Introduction
Tumors utilize a variety of mechanisms to impair the
functionality of tumor-specific immune cells, T cells, macrophages
and other cells associated with the immune response (1,2). These
mechanisms include the expression of ligands which bind to
inhibitory receptors expressed on T cells and suppressing the
function of T cell stimulatory receptors, such as T-cell receptor
(TCR)/CD3 and CD28 (3,4). In general, T cells are activated by the
interaction of the TCR/CD3 complex with an antigen and
co-activation of CD28 (5).
Co-stimulation of the TCR with CD28 and an antigen promotes the
initial phosphorylation events of signal transduction from the TCR
and enhances immune support functions (6). In addition to the foremost activation
pathways, a number of immune checkpoints have been discovered to
regulate the immune system. These pathways are crucial for
self-tolerance and innate immunity and prevent the immune system
from attacking cells indiscriminately (2). Immune checkpoints consist of
stimulatory checkpoint molecules and inhibitory checkpoint
molecules (7,8). Inhibitory checkpoint molecules have
been considered important targets for cancer immunotherapy
(9). Currently, several checkpoint
inhibitors which block cytotoxic T-lymphocyte associated protein 4
(CTLA4), programmed cell death-1 (PD-1) and programmed death
ligand-1 (PD-L1) have been approved for clinical use (10).
The immune system regulates tumor biology, and,
depending on the tumor, can either support or inhibit tumor
development, growth, invasion and metastasis (11,12).
Certain tumors may evade immune detection through recruitment of
immunosuppressive leukocytes, which create a microenvironment that
blocks the antitumor immune response. Several mechanisms, including
defects in antigen-presenting cells, negative immune regulation by
suppressive cells and defective antitumor T cells have been
hypothesized and demonstrated to explain evasion or tolerance of
the immune response in different types of cancer (11). Jurkat cells are an immortalized line
of human T lymphocyte cells that have been used to study acute
T-cell leukemia and T-cell signaling (13). Jurkat cells have been used in a
diverse array of molecular investigations, some of which underpin
our current understanding of multiple signaling pathways (13). Evidence suggests that
CD3/CD28-costimulated Jurkat T cells and co-engagement of TCR/CD3
and CD28 results in interleukin (IL)-2 production and activation of
extracellular signal regulated kinase (ERK)/c-Jun N-terminal kinase
and NF-κB inhibitor β kinase, which is frequently used as a
functional readout of activation of Jurkat cells (14).
As an immunosuppressive molecule receptor, PD-1 can
inhibit the activation of T lymphocytes and play an important role
in immune escape. PD-1 belongs to the CD28/CTLA-4 family of
molecules, and it negatively regulates PD-1 signaling. When two
PD-L1 or PD-L2 ligands are concomitantly bound to PD-1, a protein
tyrosine phosphatase, tyrosine-protein phosphatase non-receptor
type 11 (SHP-2) is recruited intracellularly (15,16).
PD-L1 also termed B7H1 or CD274, is primarily expressed by tumor
cells and tumor-infiltrating immune cells (17), whereas PD-L2, also known as B7-DC or
CD273, is expressed mainly by dendritic cells and macrophages
(18). In addition to, PD-L1, but
not PD-L2, undergoes a conformational change upon binding, which
delays its interaction and thus activation (19,20).
Following PD-L1 biding to its receptor, SHP-2, dephosphorylates
downstream effector molecules such as Syk and PI3K in B cells, and
tyrosine-protein kinase ZAP70 (ZAP70) and CD3 in T cells (21,22).
PD-L1 is expressed in a variety of tumors (17,23).
PD-1/PD-L1 interaction activates a signal which inhibits
TCR-mediated T-cell activation and proliferation, suppresses
secretion of cytokines, such as interferon-γ (IFN-γ) and
interleukin-2, and promotes cytotoxic T-cell apoptosis and
regulatory T-cell differentiation (24,25). A
number of pathways involved in T-cell activation, including major
histocompatibility complex (MHC)-TCR-ZAP70-RAS-GTPase (RAS)-ERK and
CD80-CD28-PI3K protein kinase B (AKT) pathways, have been reported
to be regulated by PD-1/PD-L1 interaction.
Although several PD-1/PD-L1 inhibitors have been
approved for cancer therapy, the effectiveness of these inhibitors
appears to be tumor specific (26).
Therefore, the aim of the present study was to determine whether
the expression levels of PD-L1 on tumor cells affected its
immuno-suppressive activity and thus, the therapeutic effects of
PD-1/PD-L1 inhibitors. The immuno-suppressive effects of a number
of tumor cell lines were assessed. It was demonstrated that MCF-7
and MDA-MB-231 cells expressed the highest levels of PD-L1 and also
displayed the highest degree of immune suppression. Additionally,
the tumor cells with increased levels of PD-L1 expression exhibited
suppression of the pathways involved in T-cell activation,
including TCR-ZAP70-RAS-ERK and CD28-PI3K-AKT pathways.
Materials and methods
Plasmid and cell lines
A PGL3-nuclear factor of activated T-cells
(NFAT)-TA-Luciferase plasmid containing the full-length luciferase
gene under the control of an NFAT-driven promoter, was used in the
present study. NFAT is a nuclear factor of activated T cells that
synergizes with activator protein 1 transcription factors at
composite sites that are located in the promoters and enhancers of
a number of cytokine genes. This indicates that the NFAT promoter
an important factor in the immune response (27). The PGL3-NFAT-TA-Luciferase plasmid
was generously provided by Dr Jia Li (Shanghai Institute of Materia
Medica Chinese Academy of Science). The human embryonic kidney cell
line 293, human breast cancer cell line MCF-7, human melanoma cell
line A375, human cervical cancer cell line HeLa and human liver
cancer cell line HepG2 were obtained from the American Type Culture
Collection (ATCC) and maintained in DMEM, supplemented with 10%
FBS. The human breast cancer cell line MDA-MB-231, was obtained
from ATCC and maintained in Leibovitz's L-15 medium, supplemented
with 10% FBS. The human lung cancer cell line A549, was obtained
from ATCC and maintained in DMEM, supplemented with 10% FBS, 1%
GlutaMAX™. The human T lymphocyte cell line Jurkat, was obtained
from ATCC and maintained in RPMI-1640 medium, supplemented with 10%
FBS, 1% GlutaMAX™ and 0.1% 2-mercaptoethanol. Human peripheral
blood mononuclear cells (PBMCs) were isolated by density gradient
centrifugation using a Ficoll-Paque solution (GE Healthcare) from
heparinized peripheral blood samples.
Reagents
DMEM, RPMI-1640 medium, Leibovitz 15 medium, FBS,
GlutaMAX™ and 2-mercaptoethanol were purchased from Gibco; Thermo
Fisher Scientific, Inc. Penicillin-streptomycin was purchased from
Sigma-Aldrich (Merck KGaA). FuGENE® HD Transfection
Reagent was purchased from Promega Corporation and
TRIzol® reagent was purchased from Invitrogen (Thermo
Fisher Scientific, Inc). Anti-CD3 and anti-CD28 were purchased from
BD Biosciences. Allophycocyanin-conjugated anti-human-CD274/PD-L1
antibody were purchased from eBioscience (Thermo Fisher Scientific,
Inc.). Rabbit anti-Erk1/2, rabbit anti-phospho-Erk1/2, rabbit
anti-AKT, rabbit anti-GAPDH and horseradish peroxidase-conjugated
Goat anti-rabbit immunoglobulin G were purchased from Cell
Signaling Technology, Inc. Human IFN-γ ELISA kits and human IL-2
ELISA kits were purchased from Cisbio (PerkinElmer, Inc.). A0-L (a
PD-1 inhibitor; patent no. WO 2015/034820 A1; molecular weight,
475.58) was synthesized by Dr Wei Lv of East China Normal
University. The company name and catalog number for ELISA kits and
all antibodies are listed in Table
SI.
Preparation of conditioned medium
A total of 2×105 293, MCF-7, A375, A549,
HeLa or HepG2 cells were cultured per well in 6-well cell culture
plates with DMEM, supplemented with 10% FBS at 37°C for 24 h. In
the A549 cells, 1% GlutaMAX™ was added to the culture medium.
MDA-MB-231 (2×105 cells/well) were cultured in 6-well
cell culture plates with Leibovitz's L-15 Medium, supplemented with
10% FBS and 1% GlutaMAX™ at 37°C for 24 hours. The culture media
was collected and centrifuged at 12,000 × g for 10 min using a
pre-chilled centrifuge set to 4°C. The supernatant was collected
and termed ‘conditioned medium’.
Co-culture of tumor cells with Jurkat
cells
Jurkat cells were transfected with 3.3 µg
PGL3-NFAT-TA-Luciferase using FuGENE® HD Transfection
Reagent. After 16 h, 2×105 MCF-7, MDA-MB-231, A549,
A375, HeLa or HepG2 cells were seeded into wells with their
respective growth medium, and 2×104 Jurkat cells
transfected with PGL3-NFAT-TA-Luciferase were added to the wells.
The conditioned media was collected from the cultures after 24 h.
The conditioned media was added to 2×104 Jurkat cells
transfected with PGL3-NFAT-TA-Luciferase. After 30 min, anti-CD3 (1
µg/ml) and anti-CD28 (1 µg/ml) were added to the culture systems:
Tumor cells; Jurkat cells co-cultured with tumor cells in normal
media; Jurkat cells alone in conditioned media. After 24 h of
co-culture, luciferase activities were measured using the
Luciferase system (Promega Corporation) and EnVision multiplate
reader (PerkinElmer, Inc.) according to the manufacturer's
protocol.
Isolation of human PBMCs
PBMCs derived from healthy volunteers were provided
by the Shanghai Blood Center. PBMCs were isolated using a
Ficoll-Paque gradient. To separate PBMCs, 20 ml Ficoll was placed
in a 50 ml conical centrifuge tube and an equal volume of whole
blood diluted 1:1 with PBS was layered on top. The 50-ml tubes were
centrifuged at 2,000 × g for 30 min at room temperature with a low
acceleration speed. The PBMCs at the interface between the Ficoll
and the plasma were gently collected by aspiration using a Pasteur
pipette and placed in a 15 ml conical tube. Subsequently, the PBMCs
were washed twice with 10 ml PBS and centrifuged at 500 × g for 5
min at 4°C (28). PBMCs were
cultured for 6 h in T25 flasks in complete RPMI-1640 media (fresh
RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 100
IU/ml penicillin and 100 µg/ml streptomycin and 0.1%
2-mercaptoethanol) at 37°C in a humidified atmosphere containing 5%
CO2. The use of human PBMCs was specifically approved by
The Medical Ethics Committee of Shanghai Blood Center, (Shanghai,
China). Prior to donating blood, the volunteers were informed and
provided written informed consent for the scientific research use
of blood samples.
Co-culture of PBMCs with or without
tumor cells
A total of 2×105 MCF-7, MDA-MB-231, A549,
A375, HeLa, or HepG2 cells were seeded per a well in their
respective growth medium for 30 min and then 2×104 PBMCs
were added to each well. Tumor cell conditioned media was collected
from the cultures after 24 h. PBMCs were exposed to tumor cell
conditioned media. After 30 min, anti-CD3 (1 µg/ml) and anti-CD28
(1 µg/ml) were added to the tumor cells/PBMCs or tumor cell
conditioned media/PBMCs co-culture system. After 48 h of
co-culture, cell culture supernatants were collected and analyzed
for IL-2 and IFN-γ using the HTRF kit (Cisbio; PerkinElmer, Inc.)
according to the manufacturer's protocol.
Reverse transcription-quantitative
PCR
Total RNA was extracted using TRIzol®
reagent according to the manufacturer's protocol (Invitrogen;
Thermo Fisher Scientific, Inc.). RNA (1 µg) was used to synthesize
cDNA using a PrimeScript RT Reagent kit (Takara Bio, Inc.)
according to the manufacturer's protocol. qPCR was performed for
PD-L1, PD-L2, CD80, CD86, herpesvirus entry mediator (HVEM), CD70,
CD137, OX40L and GAPDH. The sequences of the primer pairs used are
shown in Table I. The thermocycling
conditions were: 95°C for 10 min; followed by 40 cycles of 95°C for
30 sec, 60°C for 30 sec and 72°C for 30 sec. The fold changes of
each gene were calculated using the ΔΔCq (quantification cycle)
method, and gene expression levels were normalized to GADPH
(29).
 | Table I.Forward and reverse primers used for
all RT-qPCR analyses. |
Table I.
Forward and reverse primers used for
all RT-qPCR analyses.
| Gene | Forward |
Reverse |
|---|
| GAPDH |
AGCCGCATCTTCTTTTGCGT |
TGACGAACATGGGGGCATCA |
| PDL1 |
GCTGCACTAATTGTCTATTGGGA |
AATTCGCTTGTAGTCGGCACC |
| IDO1 |
GCGCTGTTGGAAATAGCTTC |
ATGTCCTCCACCAGCAGTC |
| PDL2 |
CAGCAATGTGACCCTGGAAT |
GGACTTGAGGTATGTGGAACG |
| TIM3 |
GGAATACAGAGCGGAGGTCG |
AGGGACACATCTCCTTTGCG |
| LAG3 |
ACCCCATCCCAGAGGAGTTT |
GTCGCCACTGTCTTCTCCAA |
| CTLA4 |
CCGTGCCCAGATTCTGACTT |
ACATTCTGGCTCTGTTGGGG |
| CD80 |
TCTGTTCAGGTGTTATCCACG |
GGGCGTACACTTTCCCTTCT |
| CD86 |
ATTCGGACAGTTGGACCCTG |
CCAAGGAATGTGGTCTGGGG |
| CD28 |
ACACCTTTGTCCAAGTCCCC |
AGCAGTGCTGCTTCTCTTACC |
| ICOS |
TTGAACACTGAACGCGAGGA |
AAAACTGGCCAACGTGCTTC |
| HVEM |
GTCTTGAGGCTGGTGCTGTA |
TGGTCTGGTGCTGACATTCC |
| BTLA |
GACCCTCCAAGGACGAAGTG |
TTCTCAGGCAGCAGAACAGG |
| CD137L |
CGCAGTCTCTCGTCATGGAA |
CCTCTTTGTAGCTCAGGCCC |
| CD70 |
GACACACTCTGCACCAACCT |
TAATCAGCAGCAGTGGTCAGG |
| OX40L |
AGGCCAAGATTCGAGAGGAAC |
CAGTGGTGCATCTTACCTGAA |
FACS of tumor cells
Cells were incubated with allophycocyanin-conjugated
anti-human-CD274 antibody (1:100) at 4°C for 30 min in the dark for
flow cytometry analysis using a Guava® easyCyte Benchtop
flow cytometer and FlowJo software (FlowJo™; version 10.6.1; FlowJo
LLC) was used to analyze the data.
Immunoblot analysis
MCF-7 and MDA-MB-231 cells were plated in 6 well
plates at a density of 1×105 cells/ml. The cells were
co-cultured with Jurkat cells (2×105) in serum-free
medium, and treated with anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml)
for 5, 15 and 30 min, respectively. After the treatment, Jurkat
cells were washed in PBS and lysed with RIPA lysis buffer (CoWin
Biosciences). Protein concentrations were determined using a
bicinchoninic acid assay kit (Thermo Fisher Scientific, Inc.).
Western blot analysis was performed as previously described
(30). Rabbit polyclonal antibodies
against human phospho-AKT, AKT, phospho-ERK, ERK and GAPDH were
used at a dilution of 1:1,000 at 4°C in the dark overnight. A
horseradish peroxidase-conjugated secondary goat antibody against
rabbit immunoglobulin G was used at a dilution of 1:5,000 at room
temperature for 1 h. Signals were visualized using Pierce Western
Blotting Substrate Plus (Thermo Fisher Scientific, Inc.) and a
ChemiDocXRSþ system (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data were analyzed with GraphPad Prism version 5.0
(GraphPad Software, Inc.). The results were analyzed using a
two-way ANOVA followed by post-hoc Bonferroni's tests or a one-way
ANOVA followed by a post-hoc Newman-Keuls test. All data are
presented as the mean ± standard error of the mean. P<0.05 was
considered to indicate a statistically significant difference.
Results
Tumor cells inhibit the activation of
Jurkat cells
NFATs are a family of transcription factors which
serve important roles in the immune response (31). The human T lymphocyte-based Jurkat
cell line expressing luciferase gene under the control of NFAT
response elements can be used to study NFAT activation following
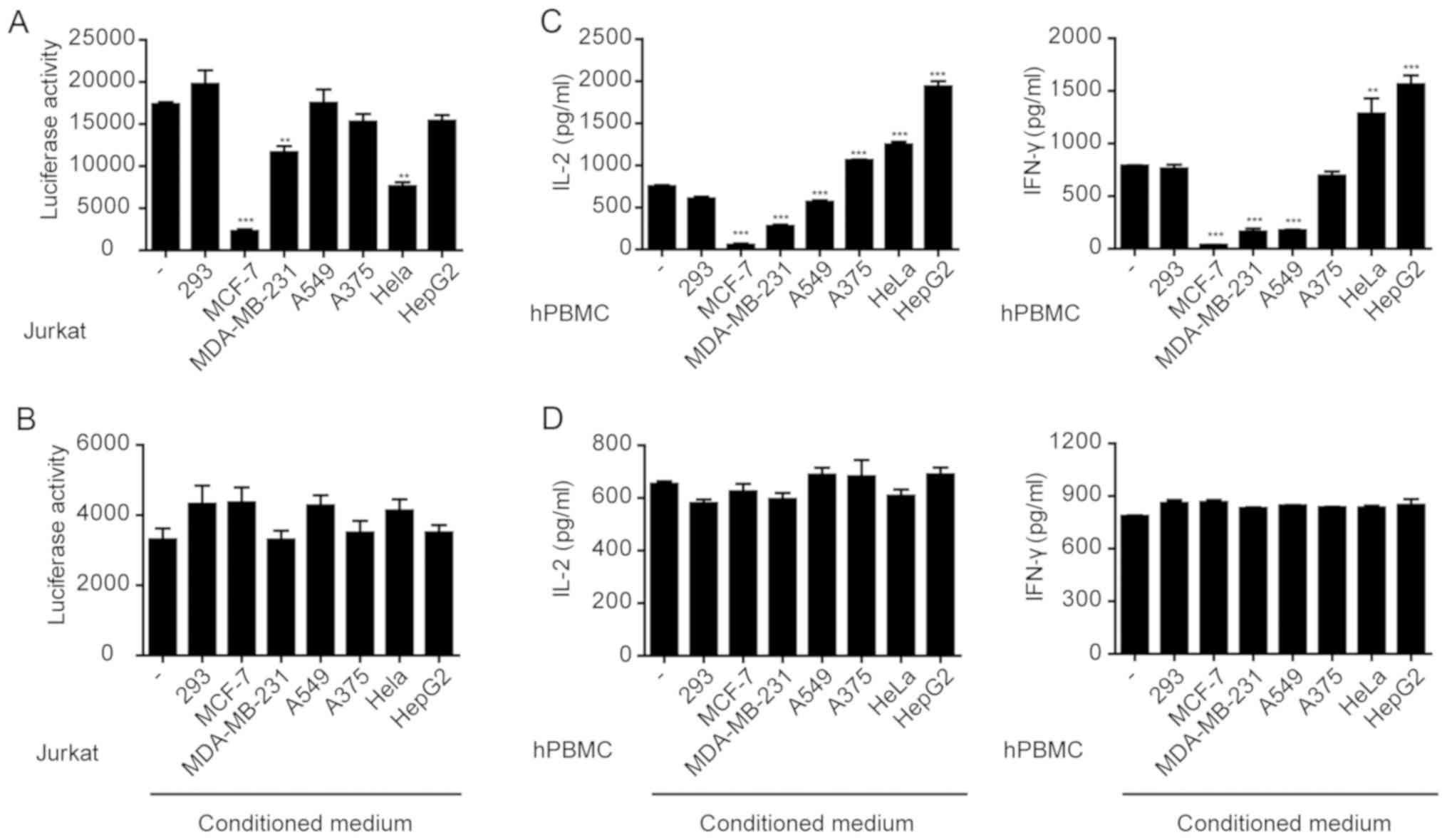
various stimuli (32) To investigate
the effects of the MCF-7, MDA-MB-231, A549, A375, HeLa and HepG2
cells on immune cell activation, these cells were co-cultured with
Jurkat cells which were transfected with PGL3-NFAT-TA-Luciferase
plasmid and stimulated with anti-CD3 and anti-CD28 antibodies. The
results showed that MCF-7, MDA-MB-231 or HeLa cells significantly
inhibited the anti-CD3/CD28-induced expression of luciferase in
Jurkat cells (Fig. 1A), whereas the
other tumor cells did not result in significant changes. However,
the conditioned media collected from any of the cell lines,
including MCF-7, MDA-MB-231 or HeLa cells, did not significantly
affect luciferase expression in Jurkat cells (Fig. 1B). These results suggest that the
direct interaction between tumor cells and Jurkat cells, rather
than the factors secreted by the tumor cells, inhibited Jurkat cell
activation.
Tumor cells inhibit cytokine secretion
from PBMCs
Jurkat is an immortalized cell line of human T
lymphocytes (13). Primary PBMCs
isolated from whole blood samples were used to investigate the
effects of tumor cells. PBMCs were co-cultured with various tumor
cell lines and stimulated with anti-CD3/CD28. The secretion of
IFN-γ and IL-2 was measured. The results showed that MCF-7,
MDA-MB-231 and A549 cells significantly inhibited IFN-γ and IL-2
secretion from PBMCs (Fig. 1C).
However, the conditioned media collected from any of the cell
lines, including MCF-7, MDA-MB-231 or A549 cell cultures had no
effect on cytokine secretion from PBMCs (Fig. 1D). Therefore, similar to the Jurkat
cells, a direct interaction between tumor cells and PBMCs resulted
in the suppression of cytokine secretion.
Expression of immune checkpoint
markers in various tumor cell lines
The aforementioned results suggest that different
tumor cells have different effects on suppressing immune cell
function. Thus, whether the expression levels of immune checkpoint
proteins on tumor cells affected their immune-suppressive activity
was determined. The mRNA expression levels of PD-L1 and other
immune checkpoint genes, PD-L2, CD80, CD86, HVEM, CD70, CD137 and
OX40L were measured in these tumor cells (Fig. 2A-H). RT-qPCR analysis demonstrated
that Hela, MCF-7 and MDA-MB-231 cells expressed significantly high
levels of PD-L1 compared with 293 cells (P<0.01; Fig. 2A). PD-L1 and PD-L2 expression was
significantly higher in A375 and HeLa cells (P<0.01 and
P<0.01; Fig. 2A and B,
respectively); and the expression of HVEM was significantly high in
MDA-MB-231 and HeLa cells (P<0.05; Fig. 2A and B). Notably, PD-1 was highly
expressed in Jurkat cells compared with the other immune checkpoint
receptors, although this was not significant (Fig. 2I). FACS analysis also confirmed that
the protein expression levels of PD-L1 were considerably higher in
MCF-7 and MDA-MB-231 cells compared to other tumor cells (Fig. 2J and K), consistent with the higher
mRNA expression levels in these cells. Taken together, these
findings suggest that the immune-suppressive activity of tumor
cells may be associated with the expression levels of PD-L1 in
these cells.
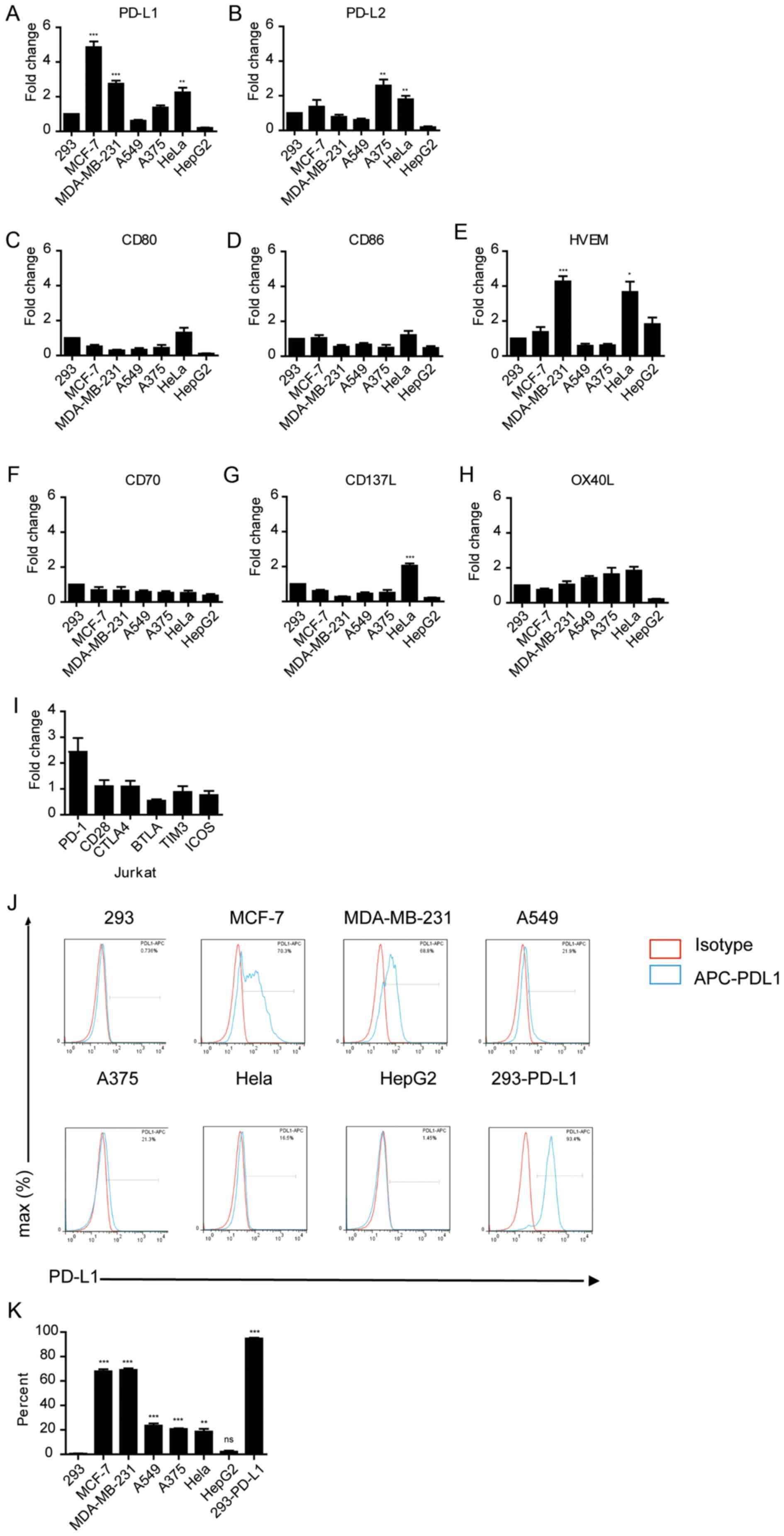 | Figure 2.Expression of immune checkpoint
markers in various cancer cell lines. mRNA expression levels of (A)
PD-L1, (B) PD-L2, (C) CD80, (D) CD86, (E) HVEM, (F) CD70, (G) CD137
and (H) OX40L in tumor cell lines. *P<0.05, **P<0.01,
**P<0.001. (I) mRNA expression levels of immune checkpoint
receptors in Jurkat cells. Gene expression was normalized to GAPDH
in the same sample. (J) FACS analysis of PD-L1 in various cancer
cell lines. APC-conjugated anti-human-PD-L1 was used as a binding
antibody to cell-surface PD-L1 protein (red line, isotype control
staining; blue line, PD-L1 staining). Cell count has been
normalized to the peak height at the mode of the distribution, such
that absolute count is presented as a percent of the total count.
(K) Quantification of the FACS analysis shown in (J). **P<0.01,
***P<0.001 vs. 293. APC, allophycocyanin. |
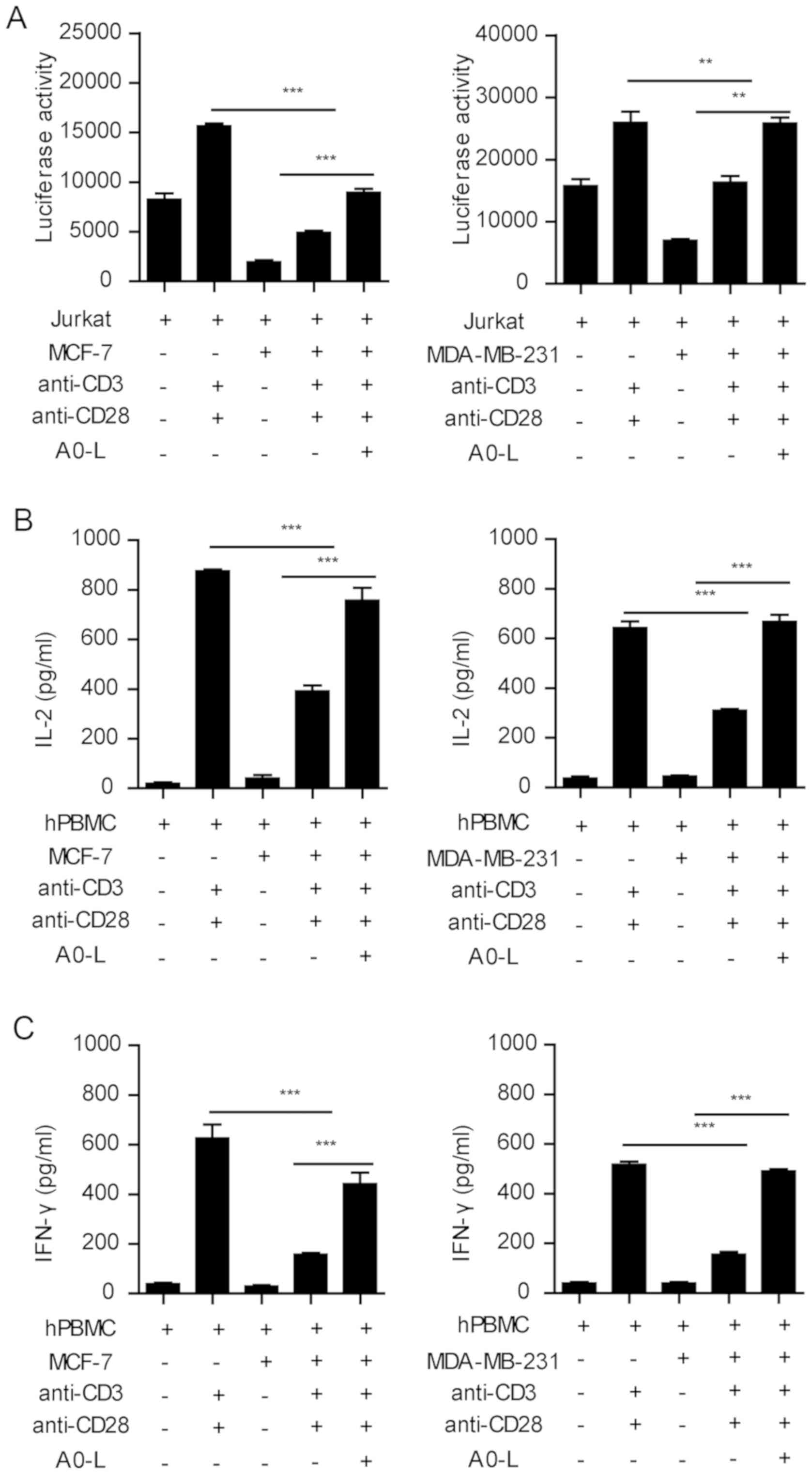
PD-1/PD-L1 inhibitor restores the
function of lymphocytes
MCF-7 and MDA-MB-231 cells had the highest level of
PD-L1 expression, and the largest inhibitory effect on T-cell
activation and cytokine secretion in the co-culture system.
Therefore, the two tumor cell lines were used to investigate the
effect of the PD-1/PD-L1 inhibitor on the function of lymphocytes.
Stimulation of Jurkat cells with anti-CD3 and anti-CD28 antibodies
significantly induced the expression of luciferase, whereas MCF-7
and MDA-MB-231 cells significantly inhibited the expression of
luciferase (Fig. 3A). In the present
study, A0-L, a small molecular inhibitor of the PD-1/PD-L1
interaction was used. A0-L significantly restored the expression of
luciferase in the tumor-Jurkat cell co-culture (Fig. 3A).
Similar results were observed in the PBMC cytokine
secretion assay. Anti-CD3 and anti-CD28 co-stimulation induced
IFN-γ and IL-2 secretion from PBMC cells, which was significantly
inhibited by the co-culture with MCF-7 and MDA-MB-231 cells
(Fig. 3B and C). Blocking the
PD-1/PD-L1 interaction between PBMCs and tumor cells with A0-L
significantly increased the secretion of IFN-γ and IL-2 (Fig. 3B and C). Therefore, these results
showed that blocking the PD-1/PD-L1 interaction effectively
abrogated the inhibition of immune cell functions by tumor
cells.
Tumor cells with high PD-L1 expression
suppress pathways involved in T cell activation
MCF-7 or MDA-MB-231 cells were co-cultured with
Jurkat cells to investigate the regulatory effects of tumor cells
on AKT and ERK1/2 phosphorylation in immune cells. Jurkat cells
were stimulated with anti-CD3 and anti-CD28 (1 µg/ml) antibodies
for various durations, and AKT and ERK1/2 phosphorylation was
assessed by western blotting. Anti-CD3 and anti-CD28 were increased
phosphorylation of AKT and ERK1/2 in a time-dependent manner
(Fig. 4A and B). By contrast, Jurkat
cells co-cultured with MCF-7 or MDA-MB-231 cells had significantly
reduced phosphorylation levels of AKT and ERK1/2 following anti-CD3
and anti-CD28 stimulation (Fig. 4A and
B). These results indicate that the tumor cells with high PD-L1
expression suppressed the pathways involved in T-cell activation,
such as the CD28-PI3K-AKT and TCR-ZAP70-RAS-ERK pathways.
Additionally, treatment with A0-L suppressed the inhibitory effects
of MCF-7 cells on the CD28-PI3K-AKT and TCR-ZAP70-RAS-ERK pathways
in Jurkat cells and restored phosphorylation levels of AKT and
ERK1/2 (Fig. 4C and D).
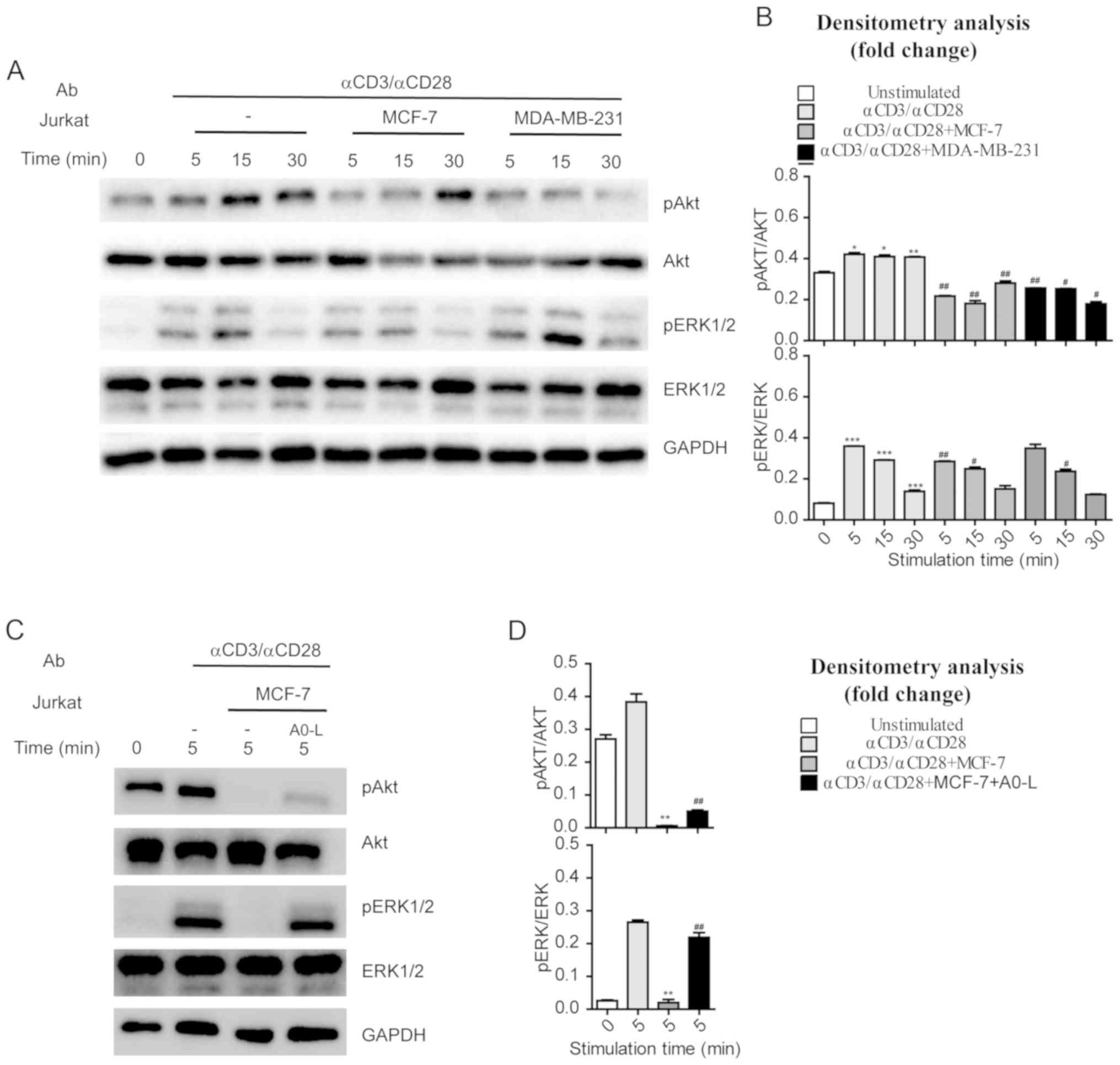 | Figure 4.Activation of AKT and ERK in Jurkat
cells co-cultured with cancer cells. (A) Jurkat cells cultured
alone or in the presence of MCF-7 or MDA-MB-231 cells were
activated by anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml). Jurkat
cells were collected and lysates were prepared, and the amounts of
the indicated proteins were determined by western blotting. (B)
Densitometry analysis of the phosphorylation of AKT and ERK
presented in (A). For each time point, fold changes in the amounts
of the indicated proteins in activated Jurkat cells that were
stimulated through anti-CD3 (1 µg/ml), anti-CD28 (1 µg/ml) were
compared with cells that were not activated. *P<0.05,
**P<0.01, ***P<0.001 vs. 0 min. unstimulated cells;
#P<0.05, ##P<0.01, ###P<0.001 vs. Jurkat cells that were
stimulated with anti-CD3 (1 µg/ml), anti-CD28 (1 µg/ml). (C) Jurkat
cells cultured with MCF-7 in the absence or presence of A0-L (10
µM) were activated by anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml).
Jurkat cells were collected and lysates were prepared, and the
amounts of the indicated proteins were examined by western
blotting. (D) Densitometry analysis of the phosphorylation of AKT
and ERK presented in (C). Fold changes in the amounts of the
indicated proteins in activated Jurkat cells that were suppressed
by MCF-7. *P<0.05, **P<0.01, ***P<0.001 vs. Jurkat cells
that were stimulated through anti-CD3 (1 µg/ml), anti-CD28 (1
µg/ml) at 5 min. unactivated Jurkat cells that were reversed by
A0-L; #P<0.05, ##P<0.01, ###P<0.001 vs. Jurkat cells that
were stimulated with anti-CD3 (1 µg/ml), anti-CD28 (1 µg/ml) and
co-cultured with MCF-7. AKT, protein kinase B; ERK,
extracellular-signal regulated kinase; p, phospho; Ab,
antibody. |
Discussion
Previous findings have shown that PD-L1 is expressed
on the surface of tumor cells in a number of different types of
cancer and could induce immunosuppression to enable the host to
evade anticancer immune responses (33,34).
PD-1, as an immunosuppressive factor and the receptor of PD-L1, is
a critical negative regulator of cancer biology with the capacity
to support cancer development, growth, invasion and metastasis
(35). PD-L1 has also been
established studied as a biomarker of a number of different types
of cancer, and several studies demonstrated that PD-L1 expression
may be used to predict the outcome of the disease. In the present
study, the results showed that PD-L1 mRNA expression levels were
upregulated in MCF-7 and MDA-MB-231 tumor cells, consistent with
the high protein expression levels of PD-L1 in these cells.
Additionally, MCF-7 and MDA-MB-231 cells significantly
downregulated T-cell activity and cytokine secretion, which was
associated with the upregulated expression of PD-L1 in these tumor
cells. Blocking PD-1/PD-L1 interaction with A0-L, a PD-1/PD-L1
inhibitor, significantly restored the activation of Jurkat cells
and the secretion of IFN-γ and IL-2 from PBMC cells, which were
significantly inhibited by MCF-7 and MDA-MB-231 cells. The results
suggest that PD-L1 is upregulated in specific tumor cells and may
downregulate T-cell activity by binding to the PD-1 receptor on T
cells.
T-cell activation is initiated by the binding of
TCRs to their physiological ligands, which are foreign peptides
bound to the MHC expressed on antigen-presenting cells (APCs)
(36). Upon activation of the TCR,
the Src family kinase tyrosine-protein kinase Lck (LCK) becomes
activated. The activated LCK phosphorylates CD3 chains, which
promote the recruitment and subsequent activation of another
tyrosine kinase, ZAP-70, and recruitment of a number of other
protein kinases involved in the activation of different signaling
cascades, such as RAS and ERK (37,38).
CD28 is a co-stimulatory molecule that promotes T-cell activation
(39). Upon ligand binding, CD28
recruits and activates PI3K, which in turn activates AKT by
phosphorylation (40). The
CD28-PI3K-AKT and TCR-ZAP70-RAS-ERK pathways are two major
functional signaling pathways involved in T cell activation. When
the TCR engages with an antigen peptide and MHC, the T cells are
activated via signal transduction, and the primary signaling
pathway involved is the TCR-ZAP70-RAS-ERK pathway (41,42).
Co-ligation of other cell surface receptors provides additional
signals required to enhance T cell activation. CD28 is a
costimulatory molecule that promotes T cell proliferation, cytokine
production, cell survival and cellular metabolism. CD28-PI3K-AKT is
the primary downstream signaling pathway of CD28 (37,38). In
the present study, it was demonstrated that tumor-T cell
interaction through PD-1/PD-L1 significantly inhibited the
above-mentioned pathways. Tumor cells with high PD-L1 expression
inhibited TCR-dependent ERK phosphorylation and CD28-dependent AKT
phosphorylation. These results suggest that PD-L1 mediates its
inhibitory effects on T cell activation by regulating TCR signaling
and CD28 signaling.
The PD-1/PD-L1 targeting strategy was a breakthrough
in immunotherapy that restores the functions of T cells (e.g.,
immune cell activation and differentiation, and cytokine secretion)
and promotes immune response (43–46).
Keytruda and Tecentriq have been approved by the FDA, their
mechanisms of action are well understood, and their clinical
efficacy and pharmacodynamics data have been determined. Although
several PD-1/PD-L1 inhibitors have been approved for cancer
therapy, they are more effective in treating certain tumors over
others (47). The results of the
present study suggest that tumor cells with a higher expression
level of PD-L1 may have higher immunosuppressive activity, and
drugs targeting the PD-1/PD-L1 interaction may have improved
therapeutic effects on tumors with higher expression levels of
PD-L1.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
National Science & Technology Major Project: Key New Drug
Creation and Manufacturing Program (Beijing, China; grant no.
2017ZX09101004-012-008); Personalized Medicines-Molecular
Signature-based Drug Discovery and Development; Strategic Priority
Research Program of the Chinese Academy of Sciences (Beijing,
China; grant no. XDA12040212), and Shanghai Commission of Science
and Technology (Shanghai, China; grant no. 16431901500).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contribution
YZ performed the majority of the experiments,
analyzed the data and wrote the manuscript. YCF performed some of
the experiments. JL supervised the study, analyzed the data and
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The use of human PBMCs was specifically approved by
The Medical Ethics Committee of Shanghai Blood Center, (Shanghai,
China). Prior to donating blood, the volunteers were informed and
provided written informed consent for the scientific research use
of blood samples. Due to ethical constraints, no clinical
information on the blood donors was obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ahmadzadeh M, Johnson LA, Heemskerk B,
Wunderlich JR, Dudley ME, White DE and Rosenberg SA: Tumor
antigen-specific CD8 T cells infiltrating the tumor express high
levels of PD-1 and are functionally impaired. Blood. 114:1537–1544.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prosser ME, Brown CE, Shami AF, Forman SJ
and Jensen MC: Tumor PD-L1 co-stimulates primary human CD8(+)
cytotoxic T cells modified to express a PD1:CD28 chimeric receptor.
Mol Immunol. 51:263–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
June CH, Ledbetter JA, Linsley PS and
Thompson CB: Role of the CD28 receptor in T-cell activation.
Immunol Today. 11:211–216. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alvarez-Vallina L and Hawkins RE:
Antigen-specific targeting of CD28-mediated T cell co-stimulation
using chimeric single-chain antibody variable fragment-CD28
receptors. Eur J Immunol. 26:2304–2309. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Louis-Dit-Sully C, Blumenthal B,
Duchniewicz M, Beck-Garcia K, Fiala GJ, Beck-García E, Mukenhirn M,
Minguet S and Schamel WW: Activation of the TCR complex by
peptide-MHC and superantigens. Exs. 104:9–23. 2014.PubMed/NCBI
|
|
6
|
Salazar-Fontana LI, Barr V, Samelson LE
and Bierer BE: CD28 engagement promotes actin polymerization
through the activation of the small Rho GTPase Cdc42 in human T
cells. J Immunol. 171:2225–2232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Passardi A, Canale M, Valgiusti M and
Ulivi P: Immune checkpoints as a target for colorectal cancer
treatment. Int J Mol Sci. 18(pii): E13242017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saad FT, Hincal E and Kaymakamzade B:
Dynamics of immune checkpoints, immune system, and BCG in the
treatment of superficial bladder cancer. Comput Math Methods Med.
2017:35730822017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma P and Allison JP: Immune checkpoint
targeting in cancer therapy: Toward combination strategies with
curative potential. Cell. 161:205–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Beatty GL and Gladney WL: Immune Escape
Mechanisms as a Guide for Cancer Immunotherapy. Clin Cancer Res.
21:687–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Janssen LME, Ramsay EE, Logsdon CD and
Overwijk WW: The immune system in cancer metastasis: Friend or foe?
J Immunother Cancer. 5:792017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bartelt RR, Cruz-Orcutt N, Collins M and
Houtman JC: Comparison of T cell receptor-induced proximal
signaling and downstream functions in immortalized and primary T
cells. PLoS One. 4:e54302009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thaker YR, Schneider H and Rudd CE: TCR
and CD28 activate the transcription factor NF-κB in T-cells via
distinct adaptor signaling complexes. Immunol Lett. 163:113–119.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong H, Zhu G, Tamada K and Chen L: B7-H1,
a third member of the B7 family, co-stimulates T-cell proliferation
and interleukin-10 secretion. Nat Med. 5:1365–1369. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Latchman Y, Wood C, Chemova T, Chaudhary
D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, et al:
PD-L2, a novel B7 homologue, is a second ligand for PD-1 and
inhibits T cell activation. Nat Immunol. 15:261–268. 2001.
View Article : Google Scholar
|
|
17
|
Kim HR, Ha SJ, Hong MH, Heo SJ, Koh YW,
Choi EC, Kim EK, Pyo KH, Jung I, Seo D, et al: PD-L1 expression on
immune cells, but not on tumor cells, is a favorable prognostic
factor for head and neck cancer patients. Sci Rep. 6:369562016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Keir ME, Liang SC, Guleria I, Latchman YE,
Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH and Sharpe
AH: Tissue expression of PD-L1 mediates peripheral T cell
tolerance. J Exp Med. 203:883–895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Butte MJ, Keir ME, Phamduy TB, Sharpe AH
and Freeman GJ: Programmed death-1 ligand 1 interacts specifically
with the B7-1 costimulatory molecule to inhibit T cell responses.
Immunity. 27:111–122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ghiotto M, Gauthier L, Serriari N, Pastor
S, Truneh A, Nunès JA and Olive D: PD-L1 and PD-L2 differ in their
molecular mechanisms of interaction with PD-1. Int Immunol.
22:651–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng P and Zhou Z: Human cancer
immunotherapy with PD-1/PD-L1 blockade. Biomark Cancer. 7 (Suppl
2):S15–S18. 2015.
|
|
22
|
Catakovic K, Klieser E, Neureiter D and
Geisberger R: T cell exhaustion: From pathophysiological basics to
tumor immunotherapy. Cell Commun Signal. 15:12017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patel SP and Kurzrock R: PD-L1 expression
as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther.
14:847–856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Palaga T, Miele L, Golde TE and Osborne
BA: TCR-mediated Notch signaling regulates proliferation and
IFN-gamma production in peripheral T cells. J Immunol.
171:3019–3024. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iwasaki M, Tanaka Y, Kobayashi H,
Murata-Hirai K, Miyabe H, Sugie T, Toi M and Minato N: Expression
and function of PD-1 in human γδ T cells that recognize
phosphoantigens. Eur J Immunol. 41:345–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang D, Lin J, Yang X, Long J, Bai Y, Yang
X, Mao Y, Sang X, Seery S and Zhao H: Combination regimens with
PD-1/PD-L1 immune checkpoint inhibitors for gastrointestinal
malignancies. J Hematol Oncol. 12:422019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Macian F: NFAT proteins: Key regulators of
T-cell development and function. Nat Rev Immunol. 5:472–484. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grievink HW, Luisman T, Kluft C, Moerland
M and Malone KE: Comparison of three isolation techniques for human
peripheral blood mononuclear cells: Cell recovery and viability,
population composition, and cell functionality. Biopreserv Biobank.
14:410–415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patsoukis N, Brown J, Petkova V, Liu F, Li
L and Boussiotis VA: Selective effects of PD-1 on Akt and Ras
pathways regulate molecular components of the cell cycle and
inhibit T cell proliferation. Sci Signal. 5:ra462012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vaeth M and Feske S: NFAT control of
immune function: New frontiers for an abiding trooper. F1000Res.
7:2602018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ponomarev V, Doubrovin M, Lyddane C,
Beresten T, Balatoni J, Bornman W, Finn R, Akhurst T, Larson S,
Blasberg R, et al: Imaging TCR-dependent NFAT-mediated T-cell
activation with positron emission tomography in vivo. Neoplasia.
3:480–488. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding ZC, Lu XY, Yu M, Lemos H, Huang L,
Chandler P, Liu K, Walters M, Krasinski A, Mack M, et al:
Immunosuppressive myeloid cells induced by chemotherapy attenuate
antitumor CD4+ T-Cell Responses through the PD-1-PD-L1 Axis. Cancer
Res. 74:3441–3453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Medina PJ and Adams VR: PD-1 Pathway
Inhibitors: Immuno-oncology agents for restoring antitumor immune
responses. Pharmacotherapy. 36:317–334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Y and Cao XT: Immunosuppressive cells
in tumor immune escape and metastasis. J Mol Med. 94:509–522. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dustin ML: The cellular context of T cell
signaling. Immunity. 30:482–492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu X, Kim JL, Newcomb JR, Rose PE, Stover
DR, Toledo LM, Zhao H and Morgenstern KA: Structural analysis of
the lymphocyte-specific kinase Lck in complex with non-selective
and Src family selective kinase inhibitors. Structure. 7:651–661.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li M, Ong SS, Rajwa B, Thieu VT, Geahlen
RL and Harrison ML: The SH3 domain of Lck modulates T-cell
receptor-dependent activation of extracellular signal-regulated
kinase through activation of Raf-1. Mol Cell Biol. 28:630–641.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beyersdorf N, Kerkau T and Hunig T: CD28
co-stimulation in T-cell homeostasis: A recent perspective.
Immunotargets Ther. 4:111–122. 2015.PubMed/NCBI
|
|
40
|
Parry RV, Riley JL and Ward SG: Signalling
to suit function: Tailoring phosphoinositide 3-kinase during T-cell
activation. Trends Immunol. 28:161–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Butler DE, Marlein C, Walker HF, Frame FM,
Mann VM, Simms MS, Davies BR, Collins AT and Maitland NJ:
Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and
compensatory Ras/Raf/MEK/ERK signalling in prostate cancer.
Oncotarget. 8:56698–56713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asati V, Mahapatra DK and Bharti SK:
PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as
anticancer agents: Structural and pharmacological perspectives. Eur
J Med Chem. 109:314–341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mahoney KM, Freeman GJ and McDermott DF:
The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in
melanoma. Clin Ther. 37:764–782. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hui E, Cheung J, Zhu J, Su X, Taylor MJ,
Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I and Vale RD: T
cell costimulatory receptor CD28 is a primary target for
PD-1-mediated inhibition. Science. 355:1428–1433. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jacquin-Porretaz C, Nardin C, Puzenat E,
Roche-Kubler B and Aubin F; Comité de suivi des effets secondaires
des immunothérapies anti-cancéreuses (CSESIAC), : Adverse effects
of immune checkpoint inhibitors used to treat melanoma and other
cancer. Presse Med. 46:808–817. 2017.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park JA and Cheung NV: Erratum to
‘limitations and opportunities for immune checkpoint inhibitors in
pediatric malignancies’ [Cancer Treat. Rev. 58C (2017) 22–33].
Cancer Treat Rev. 60:1582017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
McDermott DF and Atkins MB: PD-1 as a
potential target in cancer therapy. Cancer Med. 2:662–673.
2013.PubMed/NCBI
|