Introduction
Uterine serous carcinoma (USC) accounts for 10% of
all endometrial cancer worldwide; however, it contributes
disproportionately to endometrial cancer mortality, whereby 40% of
endometrial cancer-associated mortalities in 2016 were attributed
to its high-risk histological subtype (1). Approximately 50% of patients are
diagnosed with International Federation of Gynecology and
Obstetrics (FIGO) stage 3–4, and the 5-year disease-specific
survival rate for patients with advanced stage USC is reported to
be 33% (2). Currently, the clinical
management of advanced stage USC is based on primary cytoreductive
surgery, followed by systematic chemotherapy (3). The adjuvant platinum/taxane
chemotherapy decreases the recurrence risk and improves survival
outcomes (3). However, most patients
with advanced stage USC develop recurrent disease, which is
resistant to chemotherapy and lethal in the majority of cases
(4). Thus, it remains crucial to
elucidate how USC becomes chemotherapy-resistant, and to develop
novel treatment for recurrent USC, which are more effective
compared with that in current therapies.
Prooxidant cancer therapies, including ionizing
radiation and chemotherapy induce the production of reactive oxygen
species (ROS) in cancer cells, and the addition of an additional
oxidant stimulus to the constitutive oxidative stress in cancer
cells causes the collapse of the antioxidant systems and thereby
leads to cell death (5). Glutathione
(GSH) is a primary antioxidant, which protects cells against ROS,
and high levels of GSH have been associated with increasing cancer
cell survival and resistance to chemotherapy in ovarian and
prostate cancer cells (6,7). Thus, the intracellular redox balance
can determine the sensitivity of cancer cells to chemotherapeutic
agents (8). Recently, metabolomics
analyses demonstrated that GSH levels in a paclitaxel-resistant USC
cell line were higher compared with that in a paclitaxel-sensitive
cell line (9), which suggests that
increased GSH levels are associated with the resistance to
paclitaxel in USC. Intracellular GSH synthesis requires cystine
uptake, and this involves the xc− system, a
cystine-glutamate exchange transporter composed of xCT (the
light-chain and active subunit) and F42hc (the heavy-chain
subunit); xCT expression is essential for the cystine uptake
necessary for intracellular GSH synthesis, and thus xCT can
determine the intracellular redox balance (10,11).
Notably, xCT inhibition has been extensively demonstrated to
overcome GSH-mediated resistance to chemotherapeutic agents in head
and neck squamous carcinoma, lung cancer and colorectal cancer
(12–14). However, whether xCT inhibition can
overcome paclitaxel resistance in USC remains unknown.
Sulfasalazine (SAS), an anti-inflammatory drug,
which is clinically used for treating bowel disease and rheumatoid
arthritis (15), is a specific
inhibitor of xCT-mediated cystine transporter (10,16). SAS
can scavenge ROS, as an anti-inflammatory drug (17), inhibit leukocyte motility and
interleukin (IL)-1 and IL-2 production (18), and inhibit nuclear factor κ B (NFκB)
(19). SAS has also been reported to
effectively induce GSH depletion (90%) and arrest growth, and to
enhance sensitivity to chemotherapeutic agents in pancreatic,
prostate and mammary cancer (20–23). As
SAS inhibits a cystine-glutamate transporter, it can promote
ferroptosis, a non-apoptotic form of cell death (24,25).
This lethal process is defined by iron-dependent accumulation of
lipid ROS (24,25), and cancer cells exhibiting high
levels of Ras activity or p53 expression may be sensitized to this
process (24).
The present study aimed to investigate whether the
xCT inhibitor, SAS, induced cytotoxicity and cell death using
cytotoxicity and cell death assays in the paclitaxel-sensitive
USPC1 and -resistant PTX1 cell lines. Furthermore, the molecular
mechanism by which SAS induces cell death in cancer cells was
assessed.
Materials and methods
Cell culture
Human uterine serous papillary carcinoma-1 (USPC1)
cell lines were established by Dr Santin at the Department of
Obstetrics and Gynecology, Division of Gynecologic Oncology, Yale
University (New Haven, United States) (26), from patients who experienced rapid
tumor progression during adjuvant chemotherapy following primary
surgery. The paclitaxel-resistant cell line, PTX1 was established
from paclitaxel-sensitive USPC1 cells. To generate
paclitaxel-resistant cells, USPC1 cells were continuously exposed
to doses of paclitaxel half-maximal inhibitory concentration (IC50)
values. Cells were split every 2 weeks and cultured in RPMI-1640
(Thermo Fisher Scientific, Inc.) medium supplemented with 10% FBS
(Cytiva), an antibiotic-antimycotic mixture (100 U/ml penicillin,
100 µg/ml streptomycin and 250 ng/ml amphotericin B; Thermo Fisher
Scientific, Inc.) and GlutaMax supplement (2 mM L-glutamine; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere with 5%
CO2, with recalculated paclitaxel IC50 values for 3
months (9).
UCPC1 and PTX1 cells were provided by Dr Yaegashi at
the Department of Gynecology and Obstetrics, Tohoku University
Graduate School of Medicine (Sendai, Japan). Cells were cultured in
RPMI-1640 medium supplemented with 10% FBS, an
antibiotic-antimycotic mixture (100 U/ml penicillin, 100 µg/ml
streptomycin and 250 ng/ml amphotericin B) and GlutaMax supplement
(2 mM L-glutamine), at 37°C in a humidified atmosphere of 95% air
and 5% CO2.
The human ovarian cancer SKOV3 cell line was
obtained from the American Type Culture Collection. Cells were
cultured in M199:105 (Sigma-Aldrich; Merck KGaA) medium with 10%
FBS and 1% penicillin-streptomycin (Sigma-Aldrich; Merck KGaA) at
37°C in a humidified atmosphere with 5% CO2.
Antibodies and reagents
The following antibodies were purchased against: xCT
(1:1,000; cat. no. ab37185; Abcam), cleaved-PARP (1:1,000; cat. no.
9541; Cell Signaling Technology, Inc.), Akt (1:1,000; cat. no.
9272; Cell Signaling Technology, Inc.), phosphorylated (p)-Akt
(1:1,000; cat. no. 4058; Cell Signaling Technology, Inc.), p44/42
MAPK (Erk1/2; 1:1,000; cat. no. 4695; Cell Signaling Technology,
Inc.), p-p44/42 MAPK (1:1,000; cat. no. 9106; Cell Signaling
Technology, Inc.), p-SAPK/JNK (1:1,000; cat. no. 9251; Cell
Signaling Technology, Inc.), JNK2 (1:200; cat. no. sc7345; Santa
Cruz Biotechnology, Inc.) and β-actin (1:1,000; cat. no. A2228;
Sigma-Aldrich; Merck KGaA).
The following reagents were purchased and dissolved
in DMSO or distilled water to prepare the stock solutions: 100 mM
SAS, 10 mM 2′,7′-dichlorofluorescin diacetate (DCFH-DA) and 10 mM
ferrostatin-1 (all dissolved in DMSO and from Sigma-Aldrich; Merck
KGaA). A total of 10 mM deferoxamine mesylate (DFO) (cat. no.
ab120727; Abcam) was dissolved in water and 10 mM Z-VAD-FMK
(Peptide Institute, Inc.) in DMSO.
Cytotoxicity and cell death
assays
Following treatment at 37°C in a humidified
atmosphere with 5% CO2 with paclitaxel (0, 0.1, 1, 10,
50 or 100 nM) for 72 h, or treatment with SAS (100 or 200 µM) and
paclitaxel (0, 0.1, 1, 10, 100 or 1,000 nM) for 72 h in USPC1 and
PTX1 cells, or treatment with ferrostatin-1 (1 µM) or Z-VAD-FMK
(100 µM) for 1 h and subsequent treatment with SAS (0, 200, 400,
600, 800, 1,000 µM) for 72 h in PTX1 cells, or treatment with DFO
(1 µM) for 1 h and subsequent treatment with SAS (400 µM) for 48 h
in PTX1 cells, cell viability was assessed using the tetrazolium
compound, MTS, at 37°C for 1.5 h according to the manufacturer's
protocol (Promega Corporation). Following the MTS assay, samples
were incubated with CellTiter 96® AQueous One Solution
reagent (Promega Corporation) for 90 min at 37°C and cell viability
was subsequently analyzed at 490 nm using a Model 680 Microplate
Reader® (Bio-Rad Laboratories, Inc.). Cell viability was
calculated from the ratio of cells treated with each of the drugs
to that of untreated cells, set as 1 (mean ± SD; n=8). Untreated
cells were used as a control.
The cell death assay was conducted as previously
described (27). Following treatment
at 37°C in a humidified atmosphere with 5% CO2 with SAS
(200 µM) for 24 h in USPC1 and PTX1 cells, or treatment with
ferrostatin-1 (1 µM) for 1 h and subsequent treatment with SAS (200
µM) for 24 h in PTX1 cells, cells were incubated with 1 µg/ml
propidium iodide (PI; Invitrogen; Thermo Fisher Scientific, Inc.)
and 10 µg/ml Hoechst 33342 (Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C for 10 min to stain dead cells and the cell nuclei,
respectively. Subsequently, the numbers of PI- and Hoechst-positive
cells were scored manually under a fluorescence microscope (CKX41;
Olympus Corporation; magnification, ×100) in five fields per well,
and the rate of PI-positive cells (dead cells) against
Hoechst-positive cells (total cells) was determined. All the
experiments were carried out in quadruplicate.
GSH and ROS measurements
To confirm the increase of GSH levels in a cell
number-dependent manner, cells were plated a density of 1,000,
3,000 or 5,000 cells/well in 96-well plates. After 24 h, GSH levels
were measured using the GSH-GloTM Glutathione assay according to
the manufacturer's protocol (Promega Corporation) in USPC1 and PTX1
cells. GSH levels were measured in both cells treated with SAS (400
µM) or paclitaxel (10 µM) at 37°C in a humidified atmosphere with
5% CO2 for 24 h using the GSH-GloTM Glutathione assay.
GSH levels were calculated from the ratio of cells treated with SAS
or paclitaxel to that of untreated USPC1 cells set as 1 (mean ± SD;
n=8). For ROS measurement, cells were treated with vehicle, SAS
(200 µM), paclitaxel (10 nM) or SAS + paclitaxel at 37°C in a
humidified atmosphere with 5% CO2 for 24 h. ROS levels
were measured following treatment of cells with 10 µM DCFH-DA for
30 min at 37°C. Cells were protected from light during the
respective procedures. Cells exhibiting a signal for DCFH-DA above
the gate established using the isotype control treated without
DCFH-DA were deemed ROS-positive. These cells were subsequently
used in FACS analysis to quantify the intensity of DCF
fluorescence, using a FACSCantoTM II flow cytometer (BD
Biosciences). All the experiments were carried out in
quadruplicate.
Western blot analysis
Cells were washed twice with cold PBS and lysed
using RIPA lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1%
SDS, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride). Protein
concentration was determined using a BioPhotometer®
(Eppendorf, Hamburg, Germany). Cell lysates containing equal
amounts of protein (25 µg/well) were separated using 5% SDS-PAGE
and transferred onto nitrocellulose membranes. Blocking was
performed using 5% skimmed milk powder or 3% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) in 1X TBS at room temperature for 1 h.
The membrane was sequentially probed with the aforementioned
primary antibodies overnight at 4°C, and then with an appropriate
horseradish peroxidase (HRP)-conjugated secondary antibody at room
temperature for 1 h, according to the manufacturer's protocols. The
secondary antibodies used were: Mouse IgG HRP Linked Whole Ab
(1:5,000; cat. no. NA931-1ML) and Rabbit IgG HRP Linked Whole Ab
(1:5,000; cat. no. NA934-1ML), both purchased from Cytiva.
Immunoreactive bands were visualized using Amersham™ ECLTM prime
western blotting detection reagent (Cytiva). SKOV3, USPC1, and PTX1
cell were cultured at 37°C in a humidified atmosphere with 5%
CO2 for 24 h, and the cell lysate were probed for
anti-xCT antibody using western blot analysis. USPC1 and PTX1 cells
were treated with vehicle, SAS (200 µM), paclitaxel (10 nM) or SAS
+ paclitaxel at 37°C in a humidified atmosphere with 5%
CO2 for 24 h, and the cell lysates were probed for
anti-cleaved-PARP antibody using western blot analysis. USPC1 and
PTX1 cells were cultured at 37°C in a humidified atmosphere with 5%
CO2 for 24 h, and the cell lysates were probed for
p-AKT, AKT, p-Erk, Erk, p-JNK or JNK antibody using western blot
analysis. PTX1 cells transfected with siRNAs against JNK or with a
control siRNA were cultured at 37°C in a humidified atmosphere with
5% CO2 for 24 h, and the cell lysates were probed for
anti-p-JNK or anti-JNK antibodies using western blot analysis.
Reverse transcription (RT) PCR
Total RNA was extracted from cells using the
RNeasy® Mini kit, and cDNA was synthesized from 1 µg RNA
using the QuantiTect® Reverse Transcription kit (both
from Qiagen K.K.). RT PCR was performed on a T100TM thermal cycler
(Bio-Rad Laboratories, Inc.). The following primer sequences were
used: CD44 forward, 5′-TCCCAGACGAAGACAGTCCCTGGAT-3′ and reverse,
5′-CACTGGGGTGGAATGTGTCTTGGTC-3′; and GAPDH forward,
5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCACCCTGTTGCTGTA-3′.
The thermocycling conditions for CD44 amplification involved an
initial denaturation step at 95°C for 3 min, followed by 34 cycles
of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec
and extension at 72°C for 1 min, with a final extension step at
72°C for 5 min. The thermocycling conditions for GAPDH involved an
initial denaturation step at 95°C for 3 min, followed by 29 cycles
of denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec
and extension at 72°C for 1 min, with a final extension step at
72°C for 5 min. PCR products were electrophoresed on 2% agarose
gels containing ethidium bromide and photographed. The exons 1–5
and 16–20 that encode the constant part of CD44 are included in all
CD44 isoforms (28). Exons 6–15 that
encode the variant exons v1-v10 are either completely excluded, as
in CD44s, or are included in various combinations within the CD44
ectodomain, giving rise to the CD44 variant isoforms (CD44v)
(28). CD44v8-10 contains the
variant exons 8–10 and has more bases than CD44s. The primer for
CD44 can detect both CD44s (83 bp) and CD44v8-10 (479 bp).
Gene silencing by small interfering
(si)RNA
siRNAs against human JNK2 (cat. no. VHS40729) as
well as Medium GC Duplex #2 of Stealth RNAiTM siRNA
Negative Control Duplexes were purchased from Invitrogen (Thermo
Fisher Scientific, Inc.). Transfection with siRNAs (100 nmol/l) was
performed using Lipofectamine® RNAiMAX™ reagent (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
After 4 h, the transfection medium was removed and replaced with
culture medium. The transfected cells were cultured at 37°C in a
humidified atmosphere with 5% CO2 for 24 h, and then
were used for western blot analysis, MTS assay and cell death
assay. All of the experiments were carried out in
quadruplicate.
Subcutaneous xenograft model and
treatment in vivo
The procedures involving animals used in the present
study were approved by the Animal Care Committee of Yamagata
University (approval no. 31009) in accordance with institutional
and Japanese government guidelines for animal experiments. The
total number of mice used was 14 in all experiments. The mean
weight of the mice at the start was 19.4±0.87 g. Housing conditions
were as follows: Temperature was 37°C, humidity was 30–40%,
light/dark cycle was every 12 h, food and water were sterilized and
available ad libitum. To generate the subcutaneous xenograft
model, USPC1 (5×106 cells) or PTX1 (5×106
cells) were suspended in 200 µl of PBS following determination of
cellular viability and injected into the subcutaneous tissue of
6-week-old female Crj:SHO-PrkdcscidHrhr
hairless SCID mice (n=2) (Charles River Laboratories Inc.). Tumor
formation was visually confirmed in mice inoculated with USPC1
cells, but not in those inoculated with PTX1 cells, thus the animal
study was performed using USPC1 cells. The recipient mice were
monitored for general health status and presence of subcutaneous
tumors once a week. Tumor volume was determined by measuring tumor
diameters (measurement of 2 perpendicular axes of the tumors) and
calculated as 1/2× (larger diameter)x(smaller
diameter)2. A total of two weeks following inoculation,
one group of mice (n=6) was administered with oral PBS five times a
week (Monday to Friday) for eight weeks. A second group of mice
(n=6) was administered with SAS suspension (250 mg/kg) orally five
times a week (Monday to Friday) for eight weeks. A total of nine
weeks following initialization of treatment, mice were euthanized
with 100% CO2 at a flow rate of 20% of the chamber
volume per minute, which was used for 5 min to reach 100%
CO2 in the chamber. Mice remained in 100% CO2
for ≥10 min to ensure that they were dead.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software (version 5.0; GraphPad Software, Inc.). Data are
presented as the mean ± SD (n=4). Unpaired Student's t-test was
used to compare differences between two groups, while one-way or
two-way ANOVA, followed by Bonferroni post hoc test were used to
compare differences between multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of SAS on the proliferation of
paclitaxel-sensitive and -resistant USC cells
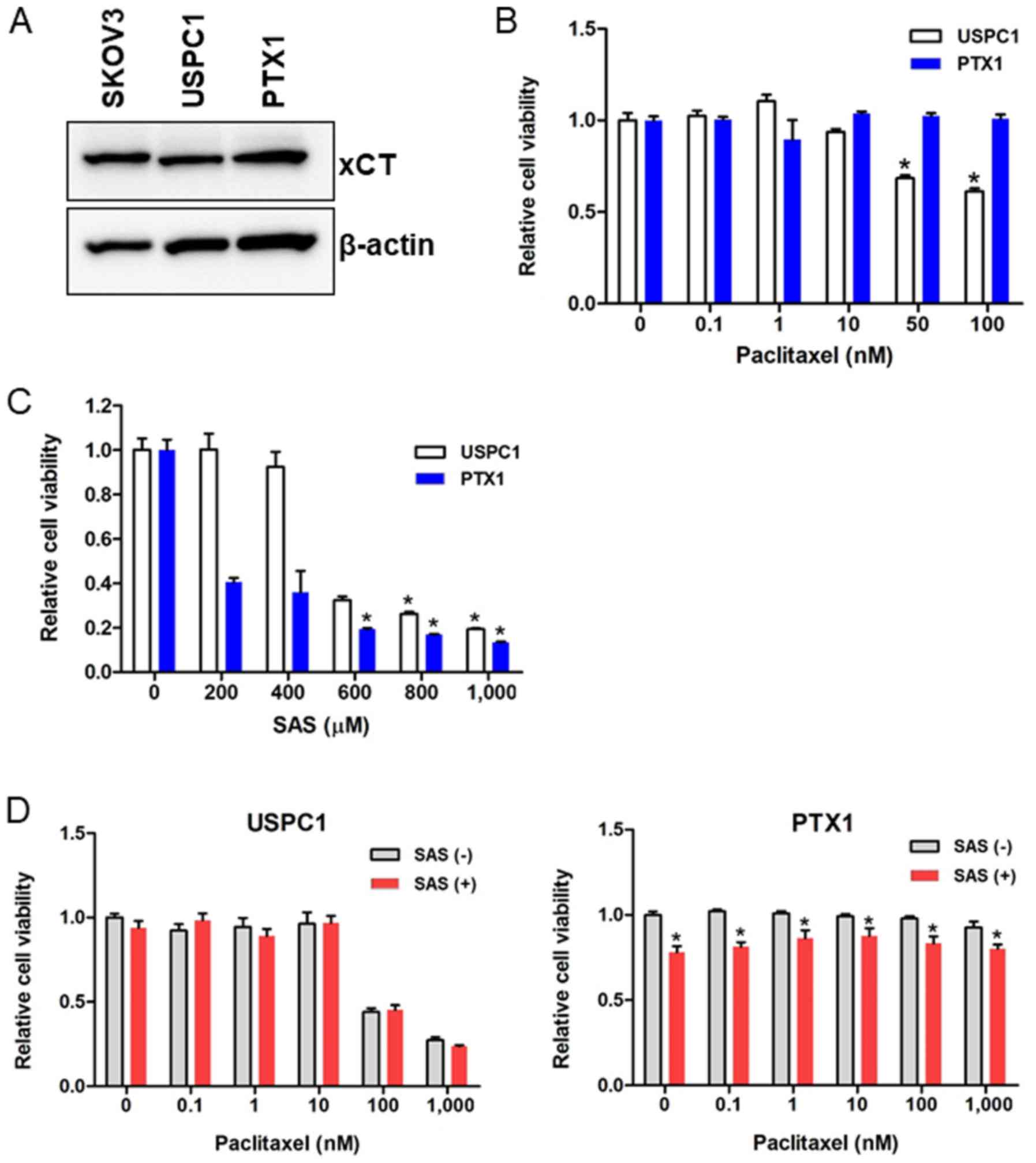
xCT expression in the USC cell lines was assessed
and the SKOV3 cells were used as the positive control, as they
express xCT. Western blot analysis demonstrated that xCT was highly
expressed in paclitaxel-sensitive USPC1 and paclitaxel-resistant
PTX1 cells (Fig. 1A). CD44, a major
extracellular matrix adhesion molecule, exists as several isoforms,
including CD44v, and is generated through mRNA splicing (29). CD44v can stabilize xCT and control
the intracellular redox status in an xCT activity-dependent manner
(30,31). RT-PCR analysis revealed that CD44s
and CD44v8-10 were expressed in USPC1 cells, but not in PTX1 cells
(Fig. S1).
Paclitaxel sensitivity in USPC1 and PTX1 cells was
subsequently investigated using the MTS assay. Cells were treated
with different concentrations of paclitaxel for 72 h, and cell
viability was significantly inhibited in paclitaxel-sensitive USPC1
cells but not paclitaxel-resistant PTX1 cells, compared with that
in the untreated control cells when >50 nM paclitaxel was used
(Fig. 1B).
To determine the effect of SAS on USC cell
viability, cells were treated with different concentrations of SAS
for 72 h. SAS significantly inhibited cell viability in PTX1 cells,
in a dose-dependent manner compared with that in the untreated
control cells, and significantly decreased cell viability in USPC1
cells when >600 µM SAS was used (Fig.
1C).
The effect of SAS on paclitaxel cytotoxicity was
also assessed by treating cells with different concentrations of
paclitaxel alone or in combination with SAS for 72 h. In USPC1
cells, combination of paclitaxel with 200 µM SAS had no significant
effect on cell proliferation compared with that in cells treated
with paclitaxel alone (Fig. 1D; left
panel). As 200 µM SAS decreased cell viability by 60% in PTX1 cells
(Fig. 1C) and the cytotoxic effect
of 200 µM SAS was too strong to detect the effect of combination
with SAS and paclitaxel, 100 µM SAS was used in combination with
different concentrations of paclitaxel in PTX1 cells, and the
results showed a significant decrease in cell viability at each
concentration of paclitaxel compared with that in cells treated
with paclitaxel alone (Fig. 1D;
right panel). These results indicated the effect of SAS, and SAS
did not enhance the efficacy of paclitaxel. Taken together, these
results suggest that SAS inhibited cell proliferation more
effectively in paclitaxel-resistant compared with that in
-sensitive cells; however, SAS failed to enhance paclitaxel
cytotoxicity in either of the USC cell lines.
GSH levels and ROS accumulation in USC
cell lines treated with SAS and/or paclitaxel
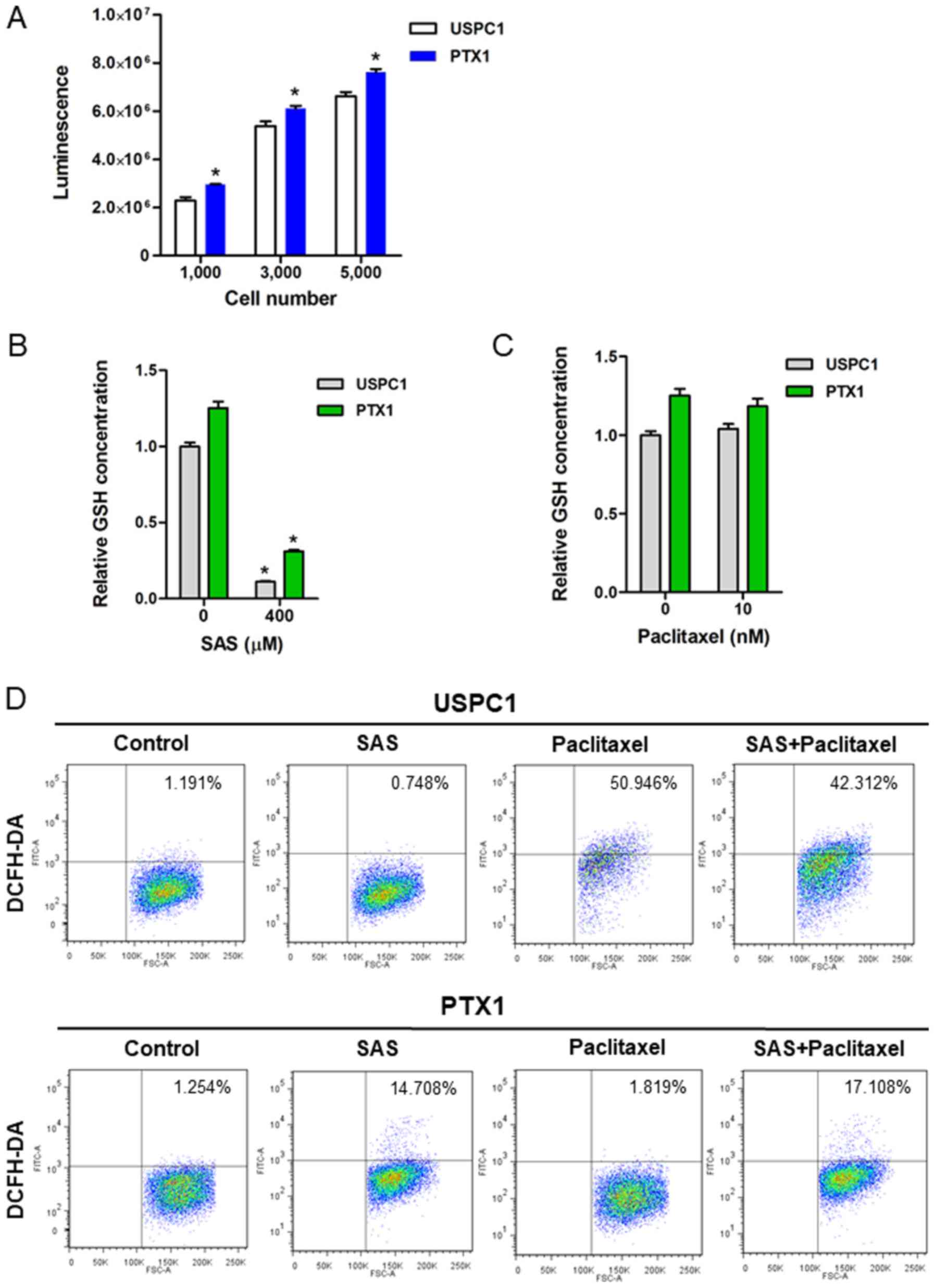
To verify the inhibitory effect of SAS on
xCT-mediated cystine transport, GSH levels and ROS accumulation in
USC cells were assessed, both prior to and following treatment with
SAS or paclitaxel. Consistent with our previous study (9), the results demonstrated that
intracellular GSH levels were significantly higher in PTX1 cells
compared with that in USPC1 cells (Fig.
2A). Furthermore, treatment with 400 µM SAS for 24 h
significantly decreased the intracellular GSH contents in both cell
lines (Fig. 2B), whereas treatment
with 10 nM paclitaxel for 24 h had no effect on the intracellular
GSH content in either cell line (Fig.
2C).
The intracellular ROS levels in USC cells treated
with SAS and/or paclitaxel were assessed using FACS analysis and
DCFH-DA. Since 200 µM SAS exhibited a marked difference on
cytotoxicity between USPC1 and PTX1 cells, the intracellular ROS
levels were analyzed in both cells treated with 200 µM SAS.
Treatment of USPC1 cells with 200 µM SAS for 24 h had no effect on
ROS levels compared with that in the vehicle-treated control,
whereas treatment with either 10 nM paclitaxel or combination of
paclitaxel with 200 µM SAS markedly increased ROS accumulation
compared with that in the vehicle-treated control (Fig. 2D; upper panels). Furthermore,
treatment with SAS increased ROS accumulation by ~12-fold in PTX1
cells compared with that in the vehicle-treated control cells,
whereas combination treatment of paclitaxel with SAS increased ROS
accumulation by ~14-fold compared with that in the vehicle-treated
control in PTX1 cells (Fig. 2D;
lower panels). However, combination treatment of paclitaxel with
SAS failed to markedly increase ROS accumulation in PTX1 cells
compared with that in cells treated with SAS alone. Collectively,
these results indicate that SAS-mediated GSH depletion markedly
affected ROS accumulation in paclitaxel- resistant cells, but not
in -sensitive cells.
SAS kills USC cells by inducing a form
of non-apoptotic cell death
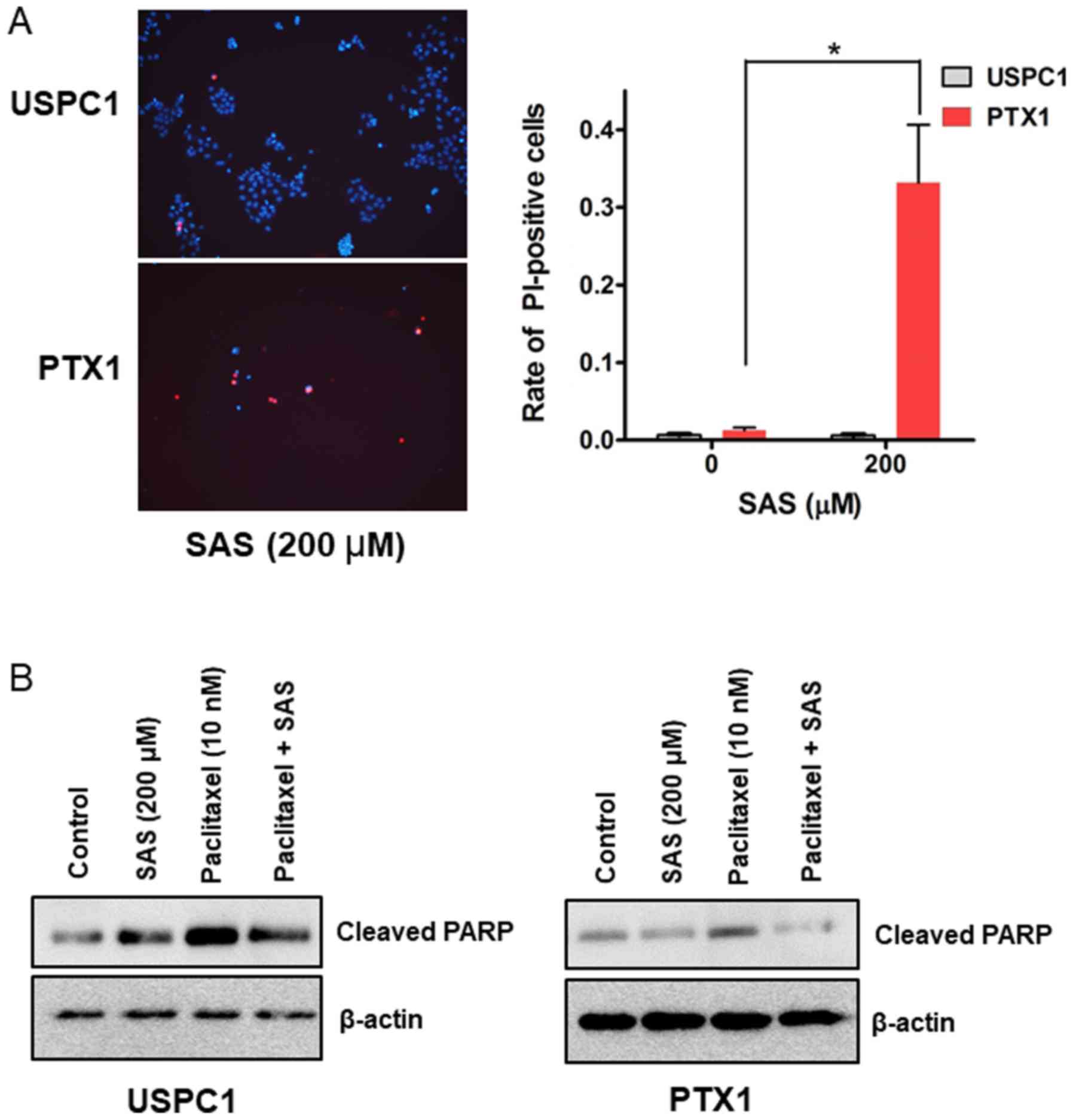
The cell death assay was performed to determine
whether SAS induces USC cell death. PI staining revealed that 200µM
SAS significantly induced cell death in PTX1 cells but not in USPC1
cells (Fig. 3A). SAS has been
reported to induce cell death and inhibit tumor growth through
apoptosis in colorectal cancer (14), and head and neck squamous cell
carcinoma (12). Thus, the
expression levels of cleaved-PARP, an apoptotic maker (32) were investigated. Western blot
analysis demonstrated that treatment with SAS alone increased the
levels of cleaved-PARP in USPC1 but not in PTX1 cells, compared
with that in the vehicle-treated control (Fig. 3B). Furthermore, combination treatment
of paclitaxel with SAS decreased the levels of cleaved-PARP in both
cell lines compared with that in cells treated with paclitaxel
alone (Fig. 3B). These results were
in contrast to the cell death and cytotoxicity assays, suggesting
that SAS induces cell death and cytotoxicity through a
non-apoptotic pathway in USC cells.
SAS inhibits cell proliferation and
induces cell death through ferroptosis in paclitaxel-resistant USC
cells
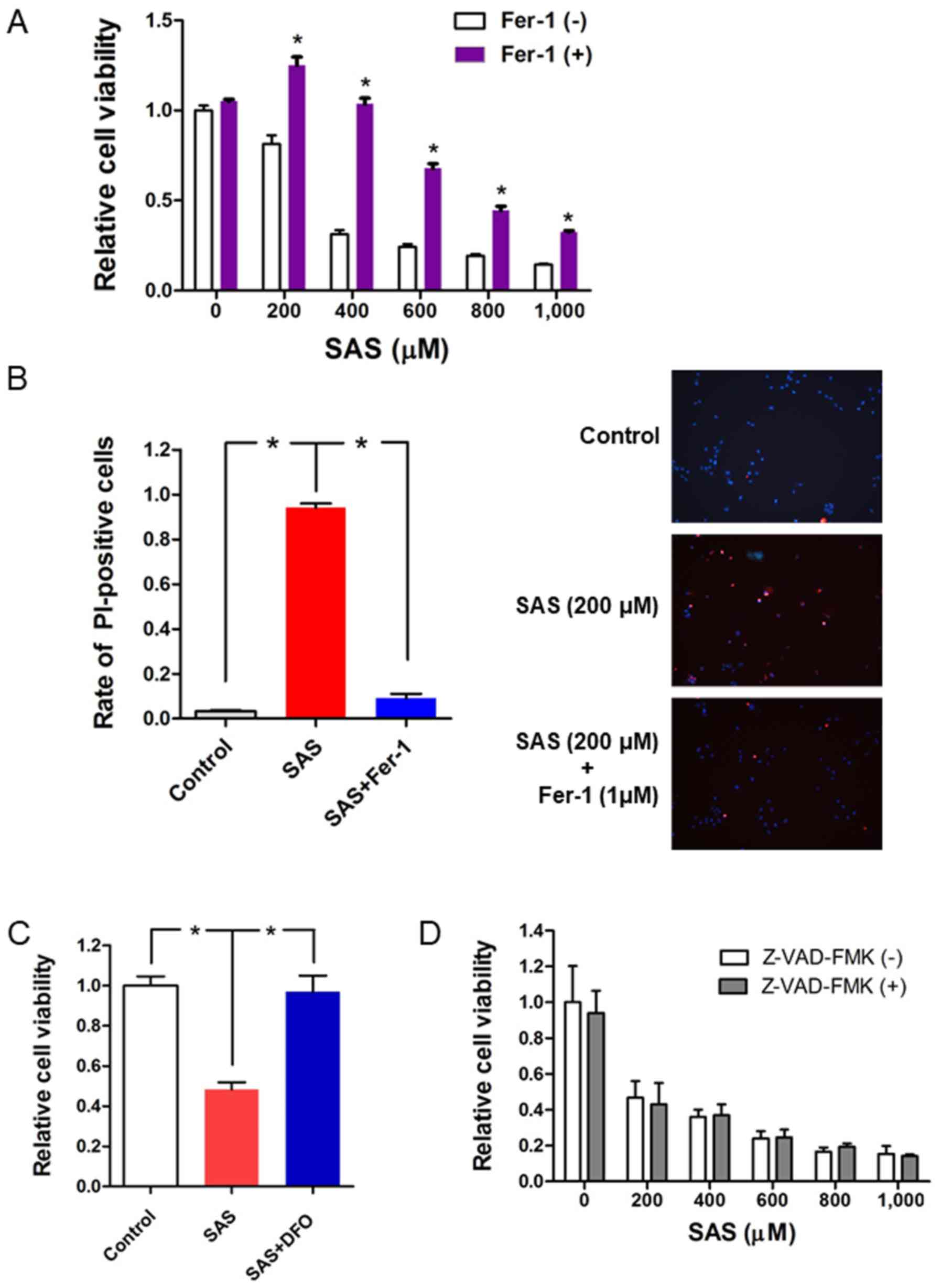
The potential molecular mechanisms by which SAS
induces cell death in paclitaxel-resistant USC cells were
investigated. SAS has been reported to induce ferroptotic cell
death in glioma cells (33). Thus,
the present study investigated whether the ferroptosis inhibitor,
ferrostatin-1, reverses the effects of SAS on inducing cytotoxicity
and cell death in PTX1 cells. Briefly, PTX1 cells were treated with
SAS alone or a combination of ferrostatin-1 with SAS for 72 h. The
results demonstrated that combination treatment of ferrostatin-1
with SAS significantly increased cell viability compared with that
in cells treated with SAS alone (Fig.
4A). Furthermore, PI staining demonstrated that combination
treatment of ferrostatin-1 with SAS reversed SAS-induced cell death
(Fig. 4B). Cell viability
significantly increased following combination treatment of the iron
chelator, DFO with SAS compared with that in cells treated with SAS
alone (Fig. 4C). To further confirm
that SAS-induced cell death occurred through ferroptosis and not
apoptosis, cells were co-treated with SAS and the apoptotic
inhibitor, Z-VAD-FMK (34). The
results indicated that Z-VAD-FMK had no effect on the inhibitory
effect of SAS on cell proliferation (Fig. 4D). Taken together, these results
suggest that ferroptosis plays a key role in SAS-induced cell
proliferation inhibition and cell death in paclitaxel-resistant USC
cells.
Knockdown of JNK results in loss of
SAS-induced cell proliferation inhibition and cell death
Ferroptosis was originally characterized in a
previous study investigating compounds which are selectively lethal
to RAS-mutant tumor cells (24).
Thus, it was hypothesized that the effect of SAS on cell death
depends on Ras activity in USC cells. The present study
investigated the expression and activation of the Ras effectors,
AKT, Erk and JNK before treatment with SAS in USPC1 and PTX1 cells.
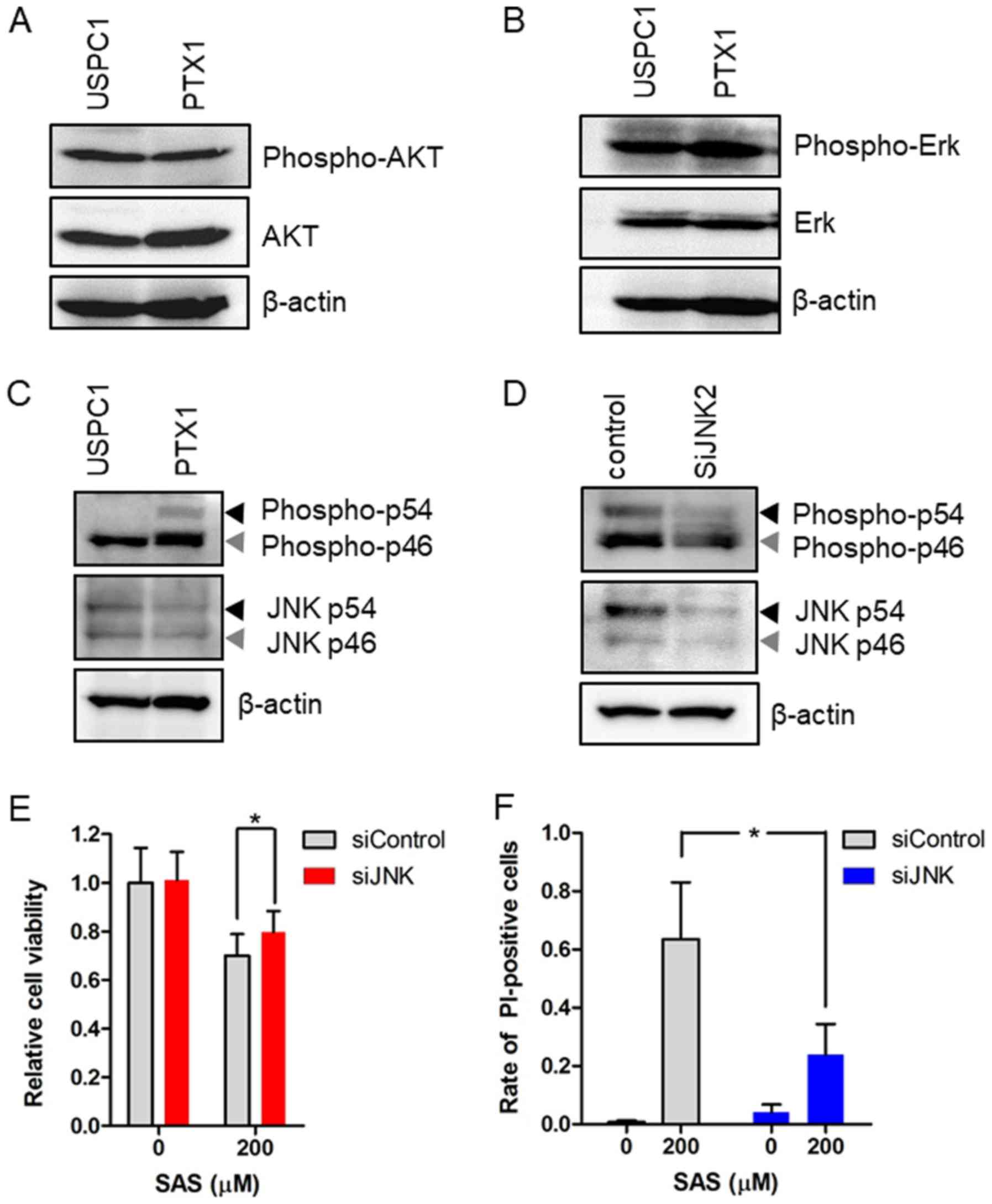
Western blot analysis demonstrated no differences in the levels of
phosphorylated AKT and Erk levels between USPC1 and PTX1 cells
(Fig. 5A and B), whereas
phosphorylated JNK levels were higher in PTX1 cells compared with
that in USPC1 cells (Fig. 5C). To
verify the association between SAS and the JNK signaling pathway,
the effect of JNK knockdown on SAS-induced cell proliferation
inhibition and cell death was investigated. PTX1 cells were
transiently transfected with siRNA against JNK to effectively
inhibit JNK activity. Western blot analysis demonstrated that
transfection with siRNA-JNK decreased the expression levels of
phosphorylated JNK and total JNK compared with that in the control
cells (Fig. 5D). Furthermore,
knockdown of JNK reversed SAS-induced cell proliferation inhibition
(Fig. 5E) and cell death (Fig. 5F). Taken together, these results
suggest that activation of the JNK signaling pathway, which is
downstream from Ras (35), enhances
sensitivity to SAS in paclitaxel-resistant USC cells.
The effect of SAS on tumor growth
inhibition
SAS has been widely demonstrated to inhibit tumor
growth via xenograft animal models for human glioma, pancreatic and
lung cancer (36–38). The present study investigated whether
SAS inhibited tumor growth in vivo using USPC1 cells. Two
weeks after inoculation, one group of mice (n=6) was administered
with oral PBS, while a second group of mice (n=6) was administered
with SAS suspension (250 mg/kg). In the second group, two mice were
excluded since one died of subcutaneous emphysema after SAS
administration and the other exhibited no tumor formation due to
failure of inoculation. Treatment with SAS had no significant
effect on inhibiting tumor growth compared with the control treated
with PBS (Fig. S2).
Discussion
The results of the present study demonstrated that
the xCT inhibitor, SAS suppressed cell proliferation in the USC
cell lines, and that the cytotoxic effect of SAS was stronger in
paclitaxel-resistant cells compared with that in -sensitive cells.
Furthermore, the results indicated that SAS-mediated cell death was
induced through ferroptosis rather than apoptosis, in a
JNK-dependent manner in paclitaxel-resistant cells. Collectively,
these results suggest that xCT inhibition may be an effective
treatment strategy for patients with recurrent paclitaxel-resistant
USC.
Intracellular GSH levels were higher in
paclitaxel-resistant USC cells compared with that in
paclitaxel-sensitive cells. These results were consistent with
previous findings, which suggest that high levels of GSH are
observed in ovarian and prostate cancer and promote resistance to
chemotherapy (6,7). The xc− system, which is
composed of xCT and F42hc, is essential for cystine uptake which is
necessary for intracellular GSH synthesis, thus this system may
also contribute to drug resistance in ovarian and lung cancer cells
(39,40). In the present study, the
intracellular GSH levels were significantly higher in
paclitaxel-resistant cells compared with those in
paclitaxel-sensitive cells. However, the results of the present
study exhibited no significant difference in xCT expression between
paclitaxel-sensitive and -resistant cells. CD44v can enhance the
capacity of GSH synthesis by stabilizing xCT (29); therefore, the difference in CD44v
expression between paclitaxel-sensitive and -resistant cells was
analyzed. Although GSH levels were significantly higher in PTX1
cells compared with those in USPC1 cells, CD44v was not expressed
in PTX1 cells. These results suggested that CD44v was not
associated with intracellular GSH levels in USC cells. Uptake of
cystine by xCT provides the majority of cysteine into the cells,
which is converted to GSH; however, a significant percentage is
derived from methionine via the transsulfuration pathway (41). In a previous study, an xCT inhibitor
depleted GSH levels to 51% of the control, while inhibition of the
transsulfuration pathway depleted GSH to 77% of the control in
glioma cells (41). Thus,
intracellular GSH levels depend on the transsulfuration pathway in
USC cells, along with xCT expression.
SAS has been extensively demonstrated to enhance the
sensitivity of different types of cancer, such as colorectal,
pancreatic, prostate and breast cancer, to chemotherapeutic agents
by inducing GSH depletion, ROS accumulation and apoptosis (14,20–23). It
was initially hypothesized that the xCT inhibitor, SAS, induced
cytotoxicity and enhanced the efficacy of paclitaxel via inducing
GSH depletion, ROS accumulation and apoptosis (14,20–23). The
results of the present study demonstrated that SAS induced
cytotoxicity; however, it did not enhance the efficacy of
paclitaxel in USC cells. Combination treatment of paclitaxel with
SAS had no marked effect on ROS accumulation compared with that in
cells treated with SAS alone, thus, SAS did not enhance
paclitaxel-induced cytotoxicity in both USC cell lines.
Furthermore, SAS-mediated GSH depletion did not increase ROS
accumulation and cell death in paclitaxel-sensitive USPC1 cells.
The molecular mechanism underlying the association between GSH
levels and ROS accumulation in USPC1 cells currently remains
unclear and may involve additional antioxidants against ROS. SAS
inhibits xCT and induces ferroptotic cell death in glioma cells
(33), and head and neck cancer
(42). The results of the present
study demonstrated that SAS markedly induced GSH depletion and ROS
accumulation, and promoted ferroptotic cell death in
paclitaxel-resistant PTX1 cells. The use of a small-molecule
activator of ferroptosis may enable selective elimination of cancer
cells in which the Ras-RAF-MEK signaling pathway is activated,
although this remains debated (43–45). Ras
expression increases ROS production (46), and the interaction between ROS
accumulation and Ras activation induces a synthetic lethality in
cancer cells (47). The present
study demonstrated that SAS increased ROS production in
paclitaxel-resistant PTX1 cells, and that JNK, which is one of the
downstream targets in the Ras signaling pathway (35), was activated in PTX1 cells, but not
in paclitaxel-sensitive USPC1 cells. ROS accumulation and JNK
activation may be essential for SAS-induced cytotoxicity and cell
death, through ferroptosis in paclitaxel-resistant USC cells
(Fig. S3). Previous studies have
reported that JNK activation was associated with paclitaxel
resistance in ovarian, pancreatic and lung cancer cells (48,49).
Taken together, these findings suggest that the synthetic lethal
interaction between ROS accumulation and the activation of the Ras
effector, JNK, induces ferroptotic cell death in
paclitaxel-resistant USC cells, thus explaining why the cytotoxic
effect of SAS was stronger in paclitaxel-resistant cells compared
with that in -sensitive cells. Furthermore, ferroptosis may act as
an endogenous tumor suppressive mechanism downstream of p53, which
is stabilized and activated by JNK signaling (50), and therefore, SAS may readily induce
ferroptosis in JNK-activated paclitaxel-resistant USC cells.
SAS has been widely found to inhibit tumor growth
via xenograft animal models for human glioma, pancreatic and lung
cancers (36–38). These reports demonstrate that the
anticancer effects of SAS are based on the inhibition of NFκB,
which is a transcription factor that plays a central role in the
immune response and is involved in several physiological phenomena,
such as acute and chronic inflammatory responses, cell
proliferation and apoptosis (51).
The present study demonstrated that SAS had no significant effect
on inhibiting tumor growth compared with the control in
paclitaxel-sensitive cells. The in vitro analysis
demonstrated that SAS promoted cytotoxicity more effectively in
paclitaxel-resistant cells than -sensitive cells. If
paclitaxel-resistant PTX-1 cells exhibited tumorigenic activity and
SAS demonstrated inhibition of tumor growth in mice studies, SAS
may have been considered a strong candidate target in the treatment
of patients with paclitaxel-resistant USC. A major limitation of
the present study was that all experiments were performed with only
one paclitaxel-resistant USC cell line. Thus, prospective studies
will focus on using different cell lines, to confirm the effect of
SAS on USC cells.
The xCT inhibitor, SAS is clinically used in
treating bowel disease and rheumatoid arthritis (15). However, there has been limited
positive response to xCT-targeted therapy in cancer. A phase I/II
study investigating SAS in patients (n=10) with recurrent or
progressive glioma was terminated due to a lack of clinical
response and the high frequency of grade 1–2 adverse effects,
including increased neurologic deficit, neutropenia,
thrombocytopenia, proteinuria (52).
Similar results were reported in patients with advanced and
chemotherapy-resistant gastric cancer (53,54). In
these clinical studies, patients continuously received SAS orally
between 1.5 and 6 g/day, and the results from the present study
demonstrated that SAS inhibited xCT at a dose of 200 µM in
paclitaxel-resistant USC cell lines. A total of 200 µM SAS is
equivalent to a SAS dose of 5.6 g/day, and this dose was ~1.6 folds
higher compared with that routinely used to treat Crohn's disease
(55). Thus, the use of SAS as a
novel candidate for recurrent USC chemotherapy-resistant treatment
may be questionable, and the development of more potent xCT
inhibitors are required for the clinical treatment of patients with
recurrent USC. xCT inhibition induces cell death in cancer stem
cells (29), and normalizes the
tumor microenvironment by decreasing tumor-derived edema in glioma
(32). Thus, targeting xCT
inhibition may be used to maintain therapy for patients with
recurrent chemotherapy-resistant USC.
In summary, the effect of the xCT inhibitor, SAS on
cytotoxicity was stronger in paclitaxel-resistant USC cells
compared with that in paclitaxel-sensitive USC cells. Furthermore,
the synthetic lethal interaction between the accumulation of ROS
and the activation of the Ras effector, JNK, induced
cell-proliferation inhibition and ferroptotic cell death in
paclitaxel-resistant USC cells. However, previous studies on SAS
have reported a lack of clinical response and frequent adverse
effects. Thus, the development of more potent xCT inhibitors are
required for clinical application. Collectively, the results of the
present study suggest that xCT inhibition may be an effective
treatment for patients with recurrent paclitaxel-resistant USC.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms Chisato Tateishi
(Experimental Research Technician, Department of Obstetrics and
Gynecology, Faculty of Medicine, Yamagata University, Yamagata,
Japan) for technical assistance.
Funding
The present study was funded in part by the Japan
Society for the Promotion of Scientific KAKENHI (grant nos.
15K10699 and 17K16827).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
AS, TO and SN designed the study. AS, MS and KT
conducted the experiments. AS, TO and MO analyzed the data. AS and
TO wrote the main manuscript and prepared all figures. All authors
interpreted the results. All authors reviewed and approved the
final manuscript.
Ethics approval and consent to
participate
The procedures involving animals used in the present
study were approved by the Animal Care Committee of Yamagata
University (approval no. 31009) in accordance with institutional
and Japanese government guidelines for animal experiments.
Patient consent for publication
Not applicable.
Competing interests
S. Nagase received lecture fees from Chugai
Pharmacetical Co. Ltd., and AstraZeneca.
Glossary
Abbreviations
Abbreviations:
|
GSH
|
glutathione
|
|
ROS
|
reactive oxygen species
|
|
USC
|
uterine serous carcinoma
|
|
SAS
|
sulfasalazine
|
References
|
1
|
Menderes G, Clark M and Santin AD: Novel
targeted therapies in uterine serous carcinoma, an aggressive
variant of endometrial cancer. Discov Med. 21:293–303.
2016.PubMed/NCBI
|
|
2
|
Hamilton CA, Cheung MK, Osann K, Chen L,
Teng NN, Longacre TA, Powell MA, Hendrickson MR, Kapp DS and Chan
JK: Uterine papillary serous and clear cell carcinomas predict for
poorer survival compared to grade 3 endometrioid corpus cancers. Br
J Cancer. 94:642–646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boruta DM II, Gehrig PA, Fader AN and
Olawaiye AB: Management of women withuterine papillary serous
cancer: A society of gynecologic oncology (SGO) review. Gynecol
Oncol. 115:142–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goff BA, Kato D, Schmidt RA, Ek M, Ferry
JA, Munts HG, Cain JM, Tamimi HK, Figge DC and Greer BE: Uterine
papillary serous carcinoma: Patterns of metastatic spread. Gynecol
Oncol. 54:264–268. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lecane PS, Karaman MW, Sirisawad M,
Naumovski L, Miller RA, Hacia JG and Magda D: Motexafin gadolinium
and zinc induce oxidative stress responses and apoptosis in B-cell
lymphoma lines. Cancer Res. 65:11676–11688. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Godwin AK, Meister A, O'Dwyer PJ, Huang
CS, Hamilton TC and Anderson ME: High resistance to cisplatin in
human ovarian cancer cell lines is associated with marked increase
of glutathione synthesis. Proc Natl Acad Sci USA. 89:3070–3074.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mulcahy RT, Untawale S and Gipp JJ:
Transcriptional up-regulation of gamma-glutamylcysteine synthetase
gene expression in melphalan-resistant human prostate carcinoma
cells. Mol Pharmacol. 46:909–914. 1994.PubMed/NCBI
|
|
8
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seino M, Ohta T, Sugiyama A, Sakaki H,
Sudo T, Tsutsumi S, Shigeta S, Tokunaga H, Toyoshima M, Yaegashi N
and Nagase S: Metabolomic analysis of uterine serous carcinoma with
acquired resistance to paclitaxel. Oncotarget. 9:31985–31998. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lo M, Wang Y and Gout PW: The x(c)-
cystine/glutamate antiporter: A potential target for therapy of
cancer and other diseases. J Cell Physiol. 215:593–602. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aquilano K, Baldelli S and Ciriolo MR:
Glutathione: New roles in redox signaling for an old antioxidant.
Front Pharmacol. 5:1962014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshikawa M, Tsuchihashi K, Ishimoto T,
Yae T, Motohara T, Sugihara E, Onishi N, Masuko T, Yoshizawa K,
Kawashiri S, et al: xCT inhibition depletes CD44v-expressing tumor
cells that are resistant to EGFR-targeted therapy in head and neck
squamous cell carcinoma. Cancer Res. 73:1855–1866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu WJ, Zhang Y, Zeng ZL, Li XB, Hu KS, Luo
HY, Yang J, Huang P and Xu RH: β-phenylethyl isothiocyanate
reverses platinum resistance by a GSH-dependent mechanism in cancer
cells with epithelial-mesenchymal transition phenotype. Biochem
Pharmacol. 85:486–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song
M, Yang J, Wen S, Xu RH, Hu Y and Huang P: Xc- inhibitor
sulfasalazine sensitizes colorectal cancer to cisplatin by a
GSH-dependent mechanism. Cancer Lett. 368:88–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zenlea T and Peppercorn MA:
Immunosuppressive therapies for inflammatory bowel disease.
20:3146–3152. 2014.PubMed/NCBI
|
|
16
|
Zhang W, Trachootham D, Liu J, Chen G,
Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W,
et al: Stromal control of cystine metabolism promotes cancer cell
survival in chronic lymphocytic leukaemia. Nat Cell Biol.
14:276–286. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nielsen OH, Verspaget HW and Elmgreen J:
Inhibition of intestinal macrophage chemotaxis to leukotriene B4 by
sulphasalazine, olsalazine, and 5-aminosalicylic acid. Aliment
Pharmacol Ther. 2:203–211. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wahl C, Liptay S, Adler G and Schmid RM:
Sulfasalazine: A potent and specific inhibitor of nuclear factor
kappa B. J Clin Invest. 101:1163–1174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lo M, Ling V, Low C, Wang YZ and Gout PW:
Potential use of the anti-inflammatory drug, sulfasalazine, for
targeted therapy of pancreatic cancer. Curr Oncol. 17:9–16.
2010.PubMed/NCBI
|
|
21
|
Doxsee DW, Gout PW, Kurita T, Lo M,
Buckley AR, Wang Y, Xue H, Karp CM, Cutz JC, Cunha GR and Wang YZ:
Sulfasalazine-induced cystine starvation: Potential use for
prostate cancer therapy. Prostate. 67:162–171. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nnrang VS, Pauletti GM, Gout PW, Buckley
DJ and Buckley AR: Sulfasalazine-induced reduction of glutathione
levels in breast cancer cells: Enhancement of growth-inhibitory
activity of Doxorubicin. Chemotherapy. 53:210–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kagami T, Wang Y, Tien A, Watahiki A, Lo
M, Xue H, Gout P and Wang ZY: Sulfasalazine enhances
growth-inhibitory activity of doxorubicin: Potential use in
combination therapy of advanced prostate cancer. Proc 98th Ann
Assoc Cancer Res (Ros Angeles, CA). 2007.
|
|
24
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Santin AD, Bellone S, Gokden M, Palmieri
M, Dunn D, Agha J, Roman JJ, Hutchins L, Pecorelli S, O'Brien T, et
al: Overexpression of HER-2/neu in uterine serous papillary cancer.
Clin Cancer Res. 8:1271–1279. 2002.PubMed/NCBI
|
|
27
|
Sakaki H, Okada M, Kuramoto K, Takeda H,
Watarai H, Suzuki S, Seino S, Seino M, Ohta T, Nagase S, et al:
GSKJ4, a selective Jumonji H3K27 demethylase inhibitor, effectively
targets ovarian cancer stem cells. Anticancer Res. 35:6607–6614.
2015.PubMed/NCBI
|
|
28
|
Orian-Rousseau V: CD44 acts as a signaling
platform controlling tumor progression and metastasis. Front
Immunol. 6:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tanabe KK, Nishi T and Saya H: Novel
variants of CD44 arising from alternative splicing: Changes in the
CD44 alternative splicing pattern of MCF-7 breast carcinoma cells
treated with hyaluronidase. Mol Carcinog. 7:212–220. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ishimoto T, Nagano O, Yae T, Tamada M,
Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al:
CD44 variant regulates redox status in cancer cells by stabilizing
the xCT subunit of system xc(−) and thereby promotes tumor growth.
Cancer Cell. 19:387–400. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yae T, Tsuchihashi K, Ishimoto T, Motohara
T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H,
et al: Alternative splicing of CD44 mRNA by ESRP1 enhances lung
colonization of metastatic cancer cell. Nat Commun. 3:8832012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shem T, Fan Z, Ghoochani A, Rauh M,
Engelhorn T, Minakaki G, Dörfler A, Klucken J, Buchfelder M,
Eyüpoglu IY and Savaskan N: Sulfasalazine impacts on ferroptotic
cell death and alleviates the tumor microenvironment and
glioma-induced brain edema. Oncotarget. 7:36021–36033. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Misaghi S, Korbel GA, Kessler B, Spooner E
and Ploegh HL: z-VAD-fmk inhibits peptide:N-glycanase and may
result in ER stress. Cell Death Differ. 13:163–165. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Enomoto M, Kizawa D, Ohsawa S and Igaki T:
JNK signaling is converted from anti- to pro-tumor pathway by
Ras-mediated switch of Warts activity. Dev Biol. 403:162–171. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Robe PA, Bentires-Alj M, Bonif M, Rogister
B, Deprez M, Haddada H, Khac MT, Jolois O, Erkmen K, Merville MP,
et al: In vitro and in vivo activity of the nuclear factor-kappaB
inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res.
10:5595–5603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Müerköster S, Arlt A, Witt M, Gehrz A,
Haye S, March C, Grohmann F, Wegehenkel K, Kalthoff H, Fölsch UR
and Schäfer H: Usage of the NF-kappaB inhibitor sulfasalazine as
sensitizing agent in combined chemotherapy of pancreatic cancer.
Int J Cancer. 104:469–476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lay JD, Hong CC, Huang JS, Yang YY, Pao
CY, Liu CH, Lai YP, Lai GM, Cheng AL, Su IJ and Chuang SE:
Sulfasalazine suppresses drug resistance and invasiveness of lung
adenocarcinoma cells expressing AXL. Cancer Res. 67:3878–3887.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Okuno S, Sato H, Kuriyama-Matsumura K,
Tamba M, Wang H, Sohda S, Hamada H, Yoshikawa H, Kondo T and Bannai
S: Role of cystine transport in intracellular glutathione level and
cisplatin resistance in human ovarian cancer cell lines. Br J
Cancer. 88:951–956. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang Y, Dai Z, Barbacioru C and Sadée W:
Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity
and chemoresistance. Cancer Res. 65:7446–7456. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kandil S, Brennan L and McBean GJ:
Glutathione depletion causes a JNK and p38MAPK-mediated increase in
expression of cystathionine-gamma-lyase and upregulation of the
transsulfuration pathway in C6 glioma cells. Neurochem Int.
56:611–619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roh JL, Kim EH, Jang HJ, Park JY and Shin
D: Induction of ferroptotic cell death for overcoming cisplatin
resistance of head and neck cancer. Cancer Lett. 381:96–103. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Irani K, Xia Y, Zweier JL, Sollott SJ, Der
CJ, Fearon ER, Sundaresan M, Finkel T and Goldschmidt-Clermont PJ:
Mitogenic signaling mediated by oxidants in Ras-transformed
fibroblasts. Science. 275:1649–1652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garama DJ, Harris TJ, White CL, Rossello
FJ, Abdul-Hay M, Gough DJ and Levy DE: A synthetic lethal
interaction between glutathione synthesis and mitochondrial
reactive oxygen species provides a tumor-specific vulnerability
dependent on STAT3. Mol Cell Biol. 35:3646–3656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Seino M, Okada M, Sakaki H, Takeda H,
Watarai H, Suzuki S, Seino S, Kuramoto K, Ohta T, Nagase S, et al:
Time-staggered inhibition of JNK effectively sensitizes
chemoresistant ovarian cancer cells to cisplatin and paclitaxel.
Oncol Rep. 35:593–601. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Okada M, Kuramoto K, Takeda H, Watarai H,
Sakaki H, Seino S, Seino M, Suzuki S and Kitanaka C: The novel JNK
inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo.
Oncotarget. 7:27021–27032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Robe PA, Martin DH, Nguyen-Khac MT, Artesi
M, Deprez M, Albert A, Vanbelle S, Califice S, Bredel M and Bours
V: Early termination of ISRCTN45828668, a phase 1/2 prospective,
randomized study of sulfasalazine for the treatment of progressing
malignant gliomas in adults. BMC Cancer. 9:3722009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shitara K, Doi T, Nagano O, Imamura CK,
Ozeki T, Ishii Y, Tsuchihashi K, Takahashi S, Nakajima TE, Hironaka
S, et al: Dose-escalation study for the targeting of CD44v+ cancer
stem cells by sulfasalazine in patients with advanced gastric
cancer (EPOC1205). Gastric Cancer. 20:341–349. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shitara K, Doi T, Nagano O, Fukutani M,
Hasegawa H, Nomura S, Sato A, Kuwata T, Asai K, Einaga Y, et al:
Phase 1 study of sulfasalazine and cisplatin for patients with
CD44v-positive gastric cancer refractory to cisplatin (EPOC1407).
Gastric Cancer. 20:1004–1009. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sandborn WJ and Feagan BG: Review article:
Mild to moderate Crohn's disease-defining the basis for a new
treatment algorithm. Aliment Pharmacol Ther. 18:263–277. 2003.
View Article : Google Scholar : PubMed/NCBI
|