Introduction
Chronic hepatitis B virus (HBV) infection is the
leading cause of chronic hepatitis, cirrhosis and hepatocellular
carcinoma (HCC) (1). At present, the
mechanism by which HBV causes chronic hepatitis and liver cancer
remains unclear; however, the hepatitis B virus × (HBx) protein may
serve a role in this process (1).
HBx is a multifunctional regulatory protein composed of 154 amino
acids, with a molecular weight of 17 kDa (1). HBx is involved in a variety of signal
transduction pathways affecting cell cycle progression, apoptosis
and cancer progression (1). HBx can
also affect mitochondria by altering the permeability of
mitochondrial membranes, as HBx disrupts oxidative phosphorylation,
interferes with the mitochondrial respiratory chain and inhibits
ATP synthesis, leading to liver cell damage (2–6). Rahmani
et al (2) have reported that
HBx may be localized in the mitochondria, where it binds to the
voltage-dependent anion channel 3 on the outer mitochondrial
membrane, thus changing the mitochondrial membrane potential. Lee
et al (3) have demonstrated
that HepG2 cells that stably express HBx, causing the proton
transfer to be blocked, induces the mitochondria of hepatocellular
carcinoma cells in a sensitive state, thus leading to the formation
of reactive oxygen species (ROS) and lipid peroxidation. Excessive
ROS production then affects cell proliferation and differentiation,
inducing apoptosis and gene mutations, thus promoting the
occurrence of HCC. HBx can also induce the opening of the
mitochondrial permeability transition pore and swelling of the
mitochondrial matrix, mitochondrial membrane potential
depolarization-induced release of cytochrome c
apoptosis-inducing factor and calcium ions into the cytoplasm, and
apoptosis by activation of the caspase signaling cascade (4–6). The
mitochondrial permeability transition pore and swelling of the
mitochondrial matrix induce apoptosis or necrosis, lead to
cytoplasmic calcium overload, enhance HBV replication and
contribute to liver inflammation (4–6). In
addition, HBx can also induce liver disease by activating
autophagy, mitochondria-dependent apoptosis pathways, mitochondrial
division and fusion damage (7,8).
During the development of chronic hepatitis and
cirrhosis, the mitochondrial respiratory chain is damaged and its
function is significantly decreased (9). Cytochrome c oxidase (COX),
composed of 13 subunits including COXI, COXII and COXIII, which are
encoded by mitochondrial DNA, serves a key role in oxidative
phosphorylation (10,11). The lack of functional order in COXIII
hinders proton transfer and the accumulation of excess electrons
and oxygen molecules leads to decreased ATP synthesis and mass ROS
production (10,11) during chronic hepatitis and cirrhosis.
It has been reported that intracellular HBx is primarily localized
in mitochondria (12,13) and that HBx binds to COXIII, thereby
upregulating ROS production (14–16).
Alterations in the levels of ROS in the mitochondria
are common factors in the pathogenesis of inflammatory diseases and
tumors (17). In acute liver
inflammation, ROS primarily induces mitochondrial dysfunction
through intracellular oxidative stress (17). Previous studies have demonstrated
that the C-terminus of HBx causes mitochondrial DNA damage,
resulting in the increase in ROS levels in liver cells.
Accumulation of ROS can also upregulate HBx expression levels
(13,18), indicating that mitochondria are the
primary targets of ROS.
The NF-κB/AKT signaling pathway is a key
inflammatory pathway involved in the development of cancer.
Abnormal activation of NF-κB/AKT signaling in liver tissues can
inhibit apoptosis and promote liver cell survival, which may lead
to the development of liver cancer (19,20). It
has been reported that the NF-kB signaling pathway serves a role in
ROS-mediated liver injury (21). In
addition, previous studies have demonstrated that the PI3K/AKT
signaling pathway is involved in HBx-induced liver cancer formation
and also serves a role in the regulation of antioxidant genes
(22,23).
Inflammatory mediators are the primary signal
transducers in the tumor microenvironment and have been
demonstrated to be involved in the development and progression of
liver cancer. Numerous in vivo and in vitro studies
have confirmed that the levels of inflammatory mediators, such as
interleukin (IL)-6, IL-1β, IL-10, tumor necrosis factor-α (TNF-α)
and transforming growth factor-β (TGF-β) are often increased in the
tissues and sera of patients with liver cancer (24–26).
Pro-inflammatory IL-18 is involved in immune response regulation,
and excessive IL-18 expression levels can lead to immune system
deregulation, causing inflammatory damage to tissues and organs
(27). The pro-inflammatory
mediators (IL-6, IL-1β, IL-10, TNF-α, TGF-β and IL-18) can promote
tumor growth, inhibit apoptosis, induce epithelial-to-mesenchymal
transition and ultimately promote the invasion and metastasis of
liver cancer.
Increasing attention has drawn to the relationship
between malondialdehyde (MDA), superoxide dismutase (SOD) and
mitochondria. Several studies on drugs (such as the impairment of
mitochondrial function in mice with a low or excessive selenium
diet) and the pathogenesis of diseases (for example, liver
dysfunction) have been conducted by measuring the expression levels
of MDA and SOD and observing the structural changes in mitochondria
and demonstrating their association (28,29).
Paradies et al (30) reported
that decreased COX activity of myocardial mitochondria in rats was
associated with increased lipid oxidation levels caused by
increased MDA activity. Therefore, changes in MDA and SOD levels in
the serum may reflect the state of mitochondria and indicate the
degree of liver cell damage.
In the present study, a mouse model expressing HBx
was established to observe acute hepatocyte injury in mice, and the
levels of IL-6, IL-1β, IL-18, MDA and SOD and COX activity were
determined. Changes in ROS levels, NF-κB, AKT and phosphorylated
(p-) AKT were measured. The mechanism by which HBx affects
mitochondrial function in hepatocytes, induces inflammation and
damages hepatocytes was explored in vivo, providing new
insight into the mechanism underlying HBV-related chronic hepatitis
and hepatocellular carcinoma.
Materials and methods
Animals
The present study was approved by The Institutional
Animal Care and Use Committee of Fujian Medical University (Fujian,
China). Mice were euthanized using an intraperitoneal injection of
2% sodium pentobarbital at a dose of 100 mg/kg and death was
confirmed by observing the ventilation, complete cardiac arrest and
loss of reflexes. A total of 30 male ICR mice, aged 6–8 weeks and
weighing 19–22 g, were purchased from Shanghai SLAC Laboratory
Animal Technology Co. Ltd. The mice were maintained at 25°C with a
12:12 h light/dark cycle and were provided with food and water, and
access to food and water was arbitrary. The mice were randomly
divided into 3 groups (n=10 per group) based on the administered
treatment: i) Experimental; ii) null-plasmid control; and iii)
blank control (plasmid solvent) groups. The solutions were injected
into the caudal vein under high pressure.
Preparation of competent bacteria
E. coli DH5α cells from Tiangen Biotech Co.,
Ltd. were cultured on agar plates at 37°C in an incubator
overnight. A single colony was collected from the plate and the
cells were cultured further with agitation at 37°C and 250 rpm for
12 h. Subsequently, the bacteria were incubated on ice for 30 min
followed by centrifugation at 4,000 × g and for 10 min at 4°C. The
supernatant was discarded, and 1 ml precooled 0.1 mol/l
CaCl2 was added to the obtained pellet. The mixture was
mixed with a pipette.
Plasmid transformation and
extraction
pcDNA3.1-HBx plasmid (stored in the laboratory) was
added to a 200-µl suspension of susceptible E. coli DH5α
cells, mixed by shaking gently, placed on ice for 30 min, subjected
to heat shock at 42°C for 2 min, and quickly moved to ice for 3–5
min. The cells were added to 800 µl LB medium and cultured with
agitation at 37°C and 250 rpm for 1 h. Bacterial cells were then
streaked onto liquid culture medium using inoculation loops,
followed by incubation of the plates at 37°C for 18–24 h. The
transformed bacteria were followed by plasmid extraction using an
EndoFree Plasmid Maxi kit (Qiagen, Inc.).
RNA extraction and reverse
transcription PCR analysis
Total RNA from mouse liver tissue was extracted
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA (2 µg) was reverse-transcribed into
cDNA using a RevertAid First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.). The conditions for the RT of RNA into DNA
were as follows: 60 min at 42°C, 5 min at 70°C, and storage at 4°C.
The primer sequences of each gene are listed in Table I. The thermo cycling conditions of
PCR were as follows: Pre-denaturation at 95°C for 5 min; followed
by 30 cycles of denaturation at 95°C for 30 sec, annealing at 62°C
for 30 sec and 72°C for 1 min; and a final extension step at 72°
for 7 min. PCR was performed using a Taq PCR Master mix (Thermo
Fisher Scientific, Inc.). A total of 10 µl PCR product with 5 µl
marker (cat. no. MD110; Tiangen Biotech Co., Ltd.) was loaded onto
a 2% agarose gel containing 1.5 µl gold view (Beijing Solarbio
Science & Technology Co., Ltd.). Gel electrophoresis was
performed at 100 V for 15–30 min. The results of the
electrophoresis were scanned using a UVP scanner with Grab-IT (Gel
Doc 100; Bio-Rad Laboratories, Inc.) and analyzed by Gelpro32
(Media Cybernetics, Inc.).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Sequences
(5′→3′) |
|---|
| HBx-flag |
|
|
Forward |
ATGCAAGCTTATGGCTGCTAGGCTGTACTG |
|
Reverse |
TGCGAATTCTTAGGCAGAGGTGAAAAAGTT |
| β-actin |
|
|
Forward |
GGCATCGTGATGGACTCCG |
|
Reverse |
GCTGGAAGGTGGACAGCGA |
Western blot analysis
Liver tissues were lysed in RIPA buffer (Beyotime
Institute of Biotechnology) supplemented with protease inhibitors
(Beyotime Institute of Biotechnology) and kept on ice for 30 min,
followed by centrifugation at 12,000 × g for 15 min at 4°C. Protein
concentration was measured using a Bicinchoninic Acid (BCA) Protein
Assay kit (Beyotime Institute of Biotechnology). Total protein
samples (60 µg) were loaded onto a 12% gel, resolved using
SDS-PAGE, transferred to a nitrocellulose filter membrane and
blocked with 5% non-fat milk in Tris-buffered saline containing
0.1% Tween-20 (TBST) for 60 min at room temperature. The membranes
were incubated overnight at 4°C with the following primary
antibodies: Rabbit anti-human HBx (1:1,000; cat. no. ab39716;
Abcam), rabbit anti-mouse NF-κB (1:1,000; cat. no. 8242; Cell
Signaling Technology, Inc.), rabbit anti-mouse AKT (1:1,000; cat.
no. 4685; Cell Signaling Technology, Inc.), rabbit anti-mouse p-AKT
(1:500; cat. no. 4060; Cell Signaling Technology, Inc.) and rabbit
anti-mouse GAPDH (1:1,000; cat. no. 2118; Cell Signaling
Technology, Inc.). The membrane was then washed three times (10 min
each) with TBST and incubated with a goat anti-rabbit horseradish
peroxidase-conjugated secondary antibody (1:5,000; cat. no.
ZB-2301; OriGene Technologies, Inc.) for 60 min at room
temperature. After washing with TBST, the membrane was incubated
with an ECL chemiluminescence kit (1:1; OriGene Technologies, Inc.)
for 1 min. A auto-exposure (ChemiDoc MP System; Bio-Rad
Laboratories, Inc.) was used to expose the target strip and
analyzed by Gelpro32.
Construction and identification of a
HBx expression mouse model
Hydrodynamics-based transfection is an in
vivo gene transfection method based on the principle of making
the venous system and hepatic sinus of mice hyperemic and
generating high venous pressure, which permits the transfer of
plasmids into liver cells by rapidly injecting a large volume of
plasmid solution into the tail vein (31,32). The
present study used this method to transfect mice with pcDNA3.1-HBx,
empty pcDNA3.1 plasmids and plasmid solvent, according to the
manufacturer's protocol using a TransIT In Vivo Gene
Delivery system (Mirus Bio). A total of ~250 µg of plasmid was
added to the in vivo polymer solution in a 15-ml centrifuge
tube that does not contain nucleotide enzymes, and the tube was
allowed to stand at room temperature for 5 min. The solution was
then injected into the mice via the caudal vein at a constant speed
within 4–8 sec. After 24 h, the mice were euthanized and eyeball
blood and liver tissues were collected; eyeball blood was stored at
−20°C, and liver tissue was cryopreserved at −70°C.
Isolation of mitochondria and
measurement of COX activity
Mitochondria were isolated using the Mitochondrial
Isolation kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. Following lysis of ~100 mg of
liver tissue, cell debris and nuclei were removed by centrifugation
at 700 × g for 10 min at 4°C, followed by centrifugation at 3,000 ×
g for 15 min at 4°C to pellet the mitochondria-enriched fraction
and then at 12,000 × g for 5 min at 4°C to pellet the isolated
mitochondria. Protein concentrations were measured using the BCA
Assay kit (Beyotime Institute of Biotechnology) and adjusted to 1
µg/µl. COX activity was determined using the Cytochrome c
Oxidase Assay kit (Genmed Scientifics Inc,) according to the
manufacturer's instructions and measured by an ELx800 microplate
reader (BioTek Instruments Inc.) at 550 nm.
In situ fluorescence assay for
reactive oxygen species measurement
In situ fluorescence staining was used to
detect the ROS levels in the frozen sections of mouse liver tissue.
Frozen sections (10 mm) were stained using a ROS in situ
fluorescence staining kit (GenMed) and placed at room temperature.
Briefly, the section surface was covered with 500 µl pre-cooled
cleaning solution, which was then carefully removed. Subsequently,
200 µl staining and diluent dye solutions were carefully added to
cover the section surface, and the sections were incubated at 37°C
for 30 min. The sections were treated again with 500 µl of
pre-cooled cleaning solution after removal of the other reagents,
followed by removal of the pre-cooled cleaning solution. The
sections were then transferred to glass slides, covered and
observed under a Nikon Eclipse TE 2000-U inverted fluorescence
microscope at ×100 magnification (Nikon Corporation); enhanced
green fluorescence was observed at 499 nm excitation and 515 nm
emission wavelengths.
H&E staining
The control group and the experimental group model
mice were injected with pcDNA3.1-HBx, empty pcDNA3.1 plasmids and
plasmid solvent solution into the tail vein for 24 h, and liver
tissues sized 1.0×1.0×0.3 cm were obtained, fixed with 4%
paraformaldehyde for 24 h at room temperature. Subsequently, the
tissue sections were dehydrated in 80, 90, 95 and 100% gradient
ethanol for 2–4 h. The embedded liver tissue was cut into 3-µm
sections, using the microtome (Thermo Fisher Scientific, Inc.), and
dried for H&E staining. Following 20 min of baking at 60°C,
xylene I and II were used to dewax the tissues for 10 min each.
Subsequently, tissues were incubated in 100, 95, 85, 75% gradient
ethanol for 5 min each, and in distilled water for 5 min to
complete the dewaxing process. Hematoxylin (cat. no. C0390; Beijing
Noblelight Technology Co., Ltd.) staining was performed for 5 min
at room temperature, after washing with water. Eosin staining (cat.
no. C0390, NobleRyder) was conducted for 5 sec, and the sections
were subsequently fully washed (33). Sections were then dehydrated using
75, 85, 95 and 100% ethanol (2 min each). Xylene I, II transparent
for 5 min neutral gum seal is intended for fixation. Sections were
observed and images were captured using a Leica DM2000 light
microscope (Leica Microsystems, Inc.; magnification, ×20).
Detection of MDA, SOD, IL-6, IL-1β and
IL-18 in serum
Inflammatory cytokine expression in mouse serum was
detected using mouse ELISA kits (Mouse MDA/SOD/IL-6/IL-1β/IL-18
ELISA kits; cat. nos. M6000B, DYC3419-2, 7625, MLB00C; R&D
Systems, Inc.). The serum was thawed at room temperature and
thoroughly mixed. A wash buffer diluted 1:20 with distilled water
was used. In the ELISA plate, the wells were divided into standard,
sample and blank wells. A total of 50 µl of the different
concentrations of the standard (10 µl of the test sample and 40 µl
of the diluent) were added to the standard and sample wells,
respectively. Nothing was added to the blank well. Subsequently,
100 µl of the horseradish peroxidase-labeled detection antibody was
added to the standard and sample wells. The reaction plate membrane
aperture was sealed, and the plate was incubated at 37°C for 60
min. After the incubation period, unbound components were
discarded, and the plate was patted dry with absorbent paper. The
wells were filled with washing liquid, and the plate was allowed to
rest for 1 min. The washing liquid was then discarded, and the
plate was patted dry with absorbent paper. The substrates (50 µl
each) were added, and the plate was incubated at 37°C in the dark
for 15 min, followed by the addition of 50 µl termination solution
to each well for 15 min. The optical density was measured at 450 nm
using an ELx800 microplate reader (BioTek Instruments, Inc.).
Statistical analysis
Data were analyzed using GraphPad Prism statistical
software version 5.0 (GraphPad Software, Inc.) and ImageJ 2×
(National Institutes of Health). Data are expressed as the mean ±
standard deviation (unless otherwise stated) from at least three
independent experiments. One-way ANOVA was used for statistical
analysis, followed by Tukey's post hoc test to assess statistical
differences among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Transfection efficiency
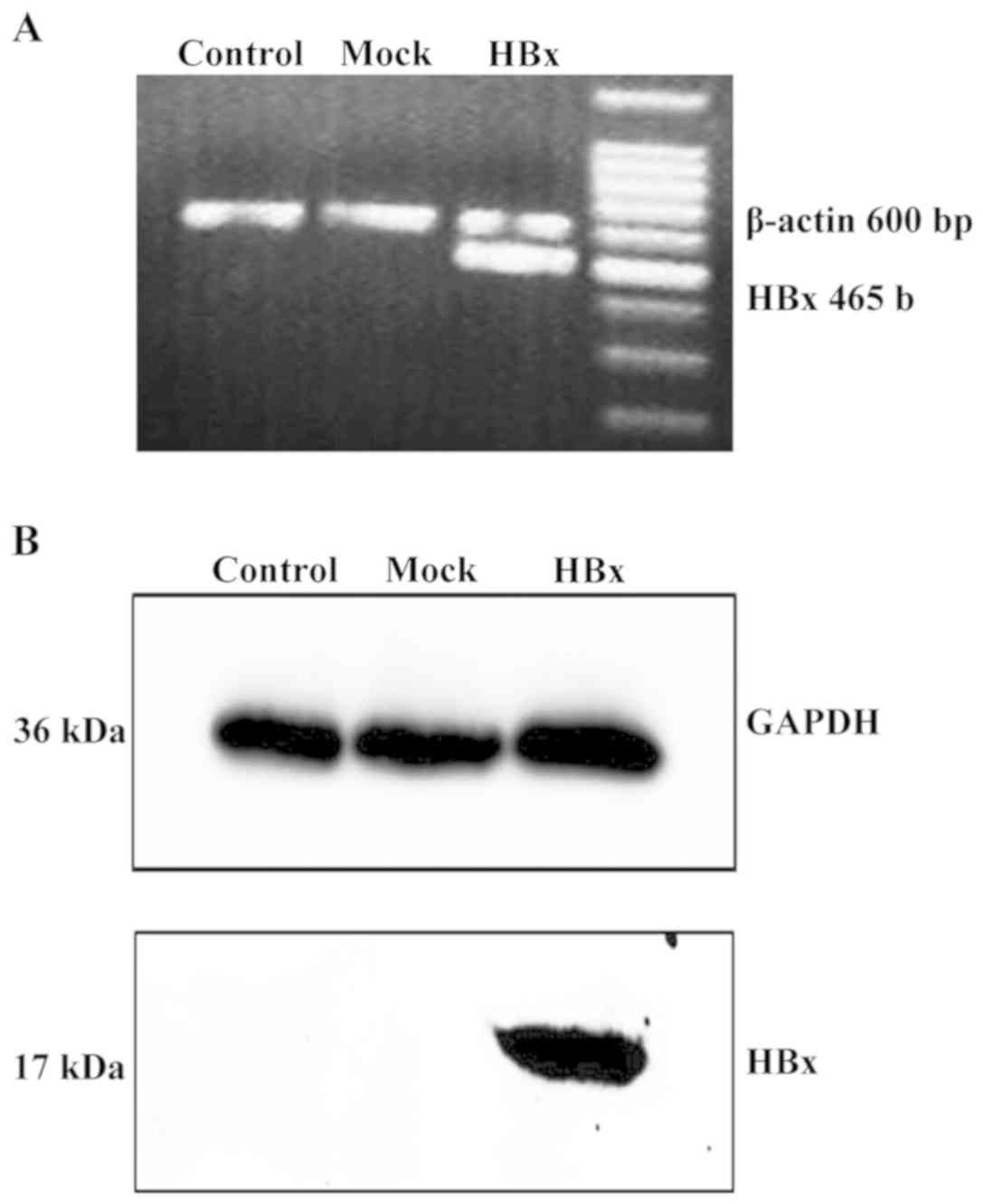
Transfection of pCDNA3.1-HBx notably increased the
mRNA (Fig. 1A) and protein (Fig. 1B) expression levels in mouse liver
tissues. HBx expression was not observed in the control or empty
vector-transfected liver tissues. These results demonstrated that
the HBx gene was successfully transfected into the livers of
experimental mice.
Effect of HBx on the mitochondrial
function in mouse hepatocytes
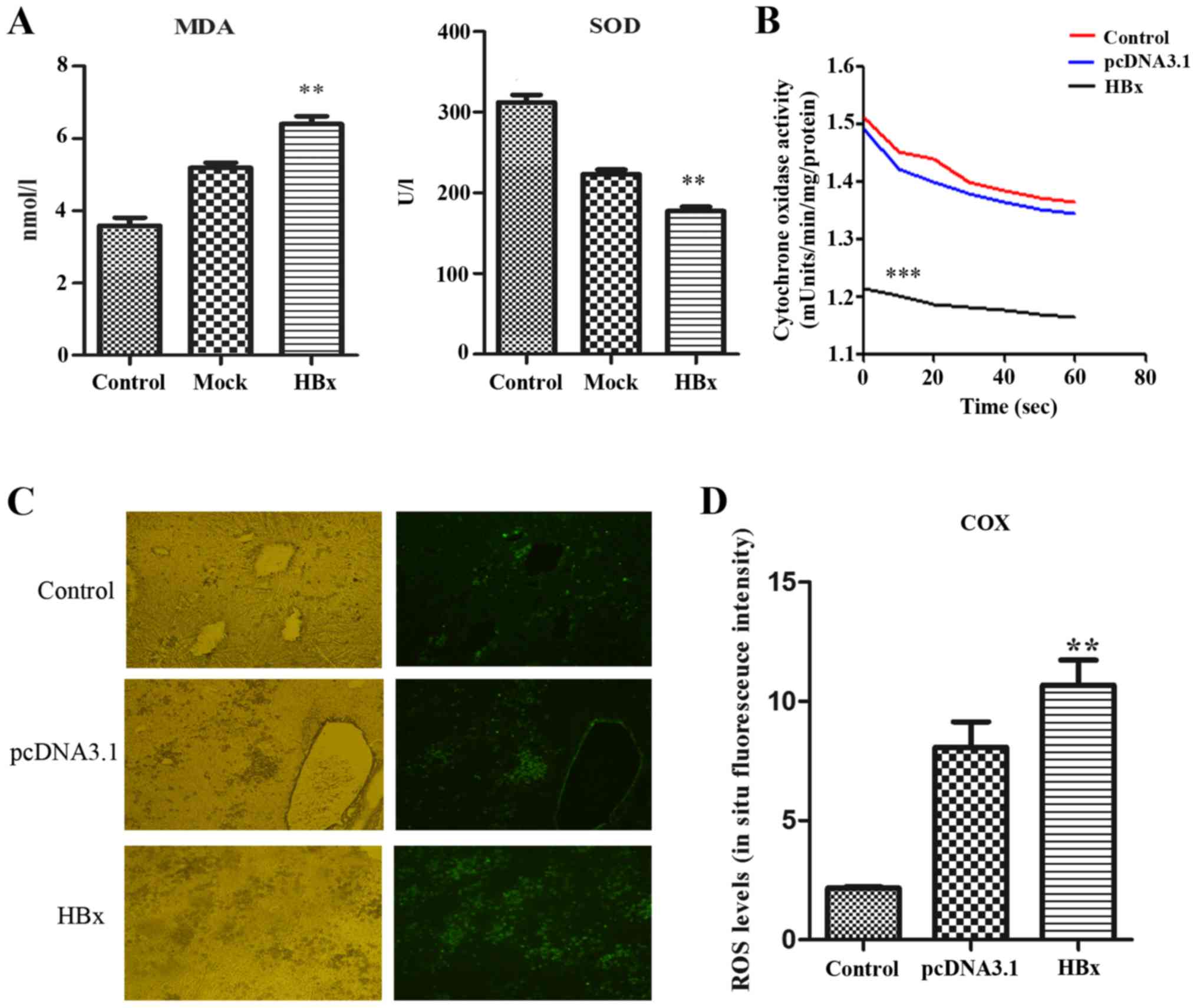
MDA levels in serum of mice in the experimental
group transfected with pcDNA3.1-HBx were significantly higher
compared with the control group and mock group, whereas SOD levels
were significantly lower compared with the control group and mock
group (P<0.01; Fig. 2A).
The detection of COX using enzymatic kinetics
demonstrated that COX activity in the experimental group
transfected with pcDNA3.1-HBx was significantly lower compared with
in the control group and mock group (P<0.001; Fig. 2B), indicating that COX activity was
reduced by HBx, which was in agreement with our previous
experimental results in HL-7702 cells stably expressing HBx
(15).
Oxidative stress was determined by measuring the ROS
levels in frozen sections using in situ fluorescence
staining. In the experimental HBx group, compared with the pcDNA3.1
plasmid-transfected group and the control group, the ROS levels in
the liver tissues were significantly increased (P<0.05; Fig. 2C and D).
ROS-mediated effects of HBx on NF-κB
and p-AKT expression in mouse hepatocytes
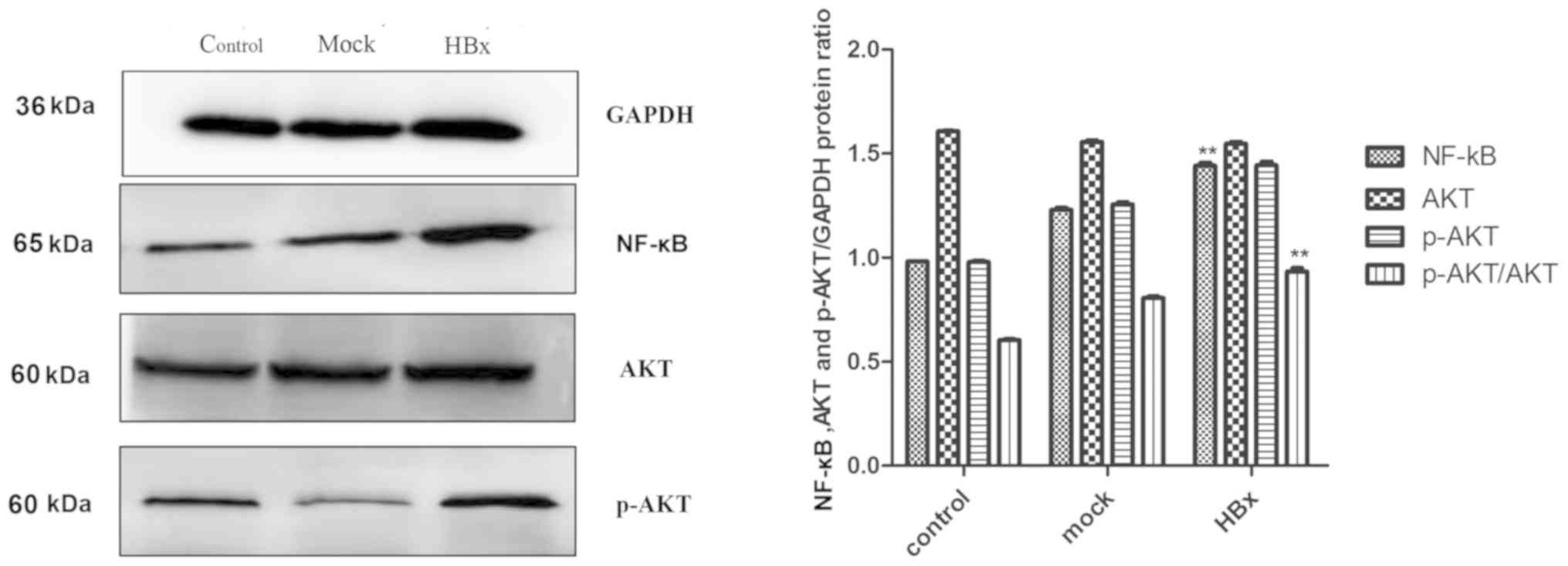
The results of western blot analysis demonstrated
that the expression levels of NF-κB and p-AKT were significantly
increased in the experimental HBx group, which was significant
compared with the empty plasmid group and the blank group
(P<0.01; Fig. 3).
Detection of HBx-induced inflammatory
damage in hepatocytes
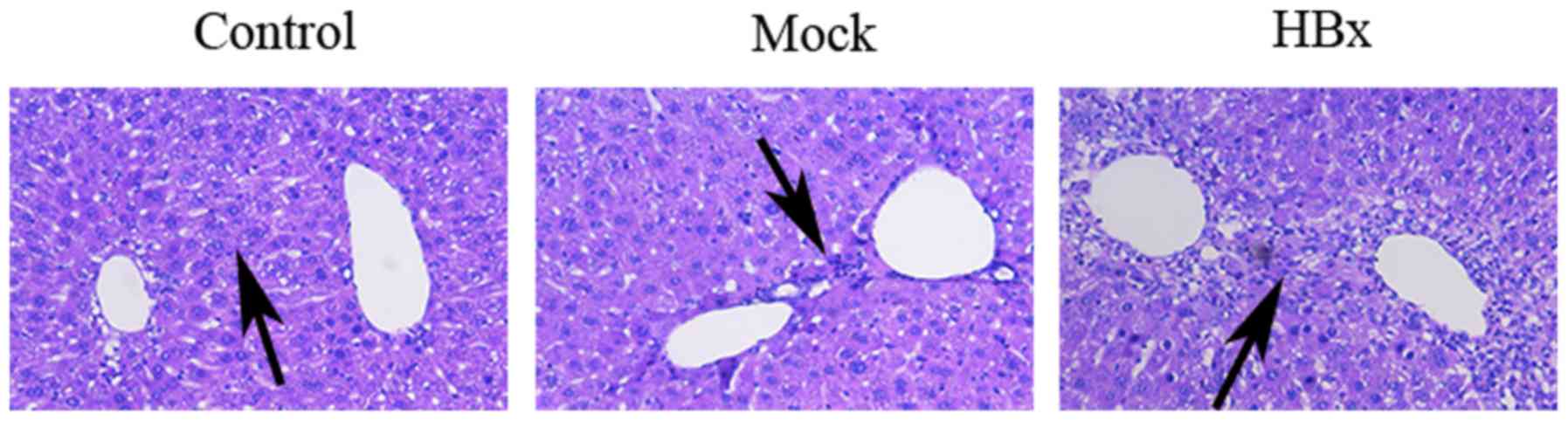
Edema and inflammatory cell infiltration were
observed in the liver tissue of the HBx group by HE staining
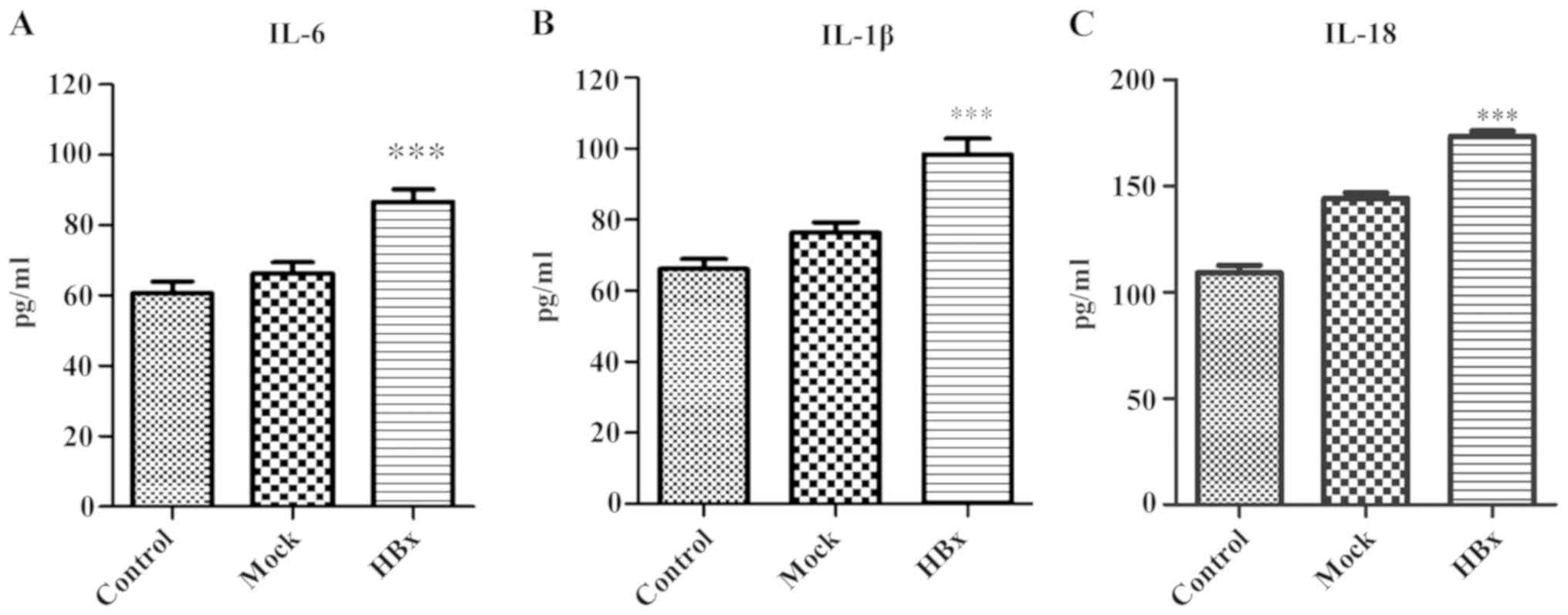
(Fig. 4). The serum levels of the
inflammatory cytokines IL-6, IL-1β and IL-18 in the experimental
group were significantly higher compared with the control group and
mock group (P<0.001; Fig. 5A-C).
This indicated that HBx may induce the synthesis and secretion of
IL-6, IL-1β and L-18, thus promoting inflammatory damage in liver
cells.
Discussion
Under normal physiological conditions, ROS levels
are in a stable state, and their production and clearance maintain
a certain dynamic balance. When the body is invaded by bacteria or
viruses, ROS are actively produced as part of the immune response.
Another major source of ROS production is the oxidative metabolism
of mitochondria. The increase in ROS levels can affect cell
signaling pathways and cell growth (34). In particular, ROS influences the
NF-κB signaling pathway and activates the MAPK and STAT3 signaling
pathways via the release of IL-1β, IL-6 and TNF-α (35–37).
However, excessive production of inflammatory cytokines induced by
HBx can stimulate cells to produce a large amount of ROS, further
stimulating NF-κB to produce inflammatory mediators (for example:
IL-1β, IL-6 and TNF-α), forming a cycle and thus accelerating the
inflammatory injury of liver cells and HBV replication (37).
Previous studies have demonstrated that the HBx
protein interacts with the mitochondrial COXIII subunit, causing
mitochondrial damage and affecting the biological activity of liver
cells (14–16). In the present study, HBx was
demonstrated to alter the mitochondrial oxidative respiratory chain
activity by reducing COX activity, that was in agreement with our
previous experimental results in HL-7702 cells stably expressing
HBx (15). This resulted in altered
intracellular ROS levels and increased expression levels of NF-κB
and p-AKT. Therefore, HBx may have increased NF-κB protein
expression levels, which subsequently promoted an increase in
hepatocyte ROS levels, thus damaging hepatocytes (37). HBx may also upregulate p-AKT protein
expression levels as a mechanism to minimize oxidative damage in
hepatocytes, although it was observed in this study that the
antioxidant effect of p-AKT was insufficient to reverse the liver
damage caused by HBx. However, HBx-induced upregulation of MAPK,
NF-κB and PI3K is considered an important factor in the development
of HCC (34,38,39), and
abnormal activation of NF-κB in liver cancer tissue has been
reported to inhibit apoptosis and promote liver cell proliferation,
contributing to cancer development (38).
Previous studies have demonstrated that the PI3K/AKT
pathway serves a role in HBx-induced HCC formation and the
activation of nuclear factor erythroid 2-related factor 2 (NRF2), a
key transcription factor regulating antioxidant genes during
oxidative stress and maintaining intracellular redox homeostasis
(40,41). In addition, SOD is a protease that
scavenges for excess oxygen free radicals, serving a role in
maintaining the generation and scavenging balance of oxygen free
radicals. Therefore, the decreased serum levels of SOD observed in
experimental mice of the present study supported the notion that
the antioxidant capacity in the HBx group was enhanced.
Previous studies have demonstrated that, under
oxidative stress, NRF2/ARE can induce the expression of cell
protective genes in HBV-positive cells (40,42).
Papaiahgari et al (40) have
confirmed that ROS activates NRF2 via the PI3K/AKT signaling
pathway. Increased ROS activates mitotic pathways through oxidative
inactivation of PTEN, a tumor suppressor that serves a role in AKT
dephosphorylation, therefore permitting AKT phosphorylation and
activation and accelerating HepG2 cell growth, which is closely
associated with the development of liver cancer in HBx transgenic
mice (43). The increased expression
of NF-κB and p-AKT in the experimental group of the present study
indicated that the NF-κB and AKT signaling pathways may serve a
role in ROS-mediated HBx-induced inflammatory hepatocyte injury.
The present study demonstrated that the expression levels of NF-κB
and p-AKT were increased in the experimental group; in addition,
the expression levels of NF-κB and p-AKT in liver cells were
significantly decreased following treatment with ROS inhibitors
(data not shown). These results suggested that HBx may activate
NF-κB and AKT signaling through multiple pathways mediated by ROS.
Therefore, ROS may serve an important role in HBx-induced
hepatocyte inflammatory injury.
During HBV infection, the immune response may lead
to liver cell injury as, HBx increases the expression of MHC-I and
MHC-II by activating MHC promoters and forming of HBx-MHC
antigen-peptide complexes, which ultimately activate cellular and
humoral immune responses, respectively (44). Lara-Pezzi et al (45,46) have
demonstrated that HBx upregulates the expression levels of TNF-α in
hepatocytes by activating the nuclear factor of activated T cells
in the cellular immune response. TNF-α mediates the activation of
CD8+ cytotoxic T cells, which eliminates infected cells
by releasing toxic particles and activating death receptor
pathways. In addition, CD8+ cytotoxic T lymphocytes may
damage the membranes of liver cells, which results in liver cell
injury and initiates apoptosis.
A previous study has demonstrated that HBx
stimulates the synthesis and secretion of IL-6, a major
pro-inflammatory cytokine, in a toll-like receptor adaptor protein
myeloid differentiation factor 88 (MYD88)-dependent manner
(47). Quétier et al
(48) reported that HBx transgenic
mice overexpressed IL-6, increased hepatic proliferation and
delaying hepatocyte regeneration. A possible explanation for this
may be that HBx also activates signaling proteins downstream of
MYD88, including NF-κB, which was upregulated in the experimental
group of the present study and may have contributed to hepatocyte
injury (47).
HBx selectively regulates other pro-inflammatory
cytokines, such as IL-1β, IL-18 and IL-23, and participates in the
regulation of immune cell interactions (49,50).
These cytokines serve a role in the development and progression of
liver cancer. IL-1β promotes neutrophil migration to the liver,
phagocytosis and pathogen elimination, regulates tumor growth and
is associated with the invasion and metastasis of liver cancer
(51,52). In addition, Chen et al
(38) have demonstrated that HBx
increases IL-1 secretion and induces NF-kB activation by
interacting with an evolutionarily-conserved signaling intermediate
in the Toll pathway. IL-18 is a pro-inflammatory cytokine that
mediates the inflammatory cascade reaction and is a factor in acute
liver injury (53). IL-18 levels in
the sera of patients with hepatocellular carcinoma are
significantly increased, which suggests that HBx may promote the
occurrence and development of hepatocellular carcinoma by
regulating IL-18 (53,54). Overall, these studies suggest that
pro-inflammatory cytokines serve a role in HBx-induced hepatic
inflammatory injury.
In the present study, H&E staining of liver
tissue and the levels of IL-6, IL-1β and IL-18 in the sera of the
experimental group demonstrated that edema and inflammatory cell
infiltration had occurred in the central part of the portal tissue.
The levels of IL-6, IL-1β and IL-18 were also significantly higher
compared with the control group. In addition, HBx increased the
expression levels of TNF-α, receptor-interacting protein kinase
(RIP)3 mRNA and RIP3 protein (date not shown). This was consistent
with the results from a previous study, which demonstrated that
TNF-α activates RIP3, inducing programmed necrosis of cells and
aggravating cellular inflammatory responses (data not shown).
In summary, the present study demonstrated that HBx
upregulated the expression levels of IL-6, IL-1β IL-18, NF-kB and
p-AKT, increased the level of oxidative stress and ultimately
contributed to an inflammatory microenvironment in the liver. HBx
reduced the activity of COX and may affect mitochondrial
respiration in liver cells, resulting in mitochondrial dysfunction
and subsequent inflammatory damage of liver cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Fujian
Natural Science Foundation (grant no. 2018J01314), The Fujian
Province Health and Family Planning Research Talent Training
Project (grant no. 2018-CX-21) and The Joint Funds for the
Innovation of Science and Technology (grant no. 2017Y9048).
Availability of data and materials
The datasets used and/or analyzed during the current
study will be provided by the corresponding author on reasonable
request.
Authors' contributions
DL designed the experiments. LL and DZ performed the
experiments and wrote the manuscript. ZZ, WX, ZC, YH and XZ
analyzed the experimental results.
Ethics approval and consent to
participate
The present study was approved by The Institutional
Animal Ethics Committee of Fujian University of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Martín-Vílchez S, Moreno-Otero R and
Sanz-Cameno P; Grupo CIBERehd de Hepatología del Hospital
Universitario de La Princesa, : Effects of hepatitis B virus X
protein on chronic hepatitis B pathophysiology. Med Clin (Barc).
140:508–513. 2012.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahmani Z, Huh KW, Lasher R and Siddiqui
A: Hepatitis B virus X protein colocalizes to mitochondria with a
human voltage-dependent anion channel, HVDAC3, and alters its
transmembrane potential. J Virol. 74:2840–2846. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee YI, Hwang JM, Im JH, Lee YI, Kim NS,
Kim DG, Yu DY, Moon HB and Park SK: Human hepatitis B virus-X
protein alters mitochondrial function and physiology in human liver
cells. J Biol Chem. 279:15460–15471. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tan C, Guo H, Zheng M, Chen Y and Huang W:
Involvement of mitochondrial permeability transition in hepatitis B
virus replication. Virus Res. 145:307–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shirakata Y and Koike K: Hepatitis B virus
X protein induces cell death by causing loss of mitochondrial
membrane potential. J Biol Chem. 278:22071–22078. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gearhart TL and Bouchard MJ: Replication
of the hepatitis B virus requires a calcium-dependent HBx-induced
G1 phase arrest of hepatocytes. Virology. 407:14–25. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim SJ, Khan M, Quan J, Till A, Subramani
S and Siddiqui A: Hepatitis B virus disrupts mitochondrial
dynamics: Induces fission and mitophagy to attenuate apoptosis.
PLoS Pathog. 9:e10037222013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao B, Cao JF, Hu GJ, Chen ZW, Wang LY,
Shangguan XX, Wang LJ, Mao YB, Zhang TZ, Wendel JF and Chen XY:
Core cis-element variation confers subgenome-biased expression of a
transcription factor that functions in cotton fiber elongation. New
Phytol. 218:1061–1075. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sato N: Central role of mitochondria in
metabolic regulation of liver pathophysiology. J Gastroenterol
Hepatol. 22 (Suppl 1):S1–S6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Varanasi L and Hosler JP: Subunit
III-depleted cytochrome c oxidase provides insight into the
process of proton uptake by proteins. Biochim Biophys Acta.
1817:545–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hosler JP: The influence of subunit III of
cytochrome c oxidase on the D pathway, the proton exit
pathway and mechanism-based inactivation in subunit I. Biochim
Biophys Acta. 1655:332–339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jung SY and Kim YJ: C-terminal region of
HBx is crucial for mitochondrial DNA damage. Cancer Lett.
331:76–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Clippinger AJ and Bouchard MJ: Hepatitis B
virus HBx protein localizes to mitochondria in primary rat
hepatocytes and modulates mitochondrial membrane potential. J
Virol. 82:6798–6811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li D, Ding J, Chen Z, Chen Y, Lin N, Chen
F and Wang X: Accurately mapping the location of the binding site
for the interaction between hepatitis B virus X protein and
cytochrome c oxidase III. Int J Mol Med. 35:319–324. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zou LY, Zheng BY, Fang XF, Li D, Huang YH,
Chen ZX, Zhou LY and Wang XZ: HBx co-localizes with COXIII in
HL-7702 cells to upregulate mitochondrial function and ROS
generation. Oncol Rep. 33:2461–2467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng BY, Fang XF, Zou LY, Huang YH, Chen
ZX, Li D, Zhou LY, Chen H and Wang XZ: The co-localization of HBx
and COXIII upregulates COX-2 promoting HepG2 cell growth. Int J
Oncol. 45:1143–1150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jaeschke H: Reactive oxygen and mechanisms
of inflammatory liver injury: Present concepts. J Gastroenterol
Hepatol. 26 (Suppl 1):S173–S179. 2011. View Article : Google Scholar
|
|
18
|
Wang JH, Yun C, Kim S, Lee JH, Yoon G, Lee
MO and Cho H: Reactive oxygen species modulates the intracellular
level of HBx viral oncoprotein. Biochem Biophys Res Commun.
310:32–39. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang B, Tang F, Wang Z, Qi G, Liang X, Li
B, Yuan S, Liu J, Yu S and He S: Upregulation of
Akt/NF-κB-regulated inflammation and Akt/Bad-related apoptosis
signaling pathway involved in hepatic carcinoma process:
Suppression by carnosic acid nanoparticle. Int J Nanomedicine.
11:6401–6420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo LH, Li DM, Wang YL, Wang K, Gao LX, Li
S, Yang JG, Li CL, Feng W and Guo H: Tim3/galectin-9 alleviates the
inflammation of TAO patients via suppressing Akt/NF-kB signaling
pathway. Biochem Biophys Res Commun. 491:966–972. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Czaja MJ: Cell signaling in oxidative
stress-induced liver injury. Semin Liver Dis. 27:378–389. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu M, Guo J, Li W, Xia H, Lu Y, Dong X,
Chen Y, Xie X, Fu S and Li M: HBx induced AFP receptor expressed to
activate PI3K/AKT signal to promote expression of Src in liver
cells and hepatoma cells. BMC Cancer. 15:3622015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Esmaeili MA, Farimani MM and Kiaei M:
Anticancer effect of calycopterin via PI3K/Akt and MAPK signaling
pathways, ROS-mediated pathway and mitochondrial dysfunction in
hepatoblastoma cancer (HepG2) cells. Mol Cell Biochem. 397:17–31.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wong VW, Yu J, Cheng AS, Wong GL, Chan HY,
Chu ES, Ng EK, Chan FK, Sung JJ and Chan HL: High serum
interleukin-6 level predicts future hepatocellular carcinoma
development in patients with chronic hepatitis B. Int J Cancer.
124:2766–2770. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang YS, Hwang SJ, Chan CY, Wu JC, Chao
Y, Chang FY and Lee SD: Serum levels of cytokines in hepatitis
C-related liver disease: A longitudinal study. Zhonghua Yi Xue Za
Zhi (Taipei). 62:327–333. 1999.PubMed/NCBI
|
|
26
|
Kakumu S, Okumura A, Ishikawa T, Yano M,
Enomoto A, Nishimura H, Yoshioka K and Yoshika Y: Serum levels of
IL-10, IL-15 and soluble tumour necrosis factor-alpha (TNF-alpha)
receptors in type C chronic liver disease. Clin Exp Immunol.
109:458–463. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee MO, Choi YH, Shin EC, Kang HJ, Kim YM,
Jeong SY, Seong JK, Yu DY, Cho H, Park JH and Kim SJ: Hepatitis B
virus X protein induced expression of interleukin 18 (IL-18): A
potential mechanism for liver injury caused by hepatitis B virus
(HBV) infection. J Hepatol. 37:380–386. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu L, Wang C, Zhang Q, Yan H, Li Y, Pan J
and Tang Z: Mitochondrial protein profile in mice with low or
excessive selenium diets. Int J Mol Sci. 17(pii): E11372016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong L, Wang X, Wu J and Cai W:
Mitochondria-initiated apoptosis triggered by oxidative injury play
a role in total parenteral nutrition-associated liver dysfunction
in infant rabbit model. J Pediatr Surg. 44:1712–1718. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Paradies G, Ruggiero FM, Petrosillo G and
Quagliariello E: Peroxidative damage to cardiac mitochondria:
Cytochrome oxidase and cardiolipin alterations. FEBS Lett.
424:155–158. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu F, Song Y K and Liu D:
Hydrodynamics-based transfection in animals by systemic
administration of plasmid DNA. Gene Ther. 6:1258–1266. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang PL, Althage A, Chung J and Chisari
FV: Hydrodynamic injection of viral DNA: A mouse model of acute
hepatitis B virus infection. Proc Natl Acad Sci USA.
99:13825–13830. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo YJ, Wang W, Sun SH, Zeng DB, Zhao GY,
Yu H, Guo Y, Tan WJ, Lu SC and Zhou YS: Immunosuppressant
dexamethasone can significantly extend the expression of hepatitis
B virus antigens in the HBV mouse model by hydrodynamic
transfection method. Bing Du Xue Bao. 26:20–26. 2010.(In Chinese).
PubMed/NCBI
|
|
34
|
Tien Kuo M and Savaraj N: Roles of
reactive oxygen species in hepatocarcinogenesis and drug resistance
gene expression in liver cancers. Mol Carcinog. 45:701–709. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cardin R, Piciocchi M, Bortolami M,
Kotsafti A, Barzon L, Lavezzo E, Sinigaglia A, Rodriguez-Castro KI,
Rugge M and Farinati F: Oxidative damage in the progression of
chronic liver disease to hepatocellular carcinoma: An intricate
pathway. World J Gastroenterol. 20:3078–3086. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heid ME, Keyel PA, Kamga C, Shiva S,
Watkins SC and Salter RD: Mitochondrial reactive oxygen species
induces NLRP3-dependent lysosomal damage and inflammasome
activation. J Immunol. 191:5230–5238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Waris G, Huh KW and Siddiqui A:
Mitochondrially associated hepatitis B virus X protein
constitutively activates transcription factors STAT-3 and NF-kappa
B via oxidative stress. Mol Cell Biol. 21:7721–7730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen WN, Liu LL, Jiao BY, Lin WS, Lin XJ
and Lin X: Hepatitis B virus X protein increases the IL-1β-induced
NF-κB activation via interaction with evolutionarily conserved
signaling intermediate in Toll pathways (ECSIT). Virus Res.
195:236–245. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Papaiahgari S, Yerrapureddy A, Hassoun PM,
Garcia JG, Birukov KG and Reddy SP: EGFR-activated signaling and
actin remodeling regulate cyclic stretch-induced NRF2-ARE
activation. Am J Respir Cell Mol Biol. 36:304–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY,
Shim JH, Cho MK, Nam HS and Lee SH: Reactive oxygen species and
PI3K/Akt signaling play key roles in the induction of Nrf2-driven
heme oxygenase-1 expression in sulforaphane-treated human
mesothelioma MSTO-211H cells. Food Chem Toxicol. 50:116–123. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang P, Gao YM, Sun X, Guo N, Li J, Wang
W, Yao LP and Fu YJ: Hepatoprotective effect of 2′-O-galloylhyperin
against oxidative stress-induced liver damage through induction of
Nrf2/ARE-mediated antioxidant pathway. Food Chem Toxicol.
102:129–142. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ha HL and Yu DY: HBx-induced reactive
oxygen species activates hepatocellular carcinogenesis via
dysregulation of PTEN/Akt pathway. World J Gastroenterol.
16:4932–4937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Su Q, Schröder CH, Hofmann WJ, Otto G,
Pichlmayr R and Bannasch P: Expression of hepatitis B virus X
protein in HBV-infected human livers and hepatocellular carcinomas.
Hepatology. 27:1109–1120. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lara-Pezzi E, Armesilla AL, Majano PL,
Redondo JM and López-Cabrera M: The hepatitis B virus X protein
activates nuclear factor of activated T cells (NF-AT) by a
cyclosporin A-sensitive pathway. EMBO J. 17:7066–7077. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lara-Pezzi E, Majano PL, Gómez-Gonzalo M,
García-Monzón C, Moreno-Otero R, Levrero M and López-Cabrera M: The
hepatitis B virus X protein up-regulates tumor necrosis factor
alpha gene expression in hepatocytes. Hepatology. 28:1013–1021.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xiang WQ, Feng WF, Ke W, Sun Z, Chen Z and
Liu W: Hepatitis B virus X protein stimulates IL-6 expression in
hepatocytes via a MyD88-dependent pathway. J Hepatol. 54:26–33.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Quétier I, Brezillon N, Duriez M, Massinet
H, Giang E, Ahodantin J, Lamant C, Brunelle MN, Soussan P and
Kremsdorf D: Hepatitis B virus HBx protein impairs liver
regeneration through enhanced expression of IL-6 in transgenic
mice. J Hepatol. 59:285–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang DY, Zou LP, Liu XJ, Zhu HG and Zhu R:
Chemokine expression profiles of human hepatoma cell lines mediated
by hepatitis B virus X protein. Pathol Oncol Res. 22:393–399. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia L, Tian D, Huang W, Zhu H, Wang J,
Zhang Y, Hu H, Nie Y, Fan D and Wu K: Upregulation of IL-23
expression in patients with chronic hepatitis B is mediated by the
HBx/ERK/NF-κB pathway. J Immunol. 188:753–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ehling J and Tacke F: Role of chemokine
pathways in hepatobiliary cancer. Cancer Lett. 379:173–183. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li XP, Yang XY, Biskup E, Zhou J, Li HL,
Wu YF, Chen ML and Xu F: Co-expression of CXCL8 and HIF-1α is
associated with metastasis and poor prognosis in hepatocellular
carcinoma. Oncotarget. 6:22880–22889. 2015.PubMed/NCBI
|
|
53
|
Tangkijvanich P, Thong-Ngam D, Mahachai V,
Theamboonlers A and Poovorawan Y: Role of serum interleukin-18 as a
prognostic factor in patients with hepatocellular carcinoma. World
J Gastroenterol. 13:4345–4349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mohran ZY, Ali-Eldin FA and Abdel Aal HA:
Serum interleukin-18: Does it have a role in the diagnosis of
hepatitis C virus related hepatocellular carcinoma? Arab J
Gastroenterol. 12:29–33. 2011. View Article : Google Scholar : PubMed/NCBI
|