Introduction
Colorectal cancer (CRC) is the third most commonly
diagnosed cancer and the second leading cause of cancer-related
death among men and women worldwide (1). Surgery and chemotherapy are the main
treatment options for CRC. A cytotoxic combination of chemotherapy
drugs, such as fluorouracil and irinotecan, and the medication
leucovorin is the first line approach to treat CRC (2,3).
However, as cancer cells usually develop mechanisms to evade cell
death induced by chemotherapy (4–6), the
effects of these treatments are still limited. Therefore, there is
an urgent need to explore novel tumor-related genes and potential
therapeutic approaches to improve the clinical outcomes of CRC.
SAR1 gene homolog B (SAR1B), a coat protein II
(COPII) component, serves a central role in the lipid metabolism
involved in vesicular COPII-dependent transport of proteins from
the endoplasmic reticulum (ER) to the Golgi apparatus (7,8).
Mutations of the SAR1a gene homolog 2 that encodes SAR1B protein
cause the rare recessive disorder chylomicron retention disease
(CMRD) (9), which is associated with
developmental problems in infancy. The characteristics of CMRD
include fat malabsorption, steatorrhea, deficiency in fat-soluble
vitamins, low blood cholesterol and a selective absence of
chylomicrons in the blood (9–11). A
Previous study demonstrated that patients with CMRD exhibit diffuse
enterocyte vacuolization, large cytosolic lipid droplets in
intestinal histological tests and accumulation of chylomicron-like
particles in enterocytes (12). As
lipid metabolism is crucial in cancer cell proliferation, survival
and other biochemical processes (12,13),
SAR1B may be associated with physiological functions in CRC.
However, limited data are available to determine the involvement
and physiological role of SAR1B in CRC.
The present study demonstrated that the mRNA
expression levels of SAR1B were the highest in SW620 cells among
three CRC cell lines. In addition, using lentivirus-mediated
infection with short hairpin (sh)RNA targeting SAR1B (shSAR1B), the
expression of SAR1B was effectively knocked down in RKO cells. The
Celigo system, MTT and colony formation assays identified the
inhibitory effects of SAR1B on proliferation of RKO cells.
Molecular mechanism analysis results suggested that knockdown of
SAR1B with shSAR1B induced significant apoptosis of RKO cells
compared with cells infected with control short hairpin (sh)RNA
(shCtrl). Together, the results of the present study indicated a
tumorigenic function and a potential mechanism of SAR1B in CRC.
Materials and methods
Public datasets analysis
The expression of SAR1B in CRC was analyzed using
GEPIA datasets (http://gepia.cancer-pku.cn/) (14). Moreover, Kaplan-Meier method
(14) was used to analyze the
association between SAR1B expression levels and overall survival
(OS) time in patients with CRC using two public datasets, GSE17536
(15) and GSE17537 (15). In order to explore the potential
functions of SAR1B in CRC, the DepMap database was analyzed
(https://depmap.org/rnai/index) (16). The function of SAR1B in cancer cell
survival was analyzed using DepMap portal (https://depmap.org/rnai/index) based on the results of
genome-wide shRNA (or small interfering RNA) inhibition of SAR1B
from the Broad (17) and Novartis
(18) datasets. A PPI network
regulated by SAR1B was constructed using the STRING database
(https://string-db.org/cgi/network.pl)
(19). The bioinformatics analysis
was conducted using the Ingenuity Pathway Analysis system
(https://www.nihlibrary.nih.gov/resources/tools/ingenuity-pathways-analysis-ipa).
Patients
A total of 15 patients with CRC were recruited for
the present study. Patients from Taizhou People's Hospital
(Taizhou, China) were pathologically diagnosed with CRC and
underwent surgical resection between July 2019 and October 2019. No
patient received chemotherapy or radiotherapy treatment prior to
surgery. The clinical information of these patients is included in
Table I. All patients provided
written informed consent, and the present study was approved by the
Committees of Taizhou People's Hospital.
 | Table I.Clinical information of patients with
colorectal cancer used in this study. |
Table I.
Clinical information of patients with
colorectal cancer used in this study.
| Patient | Age | Sex | Tumor type | Lymph node
metastasis |
|---|
| Patient 1 | 51 | Female | Rectal
protuberance-type differentiated adenocarcinoma | Negative |
| Patient 2 | 38 | Female | Rectal ulcer type
of moderately poorly differentiated mucinous adenocarcinoma | Positive |
| Patient 3 | 46 | Male | Rectal ulcer type
moderately differentiated adenocarcinoma | Positive |
| Patient 4 | 53 | Male | Rectal
protuberance-type differentiated adenocarcinoma | Positive |
| Patient 5 | 61 | Female | Colonic ulcer type
moderately differentiated adenocarcinoma | Negative |
| Patient 6 | 68 | Female | Rectal ulcer type
moderately differentiated adenocarcinoma | Positive |
| Patient 7 | 71 | Female | Colonic ulcer type
moderately differentiated adenocarcinoma | Positive |
| Patient 8 | 29 | Male | Rectal ulcer type
moderately differentiated adenocarcinoma | Negative |
| Patient 9 | 38 | Male | Colonic bulging
type of differentiated adenocarcinoma | Positive |
| Patient 10 | 46 | Female | Rectal ulcer
mucinous adenocarcinoma | Negative |
| Patient 11 | 55 | Female | Rectal ulcer type
moderately poorly differentiated adenocarcinoma | Negative |
| Patient 12 | 51 | Male | Rectal ulcer type
moderately differentiated adenocarcinoma | Negative |
| Patient 13 | 63 | Female | Rectal prolapse
type moderately differentiated adenocarcinoma | Negative |
| Patient 14 | 65 | Female | Rectal ulcer type
moderately differentiated adenocarcinoma | Positive |
| Patient 15 | 68 | Male | Sigmoid colonic
hypertrophic adenocarcinoma | Negative |
Cell lines and culture
CRC cell lines HCT116, SW620 and RKO cells were
purchased from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences and cultured in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.). Penicillin
(100 U/ml) and streptomycin (100 U/ml) were added to the medium to
prevent contamination. Cells were incubated at 37°C in a humidified
atmosphere containing 95% air and 5% CO2.
Construction of SAR1B-knockdown
lentivirus
The target DNA sequence (5′-TGGCACAGTACATTGATTA-3′)
of SAR1B was selected from the full-length sequence (NM_016103) by
Shanghai GeneChem Co., Ltd. According to the sequence of SAR1B, two
vectors, shRNA S1 and shRNA S2, were designed. The sequences were
as follows: shRNA S1,
5′-CCGGGATGGCACAGTACATTGATTACTCGAGTAATCAATGTACTGTGCCATCTTTTTG-3′;
shRNA S2,
5′-AATTCAAAAAGATGGCACAGTACATTGATTACTCGAGTAATCAATGTACTGTGCCATC-3′.
The shRNAs were annealed and ligated to the linearized GV115-GFP
lentivirus vector (Shanghai GeneChem Co., Ltd.). The plasmid was
extracted from the DH5α cells (Shanghai GeneChem Co., Ltd.) and
verified by restriction endonuclease digestion followed by Sanger
sequencing. The plasmid was extracted and verified by enzymatic
digestion and sequencing. RKO cells (ATCC) at a density of 8,000
cells/well were infected with the lentivirus. RKO Cells infected
with an empty lentiviral vector were used as a control (shCtrl).
Cells at a density of 8,000 cells/well were cultured in RPMI-1640
medium with lentiviruses at a multiplicity of infection of 10 for
24 h at 37°C. A stable SAR1B-knockdown RKO cell line was
constructed using the GV115-GFP lentivirus vector system (Shanghai
GeneChem Co., Ltd.), which could express GFP. After 72 h of
infection, the fluorescence and infection efficiency were
determined using an inverted fluorescence microscope at 200×
magnification (IX-71; Olympus Corporation). When the infection
efficiency was 80%, the expression of SAR1B was analyzed by reverse
transcription-quantitative PCR (RT-qPCR) and western blotting. The
strong and positive EGFP signals in RKO cells following infection
with a lentivirus recombined with shSAR1B or shCtrl indicated that
the efficiency of infection in RKO cells and were determined using
an inverted fluorescence microscope (magnification, ×200;
Olympus).
RT-qPCR
Total RNA from HCT116, SW620 and RKO cells and 15
paired CRC samples were extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. A total of 2 µg RNA RNA samples were
reverse transcribed using a Reverse Transcription kit (Roche
Diagnostics GmbH) to synthesize cDNA, according to the
manufacturer's protocol. The reverse transcription temperature
protocol was as follows: 65°C for 10 min, 25°C for 10 min, 50°C for
1 h and 85°C for 5 min. cDNA (1 µl) was used as a template for PCR
using the SYBR® Green qPCR kit (Takara Biotechnology
Co., Ltd.), using the ViiA™ 7 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The primers used were
as follows: SAR1B, forward 5′-TTTTCCTACGATGGAATCTGGC-3′, reverse
5′-CAGCCTGTCTCCTCGCTTTC-3′; and GAPDH, forward
5′-TGACTTCAACAGCGACACCCA-3′ and reverse
5′-CACCCTGTTGCTGTAGCCAAA-3′. The thermocycling conditions were as
follows: Initial denaturation at 95°C for 15 sec, followed by 45
cycles at 95°C for 5 sec and 60°C for 30 sec. The PCR products of
SAR1B and GAPDH were 241 and 121 bp, respectively. All samples were
analyzed by using the 2−ΔΔCq method in triplicate
(20).
Colony formation assay
shSAR1B-infected and shCtrl RKO cells were digested
with trypsin and resuspended in standard medium after reaching the
logarithmic growth phase. Cells were seeded into 6-well plates at a
density of 500 cells/well, incubated at 37°C in a 5% CO2
incubator and observed for 10 days with half of the medium changed
every 3 days. Cells were washed with PBS and fixed with 4%
paraformaldehyde (1 ml/well; Shanghai Sangong Pharmaceutical Co.,
Ltd.) for 30–60 min at room temperature. Cells were washed with
PBS, stained with 500 µl Giemsa (cat. no. ECM550; Chemicon; Thermo
Fisher Scientific, Inc.) for 20 min at room temperature and washed
with deionized water three times. Images of cell colonies were
captured using inverted light microscope (200× magnification; IX71;
Olympus, Tokyo, Japan) and counted using ImageJ software (version
4.0; National Institutes of Health).
Cell proliferation assay
An MTT assay (Sigma-Aldrich; Merck KGaA) was
performed to assess cell proliferation following after knockdown of
SAR1B as previously described. Briefly, lentivirus-infected RKO
cells were seeded in 96-well plates at 3,000 cells/well. At 1, 2,
3, 4 and 5 days following incubation, 20 µl MTT solution (1 mg/ml)
was added to each well and incubated at 37°C for 4 h. Subsequently,
the medium was carefully removed and 100 µl acidic isopropanol (10%
SDS, 5% isopropanol and 0.01 mol/l HCl) was added to each well. The
plates were read using an automated microplate reader (Molecular
Devices, LLC) at 490 nm.
Plate analysis with the adherent cell
cytometry system Celigo®
Briefly, shSAR1B- or shCtrl-infected RKO cells were
digested with trypsin and resuspended in standard medium after
achieving the logarithmic growth phase. Cells were seeded into
96-well plates at a density of 2,000 cells/well. Fluorescence was
detected every day using the Celigo system (Nexcelom Bioscience
LLC) for 5 days continuously. Following adjustments in the
parameters in analysis settings, the number of cells by scanning
green fluorescence daily for 5 days at room temperature according
to the manufacturer's instructions.
Analysis of apoptosis using flow
cytometry
Apoptosis assessment was performed using an
Annexin-V/FITC Apoptosis Detection kit (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols.
Briefly, cells were infected with shSAR1B or shCtrl. Cells were
harvested and stained with Annexin-V/FITC and propidium iodide (PI)
in binding buffer for 15 min at room temperature in the dark. The
samples were analyzed using a BD FACScan system (BD Biosciences) to
determine the percentage of cells exhibiting Annexin-V and PI
staining. Apoptotic cells were subsequently analyzed via flow
cytometry, using MoFlo XDP (Beckman Coulter, Inc.).
Western blot analysis
Cells were infected with shSAR1B or shCtrl. Protein
was extracted using RIPA lysis buffer with proteinase inhibitor
(Beyotime Institute of Biotechnology) and western blotting was
performed as previously described (12). Protein concentrations of cell lysates
were determined using the Bradford method. Equal amounts of protein
(20 µg/lane) were separated using 12% SDS-PAGE and transferred to a
PVDF membrane. The membranes were blocked using 5% BSA, at 25°C for
2 h. After blocking at room temperature, the membrane was incubated
with anti-SAR1B (1:1,000; cat. no. ab155278, Abcam) and anti-GAPDH
(1:1,000; cat. no. sc-32233, Santa-Cruz Biotechnology, Inc.)
primary antibodies at 4°C for 12 h. Membranes were washed three
times with Tris-buffered saline Tween-20 buffer (TBST; 10 mM Tris,
150 mM NaCl, 0.05% Tween-20; Beijing Solarbio Science &
Technology Co., Ltd.). Following the primary incubation, membranes
were incubated with the horseradish peroxidase-conjugated (HRP)
anti-mouse secondary antibody (1:5,000; cat. no. sc-2005;
Santa-Cruz, Inc.) and anti-rabbit secondary antibody (1:5,000; cat.
no. sc-2004; Santa-Cruz Biotechnology, Inc.) at 25°C for 2 h.
Membranes were re-washed three times with TBST buffer.
Electrochemiluminescence (cat. no. 6883; Cell Signaling Technology,
Inc.) was used for visualization of the protein bands. GAPDH was
used as the loading control and protein expression was quantified
using ImageJ Software version 1.47 (National Institutes of
Health).
Microarray analysis
The stable RKO cells after shRNA infection were used
for the microarray analysis. Whole-genome gene expression levels of
stable RKO cells following SAR1B knockdown were examined using the
gene chip PrimeView Human Gene Expression Array (Thermo Fisher
Scientific, Inc.). The sample labeling, microarray hybridization,
washing and gene normalization were performed according to the
manufacturer's protocols. Differentially expressed genes were
subsequently identified via fold-change analysis and P-values; the
threshold set for upregulated and downregulated genes was
fold-change ≥1.5 and P<0.05. Cluster 3.0 software was used to
analyze the differentially expressed genes, using hierarchical
clustering method, and gene expression correlation coefficient as
the distance, average connection. Then the Treeview software was
used to draw the DEGs cluster map.
Bioinformatics analysis
The present study used the Ingenuity Pathway
Analysis system (https://www.nihlibrary.nih.gov/resources/tools/ingenuity-pathways-analysis-ipa)
to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
(21) and Gene Ontology (GO)
(21,22) analyses of potential targets of
SAR1B.
Statistical analysis
All experiments were performed in triplicate and
were repeated at least three times. Data are presented as the mean
± SD. Statistical analysis was performed using unpaired Student's
t-test. The probability of survival was estimated using the
Kaplan-Meier method. The log-rank test was used to statistically
compare the differences in survival times. P<0.05 was considered
to indicate a statistically significant difference.
Results
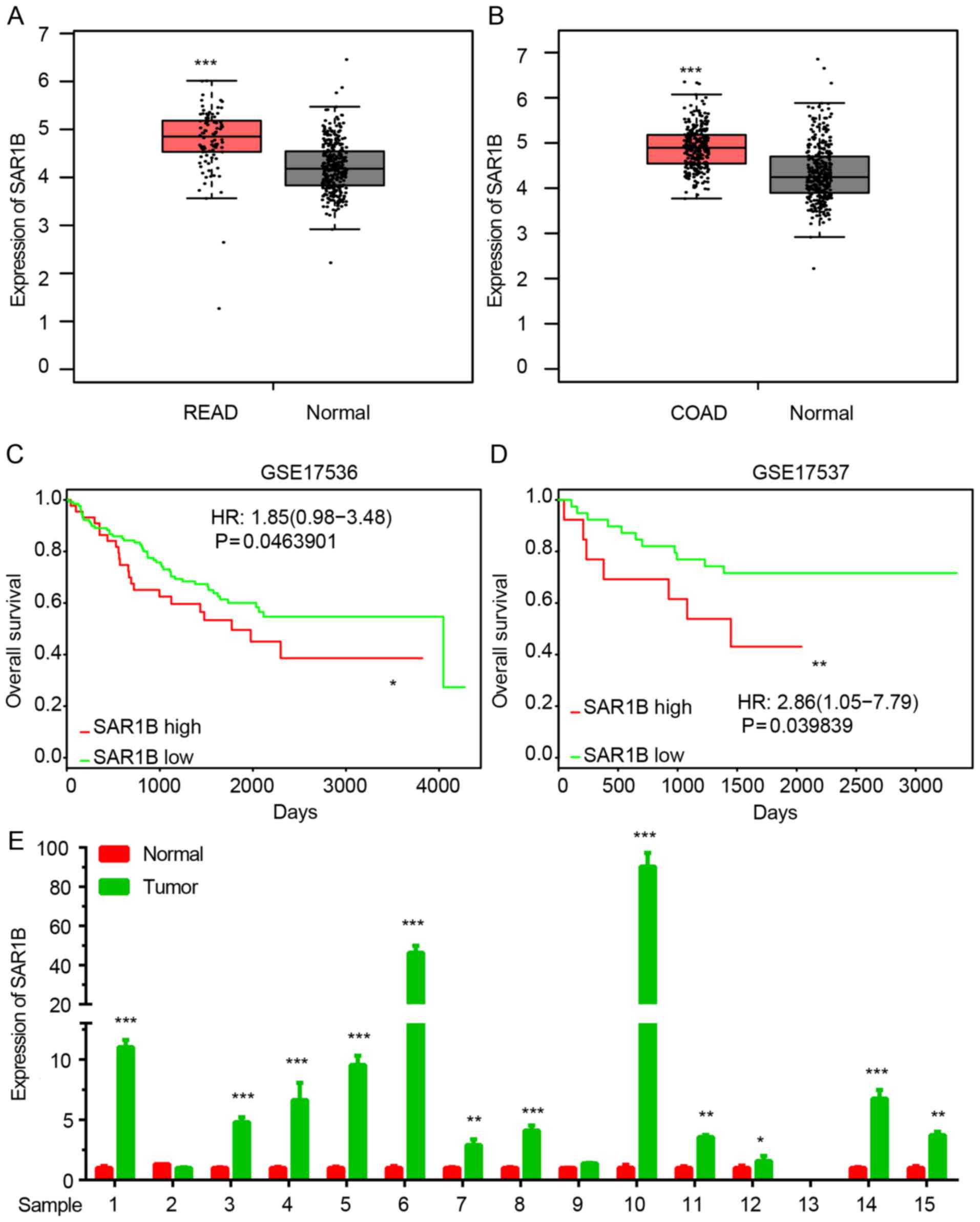
SAR1B is upregulated in CRC
The expression of SAR1B in CRC was analyzed using
GEPIA datasets. As demonstrated in Fig.
1A and B, SAR1B expression was significantly upregulated in
colon and rectum cancer samples compared with normal tissues.
Kaplan-Meier method was used to analyze the
association between SAR1B expression levels and overall survival
(OS) time in patients with CRC using two public datasets, GSE17536
and GSE17537. The results revealed that high expression of SAR1B
was significantly associated with shorter OS time in patients with
CRC (Fig. 1C and D).
In order to further validate the above analysis, 15
pairs of CRC samples were collected. As shown in Fig. 1E, RT-qPCR confirmed that SAR1B was
significantly upregulated in CRC samples compared with in normal
tissues (Fig. 1E).
Lentivirus-mediated knockdown of SAR1B
in CRC RKO cells
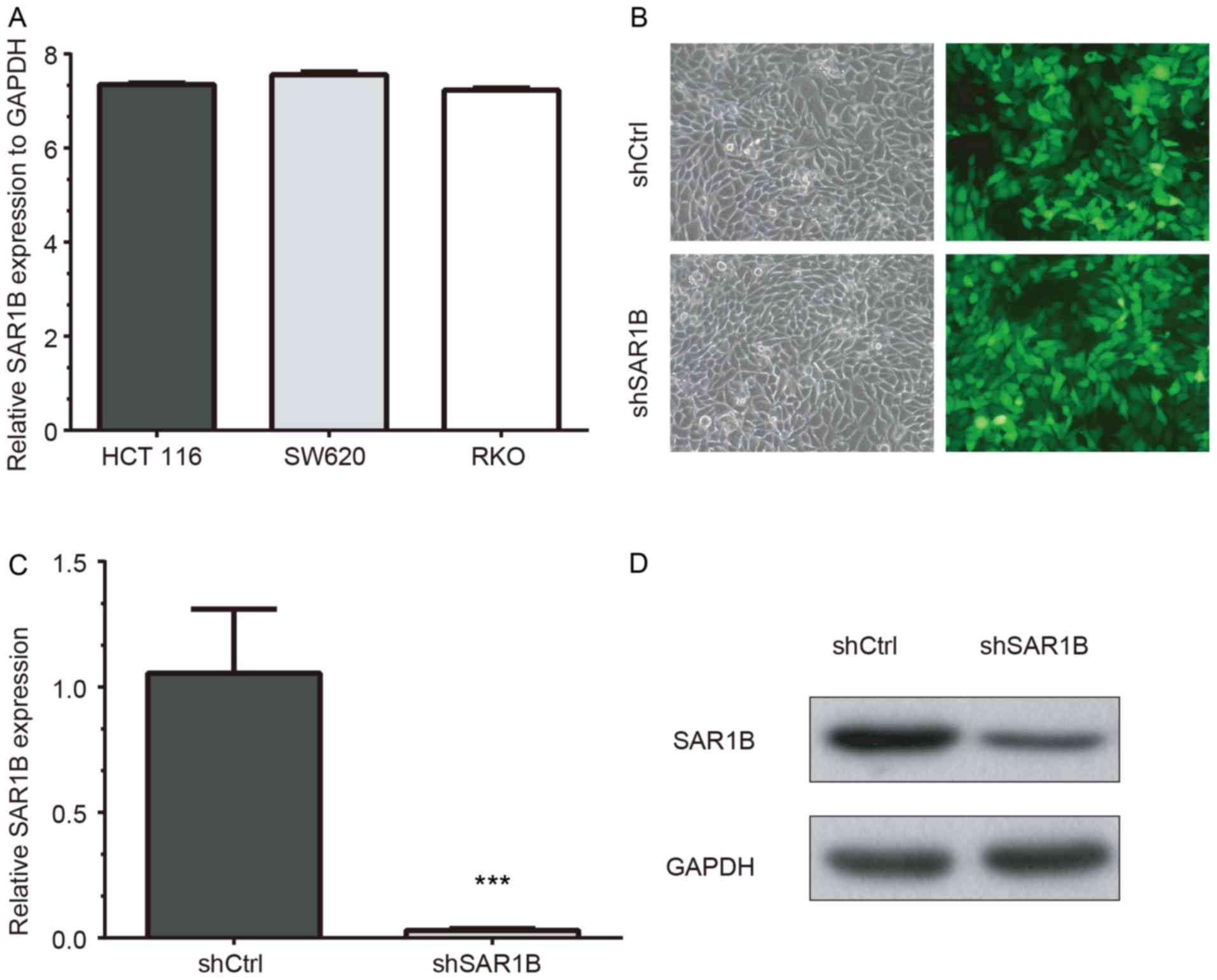
RT-qPCR was used to determine the mRNA expression
levels of SAR1B in CRC cell lines HCT116, SW620 and RKO. The
results demonstrated that SAR1B was expressed in the three cell
lines. The highest SAR1B mRNA expression was observed in SW620
cells, which was 7.56-fold higher than GAPDH expression (Fig. 2A). SAR1B mRNA expression levels were
also high in HCT116 cells (7.35-fold higher compared with GAPDH).
In addition, SAR1B mRNA expression was 7.23-fold higher than the
level of GADPH in RKO cells, which was consistent with HCT116 and
SW620 cells. The present study selected RKO cells for the
validation of functional importance of SAR1B in CRC, which has been
widely used in previous CRC studies (23–26).
To further explore the function of SAR1B in CRC,
SAR1B was silenced in CRC RKO cells using lentivirus-mediated gene
knockdown. As demonstrated in Fig.
2B, the strong and positive EGFP signals in RKO cells following
infection with a lentivirus recombined with shSAR1B or shCtrl
indicated that the efficiency of infection in RKO cells was high
and thus qualified for subsequent experiments.
RT-qPCR and western blot analysis results revealed
that knockdown of SAR1B was efficient at the mRNA and protein
levels in shSAR1B-infected RKO cells (Fig. 2C and D). The RT-qPCR results
demonstrated that the reduction efficiency reached 97% following
infection with shSAR1B, which was significantly different compared
with the shCtrl group (P<0.05; Fig.
2C). The western blotting results as also showed SAR1B protein
was reduced in shSAR1B-infected cells (Fig. 2D). These results indicated that
recombinant lentivirus targeting SAR1B effectively suppressed mRNA
and protein expression of endogenous SAR1B in CRC cells.
Knockdown of SAR1B inhibits RKO cell
proliferation
In order to explore the potential functions of SAR1B
in CRC, the DepMap database was used. The effects of SAR1B on
cancer cell survival was analyzed using the DepMap portal. The
results of genome-wide shRNA (or small interfering RNA) inhibition
of SAR1B from the Broad (Fig. S1A),
and Novartis (Fig. S1B) datasets
showed the dependency of SAR1B for cancer cell survival, thus
suggesting that SAR1B may promote the proliferation of CRC
cells.
A Celigo cell counting application, which allows for
direct imaging and counting of cells, was used to detect RKO cell
proliferation following SAR1B knockdown. The data were collected
for 5 days continually, and the results are presented in Fig. 3A. The results demonstrated that
compared with cells infected with shCtrl, the green signals in
cells infected with shSAR1B were significantly inhibited
(P<0.05). These results suggested that the downregulation of
SAR1B may inhibit cell proliferation (Fig. 3A). The average cell number was
12.76-fold higher on day 5 compared with that on day 1 in cells
infected with shCtrl, whereas the average cell number was 4.74-fold
higher on day 5 compared with that on day 1 in cells infected with
shSAR1B, which was significantly different (P<0.001).
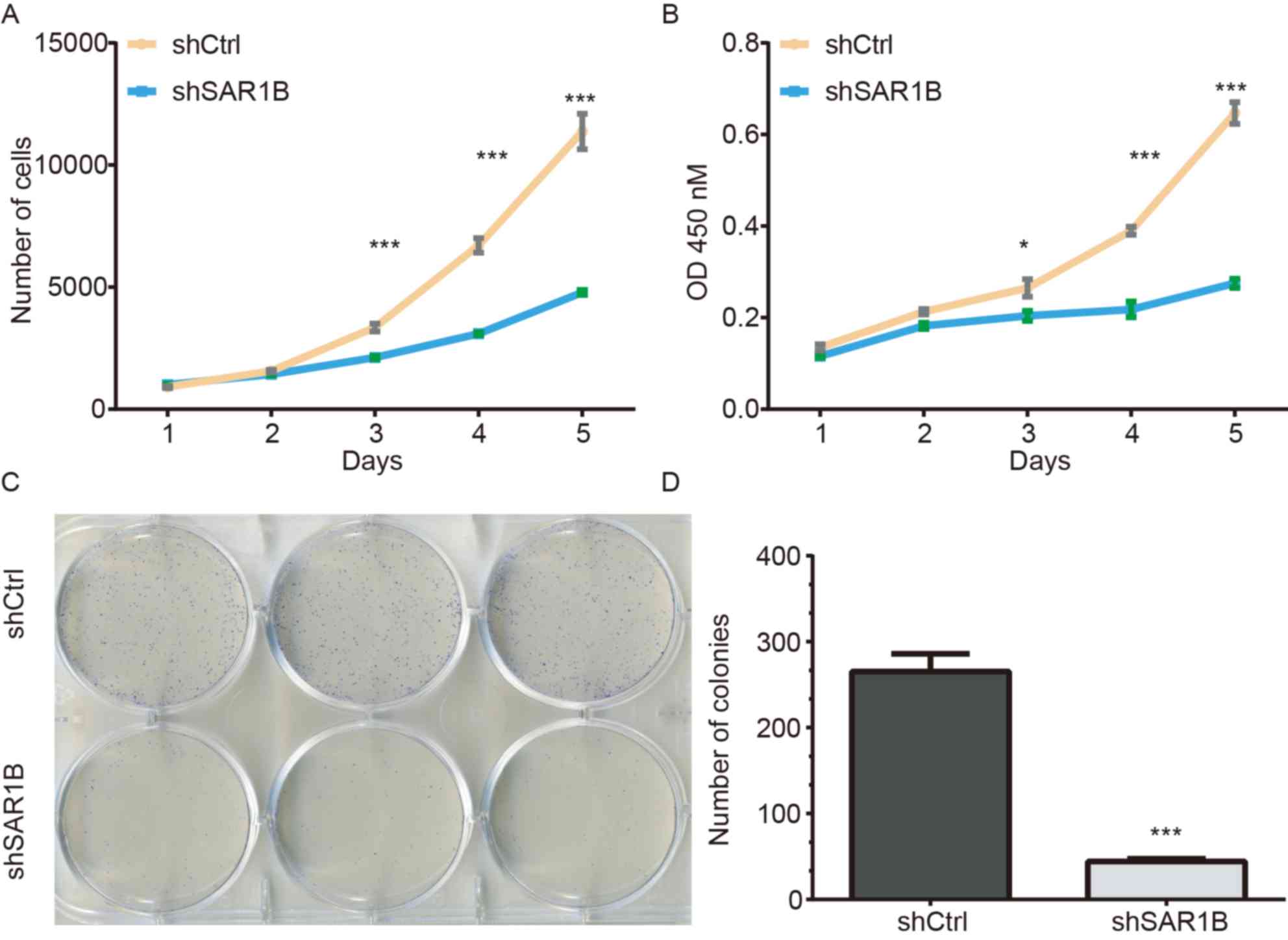 | Figure 3.Knockdown of SAR1B using shSAR1B led
to inhibition of RKO cell growth. (A) Number of RKO cells
expressing shCtrl lentivirus and shSAR1B lentivirus on days 1, 2,
3, 4 and 5, as measured by Celigo cell counting assay. (B) Cell
proliferation of RKO cells expressing shCtrl and shSAR1B on days 1,
2, 3, 4 and 5, as measured by an MTT assay. *P<0.05,
***P<0.001 vs. shSAR1B. (C) Microscope images showing colonies
of RKO cells that have been stained with Giemsa. (D) Representative
histogram showing the number of colonies in RKO cells. Each
experiment was independently performed in triplicate. ***P<0.001
vs. shCtrl. CRC, colorectal cancer; Ctrl, control; OD, optical
density; SAR1B, SAR1 gene homolog B; sh, short hairpin RNA. |
MTT and colony formation assays were used to further
investigate the potential effects of SAR1B on the proliferation of
RKO cells. A total of 2,000 cells were seeded and the cell numbers
were analyzed using an MTT assay daily for 5 days (Fig. 3B). The proliferation was reduced in
cells infected with shSAR1B compared with that in cells infected
with shCtrl (P<0.05), which suggested that the rate of cell
proliferation was significantly inhibited.
The colony formation assay also exhibited consistent
inhibition of cell proliferation in shSAR1B-infected RKO cells. As
demonstrated in Fig. 3C, the
colonies were relatively smaller and lower in number in the shSAR1B
group compared with the shCtrl group in RKO cells. Statistical
analysis further revealed that knockdown of SAR1B significantly
reduced the number of colonies formed by RKO cells (P<0.001;
Fig. 3D). These results suggested
that knockdown of SAR1B inhibited RKO cell proliferation.
Knockdown of SAR1B induces apoptosis
of RKO cells
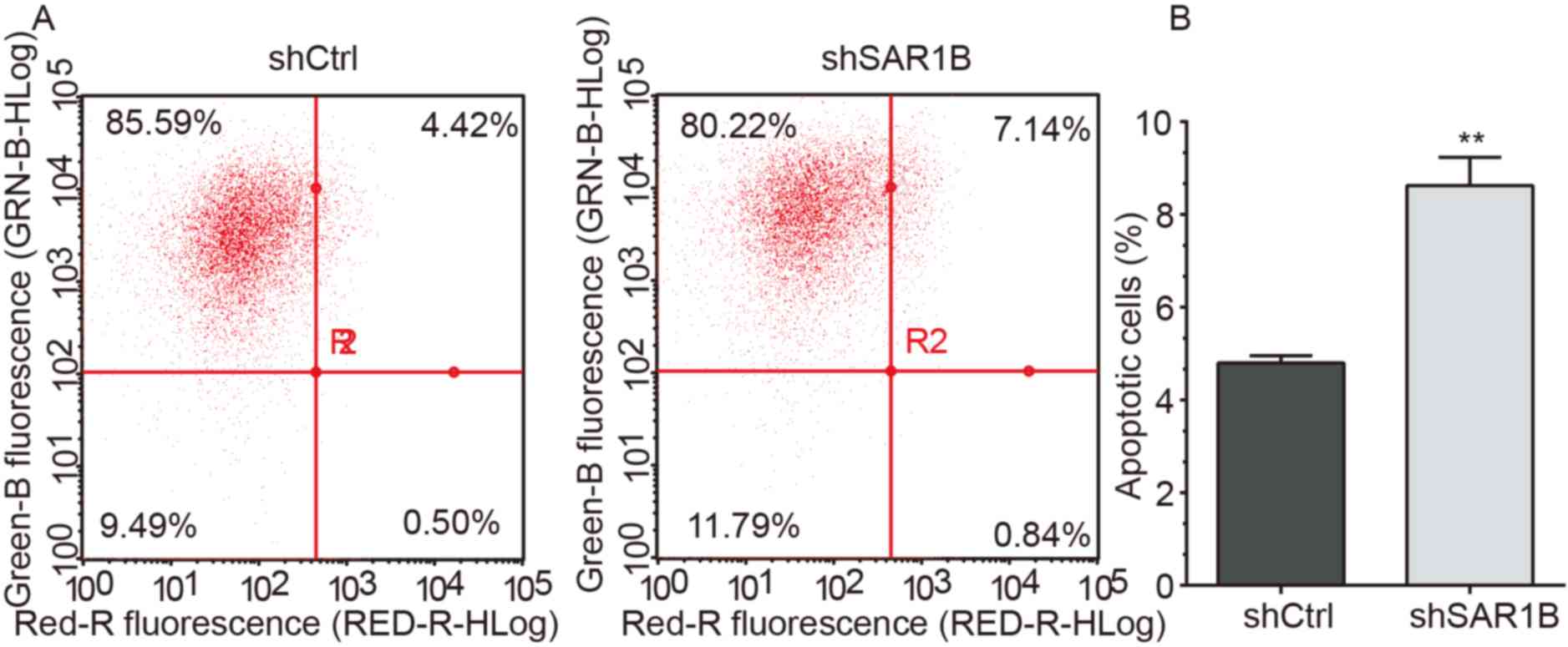
To explore the role of SAR1B in apoptosis, flow
cytometry was performed to determine the apoptotic rates in RKO
cells infected with shCtrl or shSAR1B. As demonstrated in Fig. 4A and B, apoptosis was induced
following shCtrl or shSAR1B infection. Further analysis revealed
that the apoptotic rate was significantly reduced in the shCtrl
group compared with the shSAR1B group, which was significantly
different (P<0.01). These results suggested that SAR1B knockdown
induced apoptosis of RKO cells, indicating the tumorigenic
potential of SAR1B.
Microarray analysis of the potential
roles of SAR1B in CRC
As the mechanisms underlying the effects of SAR1B on
CRC progression remain largely unknown, microarray analysis was
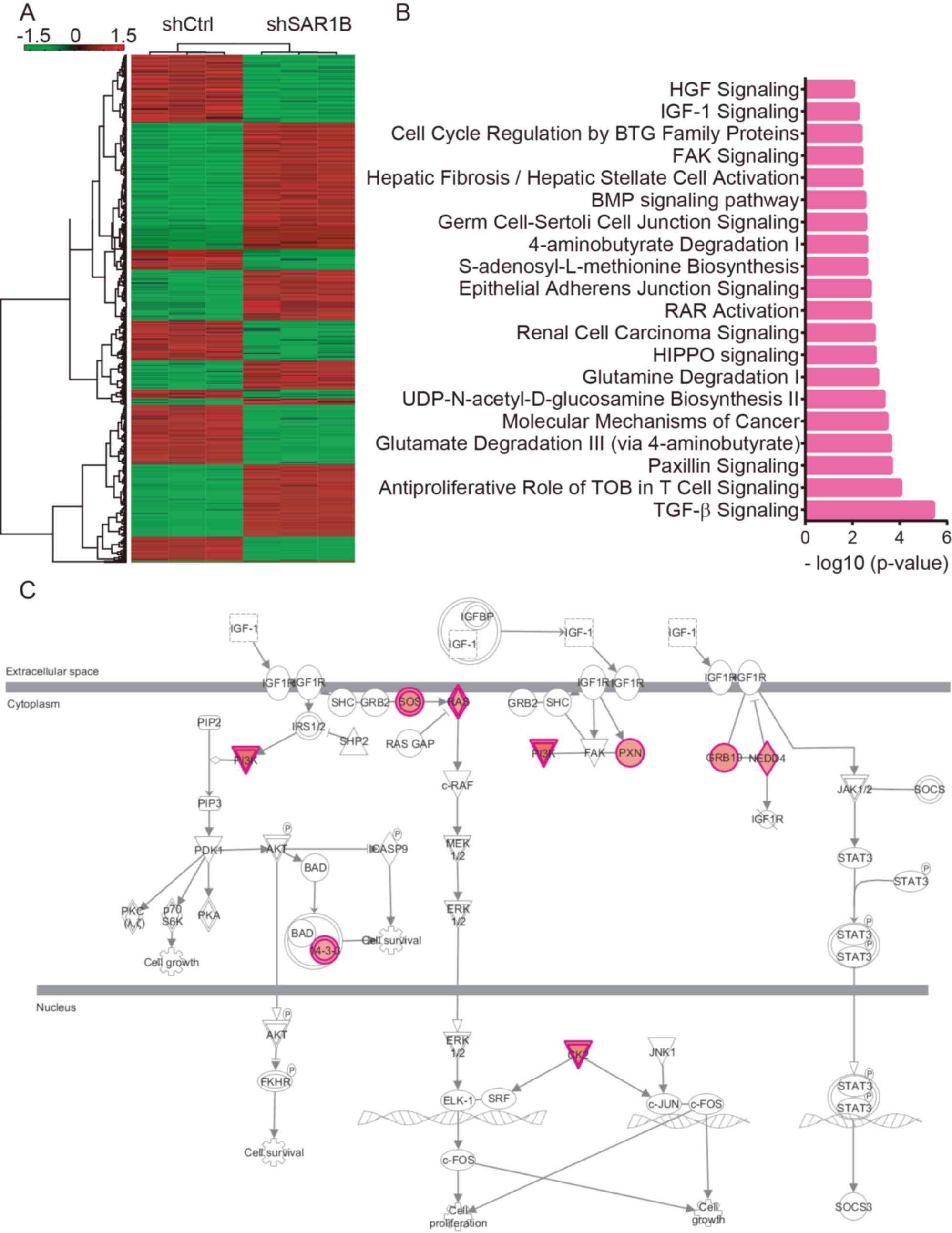
conducted. As presented in Fig. 5A,
a total of 333 genes were upregulated and 369 genes were
downregulated following SAR1B knockdown in RKO cells (Table SI).
KEGG pathway analysis revealed that SAR1B was
significantly enriched in ‘TGF-β signaling’, ‘antiproliferative
role of TOB in T cell signaling’, ‘paxillin signaling’, ‘glutamate
degradation III (via 4-aminobutyrate)’, ‘molecular mechanisms of
cancer’, ‘UDP-N-acetyl-D-glucosamine biosynthesis II’, ‘glutamine
degradation I’, ‘HIPPO signaling’, ‘renal cell carcinoma
signaling’, ‘RAR activation’, ‘epithelial adherens junction
signaling’, ‘S-adenosyl-L-methionine biosynthesis’,
‘4-aminobutyrate degradation I’, ‘germ cell-sertoli cell junction
signaling’, ‘BMP signaling pathway’, ‘hepatic fibrosis/hepatic
stellate cell activation’, ‘FAK signaling’, ‘cell cycle regulation
by BTG family proteins’ and ‘IGF-1 signaling’ (Fig. 5B). Of note, ‘IGF-1 signaling’ was
significantly activated; >9 regulators of this pathway were
upregulated following SAR1B knockdown in CRC by using Ingenuity
Pathway Analysis system (https://www.nihlibrary.nih.gov/resources/tools/ingenuity-pathways-analysis-ipa)
(Fig. 5C).
Construction of a protein-protein
interaction (PPI) network regulated by SAR1B in CRC
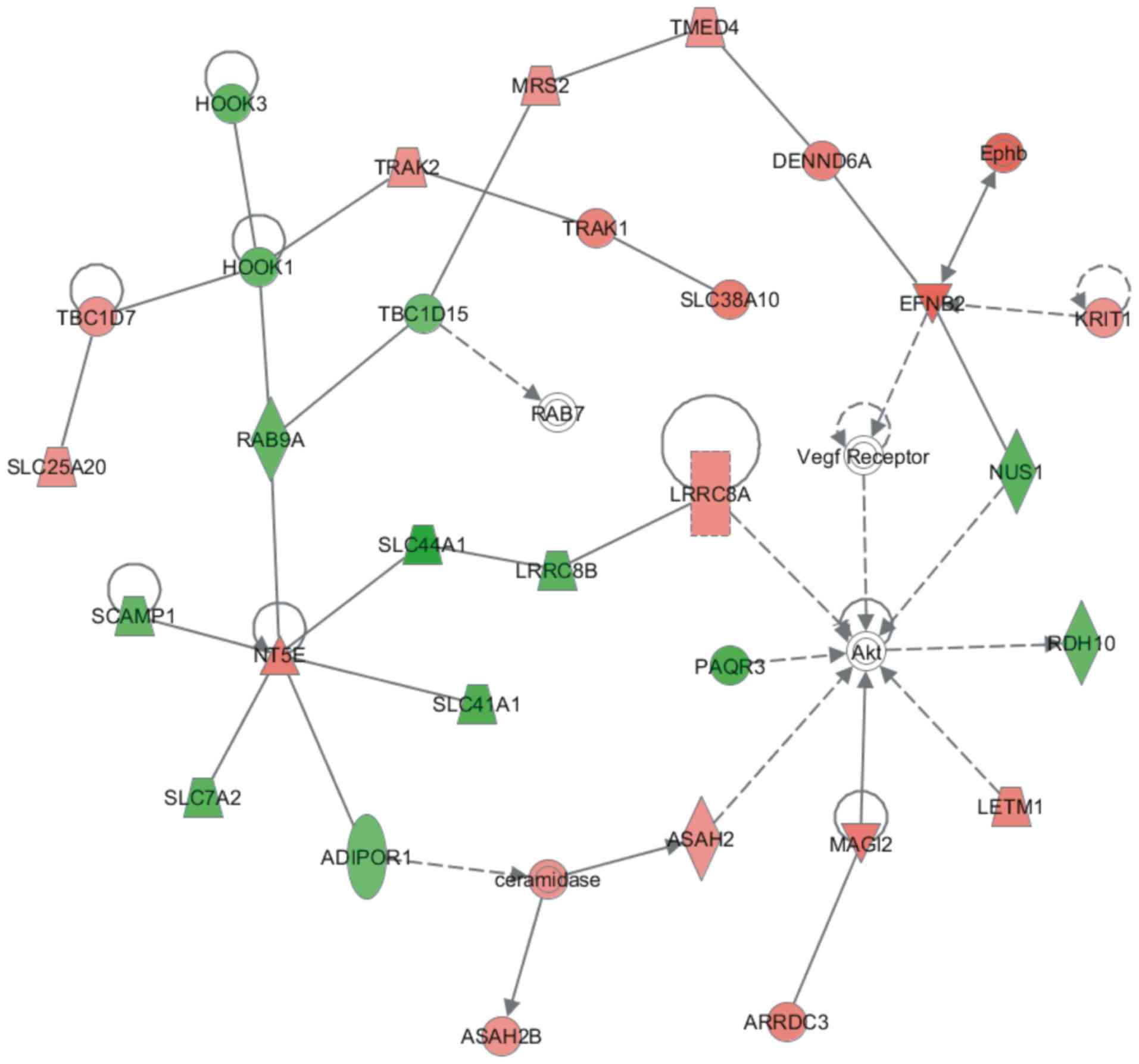
A PPI network regulated by SAR1B was constructed
using the STRING database. This network contained 35 nodes. Among
them, HOOK1, HOOK3, RAB9A, TBC1D15, SLC44A1, LRRC8B, SLC41A1,
SCAMP1, SLC7A2, ADIPOR1, PAQR3, NUS1 and RDH10 were downregulated
following SAR1B knockdown in RKO cells. Conversely, Akt, ARRDC3,
ASAH2, ASAH2B, ceramidase, DENND6A, EFNB2, Ephb, KRIT1, LETM1,
LRRC8A, MAGI2, MRS2, NT5E, RAB7, SLC25A20, SLC38A10, TBC1D7, TMED4,
TRAK1, TRAK2 and vascular endothelial growth factor receptor were
upregulated following SAR1B knockdown in RKO cells (Fig. 6).
Discussion
The results of the present study indicated that
SAR1B may be closely associated with the tumorigenesis of CRC.
SAR1B expression was determined in CRC cell lines, including
HCT116, SW620 and RKO. Functional analysis demonstrated that
knockdown of SAR1B suppressed CRC cell proliferation. Furthermore,
flow cytometry results revealed that knockdown of SAR1B induced
apoptosis of RKO cells. In addition, SAR1B downstream targets and
pathways in CRC were determined using microarray and bioinformatics
analyses.
Low molecular weight GTPases are involved in
vesicular transport associated with multiple signaling transduction
pathways that affect the regulation of cell proliferation,
differentiation and transformation (7,8). It has
been demonstrated that SAR1B GTPase is central to lipid metabolism
and regulates chylomicron secretion by the small intestine
(27,28). According to previous reports, the
expression of SAR1B was identified in cell lines (including
Caco-2/15 and McArdle-RH7777 cells), tissues and blood samples from
patients (12,29,30). In
the present study, SAR1B expression was upregulated in CRC patient
samples. By using RT-PCR, we found SAR1B was expressed in HCT116,
SW620 and RKO cell lines. These data suggested that SAR1B may be
associated with CRC tumorigenesis.
Previous metabolic studies have revealed that when
nutrients, such as lipids, proteins and nucleic acids, are
abundant, oncogenic signaling pathways directly enhance nutrient
acquisition, and facilitate cancer cell proliferation (31–33).
These results were reproduced in cultures of clear cell renal cell
carcinoma, glioblastoma and bladder cancer, suggesting that
increased lipid levels may contribute to cancer cell proliferation
(11) (9,34,35).
Considering the regulatory role of SAR1B in lipid transport, it
could be hypothesized that SAR1B may serve a role in tumorigenesis.
A lentiviral system with shSAR1B was used to knock down SAR1B in
RKO CRC cells. The effects of SAR1B on RKO proliferation were
determined by Celigo cell counting application, MTT and colony
formation assays. shSAR1B effectively suppressed mRNA and protein
expression levels of endogenous SAR1B in RKO cells. In addition,
RKO cell proliferation was reduced in cells infected with shSAR1B
compared with those infected with shCtrl. The colony formation
assay also demonstrated that knockdown of SAR1B significantly
reduced the number of colonies formed by RKO cells. The results of
the present study supported the involvement of SAR1B in CRC cell
function and demonstrated that the suppression of SAR1B inhibited
RKO cell proliferation.
Accumulating evidence has demonstrated that genes
encoding certain endocytosis-related proteins are the targets of
chromosomal rearrangement in human hematopoietic malignancies, and
abnormal expression or mutations of these proteins have been
reported in human cancer, suggesting a potential link between
endocytosis proteins and cancer (36). SAR1B is a GTPase that serves a key
role in the exportation of proteins from the ER to the Golgi
apparatus by recruiting β-COP to the ER and promoting vesicular
transport (10,37,38).
Considering the essential function of lipids in cancer, it is
possible that SAR1B may affect the molecular mechanisms involved in
cell proliferation or apoptosis. To further explore the role of
SAR1B in CRC in the present study, RKO cells were infected with
shSAR1B, and apoptosis was detected by flow cytometry. The
apoptotic rate of RKO cells infected with shSAR1B was significantly
higher compared with that in cells infected with shCtrl. These
results suggested that SAR1B knockdown induced apoptosis of RKO
cells.
Microarray analysis was performed to identify the
potential targets of SAR1B in CRC. Bioinformatics analysis revealed
that SAR1B was significantly enriched in ‘TGF-β signaling’,
‘paxillin signaling’, ‘cell cycle regulation by BTG family
proteins’ and ‘IGF-1 signaling’. Of note, >9 regulators of
‘IGF-1 signaling’ were upregulated following SAR1B knockdown in
CRC. Additionally, a SAR1B-related PPI network was constructed;
several key regulators of cancer were included in this network,
such as Akt, HOOK1 and HOOK3. HOOK1 has been demonstrated to
inhibit hepatocellular carcinoma progression and
epithelial-mesenchymal transition (EMT) (39), and high expression of HOOK3 is
associated with poor prognosis of prostate cancer (39). These reports, together with the
present results, suggested that SAR1B may serve crucial roles in
CRC by regulating these targets.
A recent study from Huang and Wang (40) revealed that SAR1B knockdown promoted
the migration and invasion of CRC cells. While this previous study
focused on the effects of SAR1B on CRC migration, invasion and EMT,
the present study focused on the effects of SAR1B on CRC
proliferation, colony formation and apoptosis. By combining these
previous results and the current findings, it can be concluded that
SAR1B may promote cancer proliferation, while suppressing cancer
metastasis in CRC. Of note, emerging studies have revealed that a
number of genes serve different roles in regulating cancer
proliferation and metastasis. For example, Wan et al
(41) revealed that patched 1
suppressed lung cancer proliferation via its coding protein;
however, it also promoted cell migration, invasion and adhesion in
non-small cell lung cancer via its 3′ untranslated region. Although
further validation into the roles of SAR1B in CRC is still
required, the present study could provide novel insights into the
functions of SAR1B.
It should be noted that there are several
limitations of the current study. Firstly, an in vivo model
should be used to further validate the roles of SAR1B in CRC, which
would be helpful to gain an understanding into the functional
importance of SAR1B in CRC. Secondly, the current study explored
the potential mechanisms of SAR1B in CRC using microarray analysis;
however, the microarray analysis was not validated in this
study.
In conclusion, the results of the present study
demonstrated that SAR1B was overexpressed in CRC cells. Inhibition
of SAR1B suppressed CRC cell proliferation and induced apoptosis of
RKO cells. Moreover, it was found that SAR1B may be involved in CRC
cell proliferation and apoptosis. These results suggested that
SAR1B could be used in the prognosis and treatment of CRC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YL and TB designed the present study. YL, SZ and RC
performed the majority of the experiments and drafted the initial
manuscript. YL and SZ performed the statistical analysis. LJ helped
perform the experiments and critically revised the manuscript for
important intellectual content. LY interpreted the data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent, and
the present study was approved by The Ethics Committee of Taizhou
Hospital of Zhejiang Province Affiliated to Wenzhou Medical
University (approval no. 2019068).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fakih MG: Metastatic colorectal cancer:
Current state and future directions. J Clin Oncol. 33:1809–1824.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Engelhardt EG, Revesz D, Tamminga HJ, Punt
CJA, Koopman M, Onwuteaka-Philipsen BD, Steyerberg EW, Jansma IP,
De Vet HCW and Coupé VMH: Clinical usefulness of tools to support
decision-making for palliative treatment of metastatic colorectal
cancer: A systematic review. Clin Colorectal Cancer. 17:e1–e12.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Advani SM, Advani P, DeSantis SM, Brown D,
VonVille HM, Lam M, Loree JM, Mehrvarz Sarshekeh A, Bressler J,
Lopez DS, et al: Clinical, pathological, and molecular
characteristics of CpG island methylator phenotype in colorectal
cancer: A systematic review and meta-analysis. Transl Oncol.
11:1188–1201. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Safa AR: Resistance to cell death and its
modulation in cancer stem cells. Crit Rev Oncog. 21:203–219. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gebremeskel S and Johnston B: Concepts and
mechanisms underlying chemotherapy induced immunogenic cell death:
Impact on clinical studies and considerations for combined
therapies. Oncotarget. 6:41600–41619. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Letai A: Cell death and cancer therapy:
Don't forget to kill the cancer cell! Clin Cancer Res.
21:5015–5020. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fryer LG, Jones B, Duncan EJ, Hutchison
CE, Ozkan T, Williams PA, Alder O, Nieuwdorp M, Townley AK,
Mensenkamp AR, et al: The endoplasmic reticulum coat protein II
transport machinery coordinates cellular lipid secretion and
cholesterol biosynthesis. J Biol Chem. 289:4244–4261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Loftus AF, Hsieh VL and Parthasarathy R:
Modulation of membrane rigidity by the human vesicle trafficking
proteins Sar1A and Sar1B. Biochem Biophys Res Commun. 426:585–589.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Charcosset M, Sassolas A, Peretti N, Roy
CC, Deslandres C, Sinnett D, Levy E and Lachaux A: Anderson or
chylomicron retention disease: Molecular impact of five mutations
in the SAR1B gene on the structure and the functionality of Sar1b
protein. Mol Genet Metab. 93:74–84. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sane AT, Seidman E, Peretti N, Kleme ML,
Delvin E, Deslandres C, Garofalo C, Spahis S and Levy E:
Understanding chylomicron retention disease through sar1b gtpase
gene disruption: Insight from cell culture. Arterioscler Thromb
Vasc Biol. 37:2243–2251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Silvain M, Bligny D, Aparicio T, Laforêt
P, Grodet A, Peretti N, Ménard D, Djouadi F, Jardel C, Bégué JM, et
al: Anderson's disease (chylomicron retention disease): A new
mutation in the SARA2 gene associated with muscular and cardiac
abnormalities. Clin Genet. 74:546–552. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levy E, Harmel E, Laville M, Sanchez R,
Emonnot L, Sinnett D, Ziv E, Delvin E, Couture P, Marcil V and Sane
AT: Expression of Sar1b enhances chylomicron assembly and key
components of the coat protein complex II system driving vesicle
budding. Arterioscler Thromb Vasc Biol. 31:2692–2699. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hannun YA and Obeid LM: Sphingolipids and
their metabolism in physiology and disease. Nat Rev Mol Cell Biol.
19:175–191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smith JJ, Deane NG, Wu F, Merchant NB,
Zhang B, Jiang A, Lu P, Johnson JC, Schmidt C, Bailey CE, et al:
Experimentally derived metastasis gene expression profile predicts
recurrence and death in patients with colon cancer.
Gastroenterology. 138:958–968. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McFarland JM, Ho ZV, Kugener G, Dempster
JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F,
Boehm JS, et al: Improved estimation of cancer dependencies from
large-scale RNAi screens using model-based normalization and data
integration. Nat Commun. 9:46102018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu C, Mannan AM, Yvone GM, Ross KN, Zhang
YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al:
High-throughput identification of genotype-specific cancer
vulnerabilities in mixtures of barcoded tumor cell lines. Nat
Biotechnol. 34:419–423. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
McDonald ER III, de Weck A, Schlabach MR,
Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E,
Gampa K, et al: Project DRIVE: A compendium of cancer dependencies
and synthetic lethal relationships uncovered by large-scale, Deep
RNAi screening. Cell. 170:577–592.e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
The Gene Ontology Consortium, . The gene
ontology resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang CY, Zhang LJ, Lu ZC, Ma CY, Ye Y,
Rahman K, Zhang H and Zhu JY: Antitumor activity of diterpenoids
from jatropha gossypiifolia: Cell cycle arrest and
apoptosis-inducing activity in RKO colon cancer cells. J Nat Prod.
81:1701–1710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garufi A, Ricci A, Trisciuoglio D, Iorio
E, Carpinelli G, Pistritto G, Cirone M and D'Orazi G: Glucose
restriction induces cell death in parental but not in
homeodomain-interacting protein kinase 2-depleted RKO colon cancer
cells: Molecular mechanisms and implications for tumor therapy.
Cell Death Dis. 4:e6392013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tansuwanwong S, Hiroyuki Y, Kohzoh I and
Vinitketkumnuen U: Induction of apoptosis in RKO colon cancer cell
line by an aqueous extract of Millingtonia hortensis. Asian Pac J
Cancer Prev. 7:641–644. 2006.PubMed/NCBI
|
|
26
|
Dang DT, Mahatan CS, Dang LH, Agboola IA
and Yang VW: Expression of the gut-enriched Kruppel-like factor
(Kruppel-like factor 4) gene in the human colon cancer cell line
RKO is dependent on CDX2. Oncogene. 20:4884–4890. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sane A, Ahmarani L, Delvin E, Auclair N,
Spahis S and Levy E: SAR1B GTPase is necessary to protect
intestinal cells from disorders of lipid homeostasis, oxidative
stress, and inflammation. J Lipid Res. 60:1755–1764. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levy E, Spahis S, Garofalo C, Marcil V,
Montoudis A, Sinnet D, Sanchez R, Peretti N, Beaulieu JF and Sane
A: Sar1b transgenic male mice are more susceptible to high-fat
diet-induced obesity, insulin insensitivity and intestinal
chylomicron overproduction. J Nutr Biochem. 25:540–548. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Magnolo L, Najah M, Fancello T, Di Leo E,
Pinotti E, Brini I, Gueddiche NM, Calandra S, Slimene NM and Tarugi
P: Novel mutations in SAR1B and MTTP genes in Tunisian children
with chylomicron retention disease and abetalipoproteinemia. Gene.
512:28–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marcil V, Seidman E, Sinnett D, Sanchez R,
Spahis S, Sané A and Levy E: Tissue distribution and regulation of
the small Sar1b GTPase in mice. Cell Physiol Biochem. 33:1815–1826.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okada T, Miyashita M, Fukuhara J, Sugitani
M, Ueno T, Samson-Bouma ME and Aggerbeck LP: Anderson's
disease/chylomicron retention disease in a Japanese patient with
uniparental disomy 7 and a normal SAR1B gene protein coding
sequence. Orphanet J Rare Dis. 6:782011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cefalu AB, Calvo PL, Noto D, Baldi M,
Valenti V, Lerro P, Tramuto F, Lezo A, Morra I, Cenacchi G, et al:
Variable phenotypic expression of chylomicron retention disease in
a kindred carrying a mutation of the Sara2 gene. Metabolism.
59:463–467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gaudet P, Livstone MS, Lewis SE and Thomas
PD: Phylogenetic-based propagation of functional annotations within
the Gene Ontology consortium. Brief Bioinform. 12:449–462. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Treepongkaruna S, Chongviriyaphan N,
Suthutvoravut U, Charoenpipop D, Choubtum L and Wattanasirichaigoon
D: Novel missense mutations of SAR1B gene in an infant with
chylomicron retention disease. J Pediatr Gastroenterol Nutr.
48:370–373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barbe L, Lundberg E, Oksvold P, Stenius A,
Lewin E, Björling E, Asplund A, Pontén F, Brismar H, Uhlén M and
Andersson-Svahn H: Toward a confocal subcellular atlas of the human
proteome. Mol Cell Proteomics. 7:499–508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu H, Tardivo L, Tam S, Weiner E, Gebreab
F, Fan C, Svrzikapa N, Hirozane-Kishikawa T, Rietman E, Yang X, et
al: Next-generation sequencing to generate interactome datasets.
Nat Methods. 8:478–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen JH, Hsieh CJ, Huang YL, Chen YC, Chen
TF, Sun Y, Wen LL, Yip PK and Chu YM: Genetic polymorphisms of
lipid metabolism gene SAR1 homolog B and the risk of Alzheimer's
disease and vascular dementia. J Formos Med Assoc. 115:38–44. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sane A, Seidman E, Spahis S, Lamantia V,
Garofalo C, Montoudis A, Marcil V and Levy E: New insights in
intestinal Sar1B GTPase regulation and role in cholesterol
homeostasis. J Cell Biochem. 116:2270–2282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun X, Zhang Q, Chen W, Hu Q, Lou Y, Fu
QH, Zhang JY, Chen YW, Ye LY, Wang Y, et al: Hook1 inhibits
malignancy and epithelial-mesenchymal transition in hepatocellular
carcinoma. Tumour Biol. 39:10104283177110982017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang M and Wang Y: Targeted quantitative
proteomic approach for probing altered protein expression of small
GTPases associated with colorectal cancer metastasis. Anal Chem.
91:6233–6241. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wan X, Kong Z, Chu K, Yi C, Hu J, Qin R,
Zhao C, Fu F, Wu H, Li Y and Huang Y: Co-expression analysis
revealed PTCH1-3′UTR promoted cell migration and invasion by
activating miR-101-3p/SLC39A6 axis in non-small cell lung cancer:
Implicating the novel function of PTCH1. Oncotarget. 9:4798–4813.
2018. View Article : Google Scholar : PubMed/NCBI
|