprotective effect of simvastatin via peroxisome
proliferator-activated receptors (PPARs) (37).
The role of Akt has been reported with cholesterol
availability and growth of certain cancer cells (18,22,37).
Downregulation of Akt signaling together with decreased
proliferation was detected in glioblastoma after statin treatment
(22). Inhibition of pAkt by
treating with the MK2206 molecule reduced CCA cell growth (50). We speculated that a decrease in CCA
cell proliferation under simvastatin would involve the Akt pathway.
The levels of Akt and pAkt (serine473) of KKU-100 cells were
analyzed. Neither total Akt nor pAkt expression level was affected
in these cells under simvastatin treatment. It had similarly shown
in pancreatic cancer cells that atorvastatin did not inhibit total
Akt levels (51). Also, in the
long-term treatment of the non-small lung cancer A549 cell line
with atorvastatin, the level of pAkt (serine473) did not alter. As
previously indicated by Miraglia and colleagues that a purinergic
receptor P2X7 was a specific target of statin and indeed only
nuclear level of Akt was downregulated by statin (52), additional analysis of nuclear and
cytoplasmic levels of those proteins in KKU-100 cells would clarify
the actual Akt expression. The ABCA1 regulation has been reported
with an Akt-dependent pathway in macrophages and hepatocytes
(23,24). Nonetheless, our results presented
otherwise that down-regulation of ABCA1 and ABCG1 was not through
the Akt pathway. Disruption in lipid rafts and ABCA1 and ABCG1
transporters could be associated with LXR pathways (49). Further investigation to demonstrate
the link between the LXR and ABCA1 and ABCG1 pathways has currently
been carried out.
To summarize, we demonstrated that ABCA1 and ABCG1
potentially play roles in cholesterol translocation in CCA cells.
Simvastatin decreased CCA cell viability, intracellular cholesterol
and ABCA1 and ABCG1 expression by an Akt-independent pathway.
However, pre-exposure of KKU-100 cells to cholesterol reduced the
effect of statins on cell viability and intracellular cholesterol
levels. Cholesterol export via ABCA1 and ABCG1 remained unaffected
in cholesterol-loaded KKU-100 cells in the presence of simvastatin.
This indicates the limitations of statin treatment in CCA patients
suffering with hypercholesterolemia.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Philip D.
Round, Department of Biology, Mahidol University for critical
manuscript reading.
Funding
The current study was supported by National Research
Council of Thailand (NRCT; grant no. 2559-A1.8). PS was awarded the
2016 NRCT postgraduate scholarship and research assistantship from
Mahidol University. TJ and RT were recipients of the Thailand
Research fund and the Medical Research Council (UK), Newton Fund
Project (grant nos. DBG 5980006 and MR/N01247X/1).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
PS, TJ, TK and SK designed the study. PS, SK and RT
performed the experiments. PS, TJ, TK, and SK analyzed the data. PS
and SK wrote the manuscript. All authors reviewed the manuscript
and approved the final version.
Ethics approval and consent to
participate
All experimental procedures were performed in
compliance with institutional requirements and were approved by the
Institutional Ethics Committee of Mahidol University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Espey DK, Wu XC, Swan J, Wiggins C, Jim
MA, Ward E, Wingo PA, Howe HL, Ries LA, Miller BA, et al: Annual
report to the nation on the status of cancer, 1975–2004, featuring
cancer in American Indians and Alaska Natives. Cancer.
110:2119–2152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Srivatanakul P, Khlat M,
Chenvidhya D, Chotiwan P, Insiripong S, L'Abbé KA and Wild CP:
Liver cancer in Thailand. I. A case-control study of
cholangiocarcinoma. Int J Cancer. 48:323–328. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dhanasekaran R, Hemming AW, Zendejas I,
George T, Nelson DR, Soldevila-Pico C, Firpi RJ, Morelli G, Clark V
and Cabrera R: Treatment outcomes and prognostic factors of
intrahepatic cholangiocarcinoma. Oncol Rep. 29:1259–1267. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lieser MJ, Barry MK, Rowland C, Ilstrup DM
and Nagorney DM: Surgical management of intrahepatic
cholangiocarcinoma: A 31 year experience. J Hepatobiliary Pancreat
Surg. 5:41–47. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead
JF and Varghese Z: Inflammatory stress exacerbates lipid
accumulation in hepatic cells and fatty livers of apolipoprotein E
knockout mice. Hepatology. 48:770–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rao RK and Samak G: Bile duct epithelial
tight junctions and barrier function. Tissue Barriers.
1:e257182013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hofmann AF: The enterohepatic circulation
of bile acids in mammals: Form and functions. Front Biosci
(Landmark Ed). 14:2584–2598. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tabibian JH, Masyuk AI, Masyuk TV, O'Hara
SP and LaRusso NF: Physiology of cholangiocytes. Compr Physiol.
3:541–565. 2013.PubMed/NCBI
|
|
9
|
Nachtergaele S, Mydock LK, Krishnan K,
Rammohan J, Schlesinger PH, Covey DF and Rohatgi R: Oxysterols are
allosteric activators of the oncoprotein smoothened. Nat Chem Biol.
8:211–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Olkkonen VM, Béaslas O and Nissilä E:
Oxysterols and their cellular effectors. Biomolecules. 2:76–103.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeng Y, Peng Y, Tang K, Wang YQ, Zhao ZY,
Wei XY and Xu XL: Dihydromyricetin ameliorates foam cell formation
via LXRα-ABCA1/ABCG1-dependent cholesterol efflux in macrophages.
Biomed Pharmacother. 101:543–552. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Jiang B, Liang P, Tong Z, Liu M, Lv
Q, Liu Y, Liu X, Tang Y and Xiao X: Nucleolin protects macrophages
from oxLDL-induced foam cell formation through up-regulating ABCA1
expression. Biochem Biophys Res Commun. 486:364–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Collins HL, Ranalletta M, Fuki IV,
Billheimer JT, Rothblat GH, Tall AR and Rader DJ: Macrophage ABCA1
and ABCG1, but not SR-BI, promote macrophage reverse cholesterol
transport in vivo. J Clin Invest. 117:2216–2224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Basso F, Freeman L, Knapper CL, Remaley A,
Stonik J, Neufeld EB, Tansey T, Amar MJ, Fruchart-Najib J, Duverger
N, et al: Role of the hepatic ABCA1 transporter in modulating
intrahepatic cholesterol and plasma HDL cholesterol concentrations.
J Lipid Res. 44:296–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng B and Tabas I: ABCA1-mediated
cholesterol efflux is defective in free cholesterol-loaded
macrophages. Mechanism involves enhanced ABCA1 degradation in a
process requiring full NPC1 activity. J Biol Chem. 277:43271–43280.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee BH, Taylor MG, Robinet P, Smith JD,
Schweitzer J, Sehayek E, Falzarano SM, Magi-Galluzzi C, Klein EA
and Ting AH: Dysregulation of cholesterol homeostasis in human
prostate cancer through loss of ABCA1. Cancer Res. 73:1211–1218.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Argmann CA, Edwards JY, Sawyez CG, O'Neil
CH, Hegele RA, Pickering JG and Huff MW: Regulation of macrophage
cholesterol efflux through hydroxymethylglutaryl-CoA reductase
inhibition: A role for RhoA in ABCA1-mediated cholesterol efflux. J
Biol Chem. 280:22212–22221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller T, Yang F, Wise CE, Meng F,
Priester S, Munshi MK, Guerrier M, Dostal DE and Glaser SS:
Simvastatin stimulates apoptosis in cholangiocarcinoma by
inhibition of Rac1 activity. Dig Liver Dis. 43:395–403. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kamigaki M, Sasaki T, Serikawa M, Inoue M,
Kobayashi K, Itsuki H, Minami T, Yukutake M, Okazaki A, Ishigaki T,
et al: Statins induce apoptosis and inhibit proliferation in
cholangiocarcinoma cells. Int J Oncol. 39:561–568. 2011.PubMed/NCBI
|
|
20
|
Lee J, Hong EM, Jang JA, Park SW, Koh DH,
Choi MH, Jang HJ and Kae SH: Simvastatin induces apoposis and
suppresses insulin-like growth factor 1 receptor in bile duct
cancer cells. Gut Liver. 10:310–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Y, Sukumaran P, Varma A, Derry S,
Sahmoun AE and Singh B: Cholesterol-induced activation of TRPM7
regulate cell proliferation, migration, and viability of human
prostate cells. Biochimica Biophys Acta. 1843:1839–1850. 2014.
View Article : Google Scholar
|
|
22
|
Yanae M, Tsubaki M, Satou T, Itoh T, Iman
M, Yamazoe Y and Nishida S: Statin-induced apoptosis via the
suppression of ERK1/2 and Akt activation by inhibition of the
geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp
Clin Cancer Res. 30:742011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lyu J, Imachi H, Iwama H, Zhang H and
Murao K: Insulin-like growth factor 1 regulates the expression of
ATP-binding cassette Transporter A1 in pancreatic beta cells. Horm
Metab Res. 48:338–344. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong F, Mo Z, Eid W, Courtney KC and Zha
X: Akt Inhibition promotes ABCA1-mediated cholesterol efflux to
ApoA-I through suppressing mTORC1. PLoS One. 9:e1137892014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholangiocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sirisinha S, Tengchaisri T, Boonpucknavig
S, Prempracha N, Ratanarapee S and Pausawasdi A: Establishment and
characterization of a cholangiocarcinoma cell line from a thai
patient with intrahepatic bile duct cancer. Asian Pac J Allergy
Immunol. 9:153–157. 1991.PubMed/NCBI
|
|
27
|
Rattanasinganchan P, Leelawat K,
Treepongkaruna SA, Tocharoentanaphol C, Subwongcharoen S,
Suthiphongchai T and Tohtong R: Establishment and characterization
of a cholangiocarcinoma cell line (RMCCA-1) from a thai patient.
World J Gastroenterol. 12:6500–6506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sankaranarayanan S, Kellner-Weibel G, de
la Llera-Moya M, Phillips MC, Asztalos BF, Bittman R and Rothblat
GH: A sensitive assay for ABCA1-mediated cholesterol efflux using
BODIPY-cholesterol. J Lipid Res. 52:2332–2340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dos Santos CR, Domingues G, Matias I,
Matos J, Fonseca I, de Almeida JM and Dias S: LDL-cholesterol
signaling induces breast cancer proliferation and invasion. Lipids
Health Dis. 13:162014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pussinen PJ, Karten B, Wintersperger A,
Reicher H, McLean M, Malle E and Sattler W: The human breast
carcinoma cell line HBL-100 acquires exogenous cholesterol from
high-density lipoprotein via CLA-1 (CD-36 and LIMPII analogous
1)-mediated selective cholesteryl ester uptake. Biochem J.
349:559–566. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lim SC, Parajuli KR, Duong HQ, Choi JE and
Han SI: Cholesterol induces autophagic and apoptotic death in
gastric carcinoma cells. Int J Oncol. 44:805–811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang YN, Lin CI, Liao H, Liu CY, Chen YH,
Chiu WC and Lin SH: Cholesterol overload induces apoptosis in
SH-SY5Y human neuroblastoma cells through the up regulation of
flotillin-2 in the lipid raft and the activation of BDNF/Trkb
signaling. Neuroscience. 328:201–209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yao PM and Tabas I: Free cholesterol
loading of macrophages induces apoptosis involving the fas pathway.
J Biol Chem. 275:23807–23813. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin CJ, Liao WC, Chen YA, Lin HJ, Feng CL,
Lin CL, Lin YJ, Kao MC, Huang MZ, Lai CH and Kao CH: Statin therapy
is associated with reduced risk of peptic ulcer disease in the
taiwanese population. Front Pharmacol. 8:2102017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang T, Seah S, Loh X, Chan CW, Hartman M,
Goh BC and Lee SC: Simvastatin-induced breast cancer cell death and
deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by
metabolic products of the mevalonate pathway. Oncotarget.
7:2532–2544. 2016.PubMed/NCBI
|
|
36
|
Istvan ES and Deisenhofer J: Structural
mechanism for statin inhibition of HMG-CoA reductase. Science.
292:1160–1164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Niesor EJ, Schwartz GG, Perez A, Stauffer
A, Durrwell A, Bucklar-Suchan G, Benghozi R, Abt M and Kallend D:
Statin-induced decrease in ATP-binding cassette transporter A1
expression via microRNA33 induction may counteract cholesterol
efflux to high-density lipoprotein. Cardiovasc Drugs Ther. 29:7–14.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sone H, Shimano H, Shu M, Nakakuki M,
Takahashi A, Sakai M, Sakamoto Y, Yokoo T, Matsuzaka K, Okazaki H,
et al: Statins downregulate ATP-binding-cassette transporter A1
gene expression in macrophages. Biochem Biophys Res Commun.
316:790–794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wong J, Quinn CM, Gelissen IC, Jessup W
and Brown AJ: The effect of statins on ABCA1 and ABCG1 expression
in human macrophages is influenced by cellular cholesterol levels
and extent of differentiation. Atherosclerosis. 196:180–189. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ando H, Tsuruoka S, Yamamoto H, Takamura
T, Kaneko S and Fujimura A: Effects of pravastatin on the
expression of ATP-binding cassette transporter A1. J Pharmacol Exp
Ther. 311:420–425. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ogunwobi OO and Beales IL: Statins inhibit
proliferation and induce apoptosis in Barrett's esophageal
adenocarcinoma cells. Am J Gastroenterol. 103:825–837. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nonaka M, Uota S, Saitoh Y, Takahashi M,
Sugimoto H, Amet T, Arai A, Miura O, Yamamoto N and Yamaoka S: Role
for protein geranylgeranylation in adult T-cell leukemia cell
survival. Exp Cell Res. 315:141–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Neufeld EB, Stonik JA, Demosky SJ Jr,
Knapper CL, Combs CA, Cooney A, Comly M, Dwyer N, Blanchette-Mackie
J, Remaley AT, et al: The ABCA1 transporter modulates late
endocytic trafficking: Insights from the correction of the genetic
defect in Tangier disease. J Biol Chem. 279:15571–15578. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Neufeld EB, O'Brien K, Walts AD, Stonik
JA, Malide D, Combs CA and Remaley AT: The human ABCG1 transporter
mobilizes plasma membrane and late endosomal
non-sphingomyelin-associated-cholesterol for efflux and
esterification. Biology (Basel). 3:866–891. 2014.PubMed/NCBI
|
|
45
|
Wang J, Mitsche MA, Lütjohann D, Cohen JC,
Xie XS and Hobbs HH: Relative roles of ABCG5/ABCG8 in liver and
intestine. J Lipid Res. 56:319–330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ramirez CM, Davalos A, Goedeke L, Salerno
AG, Warrier N, Cirera-Salinas D, Suárez Y and Fernández-Hernando C:
MicroRNA-758 regulates cholesterol efflux through
posttranscriptional repression of ATP-binding cassette transporter
A1. Arterioscler Thromb Vas Biol. 31:2707–2714. 2011. View Article : Google Scholar
|
|
47
|
Genvigir FD, Rodrigues AC, Cerda A, Hirata
MH, Curi R and Hirata RD: ABCA1 and ABCG1 expressions are regulated
by statins and ezetimibe in Caco-2 cells. Drug Metab Drug Interact.
26:33–36. 2011. View Article : Google Scholar
|
|
48
|
Janowski BA, Willy PJ, Devi TR, Falck JR
and Mangelsdorf DJ: An oxysterol signalling pathway mediated by the
nuclear receptor LXR alpha. Nature. 383:728–731. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Beyea MM, Heslop CL, Sawyez CG, Edwards
JY, Markle JG, Hegele RA and Huff MW: Selective up-regulation of
LXR-regulated genes ABCA1, ABCG1, and APOE in macrophages through
increased endogenous synthesis of 24(S),25-epoxycholesterol. J Biol
Chem. 282:5207–5216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wilson JM, Kunnimalaiyaan S,
Kunnimalaiyaan M and Gamblin TC: Inhibition of the AKT pathway in
cholangiocarcinoma by MK2206 reduces cellular viability via
induction of apoptosis. Cancer Cell Int. 15:132015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mistafa O and Stenius U: Statins inhibit
Akt/PKB signaling via P2X7 receptor in pancreatic cancer cells.
Biochem Pharmacol. 78:1115–1126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Miraglia E, Högberg J and Stenius U:
Statins exhibit anticancer effects through modifications of the
pAkt signaling pathway. Int J Oncol. 40:867–875. 2012.PubMed/NCBI
|