Introduction
Colon cancer is one of the most prevalent and severe
cancer types. Due to frequent treatment failure and a high
recurrence rate, it has been reported as the second most prevalent
malignancy and the third leading cause of cancer-associated
mortality worldwide. Globally, this results in ~500,000 mortalities
and ~1 million new diagnoses each year (1,2). Colon
adenocarcinoma (COAD) is the most prevalent histological subtype of
colon cancer and its incidence is increasing due to numerous
factors, including genetic predisposition, obesity, and dietary and
individual lifestyle choices (3–5). The
current clinical treatment strategies for COAD include surgery,
chemotherapy and dietary regulation (6).
Despite improvements in the diagnostic and
therapeutic strategies for COAD over recent decades, disease
etiology remains poorly characterized, and the prognosis for
patients with COAD remains unsatisfactory. This is primarily due to
a lack of clinically significant biomarkers, which may facilitate
early diagnosis or enable the precise prediction of resistance to
conventional treatment protocols (7,8). Colon
cancer exhibits a poor overall survival (OS) rate, particularly in
patients with advanced or metastatic COAD, and treatment options
require urgent improvement (9).
Thus, the identification of novel biomarkers may improve the early
diagnosis and treatment of COAD, consequently resulting in a
reduction in the mortality rate.
The discovery and characterization of the function
of long non-coding (lnc)RNAs (>200 nucleotides) represents an
opportunity to increase understanding of the molecular mechanisms
involved in cancer progression, and also provides novel potential
therapeutic targets (10,11). lncRNAs have been implicated in
numerous cellular processes, including cell proliferation and
differentiation, chromatin dynamics and gene expression (12–15).
Moreover, lncRNAs have been implicated in multiple epigenetic
regulatory mechanisms, and the misregulation of epigenetic markers
is associated with the inappropriate activation or inhibition of
various genes, contributing to cancer development and progression
(16,17). lncRNAs serve a critical role in the
pathogenesis of numerous malignancies, including lung (18), endometrial (19), breast (20) and hepatocellular cancer (21). Numerous studies have demonstrated
that the regulation of epigenetic modifications may contribute to
several pathological processes involved in cancer progression.
Additionally, it has been discovered that the functions of specific
lncRNAs can be altered by methylation, and could therefore
influence tumor suppressor genes and proteins, resulting in the
regulation of oncogenesis and tumor development (22).
It was discovered that hypermethylation of the
maternally expressed 3 (MEG3) promoter in patients with acute
myeloid leukemia decreased expression of lncRNA MEG3, compared with
the control group (23). Moreover,
Guo et al (24) determined
that the downregulation of lncRNA LOC100130476 was caused by the
hypermethylation of CpG sites in esophageal squamous cell carcinoma
(ESCC) cells, and that this was also associated with ESCC
progression.
In summary, the DNA methylation patterns of specific
lncRNAs may represent useful biomarkers, improving the precision of
diagnosis and prognosis in patients with COAD.
In the current study, 485,577 CpG sites located
<2 kb upstream of lncRNA transcriptional start sites (TSSs) were
identified, and subsequently, a comprehensive lncRNA expression
analysis was performed on data from patients with COAD, in order to
develop a prognostic methylation-based classifier. The least
absolute shrinkage and selection operator method (LASSO) (25), and the results of the present study,
indicated that the classifier was able to divide patients into
different risk grades corresponding to their respective OS times.
Functional enrichment analysis of lncRNA co-expressed genes was
also investigated.
Materials and methods
Dataset retrieval and processing
Datasets comprised of DNA methylation, RNA-seq and
clinical data collected from patients with COAD were downloaded
from TCGA data portal (https://portal.gdc.cancer.gov/). DNA methylation
profiling was performed using the Infinium HumanMethylation450
BeadChip (Illumina, Inc.), and RNA-seq was carried out using the
IlluminaHiSeq RNA-seq platform (Illumina, Inc.). The lncRNA
annotation file was obtained from GENCODE (https://www.gencodegenes.org/). Kyoto Encyclopedia of
Genes and Genomes (KEGG) is a database that systematically analyzes
the metabolic pathways of gene products in cells and the functions
of the gene products. In the present study, the relative database
from KEGG was obtained and analyzed using Multi-Experiment Matrix
(MEM) and The Database for Annotation, Visualization and Integrated
Discovery (DAVID; (https://david.ncifcrf.gov/). All datasets are publicly
available and the study protocol adhered to the publication
guidelines.
CpG sites of lncRNAs
DNA methylation was found to occur predominantly on
cytosine, followed by guanine residues (CpG methylation). The
methylation of CpG sites was reported as a β-value ranging from 0
(unmethylated) to 1 (completely methylated). Normalization of the
methylation β-values was conducted using the ‘minfi’ package of R
software (3.5.1). CpG sites located <2 kb upstream of an lncRNA
transcriptional start site (TSS) were selected from 485,577
possible CpG sites in the HumanMethylation450 BeadChip, according
to the annotation from TCGA. Several differentially-methylated CpG
sites between COAD and normal adjacent tissues (from the same
patients) were also selected using the ‘minfi’ package. The
Student's t-test was performed to compare the β-values of CpG sites
between COAD and normal adjacent tissues. In the present study, a
difference in the β-value of CpG sites >1 (between COAD and
normal adjacent tissues) was considered significant, and was
subsequently selected for construction of the database used for
further analysis. Inhibition of multiple genes can occur when
certain regions of DNA sequences are methylated (26). In order to screen the CpG sites that
would exhibit a negative linear correlation between methylation and
lncRNA expression level, correlation analysis was performed and an
associated P-value was calculated.
Methylation-based classifier for the
prediction od patient OS time
The association between the methylation value of
specific CpG sites selected in the previous step and the OS times
of patients was assessed using a univariate Cox regression model.
Following the identification of CpG sites with a statistically
significant association with OS, in order to develop a
methylation-based classifier to predict OS, the Least Absolute
Shrinkage and Selection Operator (LASSO) regression model was then
constructed to identify CpG site predictors using estimated
regression coefficients. As a result, a methylation-based
classifier was constructed that predicted OS times using the fitted
LASSO regression model. Lasso regression is a linear regression
model that uses shrinkage in order to improve the predictive
accuracy and interpretability of regression models, by altering the
model fitting process to select only a subset of the provided
covariates for use in the final model. Shrinkage indicates that
data values have shrunk towards a central point, such as the mean
(27).
Predictive and prognostic analysis of
methylation-based classifiers
The fitted LASSO regression model was used to
estimate patient risk scores, and the predictive accuracy of each
selected classifier was evaluated via the construction of a fitted
model for OS; this utilized time-dependent receiver operating
characteristic (ROC) analysis, and was based on the predetermined
risk scores. In order to analyze the association between certain
clinicopathological characteristics and the methylation-based
classifiers and OS, univariate and multivariate Cox regression
analyses were employed to identify predictors. The predictive
accuracy of certain clinicopathological variables, and the
methylation-based classifiers, was evaluated using the area under
the curve (AUC) of time-dependent ROC curves constructed via the
‘timeROC’ R package. High- or low-risk groups were formed according
to the median cut-off point of the risk score, and Kaplan-Meier
analysis was then performed to estimate and compare the OS times of
patients in each group.
lncRNA co-expression gene and
functional enrichment analysis
The co-expressed genes of lncRNAs were identified
using MEM, a web-based, multi-experiment gene expression query and
visualization tool. It retrieves information from several hundred
publicly available gene expression datasets that represent
different tissues, diseases and conditions. To improve
compatibility and comparability, the datasets were arranged
according to their platform type. Given a gene as an input, MEM
ranks other genes by their similarity in each individual dataset.
This is a novel rank aggregation method which identifies individual
rankings to produce a score of statistically significant
estimation, and hence a ranking across all datasets simultaneously.
The new significance score is also capable of identifying a subset
of datasets where genes exhibit significant similarity, thus
allowing elimination of datasets in which significant correlation
is missing or not detectable (27).
DAVID is a bioinformatics resource consisting of an
integrated biological knowledgebase and analytical tools,
facilitating the systematic extraction of biological meaning from
large gene/protein lists derived from genomic studies. Function
enrichment analysis of the co-expression genes was performed using
DAVID Bioinformatics Resources, and the significant enrichment
terms were visualized using the ‘ggplot2’ package (2.3.0.0) of
R.
Statistical analysis
LASSO is a compression estimation model that
constructs a penalty function, helping a model to compress the
regression coefficients, set certain coefficients to zero and to
select variables. Comparisons between ROC curves were calculated by
quantifying the AUC, and the AUC of a classifier was equivalent to
the probability that the classifier would rank higher at a randomly
chosen positive instance, compared with a randomly chosen negative
instance. Cox regression models are typically used to predict the
prognosis of cancers and chronic diseases with the following
formula: h(t/X) = h0(t) exp (β1 X1 + β2 X2 + …… + βp Xp), h0(t):
Benchmark risk function, X1, X2… Xp: Variable, β1, β2… βp:
Regression coefficient. Kaplan-Meier analysis (a product-limiting
method) was used to estimate OS, according to a probability theory
called the multiplication rule. P<0.05 was considered to
indicate a statistically significant result.
Results
Clinical characteristics of the
patient datasets
A the time of retrieval, TCGA contained records of
459 patients with COAD. However, only 293 patients had records of
both DNA methylation and OS data. Table
I exhibits the summary of the clinical characteristics of the
293 patients. Regarding the methylation status, there were a total
of 334/459 COAD samples that provided methylation data. Of these
334, 296 samples were taken from COAD tissue and 38 samples were
taken from corresponding paracancerous adjacent tissues. Regarding
RNA-seq data, there were 497 tissue samples (459 COAD and 38
adjacent paracancerous tissues). A total of 314 datasets provided
both methylation and RNA-seq data.
 | Table I.Clinical characteristics of patients
with colon adenocarcinoma. |
Table I.
Clinical characteristics of patients
with colon adenocarcinoma.
| Clinicopathological
variables | Patients, n |
|---|
| Age | 293 |
| <60
years | 98 (33.4%) |
| ≥60
years | 195 (66.6%) |
| Sex | 293 |
| Men | 158 (53.9%) |
|
Women | 135 (46.1%) |
| Vascular
invasion | 255 |
|
Present | 60 (20.5%) |
|
Absent | 195 (66.6%) |
| KRAS mutation | 44 |
| Yes | 21 (7.2%) |
| No | 23 (7.8%) |
| Pathological
stage | 283 |
| I +
II | 157 (53.6%) |
| III +
IV | 126 (43.0%) |
| Recurrence | 69 (23.5%) |
| Death | 69 (23.5%) |
Selection of CpG sites and
construction of the methylation-based classifier
Following screening, a total of 11,259 CpG sites
located <2 kb upstream of lncRNA TSS's (excluding the CpG sites
on the X and Y chromosome) were identified. Using the annotation of
HumanMethylation450 BeadChip by TCGA and the ‘minfi’ package in R,
4,876 CpG sites with differential methylation between COAD and
normal adjacent tissues were selected, 2,276 of which had a β-value
difference >0.1. Among the 2,276 CpG sites, there were 1,092
whose linear correlation between the methylation and the expression
levels of lncRNA were negative.
From the 1,092 aforementioned CpG sites, univariate
Cox regression analysis identified 24 CpG sites with a
statistically significant association with OS time. In order to
develop a methylation-based classifier to predict OS, the LASSO
regression model was then employed using the methylation data of
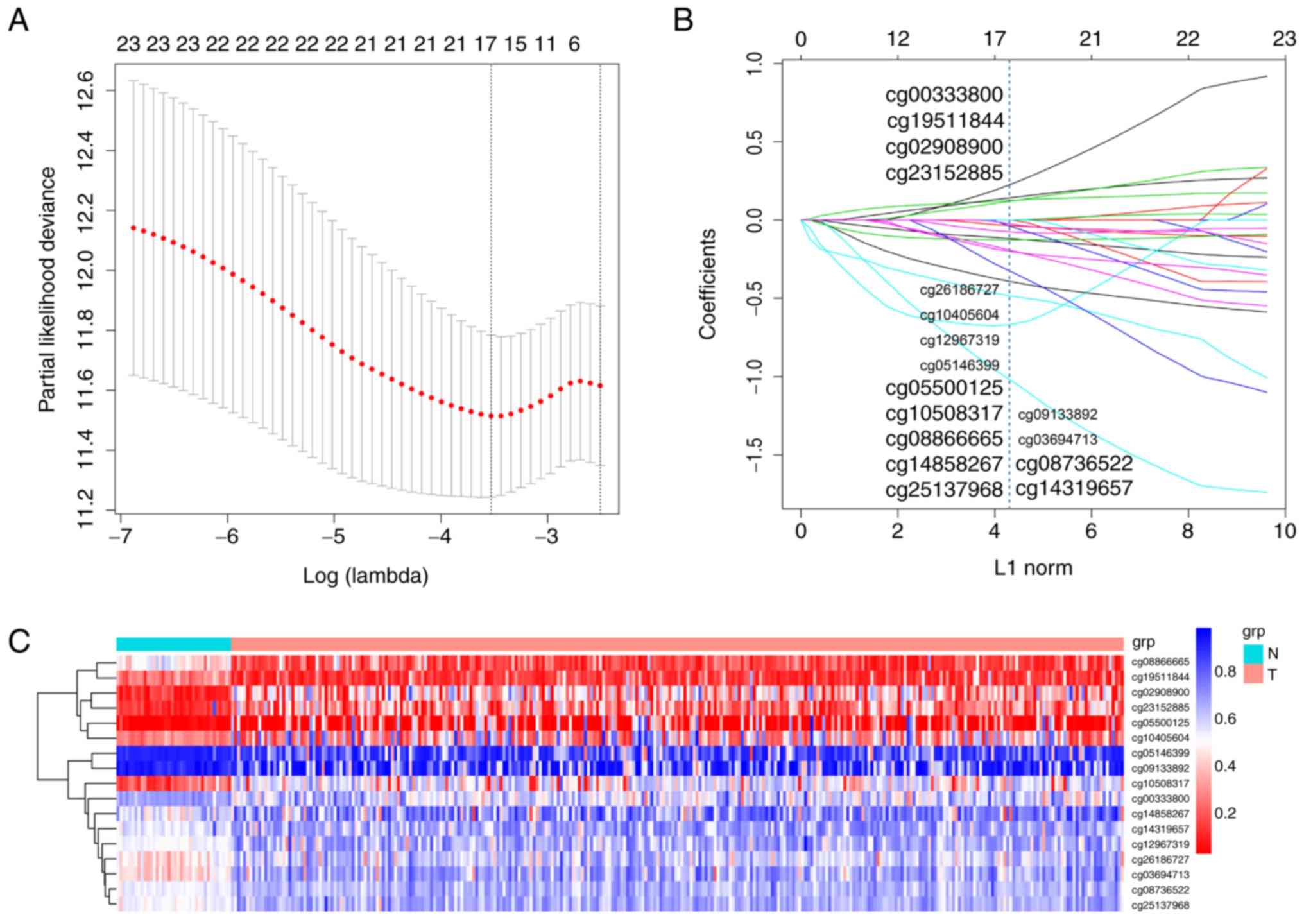
these 24 sites. The LASSO regression method was then used to
determine the regression coefficient of the 17 CpG sites, and the
statistical significance was calculated. There were 4 CpG sites
(cg00333800, cg19511844, cg02908900 and cg23152885) with a
coefficient >0, exhibiting a positive correlation. Another 13
CpG sites exhibited negative regression coefficients <0. The
corresponding coefficients of the 17 CpG sites are depicted in
Fig. 1A and B, and a risk
score-fitted model for methylation-based classifier was calculated
using the following formula: 0.231 × beta_cg00333800+0.125 ×
beta_cg02908900-0.485 × beta_cg03694713-0.076 ×
beta_cg05146399-0.120 × beta_cg05500125-0.666 ×
beta_cg08736522-0.195 × beta_cg08866665-0.395 ×
beta_cg09133892-0.036 × beta_cg10405604-0.127 ×
beta_cg10508317-0.043 × beta_cg12967319-1.028 ×
beta_cg14319657-0.202 × beta_cg14858267+0.143 ×
beta_cg19511844+0.122 × beta_cg23152885-0.332 ×
beta_cg25137968-0.027 × beta_cg26186727. Table II contains information on the
characteristics of the 17 CpG sites selected using LASSO. Table III indicates the computer-generated
risk score of the methylation-based classifier for a selection of
patients. Comparison between COAD and normal adjacent tissues
indicated that the methylation levels in 12 CpG sites were
upregulated, and downregulated in 5 CpG sites in the cancerous
tissues (Fig. S1). Additionally,
unsupervised hierarchical clustering analysis suggested that the
methylation data of these 17 CpG sites were able to accurately
discriminate between COAD and normal adjacent tissue samples
(Fig. 1C).
| Table II.Characteristics of CpG sites selected
by the least absolute shrinkage and selection operator model. CGI,
CpG island; TSS, transcriptional start site. |
Table II.
Characteristics of CpG sites selected
by the least absolute shrinkage and selection operator model. CGI,
CpG island; TSS, transcriptional start site.
| CG_ID | Gene symbol | CG chromosome
location | Position to TSS | CGI coordinate | Feature type |
|---|
| cg00333800 | CTD-2382H12.1 | chr18:
78927878-78927879 | TSS1500 |
chr18:78925187-78925397 | S Shelf |
| cg02908900 | MEOX2-AS1 | chr7:
15687196-15687197 | TSS1500 |
chr7:16399497-16399700 |
|
| cg03694713 | RP11-175E9.1 | chr8:
23706643-23706644 | TSS1500 |
chr8:23704962-23707662 | Island |
| cg05146399 | LINC00635 | chr3:
107883413-107883414 | TSS1500 |
chr3:107927623-107928094 |
|
| cg05500125 | RP11-66B24.2 | chr15:
100849818-100849819 | TSS1500 |
chr15:100849527-100850055 | Island |
| cg08736522 | RP11-108M9.3 | chr1:
16872351-16872352 | TSS1500 |
chr1:16872131-16873554 | Island |
| cg08866665 |
XXyac-YX65C7_A.3 | chr6:
169289797-169289798 | TSS1500 |
chr6:169248451-169248952 |
|
| cg09133892 | LINC01301 | chr8:
60413320-60413321 | TSS200 |
chr8:60516615-60517614 |
|
| cg10405604 | RP11-66B24.2 | chr15:
100850054-100850055 | TSS1500 |
chr15:100849527-100850055 | Island |
| cg10508317 | RP11-806H10.4 | chr17:
78359065-78359066 | TSS1500 |
chr17:78358737-78360957 | Island |
| cg12967319 | MEG3 | chr14:
100825660-100825661 | TSS1500 |
chr14:100825706-100826372 | N Shore |
| cg14319657 | LINC00898 | chr22:
47632172-47632173 | TSS1500 |
chr22:47212945-47213572 |
|
| cg14858267 | RP4-555D20.3 | chr3:
43996268-43996269 | TSS1500 |
chr3:43994915-43999612 | Island |
| cg19511844 | RP11-387H17.4 | chr17:
39927866-39927867 | TSS200 |
chr17:39926973-39927799 | S Shore |
| cg23152885 | RP11-247C2.2 | chr15:
74129941-74129942 | TSS1500 |
chr15:74127529-74130703 | Island |
| cg25137968 | RP11-439A17.4 | chr1:
121117978-121117979 | TSS1500 |
chr1:121118208-121118586 | N Shore |
| cg26186727 | RP11-676J15.1 | chr18:
72867299-72867300 | TSS1500 |
chr18:72866730-72869636 | Island |
| Table III.Samples and their relative risk score
computed are listed in the table. |
Table III.
Samples and their relative risk score
computed are listed in the table.
| Sample name | TCGA4NA93T01 |
|---|
| cg00333800 | 0.384621 |
| cg02908900 | 0.726879 |
| cg03694713 | 0.683491 |
| cg05146399 | 0.927501 |
| cg05500125 | 0.033916 |
| cg08736522 | 0.574554 |
| cg08866665 | 0.079631 |
| cg09133892 | 0.919498 |
| cg10405604 | 0.089026 |
| cg10508317 | 0.629167 |
| cg12967319 | 0.692877 |
| cg14319657 | 0.711359 |
| cg14858267 | 0.879355 |
| cg19511844 | 0.153435 |
| cg23152885 | 0.179211 |
| cg25137968 | 0.514649 |
| cg26186727 | 0.634311 |
| Score | −2.15532 |
Predictive and prognostic accuracy of
the methylation-based classifier
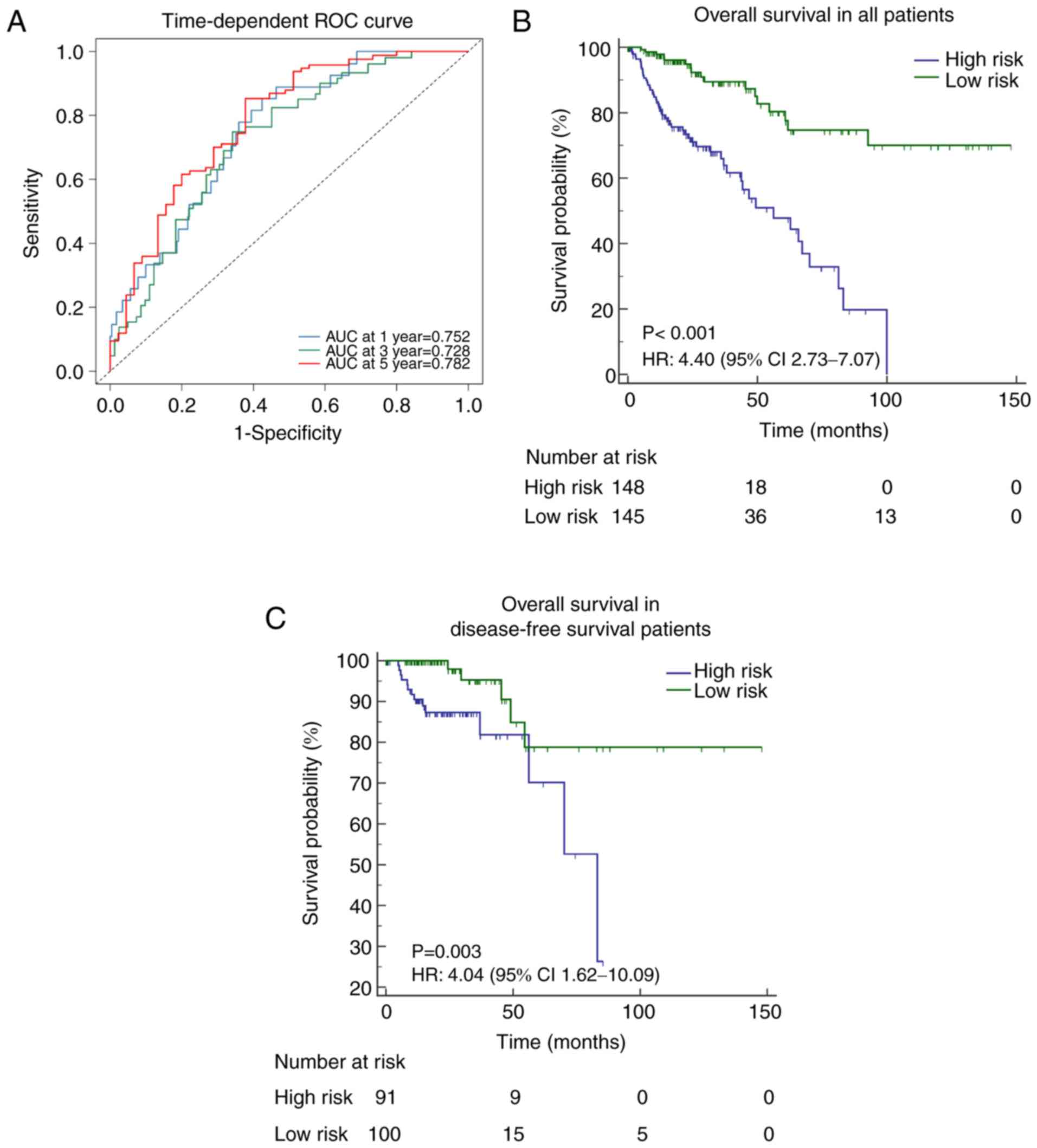
A risk score for each patient was calculated
according to the methylation-based classifier, and the predictive
accuracy of the classifier was evaluated using a time-dependent ROC
curve to predict OS times at several follow-up times; AUC at 1 year
(0.752; 95% CI, 0.668–0.836), 3 years (0.728; 95% CI, 0.640–0.816)
and 5 years (0.782; 95% CI, 0.691–0.874).
Classification into high- or low-risk groups was
defined according to the median cut-off point of the risk score.
Kaplan-Meier analysis indicated that a high risk score indicated
poorer OS [Hazard ratio (HR), 4.40; 95% CI, 2.73–7.07; P<0.001;
Fig. 2B]. A similar association was
determined following the analysis of disease-free survival (DFS)
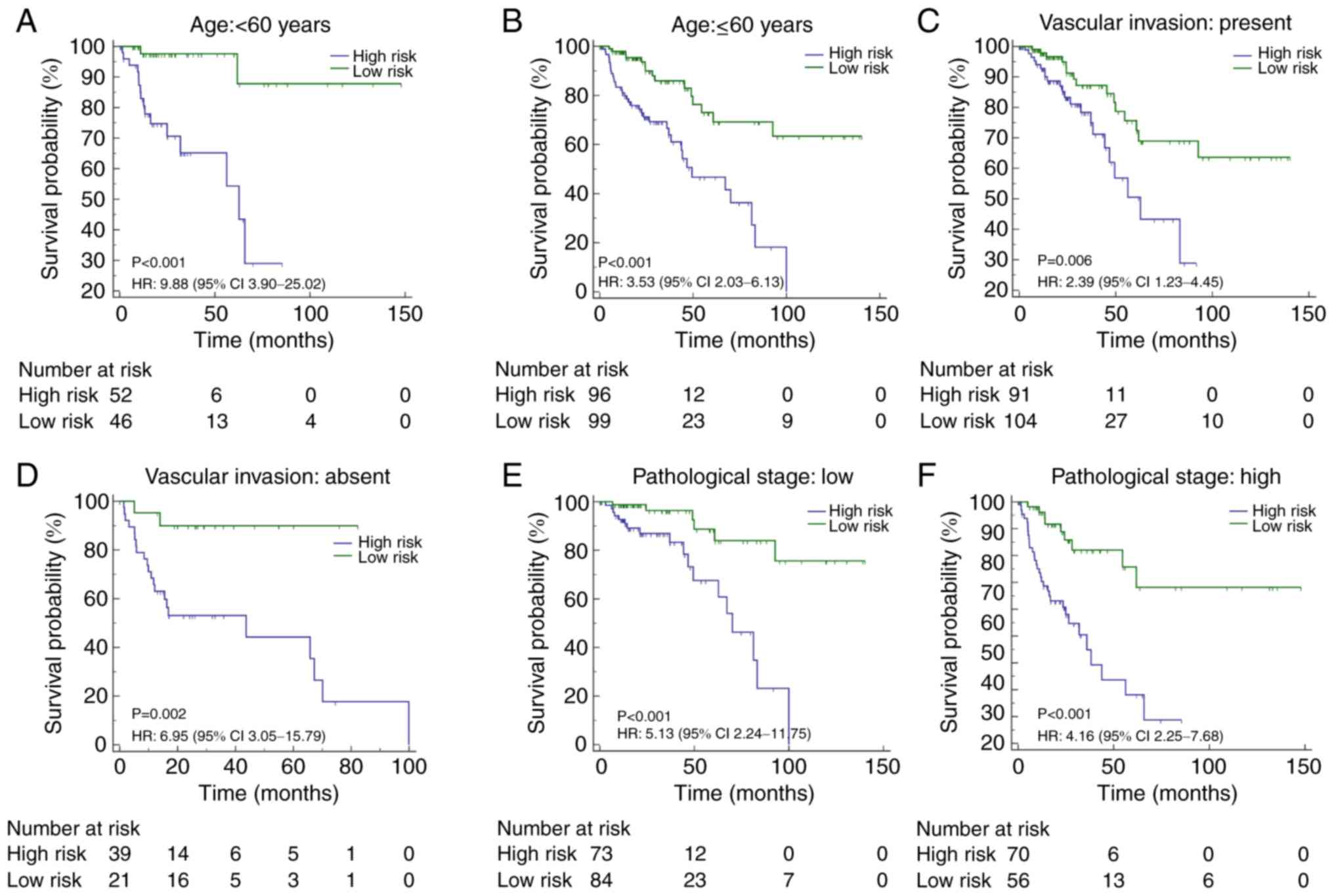
times in patients (HR, 4.04; 95% CI, 1.62–10.09; P=0.003; Fig. 2C, and also in patients stratified
according to certain clinicopathological risk factors (including
age, vascular invasion and pathological stage; Fig. 3).
In order to analyze the association between certain
clinical variables and OS, univariate Cox regression analysis was
performed and resulted in the following predictive scores for OS:
Sex (HR, 1.66; 95% CI, 1.02–2.70; P=0.043), vascular invasion (HR,
2.58; 95% CI, 1.54–4.33; P<0.001), pathological stage (HR, 2.67;
95% CI, 1.61–4.43, P<0.001) and methylation-based classifier
(HR, 4.75; 95% CI, 2.70–8.34; P<0.001). Subsequently,
multivariable adjustment of these variables was performed, and the
results indicated that the pathological stage (HR, 2.82; 95% CI,
1.64–4.86; P<0.001) and methylation-based classifier (HR, 4.08;
95% CI, 2.20–7.54; P<0.001) were both significant predictors of
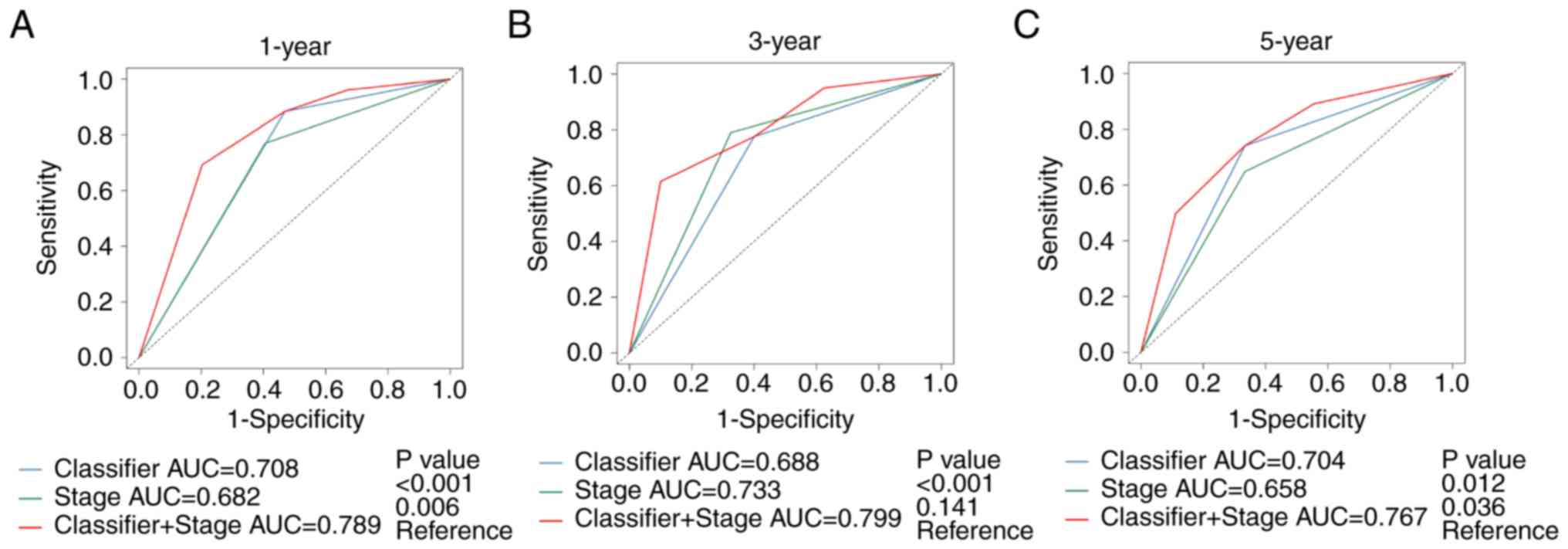
OS (Table IV). Time-dependent ROC
curve analysis determined that the methylation-based classifier,
combined with the pathological stage, provided a more accurate
prediction for OS time at 1 year (AUC, 0.789; 95% CI, 0.710–0.869),
3 year (AUC, 0.799; 95% CI, 0.716 −0.882) and 5 year (AUC, 0.767;
95% CI, 0.667–0.867) in patients with COAD (Fig. 4).
| Table IV.Univariate and multivariate analyses
of the methylation-based classifier for overall survival. |
Table IV.
Univariate and multivariate analyses
of the methylation-based classifier for overall survival.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Prognostic
parameter | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Age, ≥60 vs.
<60 | 1.36 | 0.79–2.32 | 0.267 |
|
|
|
| Sex, men vs.
women | 1.66 | 1.02–2.70 | 0.043 |
|
|
|
| Vascular invasion,
present vs. absent | 2.58 | 1.54–4.33 | <0.001 |
|
|
|
| Pathological stage,
III + IV vs. I + II | 2.67 | 1.61–4.43 | <0.001 | 2.82 | 1.64–4.86 | <0.001 |
| Methylation-based
classifier, high- vs. low-risk | 4.75 | 2.70–8.34 | <0.001 | 4.08 | 2.20–7.54 | <0.001 |
Identification of lncRNA co-expression
genes and functional evaluation
A total of 17 lncRNAs associated with the CpG sites
in the methylation-based classifier were identified (Table II). Moreover, 2,835 gene
co-expressed with the lncRNAs, were identified using MEM analysis.
DAVID was used to perform functional enrichment analysis of the
co-expressed genes, and the results indicated that the
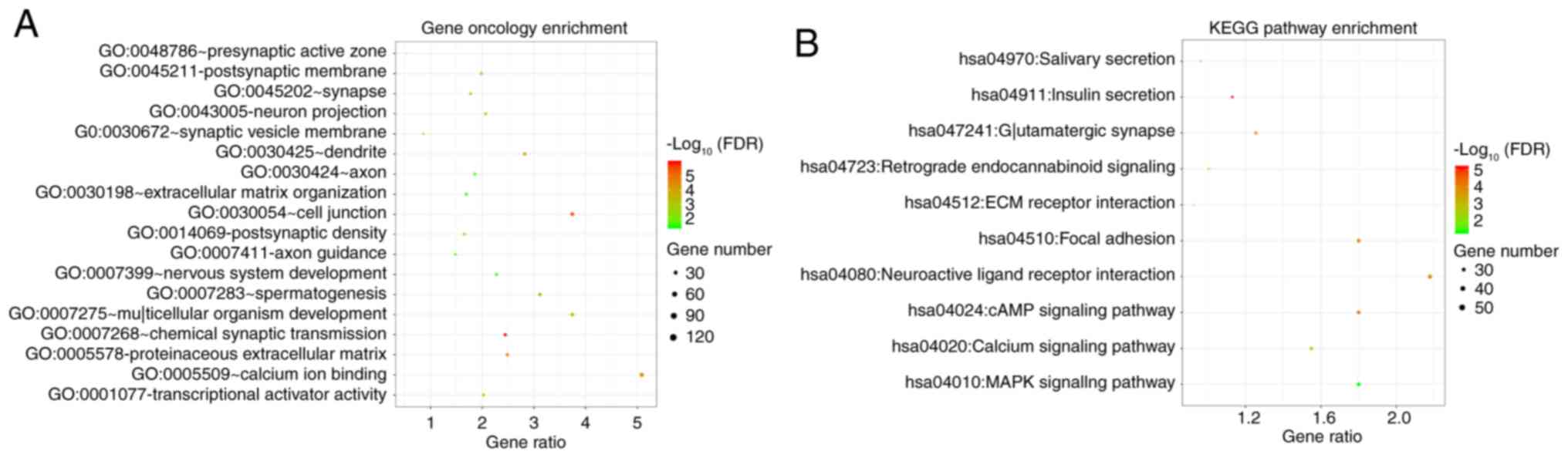
significantly enriched Gene Ontology (GO) terms were ‘extracellular
matrix organization’ (biological process), ‘cell junction’
(cellular component) and ‘transcriptional activator activity’
(molecular function). Certain KEGG pathways were also significantly
enriched, including ‘MAPK-signaling pathway’, ‘cAMP-signaling
pathway’ and ‘calcium-signaling pathway’ (Fig. 5).
Discussion
Colon cancer is the most prevalent gastrointestinal
cancer type and exhibits high incidence and mortality rates; COAD
is a colon cancer subtype that accounts for ~98% of new diagnoses
(28). Despite advances in the
diagnosis and treatment of colon cancer, due to the recognition of
various prognostic and predictive factors (including age, tumor
grade and stage, surgical margins and the number of affected local
lymph nodes), the prognosis for patients is still poor due to
limited characterization of the molecular mechanism regulating COAD
development and progression. Therefore, in order to develop novel
and effective therapeutic approaches, comprehensive research into
the molecular mechanism of COAD tumorigenesis is imperative.
Although credible biomarkers for the treatment and prognosis of
COAD have been characterized, the majority focus on the expression
and mechanistic roles of mRNAs, lncRNAs and microRNAs (29–31). The
importance of the DNA methylation profile of lncRNAs has not been
fully investigated, but it shows potential to be a key novel
biomarker, able to improve the OS of patients with COAD.
In the current study, comprehensive analysis of the
DNA methylation profile of lncRNAs was performed to investigate a
large cohort of COAD samples retrieved from TCGA, with all samples
showing altered DNA methylation patterns. Moreover, it was
discovered that numerous CpG sites exhibited significantly
different methylation statuses in COAD tissues, compared with
adjacent normal tissues; furthermore, 24 CpG sites were
significantly correlated with OS. In accordance with LASSO
regression analysis, 17 CpG sites were identified as having
statistically significant estimated regression coefficients, and
their gene symbols are denoted as: CTD-2382H12.1, MEOX2-AS1,
RP11-175E9.1, LINC00635, RP11-66B24.2, RP11-108M9.3,
XXyac-YX65C7_A.3, LINC01301, RP11-66B24.2, RP11-806H10.4, MEG3,
LINC00898, RP4-555D20.3, RP11-387H17.4, RP11-247C2.2, RP11-439A17.4
and RP11-676J15.1. All CpG sites identified in the present study
were able to significantly predict the OS times of patients with
COAD. Notably, none of the aforementioned lncRNAs had been
identified in previous studies.
Clinically, the methylation-based classifier
constructed in the present study could be employed as a diagnostic
tool, predicting OS times in patients with COAD. It was able to
predict OS time and produce high- or low-risk scores for both
patients with COAD and those in the DFS group. Moreover,
significant predictive accuracy was exhibited at several time
points during the follow-up period (according to ROC analysis),
suggesting the potential to improve and individualize clinical
decision-making regarding treatment programs. The analysis and
assessment of COAD prognosis typically includes previously reported
risk factors, including age, sex and disease stage. However,
multivariate Cox regression analysis indicated that the OS time in
patients with COAD could also be predicted using the
methylation-based classifier as the measured parameter, further
supporting the significance of methylation in disease progression.
Moreover, the accuracy OS-time prediction during the follow-up
period may be improved if the results of the methylation-based
classifier were integrated with other clinicopathological risk
factors.
Epigenetic alterations were determined to regulate
the tumorigenesis and progression of COAD. It was a complex and
intricate process, but the methylation of lncRNAs is likely to be
the access point to a more thorough understanding of the molecular
mechanism of COAD. In order to further investigate the effects of
epigenetic alterations on biological processes and pathways,
comprehensive analysis of lncRNA methylation was conducted. A total
of 2,899 genes were found to be co-expressed with aberrant
methylation of lncRNAs, and the majority are located in pivotal
cancer-signaling pathways, indicating their potential to influence
tumor biology.
A limitation of the present study was the failure to
elucidate the causality between the aberrant methylation patterns
of lncRNAs and the occurrence of tumors. Further understanding of
this mechanism may help to identify novel therapeutic targets and
improve the prognosis of patients with COAD.
In conclusion, the present study identified a
methylation-based classifier consisting of lncRNAs closely related
to the OS times of patients with COAD, and subsequently determined
that the prognosis of COAD could be accurately predicted using
altered DNA methylation patterns, as well as the involvement of
relevant genes in pivotal signaling pathways related to
oncogenesis. An advantage of the present study was that the
experiments were performed using a large population size and a
sufficient data source. Additionally, the findings indicated both
satisfactory independent prognostic value and biological relevant
pathways. To the best of our knowledge, this was the first study to
quantify the significance of the association between regulation of
DNA methylation patterns by lncRNAs and the prognosis of patients
with COAD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Conceptualization of the study was performed by QZ,
ZL and HXZ. ZL, QZ, HYZ and XB contributed the study methodology,
resources and formal analysis. Software use and initial
investigation was conducted by ZL and QZ. QZ and XB wrote the first
draft, and QZ, ZL, XB, HXZ and HYZ reviewed and edited the
manuscript. HXZ, ZL and QZ supervised the project. Project
administration was performed by HXZ. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Robertson JH, Sarkar S, Yang SY, Seifalian
AM and Winslet MC: In vivo models for early development of
colorectal liver metastasis. Int J Exp Pathol. 89:1–12. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alaiyan B, Ilyayev N, Stojadinovic A,
Izadjoo M, Roistacher M, Pavlov V, Tzivin V, Halle D, Pan H, Trink
B, et al: Differential expression of colon cancer associated
transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence.
BMC Cancer. 13:1962013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu K, Yao H, Wen Y, Zhao H, Zhou N, Lei S
and Xiong L: Functional role of a long non-coding RNA
LIFR-AS1/miR-29a/TNFAIP3 axis in colorectal cancer resistance to
pohotodynamic therapy. Biochim Biophys Acta Mol Basis Dis.
1864:2871–2880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Olthof MR, Hollman PC and Katan MB:
Chlorogenic acid and caffeic acid are absorbed in humans. J Nutr.
131:66–71. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hong J, Lu H, Meng X, Ryu JH, Hara Y and
Yang CS: Stability, cellular uptake, biotransformation, and efflux
of tea polyphenol (−)-epigallocatechin-3-gallate in HT-29 human
colon adenocarcinoma cells. Cancer Res. 62:7241–7246.
2002.PubMed/NCBI
|
|
7
|
Tsukuda K, Tanino M, Soga H, Shimizu N and
Shimizu K: A novel activating mutation of the K-ras gene in human
primary colon adenocarcinoma. Biochem Biophys Res Commun.
278:653–658. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yue B, Qiu S, Zhao S, Liu C, Zhang D, Yu
F, Peng Z and Yan D: lncRNA-ATB mediated E-cadherin repression
promotes the progression of colon cancer and predicts poor
prognosis. J Gastroenterol Hepatol. 31:595–603. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benson AB, Venook AP, Al-Hawary MM,
Cederquist L, Chen YJ, Ciombor KK, Cohen S, Cooper HS, Deming D,
Engstrom PF, et al: NCCN guidelines insights: Colon cancer, version
2.2018. J Natl Compr Canc Netw. 16:359–369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bernard D, Prasanth KV, Tripathi V,
Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L,
Coulpier F, et al: A long nuclear-retained non-coding RNA regulates
synaptogenesis by modulating gene expression. EMBO J. 29:3082–3093.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu P, Wang Y, Wu J, Huang G, Liu B, Ye B,
Du Y, Gao G, Tian Y, He L and Fan Z: lncBRM initiates YAP1
signalling activation to drive self-renewal of liver cancer stem
cells. Nat Commun. 7:136082016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nat Rev
Genet. 15:7–21. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
He Y, Meng XM, Huang C, Wu BM, Zhang L, Lv
XW and Li J: Long noncoding RNAs: Novel insights into hepatocelluar
carcinoma. Cancer Lett. 44:20–27. 2013.
|
|
15
|
Huang JL, Zheng L, Hu YW and Wang Q:
Characteristics of long non-coding RNA and its relation to
hepatocellular carcinoma. Carcinogenesis. 35:507–514. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JT: Epigenetic regulation by long
noncoding RNAs. Science. 338:1435–1439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Timp W and Feinberg AP: Cancer as a
dysregulated epigenome allowing cellular growth advantage at the
expense of the host. Nat Rev Cancer. 13:497–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu D, Yang B, Chen J, Xiong H, Li Y, Pan
Z, Cao Y, Chen J, Li T, Zhou S, et al: Upregulation of long
non-coding RNA RAB1A-2 induces FGF1 expression worsening lung
cancer prognosis. Cancer Lett. 438:116–125. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smolle MA, Bullock MD, Ling H, Pichler M
and Haybaeck J: Long non-coding RNAs in endometrial carcinoma. Int
J Mol Sci. 16:26463–26472. 2013. View Article : Google Scholar
|
|
20
|
Kim J, Piao HL, Kim BJ, Yao F, Han Z, Wang
Y, Xiao Z, Siverly AN, Lawhon SE, Ton BN, et al: Long noncoding RNA
MALAT1 suppresses breast cancer metastasis. Nat Genet.
50:1705–1715. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Y, Xiang B, Liu Y, Wang Y and Kan H:
lncRNA CDKN2B-AS1 promotes tumor growth and metastasis of human
hepatocellular carcinoma by targeting let-7c-5p/NAP1L1 axis. Cancer
Lett. 437:56–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao H, Duan M, Lin L, Wu C, Fu X, Wang H,
Guo L, Chen W, Huang L, Liu D, et al: TET2 and MEG3 promoter
methylation is associated with acute myeloid leukemia in a hainan
population. Oncotarget. 8:18337–18347. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo W, Dong Z, Shi Y, Liu S, Liang J, Guo
Y, Guo X, Shen S and Shan B: Aberrant methylation-mediated
downregulation of long noncoding RNA LOC100130476 correlates with
malignant progression of esophageal squamous cell carcinoma. Dig
Liver Dis. 48:961–969. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ranstam J and Cook JA: LASSO regression.
Br J Surg. 105:13482018. View Article : Google Scholar
|
|
26
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Adler P, Kolde R, Kull M, Tkachenko A,
Peterson H, Reimand J and Vilo J: Mining for coexpression across
hundreds of datasets using novel rank aggregation and visualization
methods. Genome Biol. 2009:R1392009. View Article : Google Scholar
|
|
28
|
Xue W, Li J, Wang F, Han P, Liu Y and Cui
B: A long non-coding RNA expression signature to predict survival
of patients with colon adenocarcinoma. Oncotarget. 8:101298–101308.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang WJ, Li HT, Yu JP, Han XP, Xu ZP, Li
YM, Jiao ZY and Liu HB: A competing endogenous RNA network reveals
novel potential lncRNA, miRNA, and mRNA biomarkers in the prognosis
of human colon adenocarcinoma. J Surg Res. 235:22–33. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang JY, Wang CL, Wang XM and Liu FJ:
Comprehensive analysis of microRNA/mRNA signature in colon
adenocarcinoma. Eur Rev Med Pharmacol Sci. 21:2114–2129.
2017.PubMed/NCBI
|
|
31
|
Chen F, Li Z and Zhou H: Identification of
prognostic miRNA biomarkers for predicting overall survival of
colonadenocarcinoma and bioinformatics analysis: A study based on
the cancer genome atlas database. J Cell Biochem. 120:9839–9849.
2019. View Article : Google Scholar : PubMed/NCBI
|