The first enhancer element was identified in a DNA
sequence of the SV40 virus in the 1980s, and was found to have
enhanced transcriptional activity of β-globin in Oryctolagus
cuniculus (1). Since then, novel
insights into the regulatory mechanism of the genome have been
accumulatively gained, due to continuous exploration (2,3). It has
been demonstrated that the transcriptional activation of genes is
controlled by cell-type-specific proximal and distal regulatory
elements, termed enhancers. As a non-coding regulatory element, an
enhancer can activate gene expression through long-range chromatin
interactions (4). Different from the
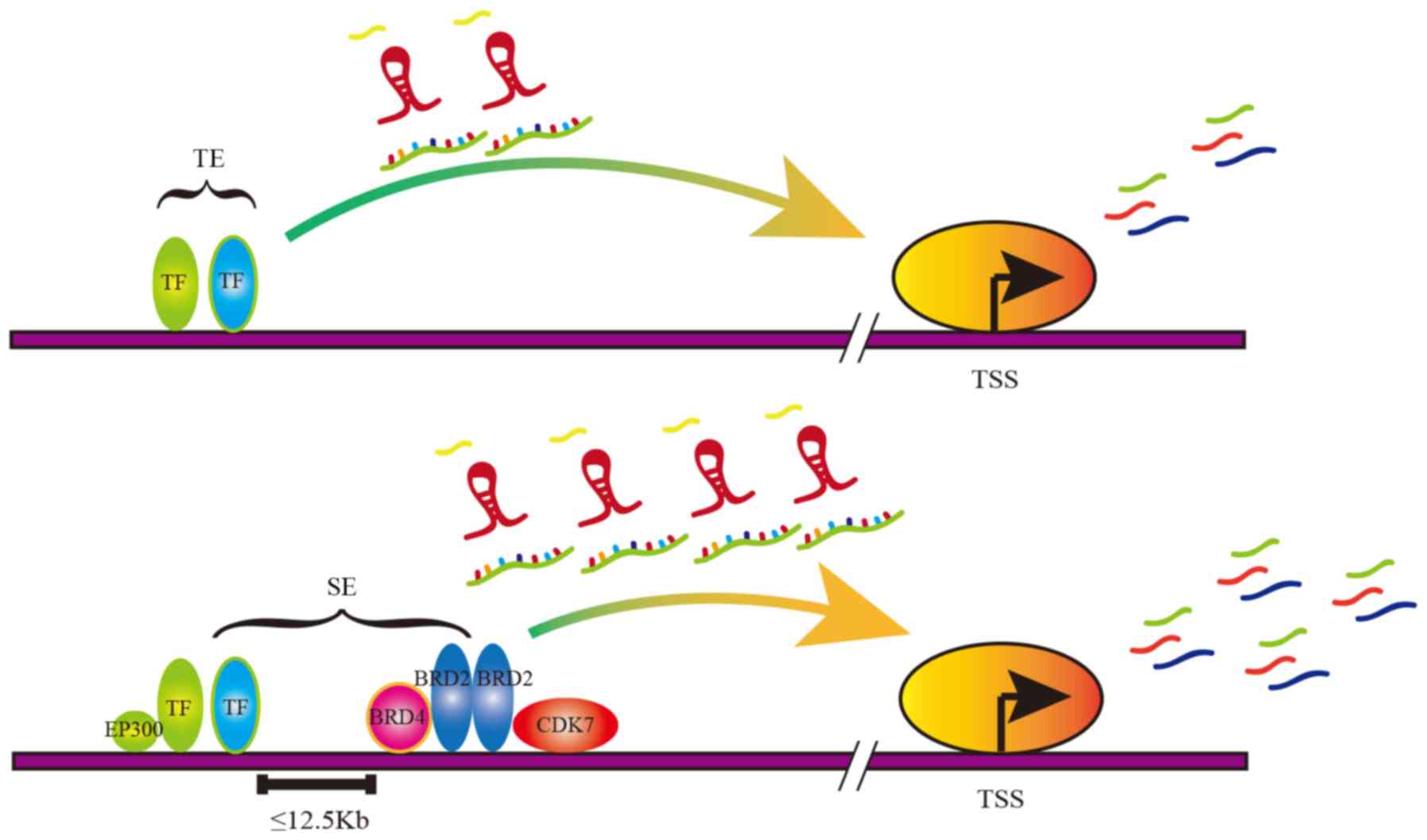
typical enhancer (TE), super-enhancer (SE) can span dozens of
kilo-base (kb) pairs compared with the dozens of base-pairs of the
TE (5).
The regions of TEs and SEs are both occupied by
enhancer-related molecules, including transcription factors (TFs),
master cofactors, mediator complexes and RNA polymerase II (pol II)
(6). However, compared with TEs, SEs
are characterized by a wider span and more aggregation of
SE-related molecules (Fig. 1)
(7,8). Therefore, the SE can drive a higher
level of gene transcription compared with TE, which is involved in
various processes of tumorigenesis and progression (9). The focus of the present review includes
the roles of the SE and its key complexes in cancer, with further
discussion on the possible new therapies involving the SE in the
targeted treatment of cancer.
Certain oncogenes exhibit low expression in normal
cells and high expression in cancer cells via SE regulation,
suggesting the significance of the SE function, particularly in the
maintenance of cancer cell growth and survival (10–13).
Compared with normal cells, cancer cells proactively construct SEs
to drive the expression of oncogenes during tumorigenesis (14). Tumor mutations generally lead to the
dysregulation of enhancers that normally control the
signal-dependent expression of growth-associated genes, resulting
in the uncontrolled proliferation of tumor cells (2). The results of genome wide association
studies demonstrated that the majority of cancer-associated genetic
variations are located outside the coding genome, and the mutation
sites are often found in the putative enhancer enrichment regions
(15). The early evidence supporting
this view comes from studies in Burkitt lymphoma in which gene
rearrangement caused the formation of MYC gene enhancer regions
leading to MYC overexpression (16–19).
Further studies have indicated that SE-driven MYC expression is
critical for maintaining cell survival and proliferation in acute
leukemia and neuroblastoma (20,21). The
inhibition of SE assembly and maintenance inhibits carcinogenic
transcription and tumor growth, which suggests that the SE is a
promising anti-cancer target (22).
The Assay for Transposase-Accessible Chromatin with
high throughput sequencing and chromatin immunoprecipitation
followed by sequencing, based on high-throughput whole-genome
sequencing, have identified a series of SE-driven oncogenes in
various tumor cells. SE can not only play a role in gene regulation
in solid tumors, such as hepatocellular, breast, esophageal and
gastric tumors, but can also elevate the expression of proteins in
non-solid tumors, such as lymphocytic leukemia and myeloid leukemia
(22). SE has been confirmed to
regulate a variety of well-defined oncogenes, such as c-MYC and
ETS-Variant Gene 6, and to affect multiple signaling pathways, such
as the MAPK and Notch signaling pathways (29–32). It
is generally believed that SE can promote tumorigenesis by
upregulating SE-associated oncogenes. The histone H3 acetyl K27
(H3K27ac) and histone H3 methyl K4 (H3K4me1) peaks have been
confirmed in the SE regions, since SE-driven gene requires an open
chromatin microenvironment (33).
However, extensive H3K4me3 peaks have also been confirmed to occur
in the regions of the tumor suppressor genes (34). It was therefore hypothesized that if
a non-specific demethylase intervenes the histone methylation
modification, it may reduce the SE-driven oncogene as well as
anti-oncogene expression. Therefore, targeting histone modification
is challenging to use as a targeted drug.
Apart from their involvement in protein-coding gene
regulation, SEs also play unique roles in RNA [including micro
(mi)RNA, enhancer (e)RNA and long non-coding (lnc)RNA] regulation.
Suzuki et al (35)
demonstrated that SE may not only promote miRNA transcription, but
also enhance the cell-specific miRNA production through
Drosha/DGCR8 recruitment and pri-miRNA processing through genome
editing, via clustered regularly interspaced short palindromic
repeats (CRISPR/Cas9). Moreover, SE-miRNAs contain many highly
cell-specific and vital miRNAs (such as ESC-, muscle-, neuron-,
hematopoietic-, skin- and inflammation-associated miRNAs) compared
with TE-miRNAs (35), indicating
that SE can drive the corresponding miRNAs and affect their
downstream pathways. Global run-on sequencing analysis revealed
that abundant eRNAs were transcribed in SE regions in
lymphoblastoid cell (LCL). In addition, the knockdown of these
eRNAs was found to result in LCL cell growth arrest, with MYC
expression significantly declining, following the knockdown of
SE-associated eRNAs of MYC (36),
illustrating that eRNA can affect not only gene expression, but
also cell growth. The SE-associated lncRNA has also been
demonstrated to play a fundamental role in the regulation of
enhancer activity and gene programs in cardiovascular pathology
(37). The disordered SE regulation
associated with lncRNA causes a serious physiological and
pathological deterioration, such as pathological stress,
remodelling and failure in the cardiovascular system (37). It is possible to alleviate
cardiovascular disease by targeting these non-coding RNAs in the
future. In hepatocellular carcinoma (HCC), the novel lncRNA HCCL5
was identified as a key oncogene, which was driven by the SE and is
significantly overexpressed in human HCC tissues (38). In addition, HCCL5 can promote the
cell proliferation, regulate the G1-S phase transition
and affect the invasive and metastatic ability of HCC cells.
Therefore, the high expression of HCCL5 may be used as a biomarker
for the poor overall survival of patients with HCC. Given the
important role of SE in RNA regulation, it may be attempted to
control the abnormal expression of RNAs by blocking the function of
SE.
In conclusion, the modifications of H3K4me1 and
H3K27ac often occur in the enhancer regions of cancer cells,
representing the formation of open chromatin structures, both of
which are absent in normal cells (39). Moreover, the SE-regulated mechanism
affects abnormal regulation at both the transcriptional and
post-transcriptional levels, resulting in malignant
transformation.
The enhanced transcriptional activity of associated
oncogenes caused by SE have been reported in various types of
cancer. Therefore, blocking the SE is a viable anticancer therapy.
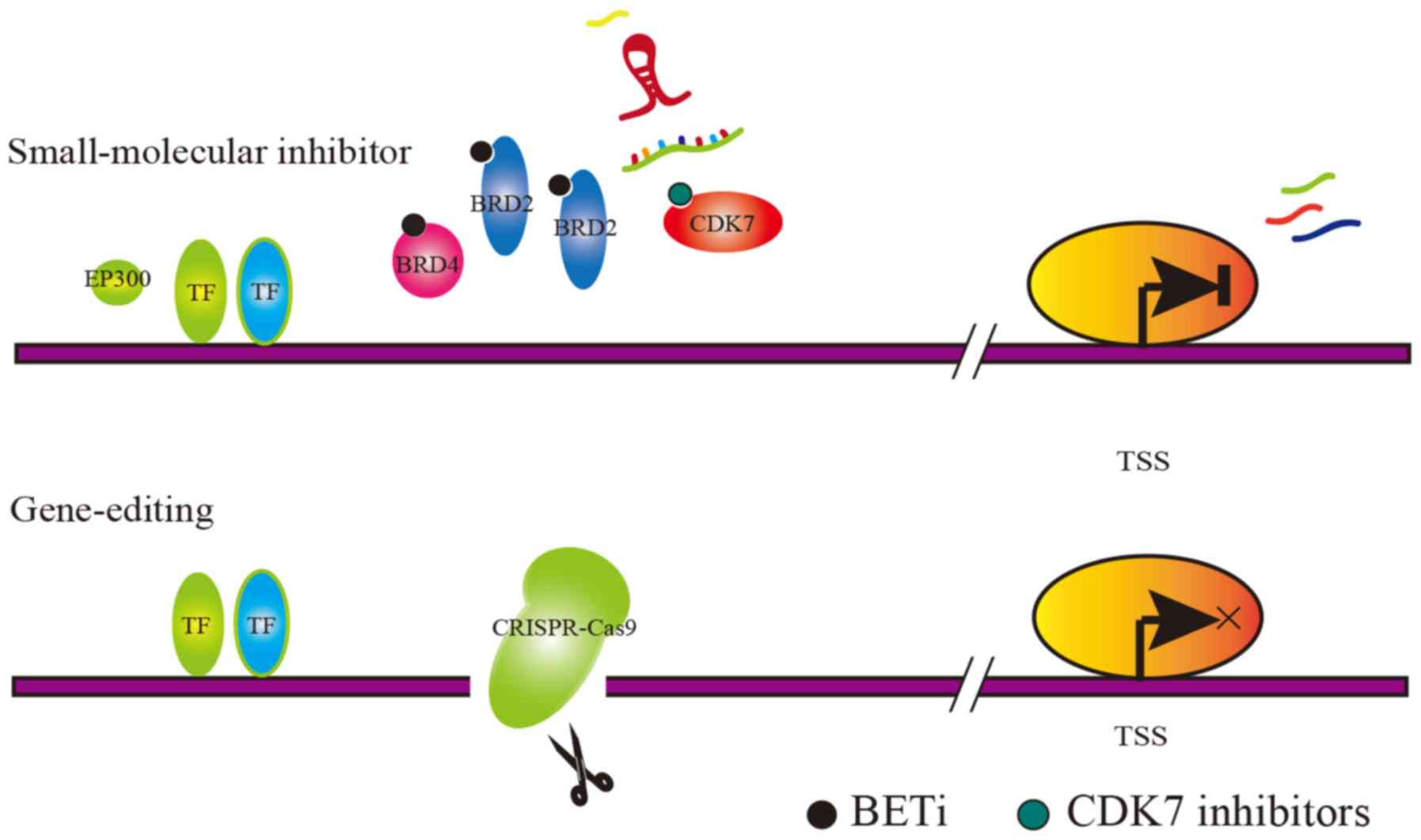
The mechanism of action that describes the potential treatments, by
targeting the SE-associated complexes, is shown in Fig. 2. However, transcription, as a
biological process that occurs universally in vivo, cannot
be inhibited as a whole, and a high degree of specificity in
clinical antitumor therapy is required to inhibit it. It is
therefore necessary to find a target for the possible intervention
among these small molecules that are involved in the SE mechanism.
Master TFs, cofactors and histone modification markers are
difficult to select as targets due to their extensive effects,
while mediator complexes such as cyclin-dependent kinase 7 (CDK7)
and BRD4 are relatively characteristic. Decreased CDK7 expression
or BRD4 expression suppresses cancer cell growth, confirming that
cancer cells are sensitive to the dosage of SE-associated mediator
complexes (40). The inhibition of
mediator complexes is considered to be a promising approach.
Studies have reported that the activity of SE must depend on the
interaction of key TFs, cofactors and mediator complexes.
Therefore, it is theoretically feasible to design inhibitors of
mediator complexes to intervene in tumor cells (41). Targeted inhibitors can specifically
block the interaction between SE regions and their corresponding
complexes, thereby rescuing the upregulated oncogene. These types
of small-molecule inhibitors have been found to exert significant
inhibitory effects in animal experiments (42–44). At
present, the small-molecule inhibitors designed for cancer
treatments include the following three types: i) CDK7 inhibitor
(THZ1); ii) BRD4 inhibitor (JQ1); and iii) other inhibitors, which
are summarized in Table I (8,21,29,35,39,45–77).
Cyclin-dependent kinases (CDKs) belong to the
serine/threonine kinase family, and play a crucial role in the
regulation of the cell cycle and transcription process (78). It has been reported that CDK7 not
only affects the cell cycle, but is also associated with the
regulation of SE-driven oncogenes (79). In addition, CDK7 has recently become
an attractive anti-cancer target, since CDK7 inhibitors (CDK7i) can
reduce the levels of oncogenic TFs that act on SE regions (51). The anticancer effects of CDK7i are
attributed to their effects on gene transcription or interference
with the function of the SE; THZ1 is one of the most effective
small-molecule inhibitors (79,80).
Despite the meaningful effects of THZ1 in a variety of tumors,
studies have shown that THZ1 inhibits myogenic differentiation,
suggesting the possible side effects of THZ1 on muscle function
during treatment (81). CDK7i may
also be combined with other anti-tumor drugs to improve efficacy
and reduce side effects. In diffuse intrinsic pontine glioma, CDK7i
has been found to disrupt transcriptional regulation in cancer
cells; however, the sensitivity of cancer cells to CDK7i is notably
increased when combined with histone deacetylase inhibitors
(79).
The bromodomain and extra terminal (BET) family of
bromodomain proteins (BRDs) consists of 4 members (BRD2, BRD3, BRD4
and BRDT) that may represent a promising new target for the
discovery of small-molecule drugs (82). BRDs can recognize histone acetylation
and promote the expression of corresponding genes, such as the MYC
oncogene (83). Specifically, BET
bromodomain inhibitor (BETi) mainly inhibits the binding function
of BRD4, one of the mediator complexes of SE, thereby suppressing
the expression of SE-driven oncogenes and attenuating the
proliferation of cancer cells (84).
However, it is still controversial whether the function of BETi
depends on the expression of the MYC gene. Certain studies found
BETi to preferentially affect the expression of the SE-driven MYC
oncogene in multiple myeloma and colon cancer, suggesting that BETi
sensitivity is significantly associated with c-MYC gene levels
(54,61). However, other studies reported
contrasting results, as they did not observe a significant
correlation between JQ1 sensitivity and c-MYC expression in colon
cancer (29). Therefore, further
research should be focused on maximizing the efficacy of BETi. In a
study on pancreatic cancer, KDM6A was reported to mediate the
abnormal activation of the SE regions of MYC and RUNX3 (58). When patients with pancreatic cancer,
accompanied by KDM6A deficiency, were treated with BETi
simultaneously, the selective sensitivity was observed. Severe BETi
side effects have been reported in multiple phase I clinical
trials, and include heart toxicity, gastrointestinal toxicity,
anemia, diarrhea, fatigue, nausea, neutropenia and thrombocytopenia
(70,85,86). For
example, a BETi, I-BET-151, may even reduce the right and left
ventricular fractional shortening, resulting in impaired heart
function (87). The cytotoxicity of
BETi caused serious side effects in clinical trials; however, the
combination of this drug with vitamin C may largely alleviate those
side effects (88).
In addition to the above two types of markedly
effective and widely applicable drugs, there are other
small-molecule drugs involved in blocking the function of SE.
Attempting to abolish the SE function, the dynamic conversion
between RACK7/KDM5C gene deletion and the accumulation of two SE
formation-dependent methylation modifications (H3K4me1 and H3K4me3)
provided a new approach to breast cancer therapy (89). Similarly, acetylation modifications
have also been reported to be involved in the course of SE action.
The IKAROS gene was usually undetectable in B cell line acute
lymphoblastic leukemia; however, it has been demonstrated that the
artificial overexpression of IKAROS gene can inhibit the expression
of the MYC gene by interfering with the H3K27ac3 modification at
the SE regions of MYC (90). PAX3
and PAX5 are the key TFs of the SE in pulmonary alveolar
rhabdomyosarcoma and chronic lymphocytic leukemia, and PAX
inhibitors have been found to exert prominent antitumor effects
(91,92). Another study proposed a novel
paradigm for NF-κB-mediated gene inhibition, in which cofactors are
redistributed and enriched through the accumulation of NF-κB at the
SE regions (93). In addition to the
key molecules mentioned above, other, novel small molecules that
are considered to be SE-dependent in chordomas have gradually
emerged, such as CDK9, CDK12 and CDK13 (50). In chordomas, the IRS4/IGF2 protein
can directly interact with the SE sequence, thereby inhibiting the
SE function detected by 4C-Seq assay (50). This phenomenon has been termed
‘enhancer hijacking’ and is widely reported in cancer. The
development of these small-molecule inhibitors also provides
promising therapeutic approaches against cancer.
Most of these drugs have expectant clinical
application prospects, and some have entered the clinical trial
phase (Table II).
Previous studies on antitumor drugs have focused on
genomics, while drug design mainly focuses on the abnormal
activation of proteins caused by mutations; however, this treatment
is not without limitations (94).
Drug resistance and low mutation frequency should not be overlooked
in cancer (95). In recent years,
with the development of epigenomics, methylation in the promoter
region has been reported to regulate gene expression. Due to the
common epigenetic modification reported in subsequent studies,
designing drugs for this epigenetic mechanism is of great
significance, which is considered as a better target (96). In the future, both universality and
specificity should be taken into consideration in the direction of
new drug design, such as the key process of SE regulation.
The abnormal base insertion, base deletion and
chromatin rearrangement in cancer cells can result in the formation
of SE. CRISPR/Cas9 technology could be applied to abolish the SE
formation caused by the aforementioned reasons. In addition, the
objective of anticancer treatment can be achieved through the
CRISPR/Cas9 system.
The CRISPR/Cas9 knockout system targets specific SE
regions to cut DNA sequences, causing non-homologous recombination
repair, thereby abolishing the SE function. RUNX1 is a
transcription factor that regulates normal and malignant blood cell
production. The disruption the SE regions of RUNX1 gene by the
CRISPR/Cas9 system was shown to increase apoptosis in acute
leukemia cells, and subsequently alter the survival of mice with
acute myeloid leukemia (AML) (97).
It was observed that, in T-cell acute lymphoblastic leukemia
(T-ALL) primary samples and cell lines, an indel mutation occurred
at the hotspot 7.5 kb upstream the transcription initiation site of
the T-cell acute leukemia 1 (TAL1) gene and contributed to the
formation of the MYB binding site and SE, thereby resulting in the
upregulation of oncogenes (98).
Following the knockout of the abnormally inserted bases by the
CRISPR/Cas9 system, SE formation and TAL1 gene overexpression were
absent in ALL (98). The
transcription activator-like effector and CRISPR/Cas9 genome
editing systems may also be used to abolish the activation of
abnormal enhancers in AML cells. Subsequent experiments confirmed
that gene-editing acting on enhancer efficiently inhibited the
expression of ecotropic viral integration site 1 and cancer growth
(99).
The CRISPR/Cas9 knock-in system is also promising in
the SE-driven oncogene expression pattern associated with
single-nucleotide polymorphisms (SNPs). It has been demonstrated
that most trait-associated SNPs occur in non-coding regions, with
64% occurring in the disease-associated SE regions defined by
H3K27ac (7). Subsequent studies
revealed one underlying mechanism through which SNPs located within
the SE regions could affect gene expression (100,101).
SNP rs6854845 is considered to be one of the risk factors for colon
cancer (102). In colon cells, it
was found that SNP rs6854845 formed in the SE region and affected
the shifted enrichment of H3K4me1 and H3K27ac at the SE regions,
further affecting the expression of SE-driven genes (103). The pathogenic SNP locus can be
verified by expression quantitative trait locus, genomic chromatin
interaction (high-throughput chromosome conformation capture),
epigenetic annotation, and a series of functional assays (104). Therefore, the site-specific genome
editing of SNPs by CRISPR/Cas9 can correct the multiple pathogenic
changes in cells by reversing the interaction between SNPs and
SE-associated genes.
Although the CRISPR technology is known for its
effectiveness and versatility, it has two major drawbacks: The
inability to arbitrarily edit bases and its off-target effects
(105). The CRISPR/Cas9 system
relies on the recognition of protospacer-adjacent motif (PAM) sites
by single guide RNA to perform DNA shear, so the system can only
edit DNA near the PAM site and cannot edit bases at any regions
(106). As an important member of
the CRISPR/Cas9 gene editing system, the Cas enzyme may cleave
non-targeting sites after its introduction into cells, causing
off-target effects (107). In
addition, CRISPR/Cas9 gene editing relies on DNA double-stranded
breaks, which may lead to an unpredictable disruption of cells
following gene editing (108).
These side effects of CRISPR/Cas9 limit their application in basic
research and medicine, and may trigger safety issues.
A new precise gene editing tool, the Prime Editor,
has been developed which can effectively convert all 12 single
bases without relying on DNA templates and accurately insert or
delete several bases (109). The
Prime Editor system acts by combining Cas9 and reverse
transcriptase into a complex, which is then brought to the specific
DNA region to insert a new DNA sequence by the prime editing guide
RNA (pegRNA) (109). This new
method aims at improving the traditional cas9 enzyme and pegRNA to
eliminate the 2 defects of the traditional CRISPR system. Through
this new system, experiments in mouse cells have successfully
repaired gene mutations that cause sickle cell anemia and Tay-Sachs
disease (109). Sickle cell anemia
is an autosomal dominant genetic disease, in which a single base
mutation of A to T occurs in a gene encoding hemoglobin, resulting
in the 6th amino acid glutamate of the hemoglobin β-peptide chain
becoming valine, which makes the sickle hemoglobin replace the
normal hemoglobin (110). Tay-Sachs
disease is an autosomal recessive disorder in which the HEXA gene
is mutated to an extra 4 bases, resulting in the inactivation of
the lipolytic enzyme encoded by the HEXA gene, which in turn leads
to the aggregation of gangliosides in the brain with ensuing
toxicity (111). These genetic
diseases could not be cured by traditional genome editing system,
while the improved prime editor may prove more effective.
Although extensive animal and clinical experiments
are required to further validate its safety and effectiveness,
gene-editing therapy is already a promising method for tumor
therapy. In conclusion, gene editing techniques are constantly
being improved, and the prospects of the clinical application of
these technologies in the near future are encouraging.
SE is a controversial topic in current clinical and
basic research, with great attention paid to its functions and
potential therapeutic prospects in the clinic. With the improvement
and breakthroughs made in next-generation sequencing technology, a
more detailed and comprehensive understanding of the genome has
emerged. By using the CRISPR/Cas9 system to interfere with the
potential SE formation, the SE can be blocked to regulate the
SE-driven oncogenes to avoid tumorigenesis. However, due to the
off-target effects of the CRIPR/Cas9 system, it is impossible to
accurately identify 100% of the human genome. Gene-editing at any
non-target sites is associated with clinical risks. To date, the
continuous improvement of CRISPR/Cas9 tools has not been effective
in resolving the problem of off-target effects, causing great
concerns regarding the safety of its clinical applications. The
activation of SE is tightly linked to the interaction of certain
key TFs. It has been reported that these key TFs play a critical
role in cancer and further interference with small-molecule
inhibitors also suppresses cancer cells (5). Since cancer cells are highly sensitive
to SE-associated complexes, the inhibition of SE-derived gene
expression is conceived as a safer and more promising therapeutic
approach to cancer. The newly improved CAPTURE-Proteomics
(ChIP-seq) technique is based on the core elements of CRISPR and
adds biotin ligase BirA to improve the sensitivity and specificity
of the experiment, which can more accurately detect the protein
complex that binds to the SE site in order to find a new target for
clinical small-molecule inhibitor therapy (112). Despite of the unclear mechanisms of
SE and SE-associated complexes regulating the genome, the targeted
therapy for SE-associated complexes remains one of the directions
of treatment. With the persistent efforts focused on SE-associated
complexes, greater breakthroughs are expected in the near
future.
Not applicable.
The present study was supported by the National
Natural Science Foundation of China (grant no. 81702789).
All data generated or analyzed during this study are
included in this published article.
CZ conceived the study and was a major contributor
to the manuscript. CZ and ML wrote the manuscript. HF revised the
manuscript for important intellectual content. CZ and ML jointly
collected relevant references. HF guided the research framework and
approved the final version for publication. All the authors have
read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Banerji J, Rusconi S and Schaffner W:
Expression of a beta-globin gene is enhanced by remote SV40 DNA
sequences. Cell. 27:299–308. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sur I and Taipale J: The role of enhancers
in cancer. Nat Rev Cancer. 16:483–493. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rickels R and Shilatifard A: Enhancer
Logic and mechanics in development and disease. Trends Cell Biol.
28:608–630. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bulger M and Groudine M: Functional and
mechanistic diversity of distal transcription enhancers. Cell.
144:327–339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whyte WA, Orlando DA, Hnisz D, Abraham BJ,
Lin CY, Kagey MH, Rahl PB, Lee TI and Young RA: Master
transcription factors and mediator establish super-enhancers at key
cell identity genes. Cell. 153:307–319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pott S and Lieb JD: What are
super-enhancers? Nat Genet. 47:8–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hnisz D, Abraham BJ, Lee TI, Lau A,
Saint-Andre V, Sigova AA, Hoke HA and Young RA: Super-enhancers in
the control of cell identity and disease. Cell. 155:934–947. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Christensen CL, Kwiatkowski N, Abraham BJ,
Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS,
Akbay EA, Altabef A, et al: Targeting transcriptional addictions in
small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell.
26:909–922. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsang FH, Law CT, Tang TC, Cheng CL, Chin
DW, Tam WV, Wei L, Wong CC, Ng IO and Wong CM: Aberrant
super-enhancer landscape in human hepatocellular carcinoma.
Hepatology. 69:2502–2517. 2019.PubMed/NCBI
|
|
10
|
Xie JJ, Jiang YY, Jiang Y, Li CQ, Lim MC,
An O, Mayakonda A, Ding LW, Long L, Sun C, et al:
Super-Enhancer-driven long non-coding RNA LINC01503, regulated by
tp63, is over-expressed and oncogenic in squamous cell carcinoma.
Gastroenterology. 154:2137–2151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng L, Jiang B, Yuan X, Qiu Y, Peng J,
Huang Y, Zhang C, Zhang Y, Lin Z, Li J, et al:
Super-Enhancer-associated long noncoding RNA HCCL5 is activated by
zeb1 and promotes the malignancy of hepatocellular carcinoma.
Cancer Res. 79:572–584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin L, Huang M, Shi X, Mayakonda A, Hu K,
Jiang YY, Guo X, Chen L, Pang B, Doan N, et al:
Super-enhancer-associated MEIS1 promotes transcriptional
dysregulation in Ewing sarcoma in co-operation with EWS-FLI1.
Nucleic Acids Res. 47:1255–1267. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cao X, Dang L, Zheng X, Lu Y, Lu Y, Ji R,
Zhang T, Ruan X, Zhi J, Hou X, et al: Targeting
super-enhancer-driven oncogenic transcription by cdk7 inhibition in
anaplastic thyroid carcinoma. Thyroid. 29:809–823. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang L and Hu G: Remodeling
super-enhancers and oncogenic transcription. Cell Cycle.
15:3157–3158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma M, Ru Y, Chuang LS, Hsu NY, Shi LS,
Hakenberg J, Cheng WY, Uzilov A, Ding W, Glicksberg BS, et al:
Disease-associated variants in different categories of disease
located in distinct regulatory elements. BMC Genomics. 16 (Suppl
8):S32015. View Article : Google Scholar
|
|
16
|
Dalla-Favera R, Bregni M, Erikson J,
Patterson D, Gallo RC and Croce CM: Human c-myc onc gene is located
on the region of chromosome 8 that is translocated in Burkitt
lymphoma cells. Proc Natl Acad Sci USA. 79:7824–7827. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Taub R, Kirsch I, Morton C, Lenoir G, Swan
D, Tronick S, Aaronson S and Leder P: Translocation of the c-myc
gene into the immunoglobulin heavy chain locus in human Burkitt
lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA.
79:7837–7841. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ar-Rushdi A, Nishikura K, Erikson J, Watt
R, Rovera G and Croce CM: Differential expression of the
translocated and the untranslocated c-myc oncogene in Burkitt
lymphoma. Science. 222:390–393. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Erikson J, Ar-Rushdi A, Drwinga HL, Nowell
PC and Croce CM: Transcriptional activation of the translocated
c-myc oncogene in burkitt lymphoma. Proc Natl Acad Sci USA.
80:820–824. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kubota S, Tokunaga K, Umezu T,
Yokomizo-Nakano T, Sun Y, Oshima M, Tan KT, Yang H, Kanai A,
Iwanaga E, et al: Lineage-specific RUNX2 super-enhancer activates
MYC and promotes the development of blastic plasmacytoid dendritic
cell neoplasm. Nat Commun. 10:16532019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chipumuro E, Marco E, Christensen CL,
Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C,
Altabef A, et al: CDK7 inhibition suppresses super-enhancer-linked
oncogenic transcription in MYCN-driven cancer. Cell. 159:1126–1139.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sengupta S and George RE:
Super-Enhancer-driven transcriptional dependencies in cancer.
Trends Cancer. 3:269–281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hnisz D, Shrinivas K, Young RA,
Chakraborty AK and Sharp PA: A Phase separation model for
transcriptional control. Cell. 169:13–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoo H, Triandafillou C and Drummond DA:
Cellular sensing by phase separation: Using the process, not just
the products. J Biol Chem. 294:7151–7159. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sabari BR, Dall'Agnese A, Boija A, Klein
IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV,
Manteiga JC, et al: Coactivator condensation at super-enhancers
links phase separation and gene control. Science. 361:eaar39582018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang JT, Smith J, Chen BC, Schmidt H,
Rasoloson D, Paix A, Lambrus BG, Calidas D, Betzig E and Seydoux G:
Regulation of RNA granule dynamics by phosphorylation of
serine-rich, intrinsically disordered proteins in C.
elegans. Elife. 3:e45912014. View Article : Google Scholar
|
|
27
|
Wippich F, Bodenmiller B, Trajkovska MG,
Wanka S, Aebersold R and Pelkmans L: Dual specificity kinase DYRK3
couples stress granule condensation/dissolution to mTORC1
signaling. Cell. 152:791–805. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Milin AN and Deniz AA: Reentrant phase
transitions and non-equilibrium dynamics in membraneless
organelles. Biochemistry. 57:2470–2477. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakamura Y, Hattori N, Iida N, Yamashita
S, Mori A, Kimura K, Yoshino T and Ushijima T: Targeting of
super-enhancers and mutant BRAF can suppress growth of BRAF-mutant
colon cancer cells via repression of MAPK signaling pathway. Cancer
Lett. 402:100–109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gunnell A, Webb HM, Wood CD, McClellan MJ,
Wichaidit B, Kempkes B, Jenner RG, Osborne C, Farrell PJ and West
MJ: RUNX super-enhancer control through the Notch pathway by
Epstein-Barr virus transcription factors regulates B cell growth.
Nucleic Acids Res. 44:4636–4650. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ke L, Zhou H, Wang C, Xiong G, Xiang Y,
Ling Y, Khabir A, Tsao GS, Zeng Y, Zeng M, et al: Nasopharyngeal
carcinoma super-enhancer-driven ETV6 correlates with prognosis.
Proc Natl Acad Sci USA. 114:9683–9688. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dave K, Sur I, Yan J, Zhang J, Kaasinen E,
Zhong F, Blaas L, Li X, Kharazi S, Gustafsson C, et al: Mice
deficient of Myc super-enhancer region reveal differential control
mechanism between normal and pathological growth. Elife.
6:e233822017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lai B, Lee JE, Jang Y, Wang L, Peng W and
Ge K: MLL3/MLL4 are required for CBP/p300 binding on enhancers and
super-enhancer formation in brown adipogenesis. Nucleic Acids Res.
45:6388–6403. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen K, Chen Z, Wu D, Zhang L, Lin X, Su
J, Rodriguez B, Xi Y, Xia Z, Chen X, et al: Broad H3K4me3 is
associated with increased transcription elongation and enhancer
activity at tumor-suppressor genes. Nat Genet. 47:1149–1157. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suzuki HI, Young RA and Sharp PA:
Super-enhancer-mediated RNA processing revealed by integrative
microrna network analysis. Cell. 168:1000–1014.e15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liang J, Zhou H, Gerdt C, Tan M, Colson T,
Kaye KM, Kieff E and Zhao B: Epstein-Barr virus super-enhancer
eRNAs are essential for MYC oncogene expression and lymphoblast
proliferation. Proc Natl Acad Sci USA. 113:14121–14126. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ounzain S and Pedrazzini T: Super-enhancer
lncs to cardiovascular development and disease. Biochim Biophys
Acta. 1863:1953–1960. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peng L, Jiang B, Yuan X, Qiu Y, Peng J,
Huang Y, Zhang C, Zhang Y, Lin Z, Li J, et al:
Super-Enhancer-associated long noncoding RNA HCCL5 Is activated by
zeb1 and promotes the malignancy of hepatocellular carcinoma.
Cancer Res. 79:572–584. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fontanals-Cirera B, Hasson D, Vardabasso
C, Di Micco R, Agrawal P, Chowdhury A, Gantz M, de
Pablos-Aragoneses A, Morgenstern A, Wu P, et al: Harnessing BET
inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene.
Mol Cell. 68:731–744.e9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pelish HE, Liau BB, Nitulescu II,
Tangpeerachaikul A, Poss ZC, Da SD, Caruso BT, Arefolov A, Fadeyi
O, Christie AL, et al: Mediator kinase inhibition further activates
super-enhancer- associated genes in AML. Nature. 526:273–276. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
See YX, Wang BZ and Fullwood MJ: Chromatin
interactions and regulatory elements in cancer: From bench to
bedside. Trends Genet. 35:145–158. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu X, Enomoto K, Zhao L, Zhu YJ,
Willingham MC, Meltzer P, Qi J and Cheng SY: Bromodomain and
extraterminal protein inhibitor JQ1 suppresses thyroid tumor growth
in a mouse model. Clin Cancer Res. 23:430–440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Augert A and MacPherson D: Treating
transcriptional addiction in small cell lung cancer. Cancer Cell.
26:783–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gerlach D, Tontsch-Grunt U, Baum A, Popow
J, Scharn D, Hofmann MH, Engelhardt H, Kaya O, Beck J, Schweifer N,
et al: The novel BET bromodomain inhibitor BI 894999 represses
super-enhancer-associated transcription and synergizes with CDK9
inhibition in AML. Oncogene. 37:2687–2701. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen D, Zhao Z, Huang Z, Chen DC, Zhu XX,
Wang YZ, Yan YW, Tang S, Madhavan S, Ni W, et al: Super enhancer
inhibitors suppress MYC driven transcriptional amplification and
tumor progression in osteosarcoma. Bone Res. 6:112018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kitazono M, Chuman Y, Aikou T and Fojo T:
Construction of gene therapy vectors targeting thyroid cells:
Enhancement of activity and specificity with histone deacetylase
inhibitors and agents modulating the cyclic adenosine
3′,5′-monophosphate pathway and demonstration of activity in
follicular and anaplastic thyroid carcinoma cells. J Clin
Endocrinol Metab. 86:834–840. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wong R, Ngoc P, Leong WZ, Yam A, Zhang T,
Asamitsu K, Iida S, Okamoto T, Ueda R, Gray NS, et al: Enhancer
profiling identifies critical cancer genes and characterizes cell
identity in adult T-cell leukemia. Blood. 130:2326–2338. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eliades P, Abraham BJ, Ji Z, Miller DM,
Christensen CL, Kwiatkowski N, Kumar R, Njauw CN, Taylor M, Miao B,
et al: High MITF expression is associated with super-enhancers and
suppressed by CDK7 inhibition in melanoma. J Invest Dermatol.
138:1582–1590. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kwiatkowski N, Zhang T, Rahl PB, Abraham
BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar
B, et al: Targeting transcription regulation in cancer with a
covalent CDK7 inhibitor. Nature. 511:616–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sharifnia T, Wawer MJ, Chen T, Huang QY,
Weir BA, Sizemore A, Lawlor MA, Goodale A, Cowley GS, Vazquez F, et
al: Small-molecule targeting of brachyury transcription factor
addiction in chordoma. Nat Med. 25:292–300. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hu S, Marineau JJ, Rajagopal N, Hamman KB,
Choi YJ, Schmidt DR, Ke N, Johannessen L, Bradley MJ, Orlando DA,
et al: Discovery and characterization of SY-1365, a selective,
covalent inhibitor of CDK7. Cancer Res. 79:3479–3491. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dooley KE, Warburton A and McBride AA:
Tandemly Integrated HPV16 Can Form a Brd4-dependent
super-enhancer-like element that drives transcription of viral
oncogenes. Mbio. 7:e01446–e01416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sengupta D, Kannan A, Kern M, Moreno MA,
Vural E, Stack BJ, Suen JY, Tackett AJ and Gao L: Disruption of
BRD4 at H3K27Ac-enriched enhancer region correlates with decreased
c-Myc expression in Merkel cell carcinoma. Epigenetics. 10:460–466.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Loven J, Hoke HA, Lin CY, Lau A, Orlando
DA, Vakoc CR, Bradner JE, Lee TI and Young RA: Selective inhibition
of tumor oncogenes by disruption of super-enhancers. Cell.
153:320–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zuber V, Bettella F, Witoelar A,
Andreassen OA, Mills IG and Urbanucci A: Bromodomain protein 4
discriminates tissue-specific super-enhancers containing
disease-specific susceptibility loci in prostate and breast cancer.
BMC Genomics. 18:2702017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chapuy B, McKeown MR, Lin CY, Monti S,
Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al:
Discovery and characterization of super-enhancer-associated
dependencies in diffuse large B cell lymphoma. Cancer Cell.
24:777–790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xiong L, Wu F, Wu Q, Xu L, Cheung OK, Kang
W, Mok MT, Szeto L, Lun CY, Lung RW, et al: Aberrant enhancer
hypomethylation contributes to hepatic carcinogenesis through
global transcriptional reprogramming. Nat Commun. 10:3352019.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Andricovich J, Perkail S, Kai Y, Casasanta
N, Peng W and Tzatsos A: Loss of KDM6A activates super-enhancers to
induce gender-specific squamous-like pancreatic cancer and confers
sensitivity to bet inhibitors. Cancer Cell. 33:512–526.e8e0. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gelato KA, Schöckel L, Klingbeil O,
Rückert T, Lesche R, Toedling J, Kalfon E, Heroult M, Lejeune P,
Mönning U, et al: Super-enhancers define a proliferative
PGC-1alpha-expressing melanoma subgroup sensitive to BET
inhibition. Oncogene. 37:512–521. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tasdemir N, Banito A, Roe JS,
Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O,
Bolden JE, Chen CC, et al: BRD4 Connects enhancer remodeling to
senescence immune surveillance. Cancer Discov. 6:612–629. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Togel L, Nightingale R, Chueh AC,
Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D,
Dhillon AS, et al: Dual targeting of bromodomain and extraterminal
domain proteins and WNT or MAPK signaling, inhibits c-MYC
expression and proliferation of colorectal cancer cells. Mol Cancer
Ther. 15:1217–1226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Drier Y, Cotton MJ, Williamson KE,
Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Sholl LM,
Afrogheh AH, et al: An oncogenic MYB feedback loop drives alternate
cell fates in adenoid cystic carcinoma. Nat Genet. 48:265–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ceribelli M, Hou ZE, Kelly PN, Huang DW,
Wright G, Ganapathi K, Evbuomwan MO, Pittaluga S, Shaffer AL,
Marcucci G, et al: A Druggable TCF4- and BRD4-Dependent
transcriptional network sustains malignancy in blastic plasmacytoid
dendritic cell neoplasm. Cancer Cell. 30:764–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu J, Duan Z, Guo W, Zeng L, Wu Y, Chen
Y, Tai F, Wang Y, Lin Y, Zhang Q, et al: Targeting the
BRD4/FOXO3a/CDK6 axis sensitizes AKT inhibition in luminal breast
cancer. Nat Commun. 9:52002018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang Z, Ma P, Jing Y, Yan Y, Cai MC,
Zhang M, Zhang S, Peng H, Ji ZL, Di W, et al: BET Bromodomain
Inhibition as a therapeutic strategy in ovarian cancer by
downregulating FoxM1. Theranostics. 6:219–230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mottok A and Gascoyne RD: Bromodomain
inhibition in diffuse large B-cell lymphoma-giving MYC a brake.
Clin Cancer Res. 21:4–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tolani B, Gopalakrishnan R, Punj V, Matta
H and Chaudhary PM: Targeting Myc in KSHV-associated primary
effusion lymphoma with BET bromodomain inhibitors. Oncogene.
33:2928–2937. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang
E, Roe JS, Suzuki Y, Pappin DJ, Joshua-Tor L and Vakoc CR:
NSD3-short is an adaptor protein that couples BRD4 to the CHD8
chromatin remodeler. Mol Cell. 60:847–859. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chan KH, Zengerle M, Testa A and Ciulli A:
Impact of target warhead and linkage vector on inducing protein
degradation: Comparison of bromodomain and extra-terminal (BET)
degraders derived from triazolodiazepine (JQ1) and
tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J Med Chem.
61:504–513. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Amorim S, Stathis A, Gleeson M, Iyengar S,
Magarotto V, Leleu X, Morschhauser F, Karlin L, Broussais F, Rezai
K, et al: Bromodomain inhibitor OTX015 in patients with lymphoma or
multiple myeloma: A dose-escalation, open-label, pharmacokinetic,
phase 1 study. Lancet Haematol. 3:e196–e204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Albrecht BK, Gehling VS, Hewitt MC,
Vaswani RG, Cote A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L,
Campbell R, et al: Identification of a Benzoisoxazoloazepine
inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET)
family as a candidate for human clinical trials. J Med Chem.
59:1330–1339. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Siu KT, Ramachandran J, Yee AJ, Eda H,
Santo L, Panaroni C, Mertz JA, Sims IR, Cooper MR and Raje N:
Preclinical activity of CPI-0610, a novel small-molecule
bromodomain and extra-terminal protein inhibitor in the therapy of
multiple myeloma. Leukemia. 31:1760–1769. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhao L, Okhovat JP, Hong EK, Kim YH and
Wood GS: Preclinical studies support combined inhibition of bet
family proteins and histone deacetylases as epigenetic therapy for
cutaneous t-cell lymphoma. Neoplasia. 21:82–92. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tripathy D, Bardia A and Sellers WR:
Ribociclib (LEE011): Mechanism of action and clinical impact of
this selective cyclin-dependent kinase 4/6 Inhibitor in various
solid tumors. Clin Cancer Res. 23:3251–3262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Geoerger B, Bourdeaut F, DuBois SG,
Fischer M, Geller JI, Gottardo NG, Marabelle A, Pearson A, Modak S,
Cash T, et al: A phase I study of the CDK4/6 inhibitor ribociclib
(LEE011) in pediatric patients with malignant rhabdoid tumors,
neuroblastoma and other solid tumors. Clin Cancer Res.
23:2433–2441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ribociclib extends survival in HR+ breast
cancer. Cancer Discov. 8:OF52018. View Article : Google Scholar
|
|
77
|
Ribociclib approved for advanced breast
cancer. Cancer Discov. 7:OF32017. View Article : Google Scholar
|
|
78
|
Cheng W, Yang Z, Wang S, Li Y, Wei H, Tian
X and Kan Q: Recent development of CDK inhibitors: An overview of
CDK/inhibitor co-crystal structures. Eur J Med Chem. 164:615–639.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nagaraja S, Vitanza NA, Woo PJ, Taylor KR,
Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al:
Transcriptional dependencies in diffuse intrinsic pontine glioma.
Cancer Cell. 31:635–652, 2017.e6. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cayrol F, Praditsuktavorn P, Fernando TM,
Kwiatkowski N, Marullo R, Calvo-Vidal MN, Phillip J, Pera B, Yang
SN, Takpradit K, et al: THZ1 targeting CDK7 suppresses STAT
transcriptional activity and sensitizes T-cell lymphomas to BCL2
inhibitors. Nat Commun. 8:142902017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ma X, Kuang X, Xia Q, Huang Z, Fan Y, Ning
J, Wen J, Zhang H, Yan J, Zhang Q, et al: Covalent CDK7 inhibitor
THZ1 inhibits myogenic differentiation. J Cancer. 9:3149–3155.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ali I, Choi G and Lee K: BET inhibitors as
anticancer agents: A patent review. Recent Pat Anticancer Drug
Discov. 12:340–364. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Henssen A, Althoff K, Odersky A, Beckers
A, Koche R, Speleman F, Schafers S, Bell E, Nortmeyer M, Westermann
F, et al: Targeting MYCN-Driven Transcription By BET-Bromodomain
inhibition. Clin Cancer Res. 22:2470–2481. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Donati B, Lorenzini E and Ciarrocchi A:
BRD4 and cancer: Going beyond transcriptional regulation. Mol
Cancer. 17:1642018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Berthon C, Raffoux E, Thomas X, Vey N,
Gomez-Roca C, Yee K, Taussig DC, Rezai K, Roumier C, Herait P, et
al: Bromodomain inhibitor OTX015 in patients with acute leukaemia:
A dose-escalation, phase 1 study. Lancet Haematol. 3:e186–e195.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Stathis A, Zucca E, Bekradda M, Gomez-Roca
C, Delord JP, de La Motte RT, Uro-Coste E, de Braud F, Pelosi G and
French CA: Clinical response of carcinomas harboring the BRD4-NUT
oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628.
Cancer Discov. 6:492–500. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Piquereau J, Boet A, Pechoux C, Antigny F,
Lambert M, Gressette M, Ranchoux B, Gambaryan N, Domergue V, Mumby
S, et al: The BET Bromodomain Inhibitor I-BET-151 induces
structural and functional alterations of the heart mitochondria in
healthy male mice and rats. Int J Mol Sci. 20:15272019. View Article : Google Scholar
|
|
88
|
Mustafi S, Camarena V, Qureshi R, Yoon H,
Volmar CH, Huff TC, Sant DW, Zheng L, Brothers SP, Wahlestedt C, et
al: Vitamin C supplementation expands the therapeutic window of
BETi for triple negative breast cancer. Ebiomedicine. 43:201–210.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z,
He C, Zheng L, Hu X, Hu Z, et al: Suppression of enhancer
overactivation by a RACK7-histone demethylase complex. Cell.
165:331–342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Katerndahl C, Heltemes-Harris LM, Willette
M, Henzler CM, Frietze S, Yang R, Schjerven H, Silverstein K,
Ramsey LB, Hubbard G, et al: Antagonism of B cell enhancer networks
by STAT5 drives leukemia and poor patient survival. Nat Immunol.
18:694–704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gryder BE, Yohe ME, Chou HC, Zhang X,
Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, et
al: PAX3-FOXO1 establishes myogenic super enhancers and confers BET
bromodomain vulnerability. Cancer Discov. 7:884–899. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ott CJ, Federation AJ, Schwartz LS, Kasar
S, Klitgaard JL, Lenci R, Li Q, Lawlor M, Fernandes SM, Souza A, et
al: Enhancer architecture and essential core regulatory circuitry
of chronic lymphocytic leukemia. Cancer Cell. 34:982–995.e7e0.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Schmidt SF, Larsen BD, Loft A, Nielsen R,
Madsen JG and Mandrup S: Acute TNF-induced repression of cell
identity genes is mediated by NF κB-directed redistribution of
cofactors from super-enhancers. Genome Res. 25:1281–1294. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Biswas S and Rao CM: Epigenetics in
cancer: Fundamentals and beyond. Pharmacol Ther. 173:118–134. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Dzobo K, Senthebane DA, Thomford NE, Rowe
A, Dandara C and Parker MI: Not everyone fits the mold: Intratumor
and intertumor heterogeneity and innovative cancer drug design and
development. Omics. 22:17–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kelly AD and Issa JJ: The promise of
epigenetic therapy: Reprogramming the cancer epigenome. Curr Opin
Genet Dev. 42:68–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mill CP, Fiskus W, DiNardo CD, Qian Y,
Raina K, Rajapakshe K, Perera D, Coarfa C, Kadia TM, Khoury JD, et
al: RUNX1 targeted therapy for AML expressing somatic or germline
mutation in RUNX1. Blood. 134:59–73. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mansour MR, Abraham BJ, Anders L,
Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan
SE, Silverman LB, et al: Oncogene regulation. An oncogenic
super-enhancer formed through somatic mutation of a noncoding
intergenic element. Science. 346:1373–1377. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gröschel S, Sanders MA, Hoogenboezem R, de
Wit E, Bouwman B, Erpelinck C, van der Velden V, Havermans M,
Avellino R, van Lom K, et al: A single oncogenic enhancer
rearrangement causes concomitant EVI1 and GATA2 deregulation in
leukemia. Cell. 157:369–381. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Marsman J, Gimenez G, Day RC, Horsfield JA
and Jones GT: A non-coding genetic variant associated with
abdominal aortic aneurysm alters ERG gene regulation. Hum Mol
Genet. 29:554–565. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kleinstern G, Yan H, Hildebrandt M, Vijai
J, Berndt SI, Ghesquieres H, McKay J, Wang SS, Nieters A, Ye Y, et
al: Inherited variants at 3q13.33 and 3p24.1 are associated with
risk of diffuse large B-cell lymphoma and implicate immune
pathways. Hum Mol Genet. 29:70–79. 2020.PubMed/NCBI
|
|
102
|
He Y, Timofeeva M, Li X, Din F, Blackmur
JP, Vaughan-Shaw P, Svinti V, Farrington SM, Campbell H, Dunlop MG,
et al: A comprehensive study of the effect on colorectal cancer
survival of common germline genetic variation previously linked
with cancer prognosis. Cancer Epidemiol Biomarkers Prev.
28:1944–1946. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cong Z, Li Q, Yang Y, Guo X, Cui L and You
T: The SNP of rs6854845 suppresses transcription via the DNA
looping structure alteration of super-enhancer in colon cells.
Biochem Biophys Res Commun. 514:734–741. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhu DL, Chen XF, Hu WX, Dong SS, Lu BJ,
Rong Y, Chen YX, Chen H, Thynn HN, Wang NN, et al: Multiple
functional variants at 13q14 risk locus for osteoporosis regulate
RANKL expression through long-range super-enhancer. J Bone Miner
Res. 33:1335–1346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Eid A, Alshareef S and Mahfouz MM: CRISPR
base editors: Genome editing without double-stranded breaks.
Biochem J. 475:1955–1964. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Leenay RT, Maksimchuk KR, Slotkowski RA,
Agrawal RN, Gomaa AA, Briner AE, Barrangou R and Beisel CL:
Identifying and visualizing functional PAM diversity across
CRISPR-Cas systems. Mol Cell. 62:137–147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Jiang F and Doudna JA: CRISPR-Cas9
structures and mechanisms. Annu Rev Biophys. 46:505–529. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kosicki M, Tomberg K and Bradley A: Repair
of double-strand breaks induced by CRISPR-Cas9 leads to large
deletions and complex rearrangements. Nat Biotechnol. 36:765–771.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Anzalone AV, Randolph PB, Davis JR, Sousa
AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A and
Liu DR: Search-and-replace genome editing without double-strand
breaks or donor DNA. Nature. 576:149–157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fernandes Q: Therapeutic strategies in
Sickle Cell Anemia: The past present and future. Life Sci.
178:100–108. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Vu M, Li R, Baskfield A, Lu B, Farkhondeh
A, Gorshkov K, Motabar O, Beers J, Chen G, Zou J, et al: Neural
stem cells for disease modeling and evaluation of therapeutics for
Tay-Sachs disease. Orphanet J Rare Dis. 13:1522018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Liu X, Zhang Y, Chen Y, Li M, Zhou F, Li
K, Cao H, Ni M, Liu Y, Gu Z, et al: In situ capture of chromatin
interactions by biotinylated dCas9. Cell. 170:1028–1043.e19e0.
2017. View Article : Google Scholar : PubMed/NCBI
|