Introduction
Despite considerable progress in cancer diagnosis
and treatment, 18.1 million new cancer cases were detected and 9.6
million cancer-associated deaths occurred worldwide in 2018
(1). The therapeutic strategies for
treating numerous types of cancer are usually ineffective,
primarily due to the lack of specific diagnostic and prognostic
markers (2). Diagnostic biomarkers
could be used for screening or early detection of cancer, while
prognostic biomarkers could be used to predict the likelihood of
recurrence or progression in patients with cancer (2). Accordingly, it is necessary to identify
novel biomarkers for potential clinical applications (3–5).
Long non-coding RNAs (lncRNAs) have crucial roles in
the progression of cancer (6–10).
Certain lncRNAs are involved in the modulation of neoplastic
proliferation, invasion and metastasis. In addition, several
studies have indicated that lncRNAs may serve as potential
cancer-specific biomarkers (2,11–13).
The gene encoding long intergenic non-protein-coding
RNA 1614 (LINC01614), also known as lung cancer-associated lncRNA
4, is located on chromosome 2q35 in between two exons and was
originally identified as being upregulated in lung cancer (14). In addition, Liu et al
(15) reported that LINC01614
knockdown inhibits cell proliferation through the microRNA
(miR)-217/forkhead box (FOX)P1 axis in lung adenocarcinoma. Another
study indicated that LINC01614 is highly expressed in breast cancer
tissue and associated with poor prognosis based on the data from
The Cancer Genome Atlas (TCGA) (16). Increasing evidence has indicated that
LINC01614 may act as an oncogene and serve as a biomarker in
several types of cancer (15–18).
However, beyond the limited information provided by these studies,
the role of LINC01614 in malignant tumors has remained elusive.
In the present study, a comprehensive pan-cancer
analysis of the data available from public databases was performed
to determine the correlation between LINC01614 expression and the
prognoses of various malignancies. In addition, bioinformatics
analyses were performed to identify the physiological functions of
this lncRNA to clarify its potential role as an oncogene.
Furthermore, the expression levels in paired normal and tumor
tissue specimens from patients with non-small cell lung cancer
(NSCLC) or colon cancer were assessed to validate the results.
Materials and methods
Malignancy-associated microarray and
RNA sequencing data in the Gene Expression Omnibus (GEO) and TCGA
databases
Microarray and RNA sequencing data on LINC01614
expression were systematically searched in the GEO database on 26th
June 2019 (http://www.ncbi.nlm.nih.gov/geo/). The search terms
were as follows: (neoplasm OR cancer OR tumor OR carcinoma) AND
(lncRNA OR long non-coding RNA). The inclusion criteria were as
follows: i) Expression data for LINC01614 were provided or it was
possible to calculate them for each sample; ii) samples in each
dataset included cancer tissues and normal tissues or cancer
tissues with prognostic information; iii) the number of samples in
each dataset was >20; and iv) the species in each dataset was
Homo sapiens. Samples based on cell lines were excluded.
Gene expression matrices were downloaded from the GEO database and
extracted using R and associated packages.
Datasets including RNA sequencing and clinical data
for 24 types of cancer from TCGA were downloaded and extracted
using the TCGA biolinks package (https://gdc-portal.nci.nih.gov/). RNA sequencing
(RNA-seq) expression data were normalized using RNA-seq by
Expectation-Maximization (19).
Comparison of LINC01614 expression in
normal and tumor tissues
LINC01614 expression in normal tissues was examined
from the RNA-seq data obtained from the Genotype-Tissue Expression
(GTEx) (https://www.gtexportal.org/home/) database, comprising
~11,600 samples from 53 tissue types. The pre-processed data were
downloaded from the portal and converted to the units of
transcripts per million (TPM) for comparison of relative expression
levels among different tissues. The RNA-seq data from TCGA for
libraries with sufficient malignant and adjacent normal tissues
were also used to evaluate the expression of LINC01614 in
malignancies. The RNA-seq expression matrices of these datasets
were normalized by log2(x+1) transformation. The relative
expression levels were statistically compared between the tumor
tissues and adjacent normal tissues using unpaired Student's
t-tests.
Receiver operating characteristic
(ROC) and summary ROC (SROC) curve analysis
Datasets with sufficient malignant and adjacent
normal tissues were used to evaluate the diagnostic value of
LINC01614 in malignancies. ROC curve analysis was performed to
evaluate the area under the curve (AUC) with 95% CIs, sensitivities
and specificities for each dataset. SROC curve analysis was
performed using Stata 14.2 (StataCorp LLC) to demonstrate pooled
sensitivity, pooled specificity and obtain the pooled AUC.
I2 and Q tests were used to assess the heterogeneity of
this meta-analysis. Meta-regression analysis was performed to
determine the potential cause of heterogeneity. Deeks' funnel plots
were used to evaluate the potential publication bias of the SROC
analysis.
Survival analysis and comprehensive
meta-analysis
The above-mentioned GEO data with survival
information and TCGA data were used to assess the prognostic value
of LINC01614. Samples in each dataset were divided into two groups
according to the cut-off value determined from ROC curves (20). Univariate Cox regression analysis was
performed to obtain hazard ratios (HRs) with 95% CIs for overall
survival (OS). Pooled HRs and 95% CIs were combined to assess the
association between LINC01614 expression and prognosis in various
malignancies. If the 95% CI of the combined HR did not overlap with
1, the results were considered significant. If I2>50%
or P≤0.05, the heterogeneity was considered significant and a
random-effects model was chosen; if not, a fixed-effects model was
used. A sensitivity analysis was also performed to assess the
stability of the combined results and a subgroup analysis was
performed to detect possible sources of heterogeneity. Begg's test
and the trim fill method were used to assess the potential
publication bias.
Tissue samples and clinical data
collection
For validation of the RNA-seq data, 74 and 78 tumor
tissue samples (alongside matched adjacent normal tissue samples)
from patients with non-small cell lung cancer (NSCLC) or colon
cancer, respectively, who underwent resection at Beijing Tongren
Hospital (Beijing, China) between October 2016 and March 2018, were
analyzed. The tissue samples were immersed in RNA later (Ambion)
and stored at −80°C until use. The clinicopathological
characteristics of each patient were obtained from their medical
records. The study was approved by the ethics committee of Beijing
Tongren Hospital (Beijing, China).
RNA extraction and RT-qPCR
Total RNA was extracted from the tissue samples with
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The RNA integrity was
detected using 1% agarose gel electrophoresis and a
spectrophotometer was used to measure its concentration and purity.
The 28S:18S ribosomal RNA ratio of RNA samples was required to be
~2:1; the A260/A280 ratios were 1.8–2.2 and the absorbance at 260
nm (A260)/A230 ratios were required to be >1.7 for the RNA
samples to be considered qualified. RNA was used to synthesize cDNA
using the SuperScript III Reverse Transcriptase kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol; the
mixture included 1 µg total RNA, 1 µl oligo(dT)20, 1 µl
dNTP Mix, 1 µl 0.1M DTT, 1 µl RNaseOUT, 1 µl SuperScript™ III RT
and enzyme-free water in a 20 µl reaction volume. The reaction
conditions used were 50°C for 60 min and 70°C for 15 min. Reverse
transcription-quantitative (RT-q)PCR was performed using the
SYBR-Green Mix (Takara Bio, Inc.) on an ABI 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: Pre-denaturation at
94°C for 2 min, 40 cycles of denaturation at 94°C for 5 sec and
annealing at 60°C for 30 sec, and a final extension step at 72°C
for 10 min. The expression levels were normalized to those of GAPDH
mRNA (16) using the
2−Δ∆Cq method (21),
where ∆Cq=(Cq of the target-Cq GAPDH) and ∆∆Cq=∆Cq tumor tissues -
∆Cq adjacent non-tumor tissues. Each sample was analyzed in
triplicate. The sequences of the primers used in the study were as
follows: LINC01614 forward, 5′-GGGACTTCAGACACGGAGAA-3′ and reverse,
5′-GGACACAGACCCTAGCACTT-3′; collagen XI α1 chain (COL11A1) forward,
5′-TAACATCGCTGACGGGAAGTG-3′ and reverse,
5′-CCGTGATTCCATTGGTATCAACA-3′; secreted protein acidic and cysteine
rich (SPARC) forward, 5′-CCCATTGGCGAGTTTGAGAAG-3′ and reverse,
5′-CAAGGCCCGATGTAGTCCA-3′; periostin (POSTN) forward,
5′-CAACGGGCAAATACTGGAAAC-3′ and reverse,
5′-TCTCGCGGAATATGTGAATCG-3′; COL5A2 forward,
5′-ACAGGGTTTACAAGGACAGCA-3′ and reverse, 5′-GGTCCAGGATCACCAGGTT-3′;
fibroblast activation protein α (FAP) forward,
5′-TCAGCTATGATGCCATTTCG-3′ and reverse, 5′-CCTCCCACTTGCCACTTGTA-3′;
and GAPDH forward, 5′-CAACTCCCTCAAGATTGTCAGCAA-3′ and reverse,
5′-GGCATGGACTGTGGTCATGA-3′.
Bioinformatics analysis of
LINC01614
To investigate the molecular pathways associated
with the upregulation of LINC01614 in malignancies, gene set
enrichment analysis (GSEA) was performed using the GSEA software
3.0 (http://software.broadinstitute.org/gsea/msigdb/index.jsp)
(22). Annotated gene sets
‘c2.cp.kegg.v6.2.symbols.gmt’ and ‘h.all.v6.2.symbols.gmt’ were
downloaded from the Molecular Signatures Database (23). The normalized enrichment score was
determined by the analysis of 1,000 permutations. A gene set was
considered significantly enriched when the false discovery rate was
<0.25. In addition, Pearson's correlation coefficient was
determined to identify the genes co-expressed with LINC01614. Genes
with Pearson's r-values >0.3 and P<0.01 were considered
significant.
Statistical analysis
Microarray and RNA-seq data were analyzed using R,
version 3.5.2, and associated packages. An unpaired Student's
t-test, the χ2 test, ROC curve analysis and univariate
Cox regression analysis were performed using SPSS Statistics v22.0
(IBM Corp.). Diagnostic and prognostic meta-analyses were conducted
using Stata 14.2 (StataCorp LLC). The measurement data were
expressed as the mean ± SD. Student's t-test and the χ2
test were used to analyze the significance of differences between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
LINC01614 expression is high in most
tumor tissues but low in most normal tissues
To determine the general distribution of LINC01614
in normal tissues, its expression was first examined across 53
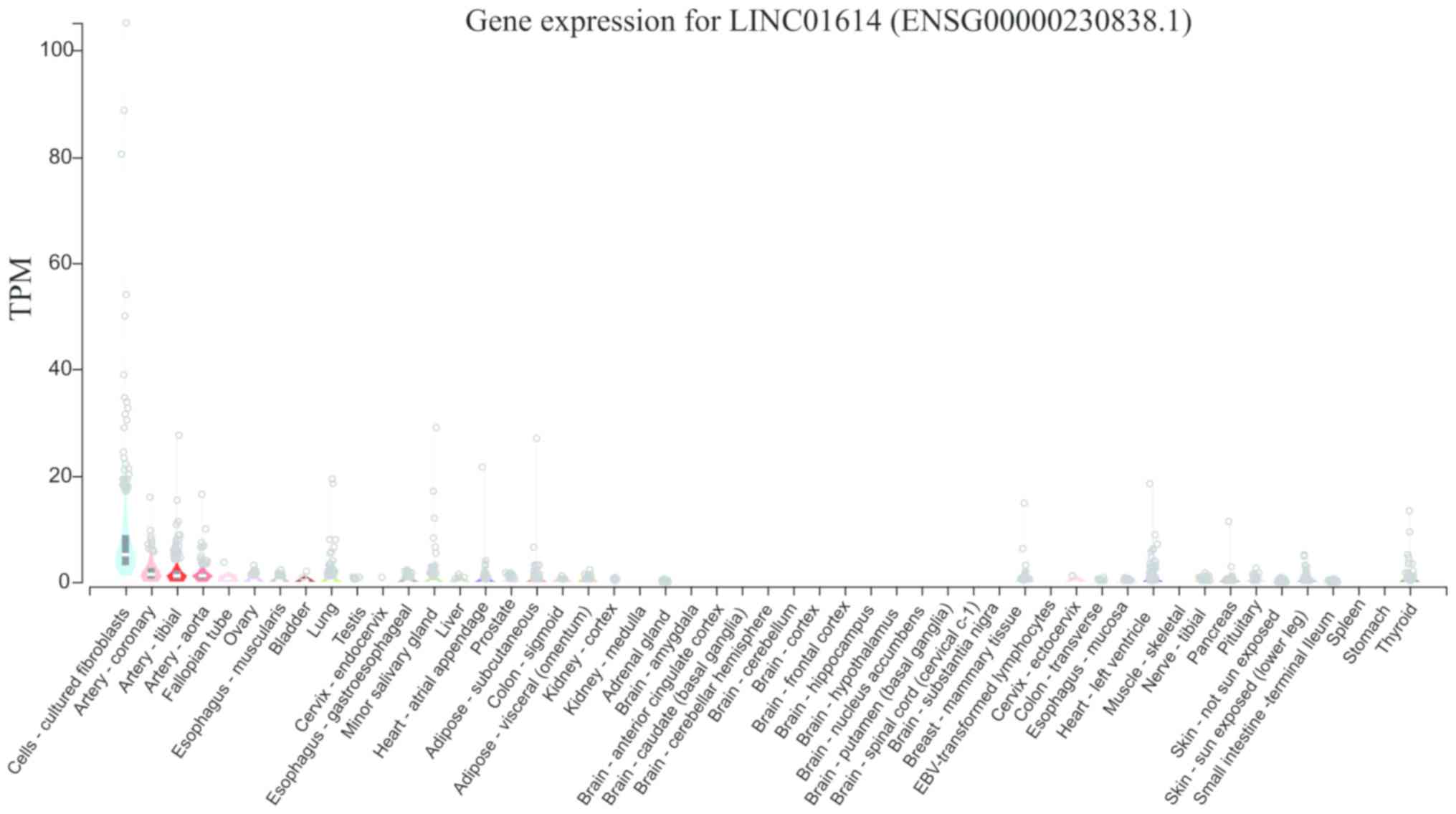
different tissues in the GTEx database. As presented in Fig. 1, the median expression levels of
LINC01614 in different tissue types ranged from a TPM of 0 (vagina)
to 5.213 (cultured fibroblasts). Except for the cultured
fibroblasts (median TPM=5.213), coronary artery (median TPM=1.496),
tibial artery (median TPM=1.012) and aorta (median TPM=1.009), all
of which exhibited relatively high levels of LINC01614, most of the
other tissues had low levels (median TPM <0.5).
After excluding the datasets that had an
insufficient amount of normal tissue samples, 18 types of cancers
with 7,043 samples from TCGA were selected to evaluate the
expression patterns of LINC01614 in cancers. As presented in
Fig. 2, most tumors (16 of them) had
higher LINC01614 levels than the normal tissues. Unpaired Student's
t-tests indicated that LINC01614 was significantly upregulated in
15 types of malignant tissues, including bladder carcinoma (BLCA),
breast invasive carcinoma (BRCA), cholangiocarcinoma, colon
adenocarcinoma, esophageal carcinoma, glioblastoma multiforme, head
and neck squamous cell carcinoma (HNSC), kidney renal clear cell
carcinoma (KIRC), kidney renal papillary cell carcinoma, liver
hepatocellular carcinoma, lung adenocarcinoma (LUAD), lung squamous
cell carcinoma (LUSC), rectum adenocarcinoma (READ), stomach
adenocarcinoma (STAD) and thyroid carcinoma (P<0.05).
Furthermore, the expression levels of LINC01614 were relatively low
in malignant prostate adenocarcinoma (PRAD) and uterine corpus
endometrial carcinoma (UCEC) tissues but not significantly
different from the levels in the adjacent normal tissues
(P>0.05). Other normal tissue types had low levels of LINC01614
when it was highly expressed in the adjacent tumor tissues.
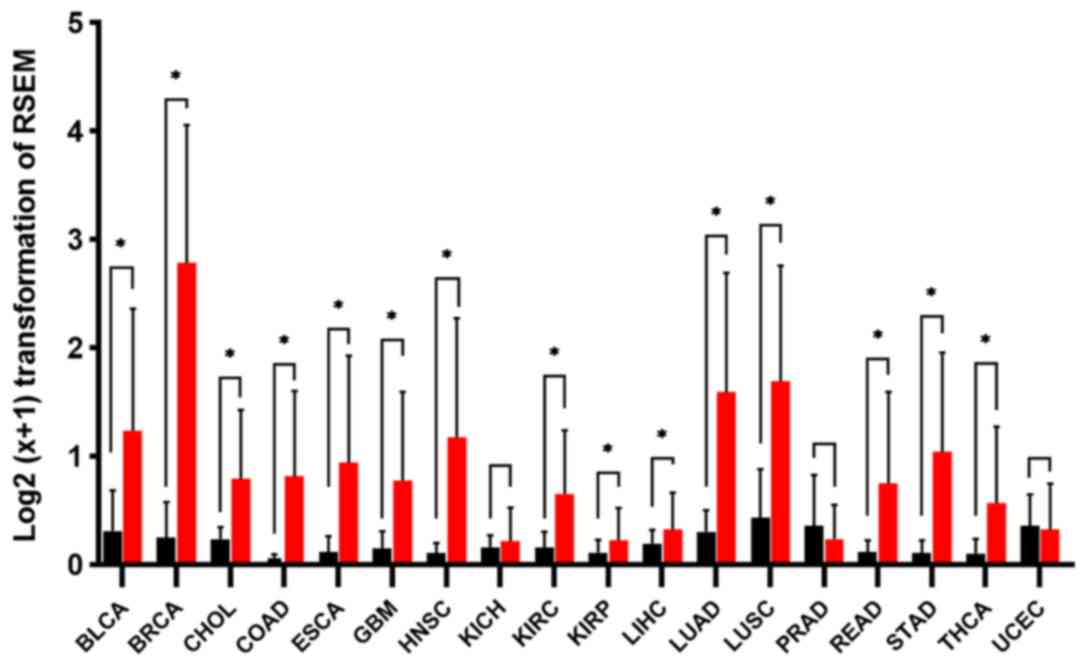 | Figure 2.Expression of long intergenic
non-coding RNA 1614 in malignancies based on The Cancer Genome
Atlas data. Significance was determined using an unpaired Student's
t-test. Values are expressed as the mean ± SD. *P<0.05. RSEM,
RNA-sequencing Expectation-Maximization; BLCA, bladder urothelial
carcinoma; BRCA, breast invasive carcinoma; CHOL,
cholangiocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal
carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck
squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney
renal clear cell carcinoma; KIRP, kidney renal papillary cell
carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung
adenocarcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate
adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach
adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus
endometrial carcinoma. |
LINC01614 may serve as a diagnostic
predictor based on GEO and TCGA databases
In total, 590 datasets were identified during the
primary search, among which 39 were downloaded later from the GEO
database after relevant assessments and evaluations. Eventually,
eight datasets were included in the analysis (GSE70880, GSE115856,
GSE61763, GSE103909, GSE104836, GSE115018, GSE53622 and GSE53624)
after screening in accordance with the inclusion criteria. Finally,
after excluding the datasets that had an insufficient amount of
normal tissue samples, 24 datasets, including 7,320 patients, from
GEO and TCGA databases were used to assess the diagnostic
predictive ability of LINC01614. The AUCs, sensitivities and
specificities of these datasets are presented in Table I. The results revealed that P<0.05
for the ROC curve in 20/27 cohorts, which indicated that LINC01614
was a suitable diagnostic marker for numerous human malignancies.
The combined sensitivities and specificities of LINC01614 for the
diagnosis of malignancies are presented in Fig. 3. Subsequently, the SROC curve
analysis was performed to determine the diagnostic value of
LINC01614 in malignancies. The combined sensitivity, specificity
and AUC of the SROC curve were 0.80 (95% CI, 0.71–0.86), 0.85 (95%
CI, 0.77–0.90) and 0.89 (95% CI, 0.86–0.92), respectively (Fig. 3A and C). The combined results
indicated that LINC01614 was suitable to diagnose human
malignancies, even though there was a marked heterogeneity among
the results. Deeks' funnel plots indicated that there was no
significant publication bias in the SROC curve analysis (P=0.22;
Fig. 3D). Thus, a meta-regression
analysis was performed to determine the cause of heterogeneity. The
results indicated that cancers of the reproductive (P<0.001) and
urinary system (P=0.01) may be a major cause of heterogeneity
(Fig. 3B). Of note, UCEC, KICH and
PRAD, which exhibited no significant differences in LINC0164
expression between tumor and normal tissues, are all cancers of the
reproductive or urinary system. Therefore, these results indicated
that except for certain cancers of these systems, LINC01614 may be
a potential diagnostic biomarker for the majority of
malignancies.
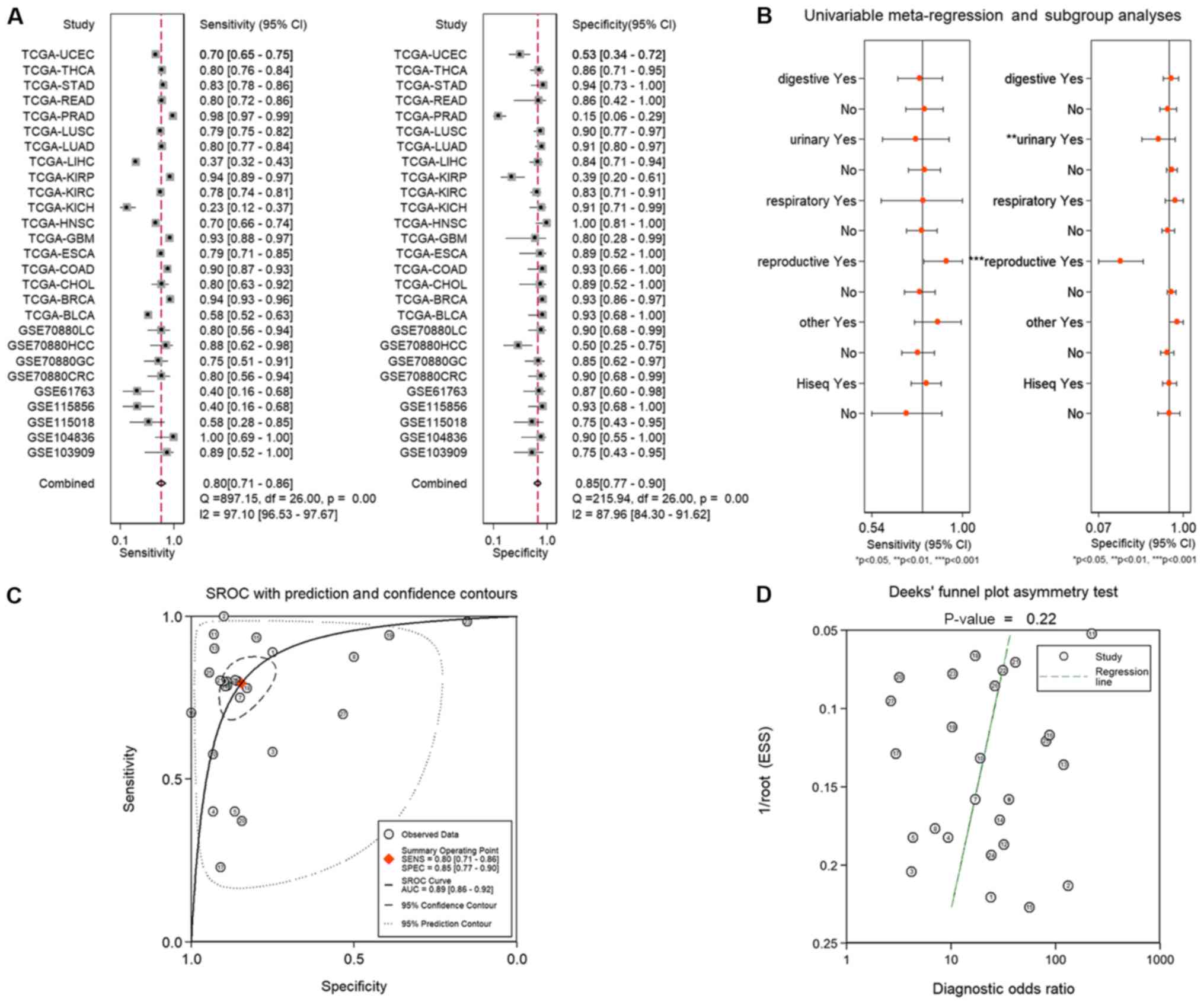 | Figure 3.(A) Combined sensitivity and
specificity of long intergenic non-coding RNA 1614 for the
diagnosis of malignancies. (B) Meta-regression analysis indicated
that reproductive system cancers (P<0.001) and urinary system
cancers (P=0.01) may be the major causes of heterogeneity. (C) SROC
curve and (D) Deeks' funnel plot analyses. The numbers in the
circles represent the number of each study in Table I. The overall diagnostic efficiency
was summarized by the regression curve. SROC, summary receiver
operating characteristic; AUC, area under curve; TCGA, The Cancer
Genome Atlas; BLCA, bladder urothelial carcinoma; BRCA, breast
invasive carcinoma; CHOL, cholangiocarcinoma; COAD, colon
adenocarcinoma; CRC, colorectal cancer; ESCA, esophageal carcinoma;
GBM, glioblastoma multiforme; GC, gastric cancer; HCC,
hepatocellular cancer; HNSC, head and neck squamous cell carcinoma;
KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma;
KIRP, kidney renal papillary cell carcinoma; LC, lung cancer; LIHC,
liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC,
lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; READ,
rectum adenocarcinoma; STAD, stomach adenocarcinoma; THCA, thyroid
carcinoma; UCEC, uterine corpus endometrial carcinoma; SENS,
sensitivity; SPEC, specificity; ESS, explained sum of square. |
 | Table I.Study characteristics in ROC and SROC
curve analyses. |
Table I.
Study characteristics in ROC and SROC
curve analyses.
| No. | Study | AUC (95% CI) | P-value | Tumor tissue
samples | Normal tissue
samples | Cut-off value | TP | FP | FN | TN | Tumor type | Detection
method |
|---|
| 1 | GSE103909 | 0.852
(0.670–1.000) | 0.007 | 9 | 12 | −2.025 | 8 | 3 | 1 | 9 | CHOL | Microarray |
| 2 | GSE104836 | 0.990
(0.962–1.000) | <0.001 | 10 | 10 | 0.049 | 10 | 1 | 0 | 9 | CRC | RNA-seq |
| 3 | GSE115018 | 0.604
(0.359–0.850) | 0.386 | 12 | 12 | −2.494 | 7 | 3 | 5 | 9 | HCC | Microarray |
| 4 | GSE115856 | 0.520
(0.290–0.750) | 0.852 | 15 | 15 | 3.332 | 6 | 1 | 9 | 14 | CRC | Microarray |
| 5 | GSE61763 | 0.520
(0.295–0.745) | 0.852 | 15 | 15 | 3.526 | 6 | 2 | 9 | 13 | RCC | Microarray |
| 6 | GSE70880CRC | 0.903
(0.808–0.997) | <0.001 | 20 | 20 | 6.148 | 16 | 2 | 4 | 18 | CRC | Microarray |
| 7 | GSE70880GC | 0.815
(0.673–0.957) | 0.001 | 20 | 20 | 4.981 | 15 | 3 | 5 | 17 | GC | Microarray |
| 8 | GSE70880HCC | 0.714
(0.533–0.897) | 0.038 | 16 | 16 | 6.662 | 14 | 8 | 2 | 8 | HCC | Microarray |
| 9 | GSE70880LC | 0.880
(0.773–0.987) | <0.001 | 20 | 20 | 8.210 | 16 | 2 | 4 | 18 | LC | Microarray |
| 10 | TCGA-BLCA | 0.772
(0.679–0.866) | <0.001 | 352 | 15 | 0.612 | 203 | 1 | 149 | 14 | BLCA | RNA-seq |
| 11 | TCGA-BRCA | 0.978
(0.970–0.988) | <0.001 | 1107 | 100 | 0.590 | 1045 | 7 | 62 | 93 | BRCA | RNA-seq |
| 12 | TCGA-CHOL | 0.851
(0.738–0.964) | 0.001 | 35 | 9 | 0.250 | 28 | 1 | 7 | 8 | CHOL | RNA-seq |
| 13 | TCGA-COAD | 0.952
(0.923–0.982) | <0.001 | 417 | 14 | 0.069 | 376 | 1 | 41 | 13 | COAD | RNA-seq |
| 14 | TCGA-ESCA | 0.870
(0.763–0.977) | <0.001 | 159 | 9 | 0.123 | 125 | 1 | 34 | 8 | ESCA | RNA-seq |
| 15 | TCGA-GBM | 0.890
(0.754–1.000) | 0.003 | 166 | 5 | 0.080 | 155 | 1 | 11 | 4 | GBM | RNA-seq |
| 16 | TCGA-HNSC | 0.915
(0.871–0.960) | <0.001 | 472 | 18 | 0.280 | 332 | 0 | 140 | 18 | HNSC | RNA-seq |
| 17 | TCGA-KICH | 0.435
(0.302–0.568) | 0.383 | 48 | 22 | 0.196 | 11 | 2 | 37 | 20 | KICH | RNA-seq |
| 18 | TCGA-KIRC | 0.865
(0.823–0.908) | <0.001 | 522 | 64 | 0.170 | 407 | 11 | 115 | 53 | KIRC | RNA-seq |
| 19 | TCGA-KIRP | 0.653
(0.524–0.781) | 0.019 | 153 | 23 | 0.025 | 144 | 14 | 9 | 9 | KIRP | RNA-seq |
| 20 | TCGA-LIHC | 0.585
(0.510–0.660) | 0.066 | 296 | 45 | 0.230 | 110 | 7 | 186 | 38 | LIHC | RNA-seq |
| 21 | TCGA-LUAD | 0.909
(0.881–0.936) | <0.001 | 532 | 56 | 0.443 | 427 | 5 | 105 | 51 | LUAD | RNA-seq |
| 22 | TCGA-LUSC | 0.874
(0.829–0.919) | <0.001 | 498 | 48 | 0.581 | 391 | 5 | 107 | 43 | LUSC | RNA-seq |
| 23 | TCGA-PRAD | 0.537
(0.445–0.630) | 0.405 | 410 | 46 | 1.026 | 403 | 39 | 7 | 7 | PRAD | RNA-seq |
| 24 | TCGA-READ | 0.879
(0.761–0.997) | 0.001 | 136 | 7 | 0.111 | 109 | 1 | 27 | 6 | READ | RNA-seq |
| 25 | TCGA-STAD | 0.915
(0.869–0.961) | <0.001 | 358 | 18 | 0.139 | 296 | 1 | 62 | 17 | STAD | RNA-seq |
| 26 | TCGA-THCA | 0.867
(0.809–0.924) | <0.001 | 473 | 37 | 0.093 | 380 | 5 | 93 | 32 | THCA | RNA-seq |
| 27 | TCGA-UCEC | 0.596
(0.489–0.702) | 0.083 | 343 | 30 | 0.232 | 240 | 14 | 103 | 16 | UCEC | RNA-seq |
Prognostic value of LINC01614 for OS
in malignancies
In total, 23 TCGA and two GEO datasets (8,213
patients) were included to analyze the association between
LINC01614 expression and survival in human cancers. The HR and 95%
CI of each dataset were obtained by univariate Cox regression model
analysis (Table II). As shown in
Fig. 4, the pooled HR from all the
datasets indicated a significant association between high LINC01614
expression and poor OS in various malignant tumors (HR=1.479; 95%
CI, 1.355–1.614; P<0.001). To assess the stability of the
combined results, a sensitivity analysis of OS was performed. The
results of the sensitivity analysis suggested that no individual
study changed the combined results; thus, the pooled results were
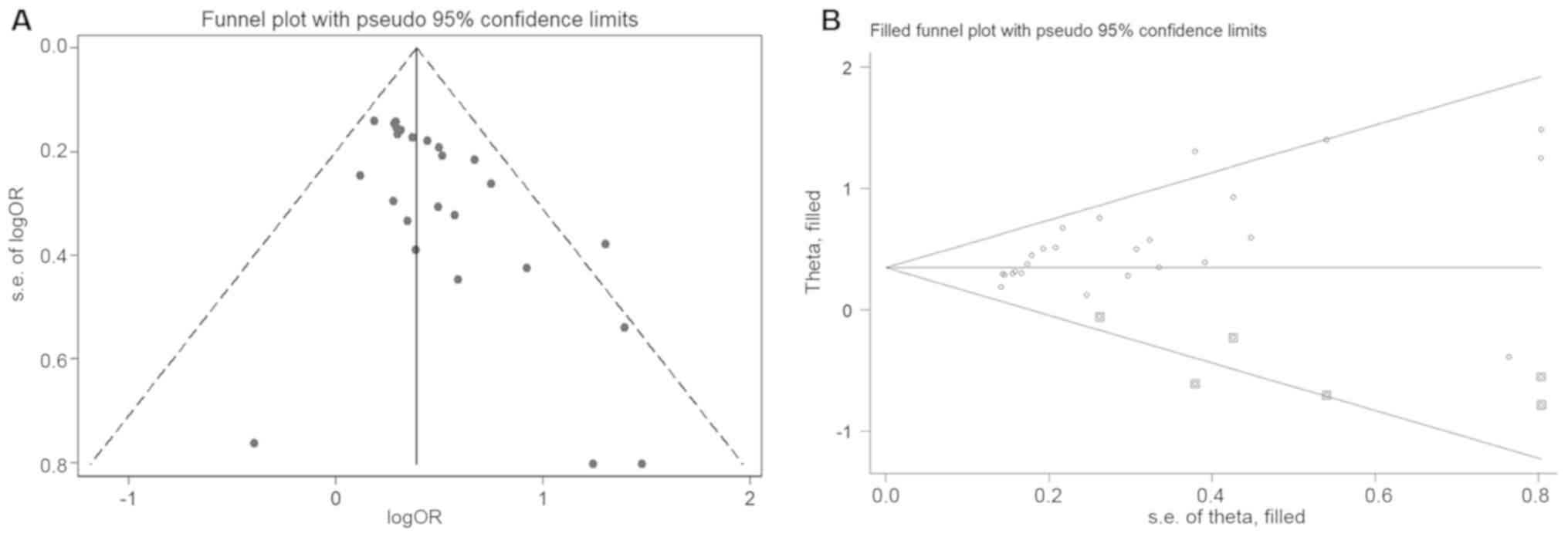
reliable (Fig. 4C). Publication bias
was assessed by Begg's test and the results suggested that there
was a considerable publication bias in this meta-analysis (P=0.001;
Fig. 5A). To further verify the
reliability of the results, a trim and fill analysis was performed
to eliminate the impact of publication bias by establishing
symmetry assumptions (24). As
indicated by the filled funnel plots in Fig. 5B, six assumed studies were filled to
eliminate the publication bias. After adjusting for filling assumed
studies, the adjusted HR and 95% CI (1.414 and 1.299–1.539,
respectively) was not significantly different compared with the
initial HR and 95% CI (1.479 and 1.355–1.614, respectively),
confirming that the results were reliable.
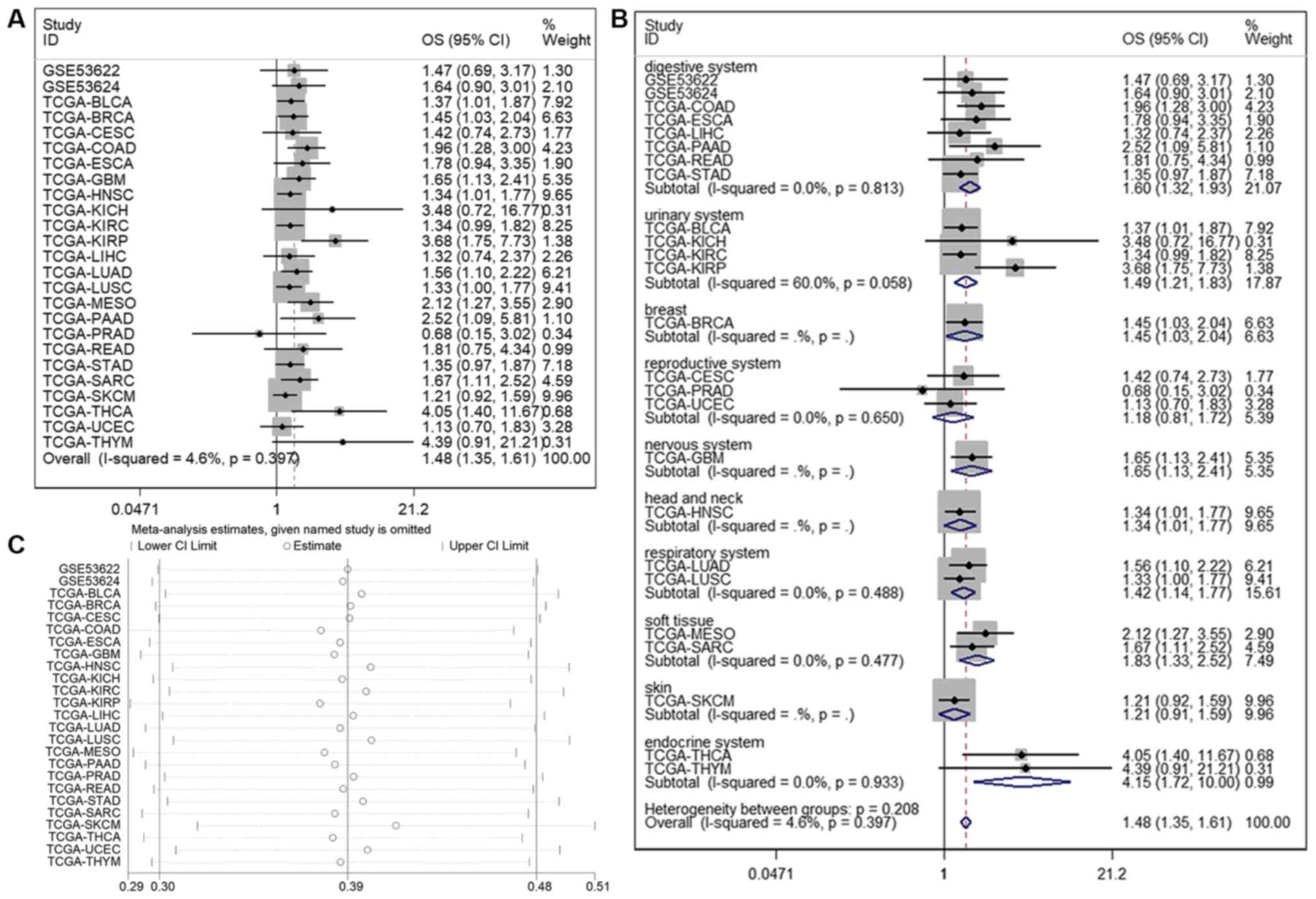 | Figure 4.(A) Forest plots for the association
between LINC01614 expression and OS. (B) Forest plots of subgroup
analysis for OS. (C) Sensitivity analysis of datasets to evaluate
the associations between LINC01614 expression and OS. LINC01614,
long intergenic non-coding RNA 1614; OS, overall survival; TCGA,
The Cancer Genome Atlas; BLCA, bladder urothelial carcinoma; BRCA,
breast invasive carcinoma; CESC, cervical squamous cell carcinoma
and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD,
colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma
multiforme; HNSC, head and neck squamous cell carcinoma; KICH,
kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP,
kidney renal papillary cell carcinoma; LIHC, liver hepatocellular
carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell
carcinoma; MESO, mesothelioma; PAAD, pancreatic adenocarcinoma;
PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC,
sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach
adenocarcinoma; THCA, thyroid carcinoma; THYM, thymoma; UCEC,
uterine corpus endometrial carcinoma. |
| Table II.Prognostic data for datasets in the
meta-analysis. |
Table II.
Prognostic data for datasets in the
meta-analysis.
| Dataset | Year | Country
measure | Outcome | HR (95% CI) | P-value | Cut-off value | Type of cancer | No. of
patients | Detection
method |
|---|
| GSE53622 | 2014 | China | OS | 1.474
(0.685–3.176) | 0.321 | 8.548 | ESCC | 120 | RNA-seq |
| GSE53624 | 2014 | China | OS | 1.645
(0.900–3.006) | 0.106 | 7.960 | ESCC | 238 | RNA-seq |
| TCGA-BLCA | 2014 | USA | OS | 1.372
(1.006–1.873) | 0.046 | 1.751 | BLCA | 352 | RNA-seq |
| TCGA-BRCA | 2014 | USA | OS | 1.452
(1.034–2.040) | 0.032 | 4.430 | BRCA | 1107 | RNA-seq |
| TCGA-CESC | 2014 | USA | OS | 1.417
(0.735–2.734) | 0.298 | 0.063 | CESC | 306 | RNA-seq |
| TCGA-COAD | 2014 | USA | OS | 1.961
(1.281–3.000) | 0.002 | 0.527 | COAD | 417 | RNA-seq |
| TCGA-ESCA | 2014 | USA | OS | 1.777
(0.942–3.350) | 0.076 | 0.126 | ESCA | 159 | RNA-seq |
| TCGA-GBM | 2014 | USA | OS | 1.650
(1.131–2.409) | 0.009 | 0.638 | GBM | 166 | RNA-seq |
| TCGA-HNSC | 2014 | USA | OS | 1.338
(1.010–1.774) | 0.043 | 1.891 | HNSC | 472 | RNA-seq |
| TCGA-KICH | 2014 | USA | OS | 3.475
(0.720–16.774) | 0.121 | 0.062 | KICH | 48 | RNA-seq |
| TCGA-KIRC | 2014 | USA | OS | 1.344
(0.991–1.822) | 0.057 | 0.372 | KIRC | 522 | RNA-seq |
| TCGA-KIRP | 2014 | USA | OS | 3.678
(1.749–7.734) | 0.001 | 0.095 | KIRP | 153 | RNA-seq |
| TCGA-LIHC | 2014 | USA | OS | 1.322
(0.739–2.366) | 0.347 | 0.046 | LIHC | 296 | RNA-seq |
| TCGA-LUAD | 2014 | USA | OS | 1.561
(1.099–2.218) | 0.013 | 0.590 | LUAD | 532 | RNA-seq |
| TCGA-LUSC | 2014 | USA | OS | 1.330
(1.000–1.769) | 0.050 | 1.370 | LUSC | 498 | RNA-seq |
| TCGA-MESO | 2014 | USA | OS | 2.122
(1.269–3.548) | 0.004 | 1.030 | MESO | 87 | RNA-seq |
| TCGA-PAAD | 2014 | USA | OS | 2.519
(1.093–5.806) | 0.030 | 0.442 | PAAD | 179 | RNA-seq |
| TCGA-PRAD | 2014 | USA | OS | 0.676
(0.151–3.021) | 0.608 | 0.074 | PRAD | 410 | RNA-seq |
| TCGA-READ | 2014 | USA | OS | 1.807
(0.751–4.345) | 0.186 | 1.040 | READ | 136 | RNA-seq |
| TCGA-STAD | 2014 | USA | OS | 1.349
(0.973–1.869) | 0.072 | 0.822 | STAD | 358 | RNA-seq |
| TCGA-SARC | 2014 | USA | OS | 1.672
(1.112–2.516) | 0.014 | 0.937 | SARC | 262 | RNA-seq |
| TCGA-SKCM | 2014 | USA | OS | 1.207
(0.915–1.593) | 0.183 | 0.462 | SKCM | 461 | RNA-seq |
| TCGA-THCA | 2014 | USA | OS | 4.047
(1.403–11.673) | 0.010 | 0.628 | THCA | 473 | RNA-seq |
| TCGA-UCEC | 2014 | USA | OS | 1.128
(0.696–1.829) | 0.624 | 0.147 | UCEC | 343 | RNA-seq |
| TCGA-THYM | 2014 | USA | OS | 4.389
(0.908–21.214) | 0.066 | 0.148 | THYM | 118 | RNA-seq |
To assess potential heterogeneity, a subgroup
analysis of OS was performed based on different organ systems. The
results suggested that upregulation of LINC01614 was significantly
associated with poor OS in malignancies from most organ systems,
including the digestive (P<0.001), urinary (P<0.001), nervous
(P=0.009), respiratory (P=0.002) and endocrine (P=0.002) systems,
in addition to the malignancies involving head and neck (P=0.043),
breast (P=0.031) and soft tissue (P<0.001), whereas there was no
association with cancers from the reproductive system (P=0.396) or
skin (P=0.183) (Table III and
Fig. 4B). Of note, only cancers of
the urinary system had significant heterogeneity within the
subgroup, suggesting that LINC01614 has distinct roles in these
four different urinary cancers. LINC01614 may be an oncogene and
prognostic marker for BLCA or KIRP, but not for KICH or KIRC.
| Table III.Subgroup analysis of overall survival
based on primary tumors of different organ systems in the
meta-analysis. |
Table III.
Subgroup analysis of overall survival
based on primary tumors of different organ systems in the
meta-analysis.
| Subgroup | No. of studies | No. of
patients | Pooled HR (95%
CI) | PHet | I2
(%) | P-value |
|---|
| Digestive
system | 8 | 1903 | 1.598 (1.321,
1.933) | 0.813 | 0.0 | <0.001 |
| Urinary system | 4 | 1075 | 1.491 (1.212,
1.833) | 0.058 | 60.0 | <0.001 |
| Reproductive
system | 3 | 649 | 1.177 (0.808,
1.716) | 0.650 | 0.0 | 0.396 |
| Respiratory
system | 2 | 1030 | 1.417 (1.136,
1.769) | 0.488 | 0.0 | 0.002 |
| Soft tissue | 2 | 349 | 1.833 (1.332,
2.524) | 0.477 | 0.0 | <0.001 |
| Endocrine
system | 2 | 591 | 4.151 (1.723,
9.998) | 0.933 | 0.0 | 0.002 |
| Breast | 1 | 1107 | 1.452 (1.034,
2.039) | – | – | 0.031 |
| Nervous system | 1 | 166 | 1.650 (1.131,
2.408) | – | – | 0.009 |
| Head and neck | 1 | 472 | 1.338 (1.010,
1.773) | – | – | 0.043 |
| Skin | 1 | 461 | 1.207 (0.915,
1.593) | – | – | 0.183 |
Bioinformatics analysis identified
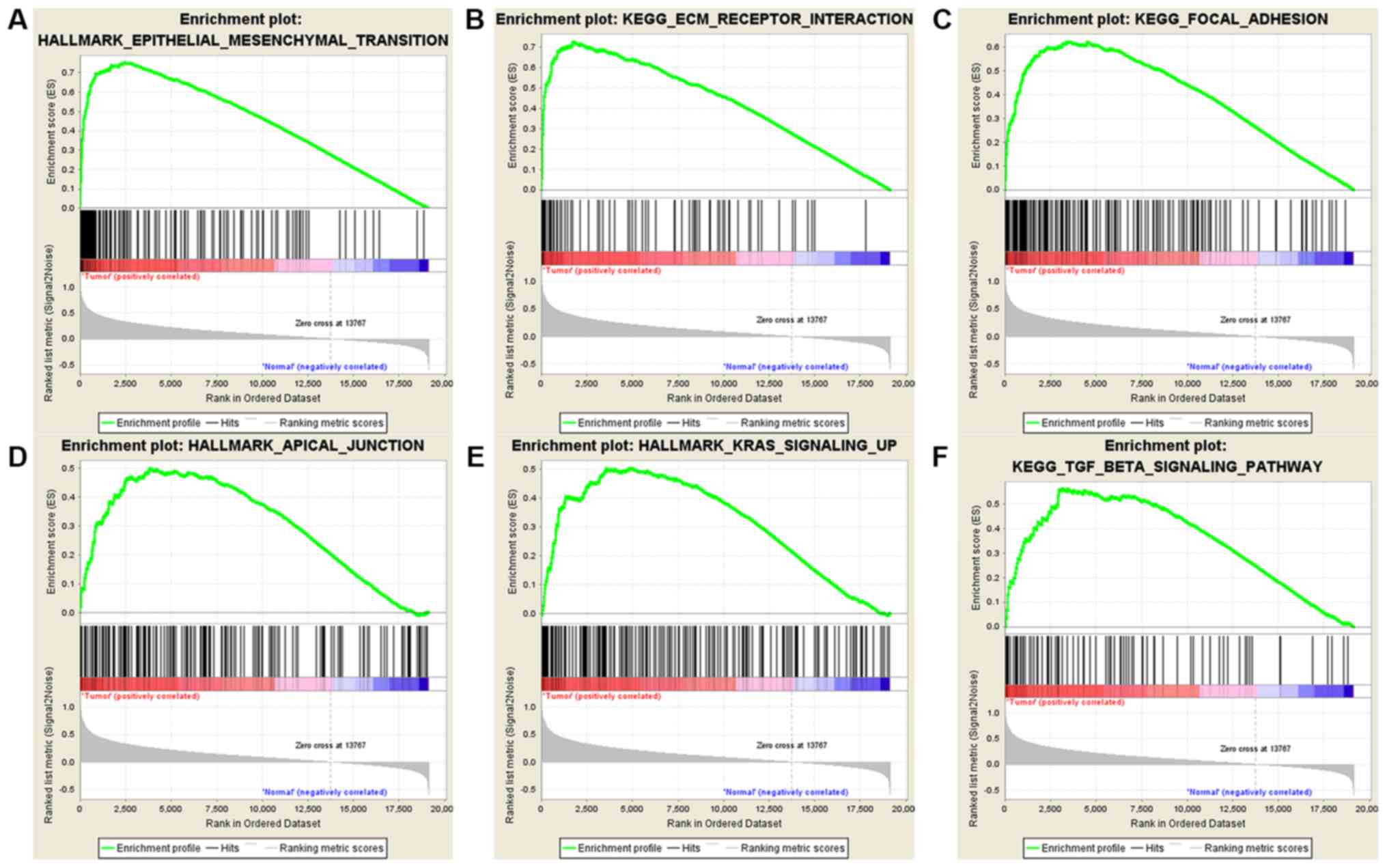
LINC01614-associated signaling pathways in human cancers
To identify the possible LINC01614-associated
signaling pathways in human cancers, GSEA was performed on eight
TCGA datasets with a weight of prognostic analyses of >5,
including BLCA, BRCA, HNSC, KIRC, LUAD, LUSC, skin cutaneous
melanoma and STAD. To eliminate the effects of tissue specificity,
the intersection between these GSEA results from different datasets
was used. As illustrated in Figs. 6,
7 and S1–7, and
Tables SI–IX, the GSEA enrichment plots indicated
that LINC01614 expression was highly associated with the
epithelial-mesenchymal transition (EMT), extracellular matrix (ECM)
receptor interaction, focal adhesion, gap junction, adherens
junction and cell adhesion molecules. Furthermore, LINC01614
expression was associated with angiogenesis, apoptosis, KRAS
signaling, transforming growth factor-β (TGF-β) signaling, tumor
necrosis factor-α (TNF-α) signaling, mitogen-activated protein
kinase (MAPK) signaling, Wnt signaling and various
cancer-associated signaling pathways.
 | Figure 7.Heatmap of gene set enrichment
analysis data, indicating the association of long intergenic
non-coding RNA 1614 expression with various signaling pathways in
all the eight datasets. KEGG, Kyoto Encyclopedia of Genes and
Genomes; ECM, extracellular matrix; TGF, transforming growth
factor; TNF, tumor necrosis factor; MAPK, mitogen-activated protein
kinase; CAMs, cell adhesin molecules; BLCA, bladder urothelial
carcinoma; BRCA, breast invasive carcinoma; HNSC, head and neck
squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma;
LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma;
SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma. |
Pearson's correlation coefficient was then used to
evaluate the genes that were co-expressed with LINC01614 in these
TCGA datasets. To obtain the most significantly co-expressed genes,
the overlapping results of eight different datasets (Pearson's
r>0.3, P<0.01) were obtained. As illustrated in Fig. 8, 36 genes were significantly
co-expressed with LINC01614 and 32 of these genes were associated
with EMT. Based on the biological roles of LINC01614 in cancers, it
was concluded that LINC01614 mainly has an important role in EMT
and associated signaling pathways in malignancies.
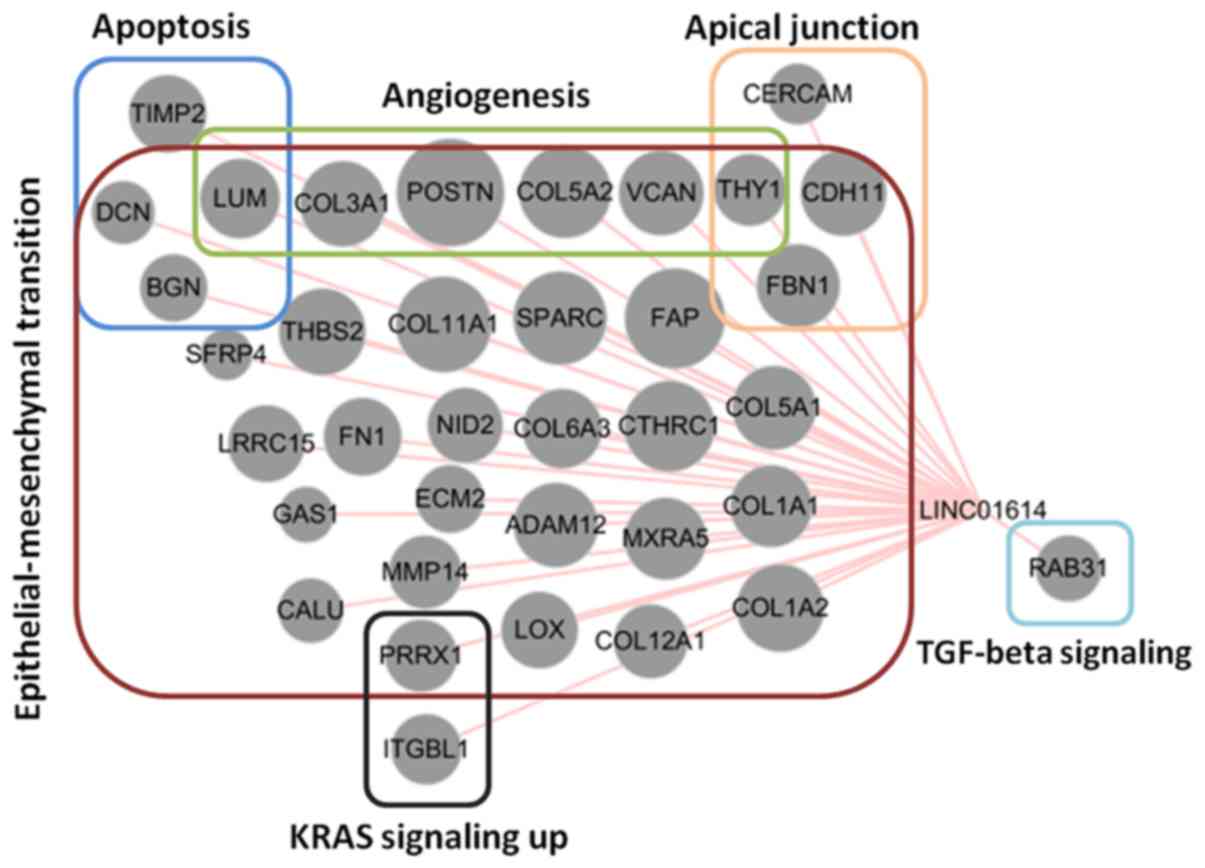 | Figure 8.Association of genes co-expressed
with LINC01614 during epithelial to mesenchymal transition based on
the overlapping results from the eight The Cancer Genome Atlas
datasets. The sizes of the nodes represent the mean values of
Pearson's correlation coefficients among the eight datasets.
LINC01614, long intergenic non-coding RNA 1614; TGF, transforming
growth factor; ADAM12, ADAM metallopeptidase domain 12; BGN,
biglycan; CALU, calumenin; CDH11, cadherin 11; CERCAM, cerebral
endothelial cell adhesion molecule; COL11A1, collagen XI α1 chain;
COL12A1, collagen type XII α1 chain; COL1A1, collagen type I α1
chain; COL1A2, collagen type I α2 chain; COL3A1, collagen type III
α1 chain; COL4A1, collagen type IV α1 chain; COL5A1, collagen type
V α1 chain; COL5A2, collagen type V α2 chain; COL5A3, collagen type
V α3 chain; COL6A2, collagen type VI α2 chain; COL6A3, collagen
type VI α3 chain; COL8A2, collagen type VIII α2 chain; COMP,
cartilage oligomeric matrix protein; CTHRC1, collagen triple helix
repeat containing 1; DCN, decorin; DPYSL3, dihydropyrimidinase like
3; ECM2, extracellular matrix protein 2; FAP, fibroblast activation
protein α; FBN1, fibrillin 1; FERMT2, fermitin family member 2;
FN1, fibronectin 1; GAS1, growth arrest specific 1; GFPT2,
glutamine-fructose-6-phosphate transaminase 2; GREM1, gremlin 1;
HTRA1, HtrA serine peptidase 1; ITGBL1, integrin subunit β like 1;
LGALS1, galectin 1; LOX, lysyl oxidase; LRP1, LDL receptor related
protein 1; LRRC15, leucine rich repeat containing 15; LUM, lumican;
MMP, matrix metallopeptidase; MXRA5, matrix remodeling associated
5; NID2, nidogen 2; NTM, neurotrimin; PDGFRB, platelet-derived
growth factor receptor β; POSTN, periostin; PRRX1, paired related
homeobox 1; RAB31, member RAS oncogene family; SFRP4, secreted
frizzled related protein 4; SPARC, secreted protein acidic and
cysteine rich; THBS2, thrombospondin 2; THY1, Thy-1 cell surface
antigen; TIMP2, TIMP metallopeptidase inhibitor 2; TPM4,
tropomyosin 4; VCAN, versican. |
RT-qPCR validation of LINC01614 and
co-expressed gene levels in cancer tissues
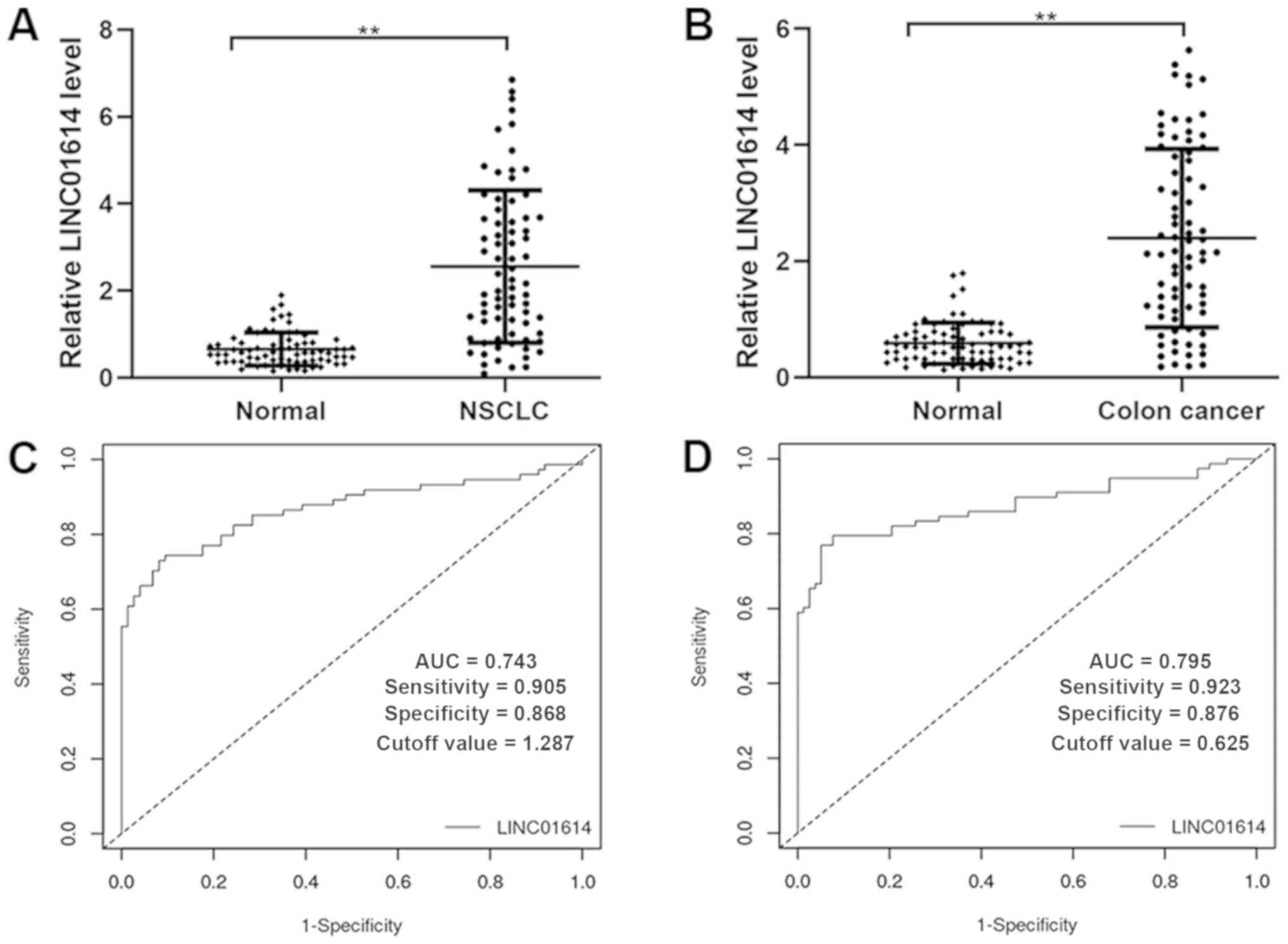
To confirm the reliability and validity of the
public RNA-seq data, the expression profiles of LINC01614 and five
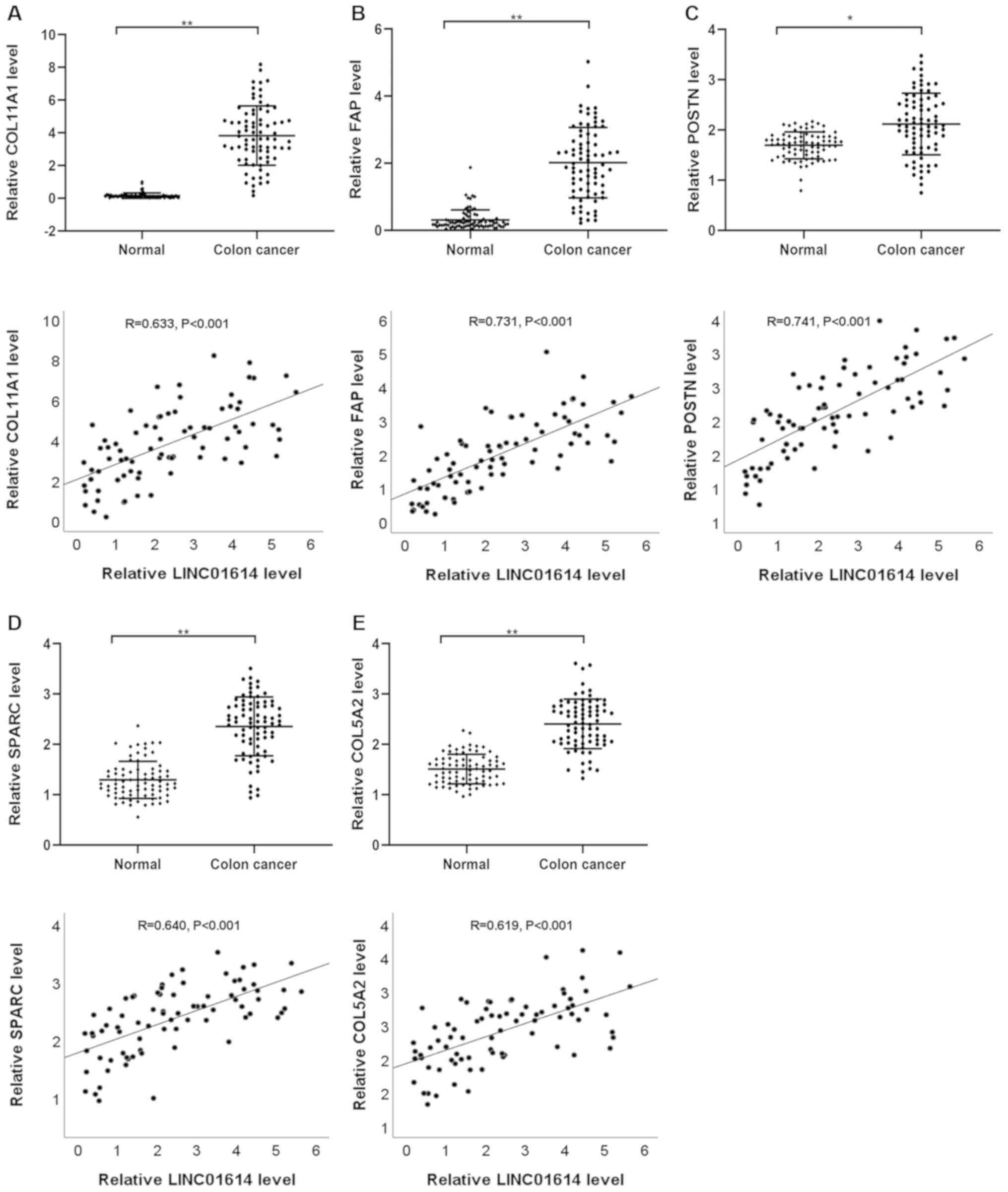
significantly co-expressed genes, including COL11A1, FAP, POSTN,
SPARC and COL5A2, were analyzed using RT-qPCR in paired samples
from 74 patients with NSCLC and 78 patients with colon cancer. The
results indicated that LINC01614 was expressed at a significantly
higher level in NSCLC and colon cancer tumor tissues than their
respective adjacent normal tissues (Fig.
9A and B). The diagnostic accuracies of LINC01614 for NSCLC and
colon cancer were investigated by ROC analyses, which indicated a
sensitivity, specificity and AUC of 0.743, 0.905 and 0.868 for
NSCLC (cutoff value =1.287) and of 0.795, 0.923 and 0.876 for colon
cancer (cutoff value =0.625), respectively (Fig. 9C and D). Further analysis of the
association between LINC01614 and clinicopathological
characteristics in the cohorts of the present study suggested that
higher LINC01614 expression was significantly associated with tumor
size, lymph node metastasis and TNM stage in patients with NSCLC,
and with lymph node metastasis and TNM stage in patients with colon
cancer (Tables IV and V). These results indicated that the
expression of LINC01614 was higher in NSCLC and colon cancer
tissues and may function as an oncogene in these cancers.
| Table IV.Association between LINC01614 and
clinicopathological characteristics of patients with non-small cell
lung cancer (n=74). |
Table IV.
Association between LINC01614 and
clinicopathological characteristics of patients with non-small cell
lung cancer (n=74).
|
|
| LINC01614 |
|---|
|
|
|
|
|---|
| Clinicopathologic
parameters | Total (n) | Low (n=37) | High (n=37) | P-value |
|---|
| Sex |
|
|
| 0.104 |
|
Male | 37 | 15 | 22 |
|
|
Female | 37 | 22 | 15 |
|
| Age (years) |
|
|
| 0.235 |
|
≤60 | 14 | 5 | 9 |
|
|
>60 | 60 | 32 | 28 |
|
| Size of tumor
(cm) |
|
|
| 0.047 |
| ≤3 | 24 | 16 | 8 |
|
|
>3 | 50 | 21 | 29 |
|
| Lymph node
metastasis |
|
|
| 0.032 |
| N0 | 45 | 27 | 18 |
|
|
N1-3 | 29 | 10 | 19 |
|
| Degree of
differentiation |
|
|
| 0.344 |
|
Moderate-well | 44 | 24 | 20 |
|
|
Poor | 30 | 13 | 17 |
|
| Histological
type |
|
|
| 0.642 |
|
Adenocarcinoma | 36 | 19 | 17 |
|
|
Squamous carcinoma | 38 | 18 | 20 |
|
| Smoking |
|
|
| 0.483 |
|
Yes | 41 | 19 | 22 |
|
| No | 33 | 18 | 15 |
|
| TNM stage |
|
|
| 0.016 |
| I | 35 | 23 | 12 |
|
| II | 25 | 7 | 18 |
|
|
III/IV | 14 | 7 | 7 |
|
| Table V.Association between LINC01614 and
clinicopathological characteristics in patients with colorectal
cancer (n=78). |
Table V.
Association between LINC01614 and
clinicopathological characteristics in patients with colorectal
cancer (n=78).
|
|
| LINC01614 |
|---|
|
|
|
|
|---|
| Clinicopathologic
parameter | Total (n) | Low (n=39) | High (n=39) | P-value |
|---|
| Sex |
|
|
| 0.174 |
|
Male | 40 | 17 | 23 |
|
|
Female | 38 | 22 | 16 |
|
| Age (years) |
|
|
| 0.645 |
|
≤60 | 32 | 15 | 17 |
|
|
>60 | 46 | 24 | 22 |
|
| T
classification |
|
|
| 0.098 |
|
T1-2 | 11 | 8 | 3 |
|
|
T3-4 | 67 | 31 | 36 |
|
| Lymph node
metastasis |
|
|
| 0.006 |
| N0 | 44 | 28 | 16 |
|
|
N1-2 | 34 | 11 | 23 |
|
| Distant
metastasis |
|
|
| 0.069 |
| M0 | 69 | 37 | 32 |
|
| M1 | 9 | 2 | 7 |
|
| Degree of
differentiation |
|
|
| 0.131 |
|
Moderate-well | 56 | 31 | 25 |
|
|
Poor | 22 | 8 | 14 |
|
| Location |
|
|
| 0.460 |
|
Right | 32 | 15 | 17 |
|
|
Transverse | 11 | 4 | 7 |
|
|
Left | 10 | 7 | 3 |
|
| Sigmoid
colon | 25 | 13 | 12 |
|
| TNM
stage |
|
|
| 0.022 |
|
I/II | 44 | 27 | 17 |
|
|
III/IV | 34 | 12 | 22 |
|
Furthermore, Pearson's correlation analysis between
LINC01614 and co-expressed genes, including COL11A1, FAP, POSTN,
SPARC and COL5A2 in NSCLC and colon cancer tissues, was performed.
The results indicated that the expression of LINC01614 was markedly
positively correlated with the levels of COL11A1, FAP, POSTN,
SPARC, and COL5A2 in NSCLC and colon cancer (Figs. 10 and 11). These results were in line with those
of the comprehensive bioinformatics analyses described above,
demonstrating the credibility of the RNA-seq data.
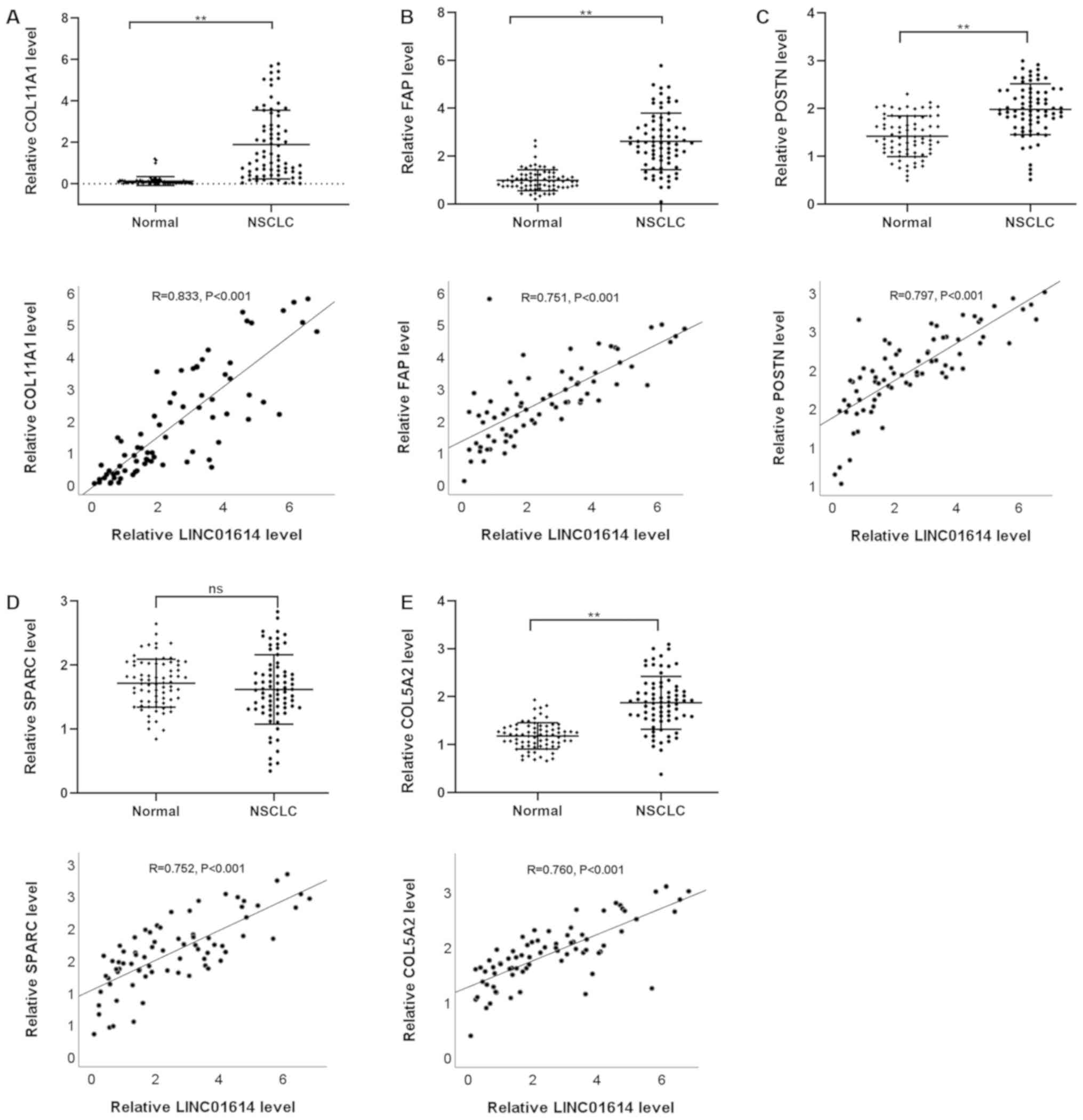 | Figure 10.Expression and Pearson's correlation
analysis between LINC01614 and (A) COL11A1, (B) FAP, (C) POSTN, (D)
SPARC and (E) COL5A2 in NSCLC. **P<0.01. ns, not significant;
LINC01614, long intergenic non-coding RNA 1614; NSCLC, non-small
cell lung cancer; COL11A1, collagen XI α1 chain; FAP, fibroblast
activation protein α; POSTN, periostin; SPARC, secreted protein
acidic and cysteine rich. |
Discussion
lncRNAs act as crucial regulators of almost all
aspects of physiological and pathological processes (25–28).
Accumulating evidence indicates that lncRNAs contribute to the
carcinogenesis and progression of various cancers (6–10).
LINC01614 was initially reported as an upregulated lncRNA in lung
cancer (14). Further studies
demonstrated that LINC01614 promotes the progression of lung
adenocarcinoma by competing with miR-217 and preventing the
miR-217/FOXP1 interaction (15).
Dysregulation of LINC01614 expression has also been reported in
breast carcinoma and indicated to be associated with poor prognosis
(16). Emerging evidence suggests
that LINC01614 acts as an oncogene and may be a potential biomarker
in malignancies. However, its expression and clinical implication
in a full spectrum of cancers have remained to be determined. To
the best of our knowledge, the present study was the first to
comprehensively assess the role of LINC01614 in cancer. Owing to
the continuous development of high-throughput sequencing, abundant
high-throughput data from human cancers have been collected in
public databases. The public data in these databases have
increasingly crucial roles in the study of cancers (29). Based on large-sample high-throughput
data from public databases, the present study aimed to investigate
the diagnostic and prognostic value of LINC01614 in
malignancies.
In the present study, comprehensive analyses across
53 normal tissue types and 24 tumor types were performed, including
data mining, differential expression analyses, ROC and SROC
analyses, survival analyses, meta-analyses and bioinformatics
analyses. Overall, the results suggested that the majority of
normal tissues have a low expression of LINC01614. Statistical
analysis (t-tests) further revealed that LINC01614 was
significantly upregulated in 15 of the 18 types of malignant
tissues compared with the expression levels in their normal
tissues. Furthermore, ROC and SROC curve analyses indicated that
LINC01614 is of suitable diagnostic value in various malignancies.
However, meta-regression analysis revealed that cancers of the
reproductive system and urinary system contributed to the
significant heterogeneity of the SROC curve analysis. In line with
this result, UCEC, KICH and PRAD, which are cancers of the
reproductive or urinary system, exhibited no significant
alterations between tumor and normal tissues. Therefore, except for
several cancers of these systems, LINC01614 was significantly
upregulated in malignant tissue samples and may be a potential
diagnostic biomarker for the majority of malignancies, including
cancers of the digestive, respiratory, nervous and endocrine
systems, in addition to the malignancies of the breast and head and
neck.
In addition, a meta-analysis was performed to
determine the prognostic predictive capacity of LINC01614 in
cancers. The pooled results indicated that upregulation of
LINC01614 was significantly associated with poor OS in malignant
tumors from most organ systems, including the digestive, urinary,
nervous, respiratory and endocrine systems, as well as of the head
and neck, breast and soft tissue with the exception of cancers of
the reproductive system or skin. In addition, only cancers of the
urinary system caused significant heterogeneity in this
subgroup.
Overall, abnormal expression of LINC01614 and its
associations with clinical outcome were highly cancer-dependent.
LINC01614 had no reliable diagnostic or prognostic value in some
cancers of the reproductive or urinary system. However, for cancers
of the digestive system, respiratory system, breast, head and neck,
nervous system and endocrine system, LINC01614 was proved to be an
oncogene and credible biomarker with diagnostic and prognostic
value.
The results of the RT-qPCR validation also indicated
that the expression of LINC01614 was higher in NSCLC and colon
cancer tissues compared with the expression levels in their
respective adjacent normal tissues; high LINC01614 expression was
associated with lymph node metastasis and tumor stage, further
supporting its role as an oncogene in these cancers. In addition, 9
of the 78 colon cancer patients had liver metastasis. In this
cohort, there were more patients with liver metastasis in the
high-expression group (7 of 38) than in the low-expression group (2
of 38), but the difference was not statistically significant
(P=0.069). This insignificant difference may be due to the limited
sample size. LINC01614 is able to promote liver and lymph node
metastases in colon cancer. These validation results demonstrated
that the comprehensive analyses were reliable.
At present, the molecular mechanisms of LINC01614 in
cancers remain to be fully elucidated. Therefore, bioinformatics
analyses were performed to discover the inherent molecular
mechanisms of LINC01614. The results indicated that LINC01614 may
have important effects on the progression of cancers by modulating
several cellular biology processes, including the EMT, ECM receptor
interaction, focal adhesion, gap junction, angiogenesis, apoptosis,
KRAS signaling, TGF-β signaling, TNF-α signaling and various
cancer-associated signaling pathways. Furthermore, most of the
genes identified to be co-expressed with LINC01614 were associated
with EMT in human cancers. Positive correlations between LINC01614
and five co-expressed genes, including COL11A1, FAP, POSTN, SPARC
and COL5A2, were identified in NSCLC and colon cancer tissues.
Relevant studies have revealed that COL11A1 and COL5A2 are
important EMT mediators and are involved in tumorigenesis and
metastasis of several cancers (30–33).
According to previous studies, FAP derived from cancer-associated
fibroblasts was able to be induced by EMT through Wnt/β-catenin
signaling (34,35). Overexpression of POSTN was able to
induce EMT through the MAPK/ERK pathway (36,37).
Furthermore, SPARC was able to increase the migration and invasion
of cancer cells by promoting EMT through the phosphorylated
(p)-FAK/p-ERK pathway (38,39). These results suggested that LINC01614
may be involved in EMT and associated signaling pathways to
influence the progression of human cancers.
During EMT, cells lose their epithelial properties,
generating migratory mesenchymal cell types with highly invasive
characteristics (40–42). In addition, cancer cells undergoing
EMT exhibit increased robustness including inhibition of apoptosis
and senescence, and acquisition of immunosuppression and drug
resistance (43,44). Metastasis is the major cause of
mortality in patients with cancer, and cancer cells that obtain
invasive characteristics through EMT have a crucial role during
metastasis (45–47). The present bioinformatics analyses
revealed that a high level of LINC01614 may be involved in EMT and
associated signaling pathways to influence cancer progression and
metastasis. In line with this, RT-qPCR validation also suggested
that high LINC01614 expression was associated with lymph node
metastasis and tumor staging in colon cancer and NSCLC. This
observation may be attributed to the influence of LINC01614 on the
EMT. The positive correlation between LINC01614 and EMT-associated
genes, including COL11A1, FAP, POSTN, SPARC and COL5A2, also
confirmed this biological process. Further studies are required to
validate the molecular mechanisms whereby LINC01614 modulates EMT
in malignancies.
There are numerous strengths to the present study.
First, it was a pan-cancer comprehensive analysis, including 53
normal tissue types and 32 cancer datasets with data from 9,091
patients, representing a complete dataset available for LINC01614
in human cancers. These large-sample data from different databases
were able to enhance the reliability of this comprehensive
analysis. Furthermore, the reliability and validity of the results
of the comprehensive analysis were confirmed with a molecular
biology technique with clinical samples obtained at our hospital.
Finally, the results of the bioinformatics analyses were validated
in eight cancer datasets. The rigorous evaluation criteria further
supported the reliability and repeatability of the results.
However, the study also has certain limitations.
First, significant heterogeneity existed in certain analyses in
this study. The heterogeneity was derived from cancers originating
from certain specific tissues that were validated by
meta-regression and subgroup analyses. Furthermore, the diagnostic
value of LINC01614 was identified in fresh solid tissues, but the
early diagnostic value requires to be validated using the blood
samples of cancer patients. In addition, due to the limited sample
size, the association between LINC01614 and certain
clinicopathological characteristics may be false-negative,
including liver metastasis in colon cancer, and this requires
further investigation with larger sample sizes and multicenter
research. Finally, the molecular mechanisms of LINC01614 were not
identified by this approach. Accordingly, further targeted studies
are required to verify these results.
In summary, LINC01614 emerged as an oncogene in most
malignancies, and its abnormal expression and clinical outcome
associations were highly cancer-dependent. For cancers of the
digestive system, respiratory system, breast, head and neck,
nervous system and endocrine system, LINC01614 is a credible
biomarker with diagnostic and prognostic value. However, for
certain cancers of the reproductive and urinary system, LINC01614
had no significant clinical association. Furthermore,
bioinformatics analysis indicated that LINC01614 may promote the
progression and metastasis of malignancies by modulating the EMT in
most malignancies. For future clinical applications, comprehensive
studies are required to assess the detailed molecular mechanisms
underlying the oncogenic role of LINC01614.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Beijing
Municipal Education Commission (grant no. KZ201910025033), Beijing
Municipal Administration of Hospitals Clinical Medicine Development
of Special Funding (grant no. ZYLX201814) and National Natural
Science Foundation of China (grant no. 81502493).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Design of the study: HL. Preparation of the
manuscript: DW. Conception of the study: HZ and XF. Editing of the
manuscript: DW and XF. Preparation of figures: XZ and HZ. Data
analysis: DW and HZ. Statistics: DW and DC. All authors contributed
toward the revision of the manuscript and agree to be accountable
for all aspects of the work. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Beijing Tongren Hospital (Beijing, China; approval no.
20180212). All patients provided written informed consent and
agreed to participate in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 6:394–424. 2018. View Article : Google Scholar
|
|
2
|
Ng L, Poon RT and Pang R: Biomarkers for
predicting future metastasis of human gastrointestinal tumors. Cell
Mol Life Sci. 70:3631–3656. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fidler MM, Gupta S, Soerjomataram I,
Ferlay J, Steliarova-Foucher E and Bray F: Cancer incidence and
mortality among young adults aged 20–39 years worldwide in 2012: A
population-based study. Lancet Oncol. 18:1579–1589. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim JL, Cho KH, Park EC and Cho WH: A
single measure of cancer burden combining incidence with mortality
rates for worldwide application. Asian Pac J Cancer Prev.
15:433–439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qin L, Chen C, Chen L, Xue R, Ou-Yang M,
Zhou C, Zhao S, He Z, Xia Y, He J, et al: Worldwide malaria
incidence and cancer mortality are inversely associated. Infect
Agent Cancer. 12:142017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dhamija S and Diederichs S: From junk to
master regulators of invasion: lncRNA functions in migration, EMT
and metastasis. Int J Cancer. 139:269–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhan A and Mandal SS: LncRNA HOTAIR: A
master regulator of chromatin dynamics and cancer. Biochim Biophys
Acta. 1856:151–164. 2015.PubMed/NCBI
|
|
8
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta. 1839:1097–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin C and Yang L: Long noncoding RNA in
cancer: Wiring signaling circuitry. Trends Cell Biol. 28:287–301.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li CH and Chen Y: Targeting long
non-coding RNAs in cancers: Progress and prospects. Int J Biochem
Cell Biol. 45:1895–1910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou M, Zhao H, Wang Z, Cheng L, Yang L,
Shi H, Yang H and Sun J: Identification and validation of potential
prognostic lncRNA biomarkers for predicting survival in patients
with multiple myeloma. J Exp Clin Cancer Res. 34:1022015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giulietti M, Righetti A, Principato G and
Piva F: lncRNA Co-expression network analysis reveals novel
biomarkers for pancreatic cancer. Carcinogenesis. 39:1016–1025.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ning L, Li Z, Wei D, Chen H and Yang C:
lncRNA, NEAT1 is a prognosis biomarker and regulates cancer
progression via epithelial-mesenchymal transition in clear cell
renal cell carcinoma. Cancer Biomark. 19:75–83. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White NM, Cabanski CR, Silva-Fisher JM,
Dang HX, Govindan R and Maher CA: Transcriptome sequencing reveals
altered long intergenic non-coding RNAs in lung cancer. Genome
Biol. 15:4292014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu AN, Qu HJ, Yu CY and Sun P: Knockdown
of LINC01614 inhibits lung adenocarcinoma cell progression by
up-regulating miR-217 and down-regulating FOXP1. J Cell Mol Med.
22:4034–4044. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vishnubalaji R, Shaath H, Elkord E and
Alajez NM: Long non-coding RNA (lncRNA) transcriptional landscape
in breast cancer identifies LINC01614 as non-favorable prognostic
biomarker regulated by TGF β and focal adhesion kinase (FAK)
signaling. Cell Death Discov. 5:1092019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Y and Ling C: Analysis of the long
non-coding RNA LINC01614 in non-small cell lung cancer. Medicine
(Baltimore). 98:e164372019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Song B, Zhu L and Zhang X: Long
non-coding RNA, LINC01614 as a potential biomarker for prognostic
prediction in breast cancer. PeerJ. 7:e79762019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo Y, Sheng Q, Li J, Ye F, Samuels DC and
Shyr Y: Large scale comparison of gene expression levels by
microarrays and RNAseq using TCGA data. PLoS One. 8:e714622013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Budczies J, Klauschen F, Sinn BV, Győrffy
B, Schmitt WD, Darb-Esfahani S and Denkert C: Cutoff Finder: A
comprehensive and straightforward Web application enabling rapid
biomarker cutoff optimization. PLoS One. 7:e518622012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liberzon A, Birger C, Thorvaldsdottir H,
Ghandi M, Mesirov JP and Tamayo P: The Molecular Signatures
Database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Duval S and Tweedie R: Trim and fill: A
simple funnel-plot-based method of testing and adjusting for
publication bias in meta-analysis. Biometrics. 56:455–463. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iwakiri J, Hamada M and Asai K:
Bioinformatics tools for lncRNA research. Biochim Biophys Acta.
1859:23–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bassett AR, Akhtar A, Barlow DP, Bird AP,
Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras
TR, Haerty W, et al: Considerations when investigating lncRNA
function in vivo. ELife. 3:e030582014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Joung J, Engreitz JM, Konermann S,
Abudayyeh OO, Verdine VK, Aguet F, Gootenberg JS, Sanjana NE,
Wright JB, Fulco CP, et al: Genome-scale activation screen
identifies a lncRNA locus regulating a gene neighbourhood. Nature.
548:343–346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shibayama Y, Fanucchi S, Magagula L and
Mhlanga MM: lncRNA and gene looping: What's the connection?
Transcription. 5:e286582014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galuschka C, Proynova R, Roth B, Augustin
HG and Muller-Decker K: Models in Translational Oncology: A public
resource database for preclinical cancer research. Cancer Res.
77:2557–2563. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim H, Watkinson J, Varadan V and
Anastassiou D: Multi-cancer computational analysis reveals
invasion-associated variant of desmoplastic reaction involving
INHBA, THBS2 and COL11A1. BMC Med Genomics. 3:512010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi JW, Liu W, Zhang TT, Wang SC, Lin XL,
Li J, Jia JS, Sheng HF, Yao ZF, Zhao WT, et al: The enforced
expression of c-Myc in pig fibroblasts triggers
mesenchymal-epithelial transition (MET) via F-actin reorganization
and RhoA/Rock pathway inactivation. Cell Cycle. 12:1119–1127. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang B, Zhang C, Yang X, Chen Y, Zhang H,
Liu J and Wu Q: Cytoplasmic collagen XIαI as a prognostic biomarker
in esophageal squamous cell carcinoma. Cancer Biol Ther.
19:364–372. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu H, Chen H, Wang J, Zhou L and Liu S:
Collagen stiffness promoted non-muscle-invasive bladder cancer
progression to muscle-invasive bladder cancer. Onco Targets Ther.
12:3441–3457. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Huang C, Peng C, Xu F, Li Y, Yutaka
Y, Xiong B and Yang X: Stromal fibroblast activation protein alpha
promotes gastric cancer progression via epithelial-mesenchymal
transition through Wnt/β-catenin pathway. BMC Cancer. 18:10992018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W,
Dang Y, Chu Y, Fan J and He R: FAP promotes immunosuppression by
Cancer-Associated fibroblasts in the tumor Microenvironment via
STAT3-CCL2 signaling. Cancer Res. 76:4124–4135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen L, Tian X, Gong W, Sun B, Li G, Liu
D, Guo P, He Y, Chen Z, Xia Y, et al: Periostin mediates
epithelial-mesenchymal transition through the MAPK/ERK pathway in
hepatoblastoma. Cancer Biol Med. 16:89–100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Choi KU, Yun JS, Lee IH, Heo SC, Shin SH,
Jeon ES, Choi YJ, Suh DS, Yoon MS and Kim JH: Lysophosphatidic
acid-induced expression of periostin in stromal cells: Prognoistic
relevance of periostin expression in epithelial ovarian cancer. Int
J Cancer. 128:332–342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi Q, Bao S, Song L, Wu Q, Bigner DD,
Hjelmeland AB and Rich JN: Targeting SPARC expression decreases
glioma cellular survival and invasion associated with reduced
activities of FAK and ILK kinases. Oncogene. 26:4084–4094. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang F, Zhang Y, Da J, Jia Z, Wu H and Gu
K: Downregulation of SPARC expression decreases cell migration and
invasion involving epithelial-mesenchymal transition through the
p-FAK/p-ERK pathway in esophageal squamous cell carcinoma. J
Cancer. 11:414–420. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
the importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nieto MA: The ins and outs of the
epithelial to mesenchymal transition in health and disease. Annu
Rev Cell Dev Biol. 27:347–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Furuya S, Endo K, Takahashi A, Miyazawa K
and Saitoh M: Snail suppresses cellular senescence and promotes
fibroblast-led cancer cell invasion. FEBS Open Bio. 7:1586–1597.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nieto MA: Context-specific roles of EMT
programmes in cancer cell dissemination. Nat Cell Biol. 19:416–418.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee SJ, Seol HJ, Lee HW, Kang WY, Kang BG,
Jin J, Jo MY, Jin Y, Lee JI, Joo KM and Nam DH: Gene silencing of
c-Met leads to brain metastasis inhibitory effects. Clin Exp
Metastasis. 30:845–854. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jie XX, Zhang XY and Xu CJ:
Epithelial-to-mesenchymal transition, circulating tumor cells and
cancer metastasis: Mechanisms and clinical applications.
Oncotarget. 8:81558–81571. 2017. View Article : Google Scholar : PubMed/NCBI
|