Introduction
Wild-type p53-induced phosphatase 1D or PPM1D (also
known as Wip1), a member of the PP2C serine/threonine phosphatase
family, functions as an oncogene in multiple different types of
cancer, including breast, esophageal, colon, thyroid, pancreatic,
gastric, liver, nasopharyngeal, bladder, prostate and ovarian
carcinoma (1–5). PPM1D exerts its oncogenic effect
primarily via dephosphorylation and increased degradation of p53
protein (6,7). Indeed, PPM1D commonly exhibits
copy number alterations and is upregulated in different types of
cancer, including breast and ovarian carcinoma (2–5). Thus,
the p53-PPM1D loop is an important mediator of the role of p53 in
genomic surveillance mechanisms.
PPM1D protein upregulation in cancer cells is
associated with a corresponding increase in mRNA expression. In
fact, it has been revealed that the RNA-binding protein RBM38 or
RNPC1 induces translation of PPM1D by binding the
3′-untranslated region (UTR) of PPM1D (8). Once translated, PPM1D protein
dephosphorylates RBM38 at serine 195 residue (8). RBM38 is phosphorylated at the serine
195 residue by glycogen synthase kinase 3β (GSK3β).
Dephosphorylated RBM38 forms a complex with the cap-binding protein
eukaryotic elongation factor 4E (eIF4E) and prevents this from
translating TP53 mRNA (9,10).
Phosphorylation by GSK3β inhibits the interaction of RBM38 with
eIF4E, resulting in translation of TP53 mRNA (9,10). Thus,
the PPM1D-RBM38-p53 axis is intricately regulated by feedback loops
of kinases and phosphatases based on specific cellular context.
Indeed, RBM38 is regulated by p53 and E2F1 (11,12).
RBM38 can also exert its pro-oncogenic roles by regulating mRNA
stability and/or alternative splicing of cyclin-dependent kinase
inhibitor 1A (encoding p21), mouse double minute 2 homolog,
ELAV-like RNA binding protein 1 (encoding human antigen R),
erythrocyte membrane protein band 4.1 and fibroblast growth factor
receptor 2 (11,13,14).
The PPM1D-RBM38-p53 axis has been mostly studied in
the context of wild-type p53. However, mutations in TP53
(both null and hot-spot point mutations) are known to exert
gain-of-function that in turn regulate both resistance to
chemotherapy and metastatic progression (15,16),
even though mutant TP53 is widely pervasive in all tumor
types, including non-small cell lung cancer (NSCLC). In the case of
T-cell lymphomagenesis, it has been demonstrated that RBM38
functions as a tumor suppressor, and genetic ablation of
RBM38 in a mice model resulted in enhanced mutant
TP53 expression (17). A
tumor is subjected to hypoxic conditions both during initial growth
and during progression. However, it is not known whether the
PPM1D-RBM38-p53 axis functions similarly under normoxic and hypoxic
conditions. Given that RBM38 regulates cellular responses to
oxidative stress by regulating translation of hypoxia-inducible
factor 1α (HIF1A) (18), and
HIF1α protein forms a complex with mutant p53 protein and induces
transcription of extracellular matrix components promoting
migration and invasion (19), it may
be possible that the PPM1D-RBM38-mutant p53 axis is regulated
differently in normoxic and hypoxic conditions. Indeed, microRNAs
(miRNAs), a class of small non-coding RNA ~22 nucleotides long,
have been shown to differentially regulate cellular response under
hypoxic and normoxic conditions (20). miR-129-1-3p and miR-129-1-5p have
been shown to function as tumor suppressors in colon, gastric,
bladder, and esophageal cancer (21–24).
However, to the best of our knowledge, it is unclear if miR-129-1
differentially regulates expression of PPM1D under normoxic or
hypoxic conditions. Hence, the objective of the current study was
to determine if PPM1D-RBM38-p53 axis is regulated similarly in
NSCLC harboring wild-type and mutant p53 gene under the conditions
of normoxia and hypoxia.
Materials and methods
Cell culture
A549 and NCI-H1770 cell lines were purchased from
the American Type Culture Collection. Both cell lines were cultured
in DMEM containing 10% fetal bovine serum and 10 ml/l
penicillin/streptomycin (all Thermo Fisher Scientific, Inc.). Cells
were incubated in normoxic conditions (21% O2, 74%
N2 and 5% CO2) at 37°C. For growth under
hypoxia cells were incubated in a hypoxic chamber (1%
O2, 94% N2 and 5% CO2; Jouan SA;
Thermo Fisher Scientific, Inc.) at 37°C for 4 h. During hypoxia
induction, HEPES (25 mM final concentration; Thermo Fisher
Scientific, Inc.) was added to prevent acidosis (25).
Cell lysate and western blot
analysis
At the end of the experimental time point, the
medium was removed and the cells were washed twice with ice-cold 1X
phosphate-buffered saline (PBS). Cells were then lysed using RIPA
buffer (20× volume of cell pellet; Thermo Fisher Scientific, Inc.),
centrifuged at 15,000 × g for 15 min at 4°C, and protein
concentration in the extracted whole cell lysate was determined
using a bicinchoninic acid assay (Thermo Fisher Scientific, Inc.).
A total of 50 µg of lysates per sample were resolved by 10%
SDS-PAGE and blotted onto PVDF membranes (Thermo Fisher Scientific,
Inc.) and processed for western blot analysis using standard
methodologies. Blots were blocked using 5% fat-free milk in TBST
buffer (0/1% Tween-20) for 30 min at room temperature before being
incubated with the following primary antibodies overnight at 4°C:
p53 (cat. no. 9282; 1:1,000; Cell Signaling Technology, Inc.),
HA-Tag (cat. no. 3724; 1:1,500; Cell Signaling Technology, Inc.),
PPM1D (cat. no. HPA022277; 1:2,000; Sigma-Aldrich; Merck KGaA),
RBM38 (cat. no. ab168455; 1:3,000; Abcam), HIF1α (cat. no. 36169;
1:1,000; Cell Signaling Technology, Inc.), p-p53 (serine 15) (cat.
no. 9284; 1:1,000; Cell Signaling Technology, Inc.), GAPDH (cat.
no. 5174; 1:4,000; Cell Signaling Technology, Inc.). Blots were
probed with anti-GAPDH antibody to ensure equivalent protein
loading across samples. After washing thrice with TBST buffer,
blots were incubated with HRP-conjugated mouse or rabbit secondary
antibody (Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Post-incubation blots were washed thrice with TBST
buffer before being developed using Pierce ECL Plus substrate
(Thermo Fisher Scientific, Inc.). Each experiment was repeated ≥
three times and densitometry analysis was conducted using ImageJ
version 2 software (National Institutes of Health) to determine the
relative changes in protein expression under different conditions.
Representative blots and quantification results from all
experiments are presented in the figures.
miRNA isolation and reverse
transcription quantitative (RT-qPCR)
Medium was aspirated off and cells were rinsed and
scrapped off in ice-cold PBS and centrifuged at 1,000 × g for 5 min
at 4°C. The cell pellet was then used to isolate miRNA using the
mirVana miRNA isolation kit (Thermo Fisher Scientific, Inc.). cDNA
synthesis was performed using a TaqMan Advanced miRNA cDNA
Synthesis kit (Thermo Fisher Scientific, Inc.). Each cDNA sample
was pre-amplified using the TaqMan PreAmp Master Mix kit (Thermo
Fisher Scientific, Inc.) then used to template the qPCR using the
TaqMan Fast Advanced Master Mix and individual TaqMan microRNA
assay probes (Thermo Fisher Scientific, Inc.). Thermocycling
conditions consisted of an initial denaturation of 20 sec at 95°C,
followed by 40 cycles of 95°C for 3 sec and 60°C for 30 sec. qPCR
was performed using TaqMan probes (Thermo Fisher Scientific, Inc.)
for miR129-1 (Assay ID: 002298; 5′-AAGCCCUUACCCCAAAAAGUAU-3′) and
RNU6B (Assay ID: 001093). RNU6B expression levels were
utilized for normalization. Post-normalization relative expression
of miR129-1 was calculated using the 2−ΔΔCq method
(26). The data were represented as
expression in hypoxic conditions relative to normoxia, or
expression following transfection of miR129-1 mimic compared with
control mimic in hypoxia [mean ± standard error of the mean (SEM)].
The RT-qPCR for the indicated genes was conducted similarly using
TaqMan probes; however, the data were normalized to GAPDH.
RT-qPCR experiments were run in triplicate.
Plasmid construction
The 3′-UTR of PPM1D was amplified from cDNA
using the following primer sequences: PPM1D forward,
5′-TGCATCTGGGAAATGAGGTT-3′ and reverse, 5′-GCCTCCTTCCAGATGACACT-3′
and cloned into the pRL-TK-CXCR4 vector (Addgene, Inc.) and called
the pRL-TK-PPM1D wild-type 3′-UTR plasmid. The miR-129-1
binding site mutant 3′-UTR (nucleotides 292–299 deleted) of
PPM1D was generated using site-directed mutagenesis
(QuickChange II kit; Agilent Technologies, Inc.) and the following
primers: Forward,
5′-GAGTCTCTGATACACAGTAATTGTGACAATATGTTTAAAGAAATCAAAAGAATCTATTA-3′
and reverse,
5′-TAATAGATTCTTTTGATTTCTTTAAACATATTGTCACAATTACTGTGTATCAGAGACTC-3′
and named the pRL-TK-ΔPPM1D 3′-UTR plasmid. The pGL3 plasmid
expressing Firefly luciferase off a CMV promoter was purchased from
Promega Corporation. The miR129-1 (hsa-miR-129-1-3p) mimic used was
the MISSION® microRNA mimic (cat. no. HMI0159;
Sigma-Aldrich; Merck KGaA) and the control mimic used was an
oligonucleotide sequence from Arabidopsis thaliana
(5′-GGUUCGUACGUACACUGUUCA-3′) with no homology to human gene
sequences (cat. no. HMC0002; Sigma-Aldrich; Merck KGaA). Lentiviral
particles of control short hairpin (sh)RNA (cat. no. sc-108080) and
RBM38 (cat. no. sc-76368-V) were obtained from Santa Cruz
Biotechnology, Inc. shRNA targeting the 3′-UTR of TP53
(V2LHS_93615) was obtained from Open Biosystems. The pCMV-Neo-Bam
p53 R248W plasmid (cat. no. 16437) was obtained from Addgene, Inc.
and was cloned into pEF-hemagglutinin (HA) vector.
Transfection and transduction
Cells were plated in respective antibiotic-free cell
culture DMEM medium. Cells were co-transfected with Renilla
luciferase reporter plasmids [pRL-TK-PPM1D wild-type 3′-UTR
or pRL-TK-ΔPPM1D 3′-UTR (nucleotides 292–299 deleted)] and
pGL3 plasmid expressing Firefly luciferase, and where indicated
along with control or miR129-1 mimic using Polyplus jetPRIME
transfection reagent (Polyplus-transfection, SA). For reporter
plasmids, cells were seeded in 24-well plates (50,000 cells/well)
and co-transfected with 0.5 µg of pRL-TK plasmid(s) and pGL3
plasmid. Luciferase assays were performed 24 h after transfection.
miRNA mimics were transfected at a final concentration of 10 nM.
Cells were harvested 48 h after transfection with the miRNA
mimics.
For transduction, NCI-H1770 cells were transduced
with either lentiviral particles containing control shRNA or shRNA
targeting RBM38, using polybrene. Cells were selected with
puromycin (2 µg/ml) for 2 weeks and successful knockdown was
verified via western blot analysis.
A549 cells were first transfected with linearized
pEF-HA-TP53 (R248W) plasmid and selected with G418 (100
µg/ml) for 2 weeks (with addition of fresh media every third day)
before being transfected with lentiviral particles containing
control shRNA or shRNA targeting 3′-UTR of TP53 using
polybrene. Cells were selected with puromycin (2 µg/ml) for 2 weeks
(with addition of fresh media every alternate day) and successful
knockdown of endogenous and overexpression of the mutant TP53
plasmids was verified by western blot analysis using p53 and HA
antibodies.
Luciferase assay
After 24 h of transfection, NCI-H1770 cells were
washed with ice-cold PBS and then incubated with 100 µl of 1X
passive lysis buffer (Promega Corporation). Plates were incubated
on a rocker at 37°C for 30 min. After incubation, 20 µl of lysates
were used to perform a luciferase assay using the Dual-Luciferase
assay kit (Promega Corporation). Firefly luciferase was used as an
internal control and used to normalize relative Renilla
luciferase expression and expressed as the mean ± SD of three
independent experiments.
Data mining
The analysis of The Cancer Genome Atlas (TCGA)
(cancer.gov/tcga) data was performed using cBioPortal for Cancer
Genomics (http://cbioportal.org) (27,28). A
total of 2,628 patients and 2,855 samples from 7 studies were
included (29–35). Analysis was carried out to determine
genome amplification, somatic mutations, association with survival
and mRNA expression. In situ prediction of miRNAs binding to
PPM1D mRNA was done using TargetScan Human Release 7.1
algorithm (36).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6.0 (GraphPad Software, Inc.). Data were expressed as the
mean ± standard deviation of three technical replicates. For
RT-qPCR, the data were expressed as the mean ± SEM of three
technical replicates. A paired Student's t-test was used to test
differences between groups. The log-rank test was used to test
whether the difference between overall survival times between two
groups was statistically significant. P<0.05 was considered to
indicate a statistically significant difference.
Results
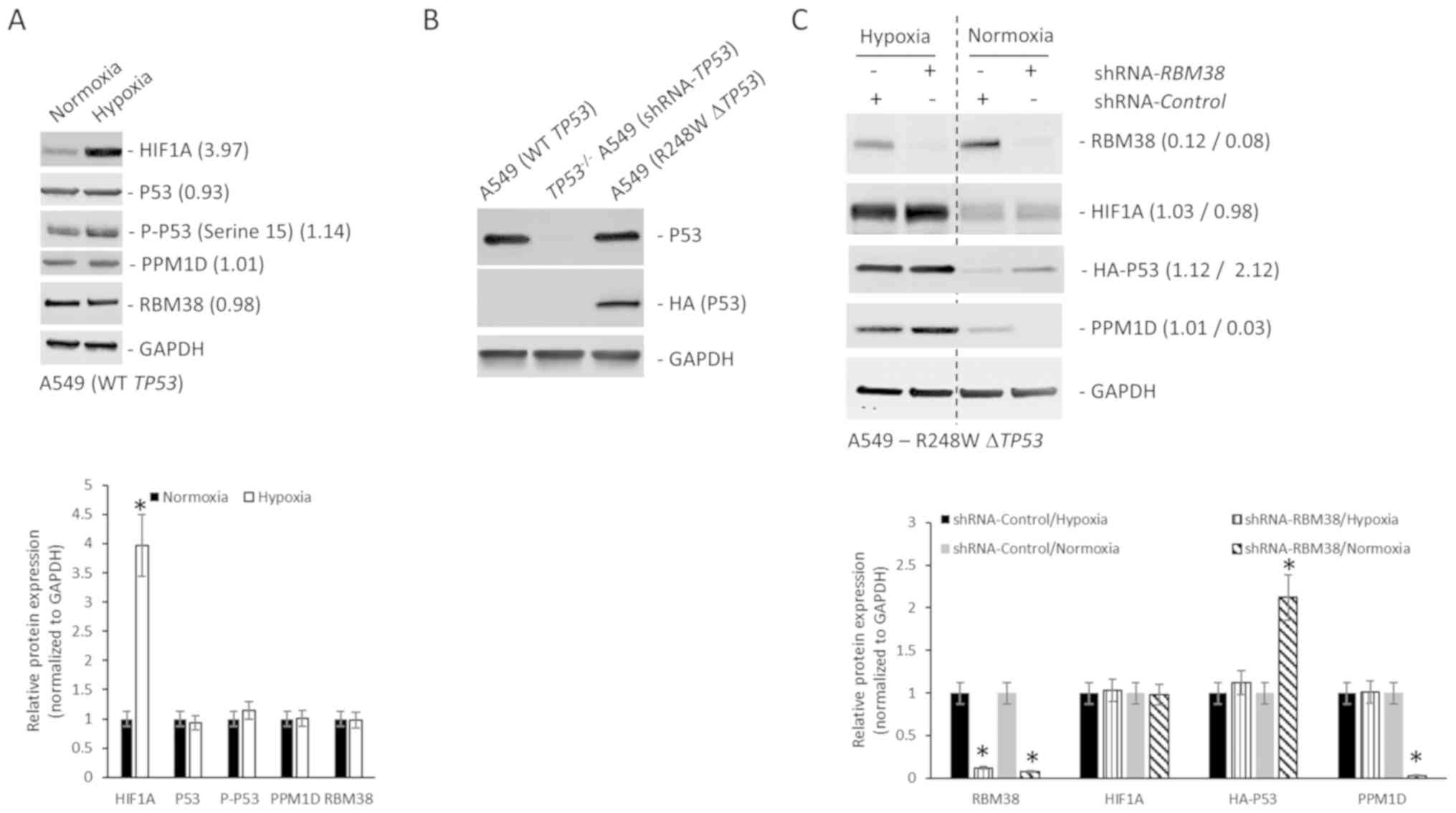
PPM1D protein expression is regulated
independent of RBM38 expression in NSCLC cell line harboring mutant
TP53
The protein expression levels of p53, PPM1D and
RBM38 were initially determined in the NSCLC cell line A549 that
harbors wild-type TP53, and in the NCI-H1770 cell line that
harbors the R248W hotspot TP53 mutation. Although RBM38 and
PPM1D expression levels were higher in the NCI-H1770 cells, p53
expression was higher in A549 cells (Fig. 1A). The higher basal expression of p53
observed in NCI-H1770 cells in comparison with the A549 cells was
opposite to what is reported by the American Type Culture
Collection (atcc.org/en/Documents/Learning_Center/~/media/
5F7B1CCACF724E3398BE56BFBEE3EFE4.ashx). Given that the objective of
the present study was to define the regulation of the
PPM1D-RBM38-p53 (wild-type/mutant) axis in the cells, this
discordance in basal p53 expression was not further
investigated.
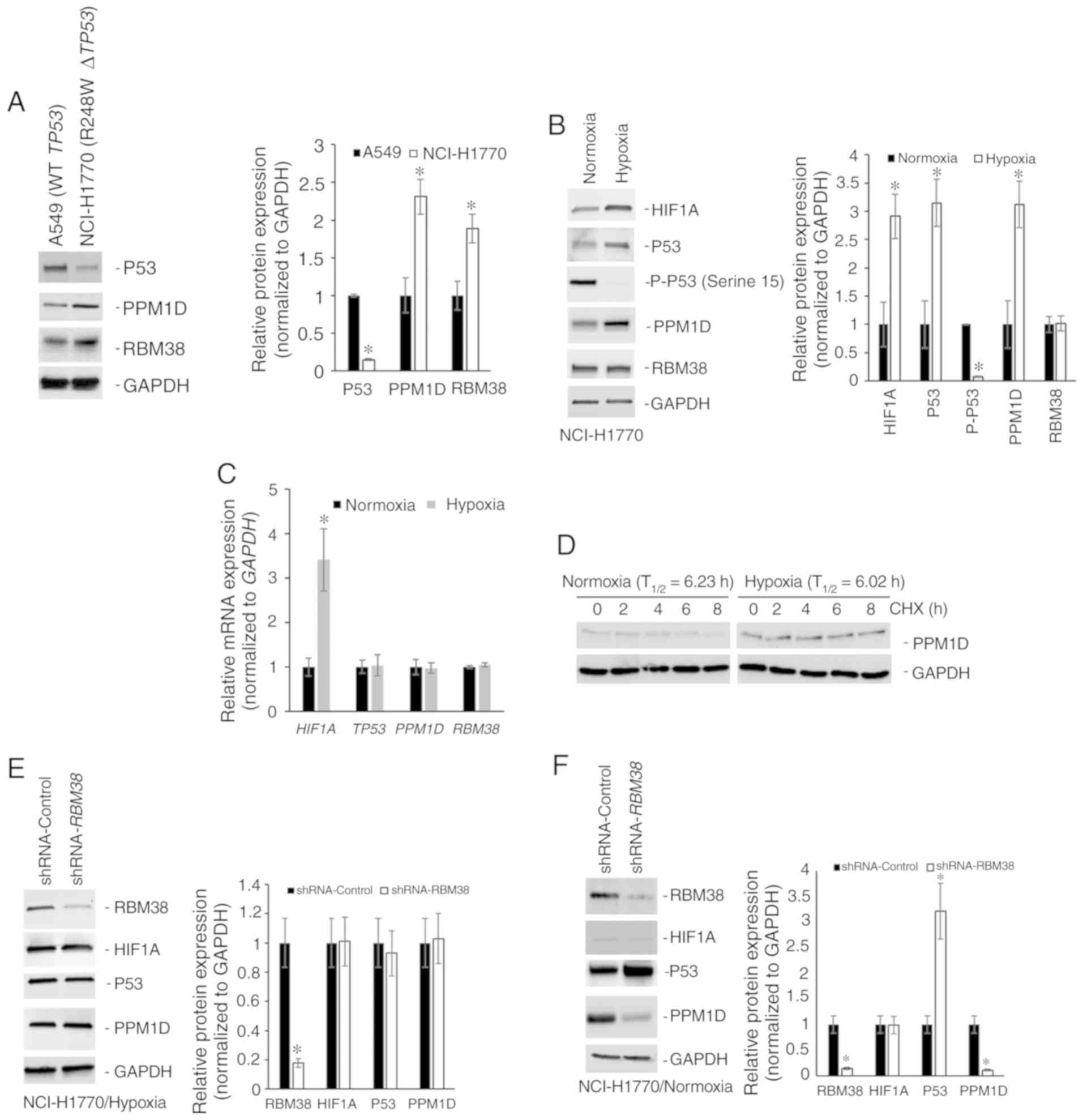 | Figure 1.PPM1D protein expression is regulated
independently of RBM38 expression in NCI-H1770 cells harboring
R248W mutant TP53. (A) Western blot analysis of indicated
proteins in A549 and NCI-H1770 cell lines maintained under normoxic
conditions. The bar graph presents the relative protein expression
levels determined by densitometry analysis (n=3) and expressed as
the mean ± standard deviation. (B) Western blot analysis of
indicated proteins in NCI-H1770 cells maintained under normoxic
conditions or subjected to hypoxia for 4 h. The bar graph presents
the relative protein expression levels determined by densitometry
analysis (n=3) and expressed as the mean ± standard deviation. (C)
Relative mRNA expression of indicated genes. Data were normalized
to GAPDH and expressed as the mean ± standard error of the
mean relative to normoxia (n=3). (D) Relative stability of PPM1D
protein in normoxic and hypoxic conditions as determined by CHX
treatment. T1/2 was calculated from three replicates and
normalized to GAPDH. Western blot analysis of indicated proteins in
NCI-H1770 cells stably transduced with either control shRNA or
shRNA targeting RBM38 and maintained under (E) hypoxia for 4 h, or
(F) normoxia. Bar graphs in indicate the relative protein
expression levels determined by densitometry analysis (n=3) and
expressed as the mean ± standard deviation. Blots in each case (A,
B, E and F) were probed with anti-GAPDH antibody to confirm equal
loading. *P<0.05. mRNA, messenger RNA; T1/2,
half-life; PPM1D, p53-induced phosphatase 1D; shRNA, short hairpin
RNA; RBM38, RNA binding motif protein 38; WT, wild type; CHX,
cycloheximide; HIF1A, hypoxia-inducible factor 1A. |
RBM38 promotes translation of mutant TP53 (9). Mutant p53 protein cooperates with HIF1α
to cause transcriptional upregulation of extracellular matrix
components (18). RBM38 can also
regulate translation of HIF1α (15).
Hence, the changes in protein expression of PPM1D, p53, and RBM38
following induction of hypoxia in the NCI-H1770 cells were
investigated. Hypoxia was induced by incubating cells at 1%
O2 for 4 h and confirmed via the induction of HIF1α
protein expression. RBM38 protein expression did not change
following the induction of hypoxia in these cells; however, both
p53 and PPM1D were upregulated following hypoxia (Fig. 1B). Phosphorylation levels of p53 at
serine 15 residue decreased following the induction of hypoxia
(Fig. 1B), which may have been
either due to decreased phosphorylation or increased phosphatase
activity, given the increase in PPM1D protein expression. The
increase in protein expression of PPM1D and p53 was not due to
transcriptional upregulation post-hypoxia, as the steady state mRNA
expression level did not differ significantly between normoxic and
hypoxic conditions (Fig. 1C). To
determine whether the increased PPM1D protein expression in hypoxia
was due to increased stability of the protein, cycloheximide chase
experiment was performed, as cycloheximide inhibits translation.
PPM1D half-life (T1/2) was not significantly different
between normoxic (6.23 h) and hypoxic conditions (6.02 h) (Fig. 1D). Cumulatively, these results
indicate that PPM1D and p53 protein expression was
post-transcriptionally upregulated during hypoxia in the NCI-H1770
cells.
Given that no change was observed in RBM38 protein
expression between normoxic and hypoxic conditions, there are two
possible explanations for the observed induction of p53 and PPM1D
proteins (Fig. 1B). The first one is
that the binding of RBM38 to PPM1D's 3′-UTR is increasing
under hypoxic conditions, which results in the increased
translation of PPM1D mRNA, and the translated PPM1D is then
dephosphorylating RBM38 protein, inducing the translation of mutant
TP53 mRNA. Alternatively, there is a secondary mechanism of
regulating PPM1D expression under hypoxic conditions. To test the
first possible explanation, NCI-H1770 variants stably expressing
either a control shRNA or shRNA targeting RBM38 were
generated. Successful knockdown was verified by western blot
analysis (Fig. 1E). Cells expressing
the control or RBM38 shRNA were subjected to hypoxia. Even
though it has been shown before that RBM38 regulates translation of
HIF1A mRNA (15), knockdown
of RBM38 did not affect HIF1α protein induction (Fig. 1E). This was not surprising given that
HIF1α is known to be regulated at multiple different levels.
RBM38 knockdown did not affect the induction in protein
expression of PPM1D or mutant p53 (Fig.
1E). However, when the same experiment was repeated under
normoxic conditions, knockdown of RBM38 resulted in a
significant decrease in PPM1D protein expression, and an increase
in p53 protein expression (Fig. 1F),
corroborating findings from other studies performed in normoxic
conditions (9,10,17).
This indicated that alternate post-transcriptional regulatory
mechanism(s) were regulating PPM1D expression under hypoxic
conditions.
Differential regulation of PPM1D in
A549 cells harboring wild-type or mutant TP53
To determine whether such a potential alternate
regulatory mechanism of PPM1D protein expression operated in NSCLC
cells harboring wild-type TP53, the A549 cells were
subjected to hypoxia. Hypoxia was induced by incubating cells at 1%
O2 for 4 h and confirmed via the induction of HIF1α
protein expression (Fig. 2A). The
protein expression of RBM38, p53, p-p53 (serine 15) and PPM1D was
not significantly different under normoxic and hypoxic conditions
(Fig. 2A), indicating a differential
response to hypoxia in NSCLC cells harboring wild-type or mutant
TP53. Given that A549 cells are epithelial cells from a lung
tumor mass, whereas the NCI-H1770 cells are neuroendocrine cells
derived from lung cancer metastasis in the lymph node, the
different origin of A549 and NCI-H1770 was then investigated for
the difference observed in A549 and NCI-H1770 cells under hypoxia.
A549 cells were transfected with a HA-tagged R428W mutant
TP53 plasmids. Stably selected cells were then transduced
with shRNA targeting the 3′-UTR of TP53. Successful knockdown of
the endogenous (>90% knockdown was achieved) and overexpression
of mutant TP53 were verified via western blot analysis
assays using p53 and HA antibodies (Fig.
2B). To determine whether the regulation of PPM1D protein
expression in the TP53-mutant A549 cells was such as that
observed in NCI-H1770 cells (Fig. 1E and
F), variants of the A549-R248W ΔTP53 cells stably expressing
either a control shRNA or shRNA targeting RBM38 were
generated. Successful knockdown was verified by western blot
analysis (Fig. 2C). Cells expressing
the control or RBM38 shRNA were either maintained under
normoxic condition or subjected to hypoxia. As in NCI-H1770 cells,
hypoxia treatment resulted in increased PPM1D and HA-p53 protein
expression (Fig. 2C, first vs. third
lane). RBM38 knockdown, as in the NCI-H1770 cells, did not
affect the protein expression of PPM1D or mutant p53 under hypoxia
(Fig. 2C). However, when the same
experiment was repeated under normoxic conditions, knockdown of
RBM38 resulted in significant decrease in PPM1D and increase
in mutant p53 protein expression levels (Fig. 2C). Cumulatively, these results
confirm that the difference observed in A549 and NCI-H1770 cells
under hypoxia was not due to the different cellular origin of the
NCI-H1770 and A549 cells. These results also confirm that in NSCLC
cells harboring R248W mutant TP53, the induction of PPM1D
protein expression is independent of RBM38 protein expression.
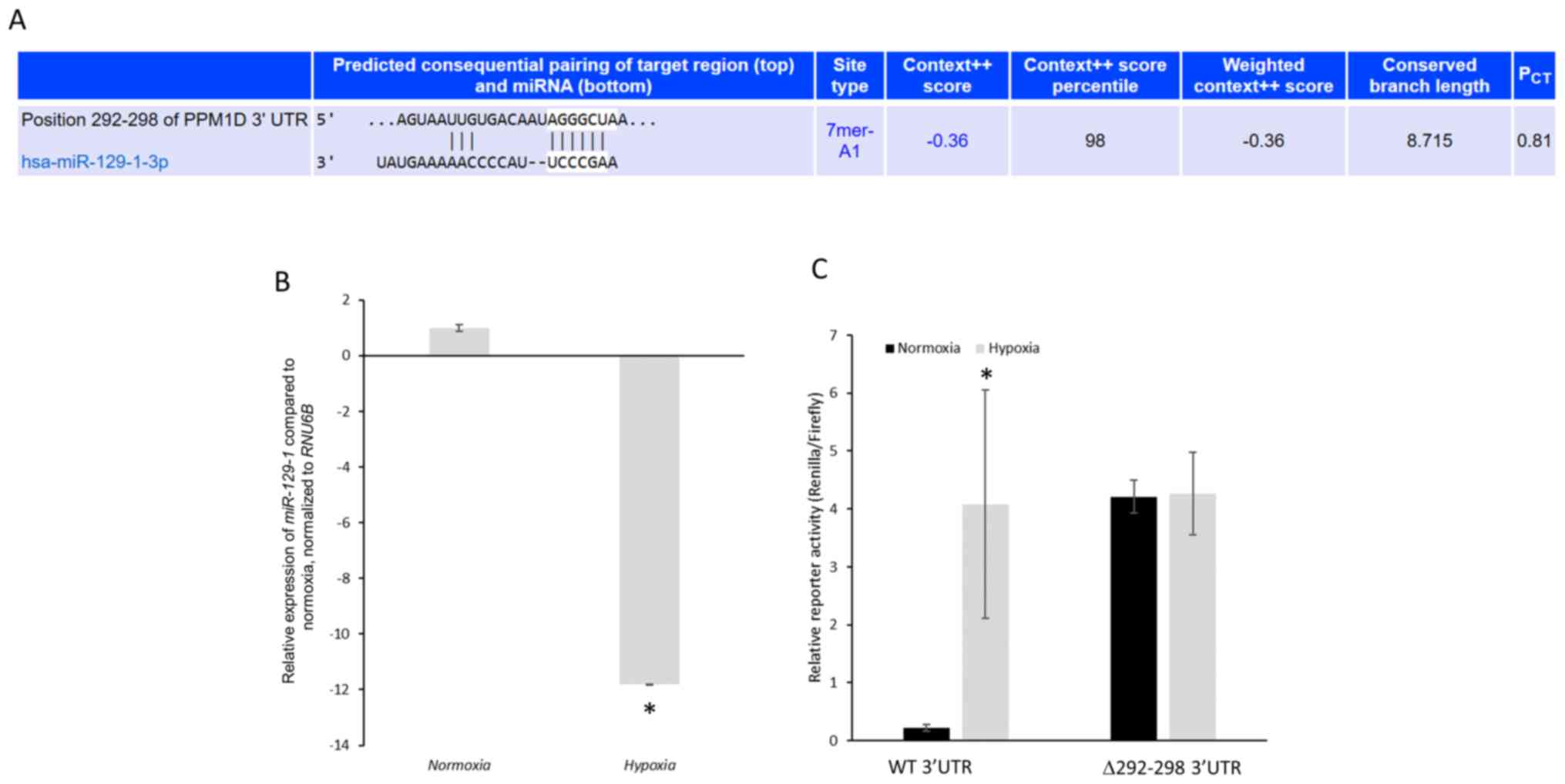
PPM1D is a putative target of
miR-129-1-3p
TargetScan algorithm (36) was used for the prediction of
potential microRNAs (miRNAs) targeting PPM1D mRNA. There was
only one conserved miRNA, miR-129-1-3p, predicted to target
nucleotides 292–298 of the 3′-UTR of PPM1D mRNA (Fig. 3A). miR-129 has previously been
demonstrated to function as a tumor suppressor in lung cancer by
regulating cell proliferation and metastatic progression (37,38).
Hence, whether PPM1D is targeted by miR-129-1-3p in lung
cancer cells harboring mutant TP53 and the effects on PPM1D
protein expression under normoxic and hypoxic conditions were
investigated.
Primarily, miR-129-1-3p expression levels in
NCI-H1770 cells under normoxic and hypoxic conditions was
determined. Hypoxia caused an 11.799±0.002-fold decrease in
miR-129-1-3p expression compared with normoxic conditions (Fig. 3B). To investigate whether
PPM1D mRNA is a direct target of miR-129-1-3p, luciferase
reporter plasmids harboring either the wild-type 3′-UTR or mutant
3′-UTR (miR-129-1-3p binding site, nucleotides 292–298, deleted)
were generated. These constructs were transfected in the NCI-H1770
cells and their expression was determined under normoxic and
hypoxic conditions. The mutant reporter was expressed significantly
higher compared with the wild-type reporter under normoxic
conditions (4.21±0.28 vs. 0.23±0.06, respectively;
P=1.19×10−5) (Fig. 3C).
However, following hypoxia induction, both the wild-type and mutant
reporters were robustly expressed without any significant
difference (4.08±1.97 vs. 4.25±0.71, P=0.89) (Fig. 3C).
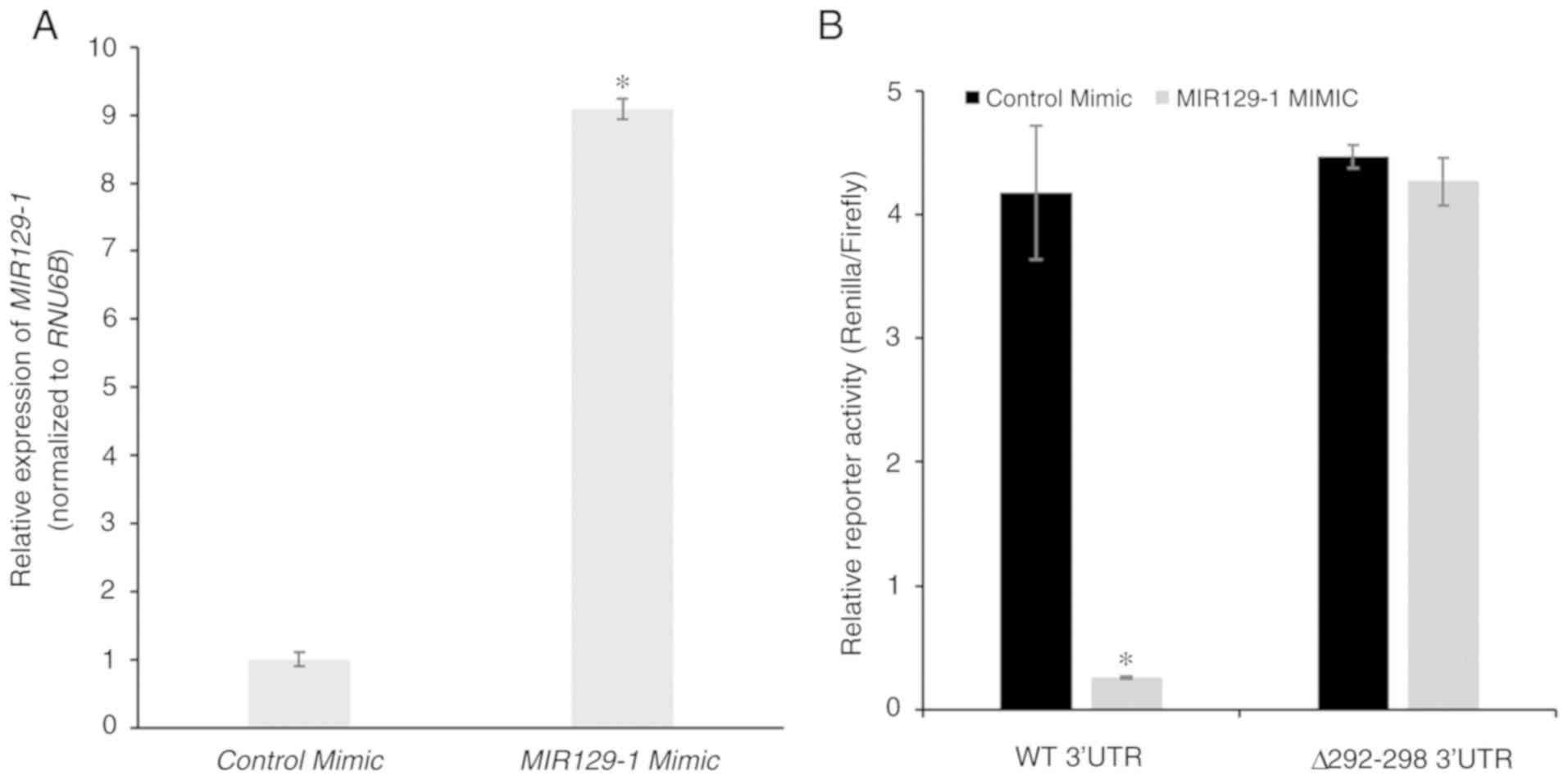
PPM1D is targeted by miR-129-1-3p in
NCI-H1770 cells under normoxia conditions
The aforementioned results indicate that the
decrease in miR-129-1-3p expression under hypoxic conditions may
explain the increase in protein expression of PPM1D. To test this
hypothesis, control or MIR129-1-3p mimic was transiently
transfected in the NCI-H1770 cells and overexpression was confirmed
via RT-qPCR (Fig. 4A). The mimic
transfected cells were co-transfected with the wild-type and mutant
luciferase reporters and subjected to hypoxia. Reporter expression
was significantly downregulated in cells transfected with
miR-129-1-3p mimic. However, this was not observed with cells
transfected with the control mimic (4.18±0.54 vs. 0.26±0.01,
respectively; P=3.32×10−6; Fig. 4B). No significant difference was
observed in the expression of the mutant reporter following
transfection of the miR-129-1-3p mimic (4.47±0.09 vs. 4.27±0.19,
P=0.41; Fig. 4B). These results
along with those presented in Fig.
2C confirm that miR-129-1-3p is targeting PPM1D mRNA in
lung cancer cells harboring mutant TP53 under normoxic
conditions.
Co-occurrence of PPM1D/RBM38 and
PPM1D/HIF1A mutations in patients with NSCLC
Based on the aforementioned observations, a TCGA
dataset on NSCLC samples was analyzed to look at genomic
alterations in PPM1D, RBM38, TP53, MIR129-1-3P and
HIF1A in 2,628 patients (2,855 samples) (Fig. S1) (29–35).
TP53 mutations were present in 61% of the cases, whereas
RBM38, PPM1D and HIF1A mutations were present in 5, 3
and 4%, respectively. Most of these were point mutations, even
though some genomic amplifications and deep deletions were also
observed (Fig. 5A).
MIR129-1-3P mutation data were missing among the datasets
selected for this study. Notably, when the tendency of
co-occurrence of mutations in the aforementioned genes was
examined, PPM1D/HIF1A and PPM1D/RBM38
exhibited significant co-occurrence (P=0.016 and 0.029,
respectively; Table SI).
RBM38 and TP53 mutations had a tendency of mutual
exclusivity, even though this was not significant. Next, the
difference in overall survival of patients with genomic alterations
in PPM1D, RBM38, HIF1A, and TP53 (n=695) and those
without any alterations in these genes (n=259) was investigated.
The median survival time was 45.4 months in patients with no
alterations vs. 43.26 months in patients with alterations (P=0.511;
Log rank t-test; Fig. 5B). These
results indicate that in a small cohort of patients with NSCLC,
PPM1D mutations tend to co-occur with RBM38 or
HIF1A; however, not both at the same time.
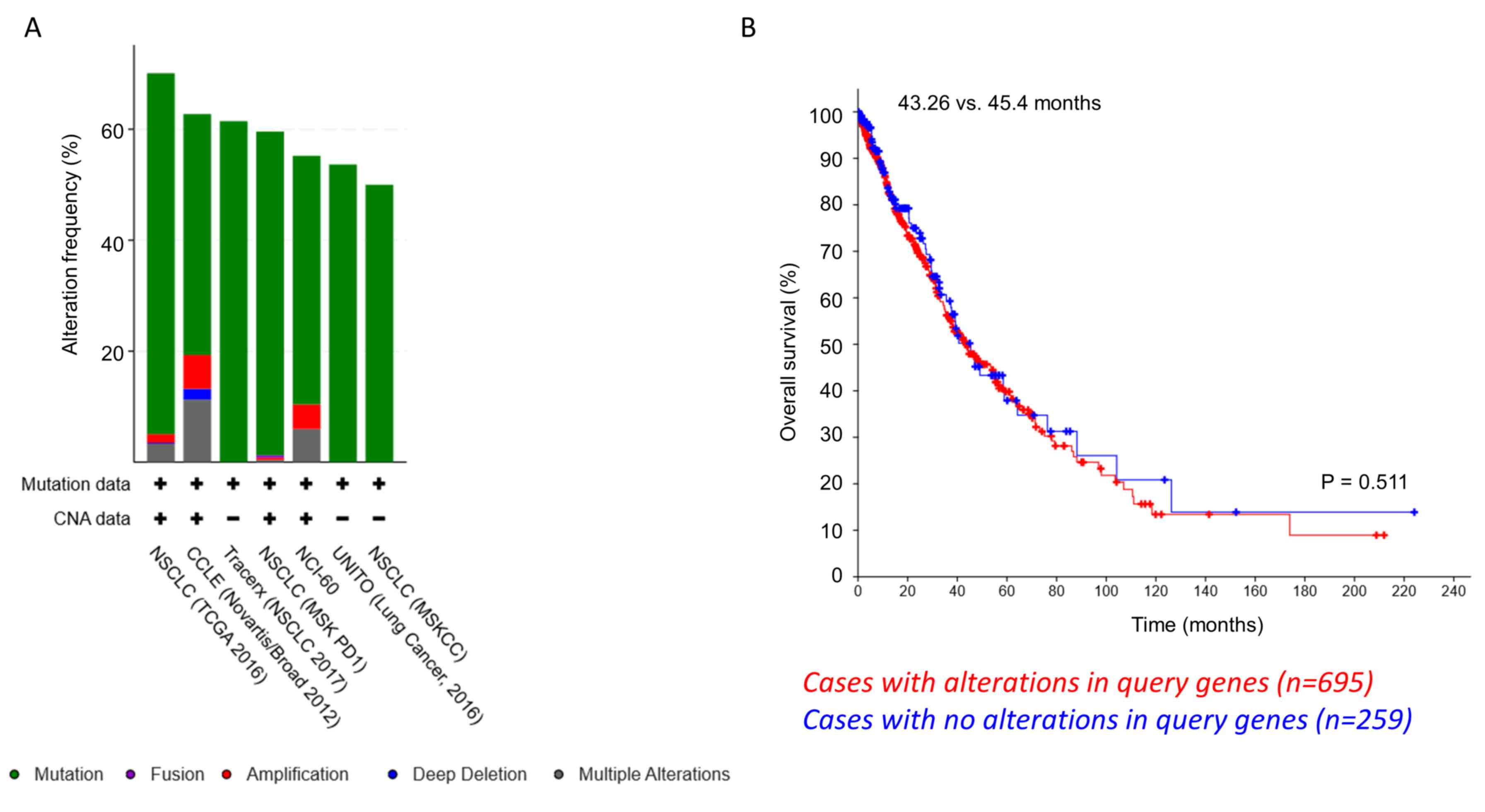 | Figure 5.Co-occurrence of PPM1D/RBM38
and PPM1D/HIF1A mutations in patients with NSCLC. (A)
Amplification and somatic mutations of TP53, PPM1D, RBM38 and HIF1A
in NSCLC cell lines and patients. Data analyzed were retrieved from
TCGA and referred to 2,628 patients (2,855 samples) from 7 studies.
The analysis based on the cancer studies is presented. (B)
Kaplan-Meier overall survival analysis curve did not reveal any
significant difference in survival times between patients with
NSCLC with (n=695 patients) and without (n=259 patients) genomic
amplification or somatic mutation in TP53, PPM1D, RBM38 and
HIF1A. P=0.551. TCGA, The Cancer Genome Atlas; PPM1D,
p53-induced phosphatase 1D; shRNA, short hairpin RNA; RBM38, RNA
binding motif protein 38; HIF1α, hypoxia-inducible factor 1α;
NSCLC, non-small cell lung cancer. |
Discussion
In the present study, miR-129-1-3p, and not RBM38,
was revealed to regulate PPM1D protein expression in mutant
p53-harboring NSCLC cells under hypoxia. Under normoxic conditions,
even though PPM1D expression was higher in the NCI-H1770 cells,
PPM1D protein was detectable in A549 cells with wild-type p53.
However, high expression of miR-129-1-3p in NCI-H1770 cells
maintained under normoxic conditions was detected. miR-129-1-3p
expression was downregulated under hypoxia resulting in a further
induction of PPM1D protein.
Even though miR-129-1-3p levels were high in
normoxic conditions in NCI-H1770 cells, there was not a complete
halt in PPM1D translation. One explanation may be that RBM38 is
present and is known to induce translation of PPM1D. Thus,
there may be a homeostatic balance between RBM38 and miR-129-1-3p
that results in decreased translation of PPM1D under
normoxic conditions.
Notably, despite the fact that RBM38 is required for
the translation of mutant TP53 and PPM1D (1,6,8,9), no
changes in the protein expression levels of p53 or PPM1D in
NCI-H1770 cells or mutant A549 following after knockdown of
RBM38 were observed. One argument can be the inefficient
knockdown of RBM38, allowing the residual protein to bind
and translate PPM1D and mutant TP53 mRNAs. However,
it is surprising that there was no decrease in PPM1D and p53
protein expression following the 80–90% decrease in RBM38 protein
expression. Thus, the mechanism that regulates PPM1D translation in
cases where RBM38 is not present needs to be determined. Of note,
RBM38 deletion is observed in tumors harboring mutant p53 (39,40). A
limitation of the present study was that the mutant A549 cells used
were not generated by CRISPR/Cas9-mediated targeted mutation which
would have been a more robust model.
Conversely, the expression level of miR-129-1-3p in
normal lung epithelial cells and lung cancer cells with wild-type
p53 needs to be determined. Even though HIF1α is known to complex
with mutant p53 alone to transcriptionally upregulate extracellular
matrix component proteins (19),
whether that complex is also downregulating miR-129-1-3p expression
and consequently inducing PPM1D protein expression is yet to be
elucidated. Finally, functional assays need to be performed to
define the significance of miR-129-1-3p-mediated PPM1D regulation
in NSCLC cells with mutant p53 under hypoxic conditions.
Lung cancer cells, both with wild-type or mutant
p53, experience hypoxic conditions, both during initial
tumorigenesis and during metastatic progression (41). PPM1D is a target of p53 and a
key modulator of the p53 genomic surveillance mechanism in normal
cells (1,6,7). RBM38
switches from a repressor of translation of p53 according to its
phosphorylation by GSK3β (10) and
dephosphorylation by PPM1D (8).
Considering that miR-129-1-3p was demonstrated in the present study
to regulate PPM1D expression under hypoxic conditions, whether and
how miR-129-1-3p expression is regulated during both tumor
initiation and progression, and whether it is a direct target of
p53 or mutant p53-HIF1α transcriptional complex, remains to be
determined.
In summary, a yet undefined mechanism of miR-129-1-
3p-mediated regulation of PPM1D protein expression in NSCLC cells
with mutant p53 under hypoxic conditions was identified. Whether a
similar mechanism exists in other tumor types with wild-type or
mutant p53 remains to be determined. In addition, the mechanism by
which co-occurring PPM1D/HIF1A and PPM1D/RBM38 mutations affect
tumor initiation and disease progression in lung cancer patients
would be of interest to investigate.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Heilongjiang
Province Applied Technology Research and Development Program (grant
no. GZ19C01).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HLY and QYL designed the experiments; HWX and QYL
performed the experiments; HLY, HWX and QYL analyzed the
experimental results. HLY wrote the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deng W, Li J, Dorrah K, Jimenez-Tapia D,
Arriaga B, Hao Q, Cao W, Gao Z, Vadgama J and Wu Y: The role of
PPM1D in cancer and advances in studies of its inhibitors. Biomed
Pharmacother. 125:1099562020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiscella M, Zhang HL, Fan SJ, Sakaguchi K,
Shen SF, Mercer WE, Vande Woude GF, O'Connor PM and Appella E:
Wip1, a novel human protein phosphatase that is induced in response
to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li J, Yang Y, Peng Y, Austin RJ, van
Eyndhoven WG, Nguyen KC, Gabriele T, McCurrach ME, Marks JR, Hoey
T, et al: Oncogenic properties of PPM1D located within a breast
cancer amplification epicenter at 17q23. Nat Genet. 31:133–134.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nannenga B, Lu X, Dumble M, Van Maanen M,
Nguyen TA, Sutton R, Kumar TR and Donehower LA: Augmented cancer
resistance and DNA damage response phenotypes in PPM1D null mice.
Mol Carcinog. 45:594–604. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan DS, Lambros MB, Rayter S, Natrajan R,
Vatcheva R, Gao Q, Marchiò C, Geyer FC, Savage K, Parry S, et al:
PPM1D is a potential therapeutic target in ovarian clear cell
carcinomas. Clin Cancer Res. 15:2269–2280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu X, Ma O, Nguyen TA, Jones SN, Oren M
and Donehower LA: The Wip1 phosphatase acts as a gatekeeper in the
p53-Mdm2 autoregulatory loop. Cancer Cell. 12:342–354. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu X, Nannenga B and Donehower LA: PPM1D
dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints.
Genes Dev. 19:1162–1174. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang M, Xu E, Zhang J and Chen X: PPM1D
phosphatase, a target of p53 and RBM38 RNA-binding protein,
inhibits p53 mRNA translation via dephosphorylation of RBM38.
Oncogene. 34:5900–5911. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Cho SJ, Shu L, Yan W, Guerrero T,
Kent M, Skorupski K, Chen H and Chen X: Translational repression of
p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev.
25:1528–1543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang M, Zhang J, Chen XL, Cho SJ and Chen
XB: Glycogen synthase kinase 3 promotes p53 mRNA translation via
phosphorylation of RNPC1. Genes Dev. 27:2246–2258. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shu L, Yan W and Chen X: RNPC1, an
RNA-binding protein and a target of the p53 family, is required for
maintaining the stability of the basal and stress-induced p21
transcript. Genes Dev. 20:2961–2972. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feldstein O, Ben-Hamo R, Bashari D, Efroni
S and Ginsberg D: RBM38 is a direct transcriptional target of E2F1
that limits E2F1-induced proliferation. Mol Cancer Res.
10:1169–1177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heinicke LA, Nabet B, Shen S, Jiang P, van
Zalen S, Cieply B, Russell JE, Xing Y and Carstens RP: The RNA
binding protein RBM38 (RNPC1) regulates splicing during late
erythroid differentiation. PLoS One. 8:e780312013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Warzecha CC, Sato TK, Nabet B, Hogenesch
JB and Carstens RP: ESRP1 and ESRP2 are epithelial
cell-type-specific regulators of FGFR2 splicing. Mol Cell.
33:591–601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olivier M, Eeles R, Hollstein M, Khan MA,
Harris CC and Hainaut P: The IARC TP53 database: New online
mutation analysis and recommendations to users. Hum Mutat.
19:607–614. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Xu E, Ren C, Yang HJ, Zhang Y,
Sun W, Kong X, Zhang W, Chen M, Huang E and Chen X: Genetic
ablation of Rbm38 promotes lymphomagenesis in the context of mutant
p53 by downregulating PTEN. Cancer Res. 78:1511–1521. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho SJ, Teng IF, Zhang M, Yin T, Jung YS,
Zhang J and Chen X: Hypoxia-inducible factor 1 alpha is regulated
by RBM38, a RNA-binding protein and a p53 family target, via mRNA
translation. Oncotarget. 6:305–316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amelio I, Mancini M, Petrova V, Cairns RA,
Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena
Acevedo JD, et al: P53 mutants cooperate with HIF-1 in
transcriptional regulation of extracellular matrix components to
promote tumor progression. Proc Natl Acad Sci USA.
115:E10869–E10878. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang WC: MicroRNAs tune oxidative stress
in cancer therapeutic tolerance and resistance. Int J Mol Sci.
20:E60942019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu X, Song H, Xia T, Han S, Xiao B, Luo L,
Xi Y and Guo J: Growth inhibitory effects of three miR-129 family
members on gastric cancer. Gene. 532:87–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bandres E, Agirre X, Bitarte N, Ramirez N,
Zarate R, Roman-Gomez J, Prosper F and Garcia-Foncillas J:
Epigenetic regulation of microRNA expression in colorectal cancer.
Int J Cancer. 125:2737–2743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Hu H, Guan X, Xiong G, Wang Y,
Wang K, Li J, Xu X, Yang K and Bai Y: CpG island methylation status
of miRNAs in esophageal squamous cell carcinoma. Int J Cancer.
130:1607–1613. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dyrskjot L, Ostenfeld MS, Bramsen JB,
Silahtaroglu AN, Lamy P, Ramanathan R, Fristrup N, Jensen JL,
Andersen CL, Zieger K, et al: Genomic profiling of microRNAs in
bladder cancer: MiR-129 is associated with poor outcome and
promotes cell death in vitro. Cancer Res. 69:4851–4860. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmaltz C, Hardenbergh PH, Wells A and
Fisher DE: Regulation of proliferation-survival decisions during
tumor cell hypoxia. Mol Cell Biol. 18:2845–2854. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Science Signal. 6:112013. View Article : Google Scholar
|
|
28
|
Unberath P, Knell C, Prokosch HU and
Christoph J: Developing new analysis functions for a translational
research platform: Extending the cBioPortal for cancer genomics.
Stud Health Technol Inform. 258:46–50. 2019.PubMed/NCBI
|
|
29
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reinhold WC, Sunshine M, Liu H, Varma S,
Kohn KW, Morris J, Doroshow J and Pommier Y: CellMiner: A web-based
suite of genomic and pharmacologic tools to explore transcript and
drug patterns in the NCI-60 cell line set. Cancer Res.
72:3499–3511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rizvi H, Sanchez-Vega F, La K, Chatila W,
Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N,
et al: Molecular determinants of response to anti-programmed cell
death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in
patients with non-small-cell lung cancer profiled with targeted
next-generation sequencing. J Clin Oncol. 36:633–641. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jamal-Hanjani M, Wilson GA, McGranahan N,
Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R,
Rosenthal R, et al: Tracking the evolution of non-small-cell lung
cancer. N Engl J Med. 376:2109–2121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vavalà T, Monica V, Lo Iacono M, Mele T,
Busso S, Righi L, Papotti M, Scagliotti GV and Novello S: Precision
medicine in age-specific non-small-cell-lung-cancer patients:
Integrating biomolecular results into clinical practice-A new
approach to improve personalized translational research. Lung
Cancer. 107:84–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rizvi NA, Hellmann MD, Snyder A, Kvistborg
P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al: Cancer
immunology. Mutational landscape determines sensitivity to PD-1
blockade in non-small cell lung cancer. Science. 348:124–128. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Campbell JD, Alexandrov A, Kim J, Wala J,
Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et
al: Distinct patterns of somatic genome alterations in lung
adenocarcinomas and squamous cell carcinomas. Nat Genet.
48:607–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Agarwal V, Bell GW, Nam J and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar
|
|
37
|
Li J, Wang H, Ke H and Ni S: MiR-129
regulates MMP9 to control metastasis of non-small cell lung cancer.
Tumour Biol. 36:5785–5790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu J, Qian J, Li C, Kwok L, Cheng F, Liu
P, Perdomo C, Kotton D, Vaziri C, Anderlind C, et al: MiR-129
regulates cell proliferation by downregulating Cdk6 expression.
Cell Cycle. 9:1809–1818. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wampfler J, Federzoni EA, Torbett BE, Fey
MF and Tschan MP: The RNA binding proteins RBM38 and DND1 are
repressed in AML and have a novel function in APL differentiation.
Leuk Res. 41:96–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leveille N, Elkon R, Davalos V, Manoharan
V, Hollingworth D, Oude Vrielink J, le Sage C, Melo CA, Horlings
HM, Wesseling J, et al: Selective inhibition of microRNA
accessibility by RBM38 is required for p53 activity. Nat Commun.
2:513–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kakkad S, Krishnamachary B, Jacob D,
Pacheco-Torres J, Goggins E, Bharti SK, Penet MF and Bhujwalla ZM:
Molecular and functional imaging insights into the role of hypoxia
in cancer aggression. Cancer Metastasis Rev. 38:51–64. 2019.
View Article : Google Scholar : PubMed/NCBI
|