Introduction
Triple-negative breast cancer (TNBC) accounts for
15–20% of all breast carcinomas and is associated with an
aggressive disease progression and a high risk of relapse (1,2). The
median survival time of relapsed patients with TNBC varies between
12 and 24 months (3). TNBC is not
efficiently treated by the currently available therapeutic
regimens; this may be attributed to the lack of estrogen,
progesterone and Erb-B2 receptor tyrosine kinase 2 (ERBB2)
receptors (4,5). Chemotherapy remains the primary
systemic treatment, and the poor prognosis of TNBC is often
ascribed to resistance to chemotherapeutic agents. After repeated
cycles of chemotherapy, enhanced tumor resistance and severe side
effects resulting from these agents worsen the clinical outcome,
often leading to therapeutic failure (6).
In recent years, various lines of research have been
performed that aimed to bypass drug resistance and improve the
sensitivity to chemotherapeutic agents in cancer cells (7,8). The
underlying mechanisms are often complicated, such as reducing the
effective drug concentration in cells, establishing abnormalities
in drug targets and altering the regulation of apoptosis (9). Various strategies, including RNA
silencing, nanopreparations, co-administration of two or more
strategies, novel cytotoxic agents and regulation of apoptosis,
have been developed to overcome drug resistance in cancer cells
(10,11).
Gemcitabine (2′,2′-difluorodeoxycytidine; dFdC) has
been evaluated for its efficacy in the treatment of TNBC in a
number of clinical trials and has been established as one of the
most efficient chemotherapeutic drugs for various types of cancer
in clinical practice (12,13). dFdC is taken up into the cells by
human equilibrative nucleoside transporters and human concentrative
nucleoside transporters (14,15).
Once inside the cell, dFdC is phosphorylated by deoxycytidine
kinase (dCK) to its monophosphorylated form, and subsequently by
nucleotide kinases to its active metabolites, dFdC diphosphate
(dFdCDP) and dFdC triphosphate (dFdCTP) (16). dFdCDP is an effective inhibitor of
ribonucleotide diphosphate reductases including
ribonucleoside-diphosphate reductase large subunit (RRM1), and
resistance to dFdC is associated with increased expression of
ribonucleotide reductase (17).
The phosphorylation induced by dFdC is the main
rate-limiting step of the anticancer effect of dFdC (18); on the other hand, dFdC can be
deactivated to its main metabolite 2′,2′-difluorodeoxyuridine
(dFdU) by cytidine deaminase (CDA) (19). The cytotoxicity of dFdC is mainly
associated with the cellular accumulation of dFdCTP (16). dFdCTP is incorporated into DNA,
causing masked chain termination by inducing a
G0/G1 and S-phase arrest in the cell cycle,
which triggers apoptosis (20). In
addition, dFdC decreases cellular deoxynucleotide (dNTP) pools and
competes with them for incorporation into DNA, which, coupled with
a decreased feedback inhibition of dCK, leads to an enhanced
incorporation of dFdC into DNA (18).
Various therapeutic approaches have been proposed to
overcome drug resistance induced by nucleoside kinase deficiency
(21–23). Previously published work from our
laboratory has demonstrated that a multisubstrate
deoxyribonucleoside kinase of Drosophila melanogaster
(Dm-dNK) may be a potential candidate suicide gene, and its
effectiveness has been investigated in a number of tumor cell
lines, including MDA-MB-231 (24),
using viral systems (retrovirus-, adenovirus- and lentivirus-based
vectors) combined with prodrugs such as araT, araC, gemcitabine and
bromovinyldeoxyuridine (BVDU) (25–31). The
effectiveness of Dm-dNK has been demonstrated to be due to its
broad substrate specificity regarding both purine and pyrimidine
nucleoside analog phosphorylation and a higher catalytic rate
compared with that of previously studied nucleoside kinases
(32–34).
Previous studies have demonstrated that
dCK-deficient cell lines display a dFdC-resistant phenotype
(35,36). Therefore, the hypothesis of the
present study was that transfection with Dm-dNK may reverse the
resistance to dFdC in TNBC cells. The present study aimed to
develop a dFdC-resistant breast cancer MDA-MB-231 cell model to
explore whether Dm-dNK may reverse dFdC resistance in TNBC and to
determine the underlying mechanisms.
Materials and methods
Lentiviral packaging and
titration
The basic plasmid was established has been
previously described (31,37). Briefly, a three-plasmid system and
lentiviral vectors were co-transfected into the packaging cells
(the 293 cell line; 5×106 cells/100-mm dish cultured for
24 h until the cell confluence rate was 70–80%) through standard
transient transfection using Lipofectamine® 2000 (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The rate of plasmid:vector:cells was 1:1:1. The medium was
collected 48 h post-transfection and filtered through a 0.45-µm
filter and diluted 2-fold with fresh medium, and then repeatedly
collected three times at 24-h intervals. Subsequently, PCR was used
to amplify the DNA fragment of Dm-dNK-3Flag and Dm-dNKmut-3Flag,
and the fragment was then ligated into a PGC-FU plasmid (Shanghai
GeneChem Co., Ltd.), which was composed of a 5′-long terminal
repeat, a cytomegalovirus promoter and a multiple cloning site in
the presence of a green fluorescent protein (GFP) sequence.
Virus-producing cells were collected by ultracentrifugation (4°C,
120,000 × g for 2 h) to collect the recombinant lentivirus,
followed by PCR identification. These lentiviruses were termed
Lv-dNK and Lv-dNKmut. Lentiviral infectivity among cell lines was
determined by dNK-GFP and dNKmut-GFP. To improve the infection
efficiency, all cell lines were transduced (37°C for 24 h repeated
three times) with lentivirus in DMEM supplemented with 6 µg/ml
polybrene (Merck KGaA). The virus titer was quantified based on the
number of GFP-positive cells and the infectious dose of the
recombinant virus with 10-fold serial dilution. The cell nuclei
were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). GFP
and DAPI fluorescence was observed using a Nikon Eclipse E600
microscope (Nikon Corporation) equipped with a SPOT RT digital
camera (Diagnostic Instruments, Inc.) at ×400 magnification in 5–6
fields per sample.
Cell culture and establishment of the
dFdC-resistant cell line
293 and MDA-MB-231 cells were obtained from The Cell
Bank of Type Culture Collection of the Chinese Academy of Sciences.
HEK293 and MDA-MB-231 cells were maintained in high-glucose
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin and 100 U/ml
streptomycin at 37°C in a humidified atmosphere containing 5%
CO2. The modified resistant strain of MDA-MB-231 was
developed as previously described (38). Briefly, MDA-MB-231 cells were
maintained in DMEM and exposed to dFdC at an initial concentration
of 1.0 µM and repeatedly cultured with increasing concentrations of
dFdC at 1.5–2-fold increments including dFdC-free intervals in
order to allow the surviving cells to recover. MDA-MB-231 cells
were seeded at a density of 3×103 cells/100-mm dish and
subcultured every 14 days with increasing dosage of dFdC treatment
from day 7 to day 12. This protocol was repeated twice. After 28
days, the cells were seeded at a density of 6×105
cells/100-mm dish and treated with dFdC after 24 h. After another
72 h, the cells were subcultured and cultured in dFdC-free medium
for 96 h to allow the surviving cells to recover. This protocol was
repeated five times. The final concentration was 80 µM.
Chemoresistant MDA-MB-231 cells were challenged with dFdC for 6
months. Prior to the experiments, chemoresistant MDA-MB-231 cells
were seeded into to the drug-free medium for 2 weeks, and these
cells were termed MDA-MB-231R cells.
Western blotting analysis
MDA-MB-231 and MDA-MB-231R cells were harvested at
72 h following lentiviral infection, and total proteins were
extracted using a lysis buffer [50 mM HEPES (pH 7.4), 250 mM NaCl,
1 mM NaF, 1 mM EDTA, 1% Triton X-100 and 1 mM DTT] supplemented
with protease inhibitors PMSF (cat. no. 8553S; Cell Signaling
Technology, Inc.). The concentration of the extracted protein was
measured using a BCA protein assay (Nanjing KeyGen Biotech Co.,
Ltd.). Equal amounts (20 µg) of the proteins were subjected to
SDS-PAGE (10% gels) and electro-transferred to PVDF membranes (EMD
Millipore). The blots were blocked in Tris-buffered saline (TBS)
containing 0.1% Tween-20 and 5% nonfat milk at room temperature for
2 h, followed by incubation with antibodies against Flag (1:1,000;
cat. no. ab205606; Abcam), dCK (1:5,000; cat. no. ab151966; Abcam),
CDA (1:300; cat. no. SAB1300717; Merck KGaA), P-gp (1:2,000; cat.
no. ab170904; Abcam) and β-actin (1:500; cat. no. sc47778; Santa
Cruz Biotechnology, Inc.) at 4°C overnight. Subsequently, the blots
were washed with TBS containing 0.1% Tween-20, followed by
incubation with an appropriate specific secondary antibody: Mouse
anti-rabbit IgG-HRP (1:5,000; cat. no. sc2357; Santa Cruz
Biotechnology, Inc.) or goat anti-mouse IgG-HRP (1:4,000; cat. no.
sc2005; Santa Cruz Biotechnology, Inc.) at room temperature for 2
h. The immunoreactive bands were visualized with ECL western
blotting substrate (Thermo Fisher Scientific, Inc.), and the
protein expression was detected using the ChemiDoc™ XRS+ Imaging
System (Bio-Rad Laboratories, Inc.). The relative band intensities
were estimated using ImageJ 1.48 software (National Institutes of
Health). β-actin was used as an internal loading control.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the MDA-MB-231 and
MDA-MB-231R cells using the AP-MN-MS-RNA-50 RNA extraction kit
(Axygen; Corning, Inc.). RT was performed using a PrimeScript RT
reagent kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer's instructions. PCR amplification of the cDNA was
performed in a 25 ml mixture containing 2 ml template cDNA, 1 ml
each forward and reverse primer (10 µmol/l) and 12.5 ml
SYBR® Premix Ex Taq II (Takara Biotechnology Co., Ltd.).
GAPDH was used as an endogenous control. The primers used were as
follows: Dm-dNK forward, 5′-ATGAGTTGCACGAGGACTGG-3′ and reverse,
5′-CTGGTACTCGGTGCCAATGT-3′; and GAPDH forward,
5′-ACAGTCCATGCCATCACTGCC-3′ and reverse,
5′-GCCTGCTTCACCACCTTCTTG-3′. The thermocycling conditions included
an initial incubation at 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec and annealing at 60°C for 30 sec.
Each experiment was performed three times. The target gene levels
were normalized to that of GAPDH as the housekeeping gene. Relative
expression levels were calculated according to the
2−ΔΔCq method (39).
Enzyme activity assays
Cellular proteins were extracted using a previously
established protocol (40) at 72 h
post-infection with Lv-dNK or Lv-dNKmut at a multiplicity of
infection (MOI) of 10. The activity of Dm-dNKmut was determined
using a 35-ml reaction mixture comprising 50 mM Tris-HCl (pH 7.6),
100 mM KCl, 2 mM DTT, 15 mM NaF, 5 mM MgCl2, 5 mM ATP,
0.5 mg/ml bovine serum albumin (GBCBIO Technologies, Inc.) and 0.6
mg protein extract. For these analyses, aliquots of the samples, in
which 2.5 mM [methyl-3H] deoxythymidine (dThd; Moravek,
Inc.) was mixed with equivalent amounts of unlabeled substrates,
were spotted onto Whatman DE-81 filter paper discs following
incubation at 37°C for 10, 20 and 30 min. Subsequently, the paper
discs were dried for 1 h, washed three times with 5 mM ammonium
formate, the elution of nucleoside monophosphates was performed
using 0.5 M KCl, and the radioactivity was subsequently determined
using a scintillation counter.
Cell viability and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The viability of MDA-MB-231 and MDA-MB-231R cells
was determined using the MTT assay. Briefly, cells were seeded in
96-well plates at a density of 104 cells/well and
treated with graded concentrations of dFdC from 0.0001 to 100 µM
for 48 h following infection with Lv-dNK or Lv-dNKmut.
Subsequently, the medium was replaced with fresh medium (DMEM
containing 10% FBS), and 20 µl MTT reagent (5 mg/ml; Promega
Corporation) was added to each well, followed by incubation for 4
h. The formazan products were dissolved by 200 µl 99%
dimethylsulfoxide (DMSO) for 10 min. The absorbance was examined by
an enzyme immunoassay instrument at a wavelength of 570 nm. Each
experiment was performed in triplicate.
Toxicity analysis with trypan blue dye
exclusion assay
Cytotoxicity was evaluated using the trypan blue dye
exclusion assay as previously described by Fraser et al
(41). Each experiment was performed
in a 35-mm tissue culture dish (three dishes per experimental
condition) with 3×104 cells per dish with 1 ml
high-glucose DMEM with 10% FBS was added. The plates were incubated
overnight in the humidified incubator at 37°C to allow the cells to
settle. The cells were subsequently infected with Dm-dNK, Dm-dNKmut
or the empty lentiviral vector at an MOI of 1. Various
concentrations of dFdC (0.01, 0.1, 1, 10 and 100 µM) were
subsequently added, and the plates were incubated at 37°C for a
further 24 h. Then, the medium was removed, followed by the
addition of 0.25 ml trypan blue dye diluted in 0.8 ml medium. After
10-min incubation at 37°C in the dark, the diluted trypan blue was
removed, and 30 fields of view with at least 20 cells in each field
were captured under a microscope and counted using ImageJ 1.48
software (National Institutes of Health).
Apoptosis assay
Induction of apoptosis was analyzed by flow
cytometry. Briefly, MDA-MB-231 and MDA-MB-231R cells were seeded in
6-well plates at 2×105 cells/well and cultured for 24 h,
followed by transduction with lentiviral vectors, Lv-dNK or
Lv-dNKmut at MOI of 10. Two days later, dFdC (1 µM) was added for
24 h, and the apoptosis assays were performed using an Annexin
V-FITC kit (cat. no. KGA105; Nanjing Keygen Biotech Co., Ltd.)
according to the manufacturer's instructions. The ratio of early
and late apoptosis was assessed by a FACScan flow cytometer
equipped with CELLQUEST and ModFITLT for Mac V1.01 software
(Becton-Dickinson and Company).
Statistical analysis
Data are presented as the means ± SD. Data were
analyzed using SPSS 23.0 (IBM Corp.). Differences between two
groups were evaluated using unpaired Student's t-test, whereas one-
and two-way ANOVA with Bonferroni post hoc test was performed for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant value.
Results
Establishment of the drug-resistant
breast cancer cell line
Parental cells MDA-MB-231 were continuously
challenged with dFdC for 6 months to generate dFdC-resistant
clones. As presented in Fig. 1A, the
MDA-MB-231R cells were less sensitive to dFdC compared with the
MDA-MB-231 cells. The mean IC50 values of MDA-MB-231R
and MDA-MB-231 were 53.72 and 4.077 µM, respectively (P<0.001),
exhibiting an ~13-fold increase in the MDA-MB-231R cells compared
with the parental MDA-MB-231 cells. Subsequently, a concentration
of 1 µM was selected for further experiments. Fig. 1B demonstrates that the cell viability
was significantly inhibited in the MDA-MB-231R cells compared with
that in the MDA-MB-231 cells after 48-h treatment with dFdC.
Furthermore, the difference in cell proliferation between the two
cell types increased over time.
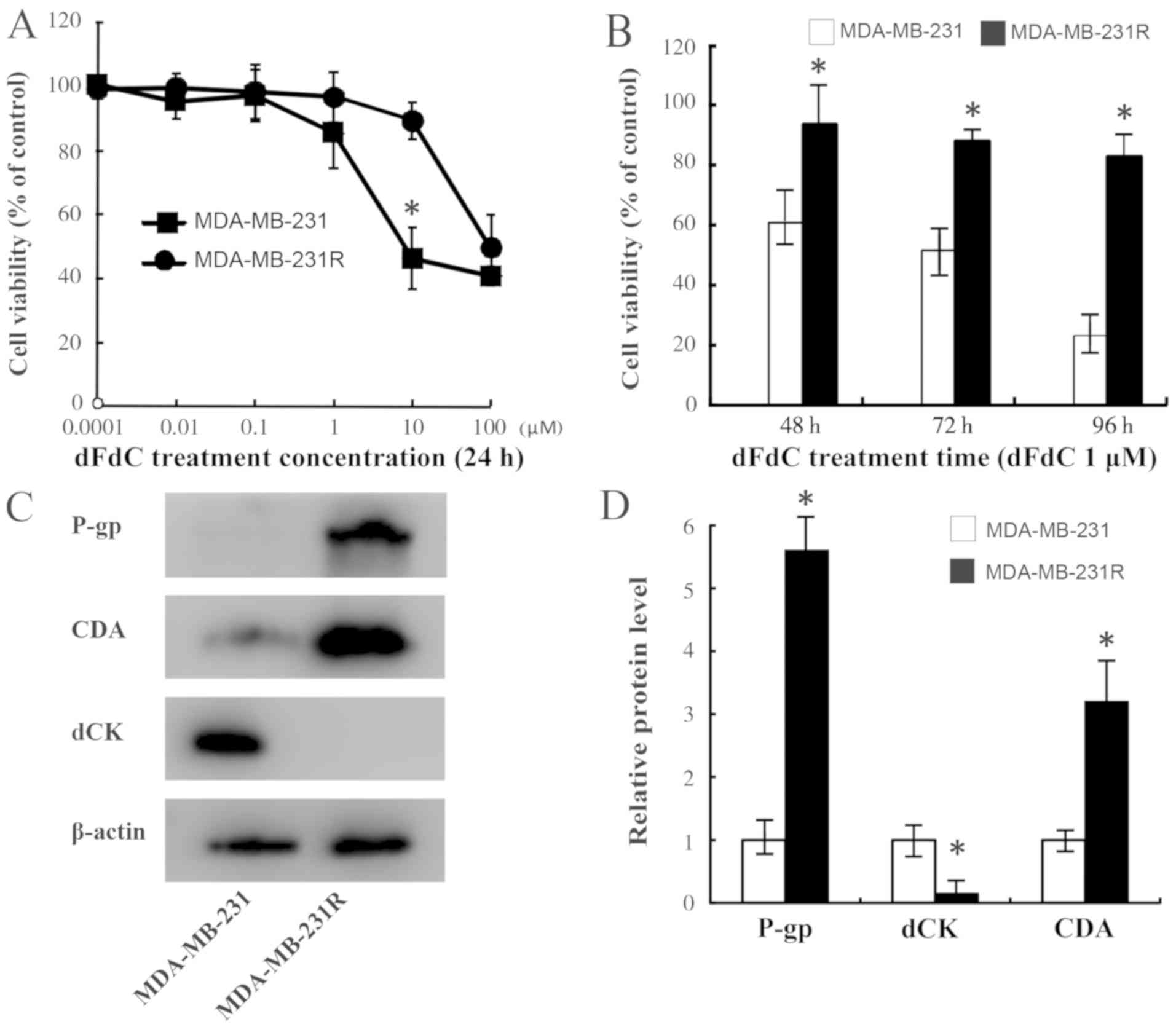 | Figure 1.Generation and identification of
dFdC-resistant cells. (A) MDA-MB-231 and MDA-MB-231R cells were
exposed to various concentrations of dFdC (0.0001, 0.01, 0.1, 1, 10
and 100 µM). Cell viability was examined using the MTT assay. The
viability of MDA-MB-231R cells was less inhibited compared with
that of MDA-MB-231 cells treated with 10 µM dFdC. (B) Cells were
exposed to 1.0 µM dFdC for 48, 72 or 96 h. The viability of
MDA-MB-231R cells was less inhibited compared with that in
MDA-MB-231 cells after 48 h, and this difference increased over
time. (C and D) The protein levels of P-gp, dCK and CDA in
MDA-MB-231 and MDA-MB-231R cells were assessed by western blotting
analysis. β-actin was used as an internal control. *P<0.05 vs.
MDA-MB-231. dFdC, gemcitabine; dCK, 2′,2′deoxycytidine kinase; CDA,
cytidine deaminase; P-gp, P-glycoprotein. |
The results also demonstrated that the levels of P
glycoprotein (P-gp) were increased in MDA-MB-231R cells compared
with those in the parental MDA-MB-231 cells (Fig. 1C). Subsequently, the protein
expression levels of dCK and CDA were evaluated and were
demonstrated to be associated with the dFdC resistance of
MDA-MB-231R cells; the protein expression level of dCK was
decreased, whereas that of CDA increased in the MDA-MB-231R cells
compared with their parental cells (Fig.
1C and D), which was consistent with the results of previous
studies (23,42), suggesting that the dFdC-resistant
cell line was successfully established.
Assessment of the enzyme activity of
Dm-dNK in the primary and drug-resistant breast cancer cell
lines
In order to visualize the subcellular localization
of the recombinant enzymes in vivo, the proteins were fused
to GFP. MDA-MB-231 and MDA-MB-231R cells were transfected with the
recombinant lentiviral vectors. Both cell lines were stably
transfected, and green fluorescence was observed in 80–90% of the
cells. Fluorescence in the nucleus was observed in cells
transfected with the vector encoding dNK-GFP, whereas it was
predominantly detected in the cytosol in cells transfected with the
vector encoding dNKmut-GFP (Fig.
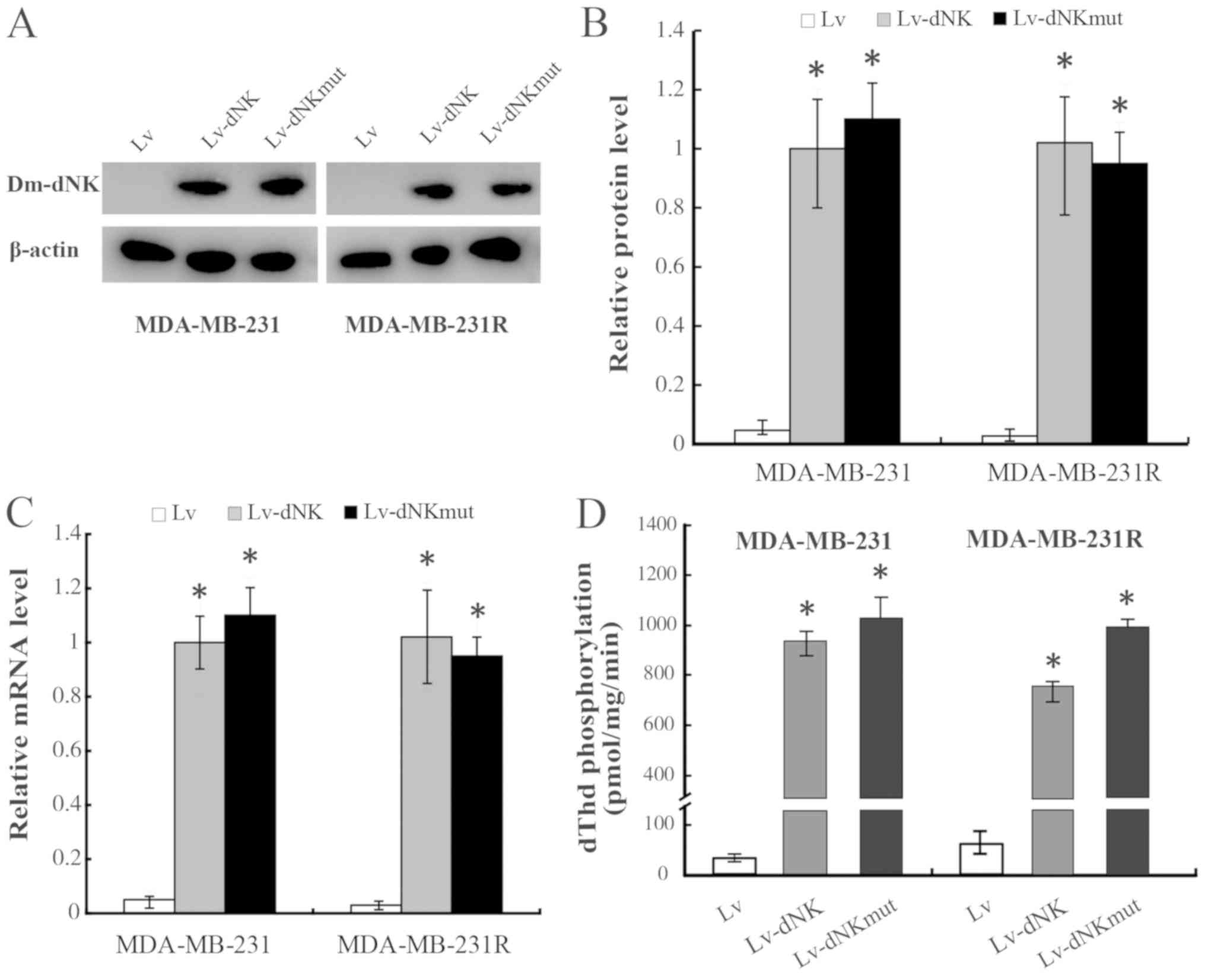
S1). RT-qPCR and western blotting analysis demonstrated similar
mRNA and protein expression levels of dNK-GFP and dNKmut-GFP in the
MDA-MB-231 and MDA-MB-231R cells, irrespective of the subcellular
localization (Fig. 2A-C).
In the present study, the levels of dThd
phosphorylation in protein extracts of MDA-MB-231 and MDA-MB-231R
cells were also evaluated to assess the enzymatic activity of the
Dm-dNK proteins, as presented in Fig.
2D. The dThd kinase activity was increased 50-fold in
MDA-MB-231 and MDA-MB-231R cells expressing nuclear Lv-dNK or
cytosolic Lv-dNKmut compared with that in the cells transduced with
lentiviral vectors. No significant differences in Dm-dNK activity
were detected between the MDA-MB-231 and MDA-MB-231R cells.
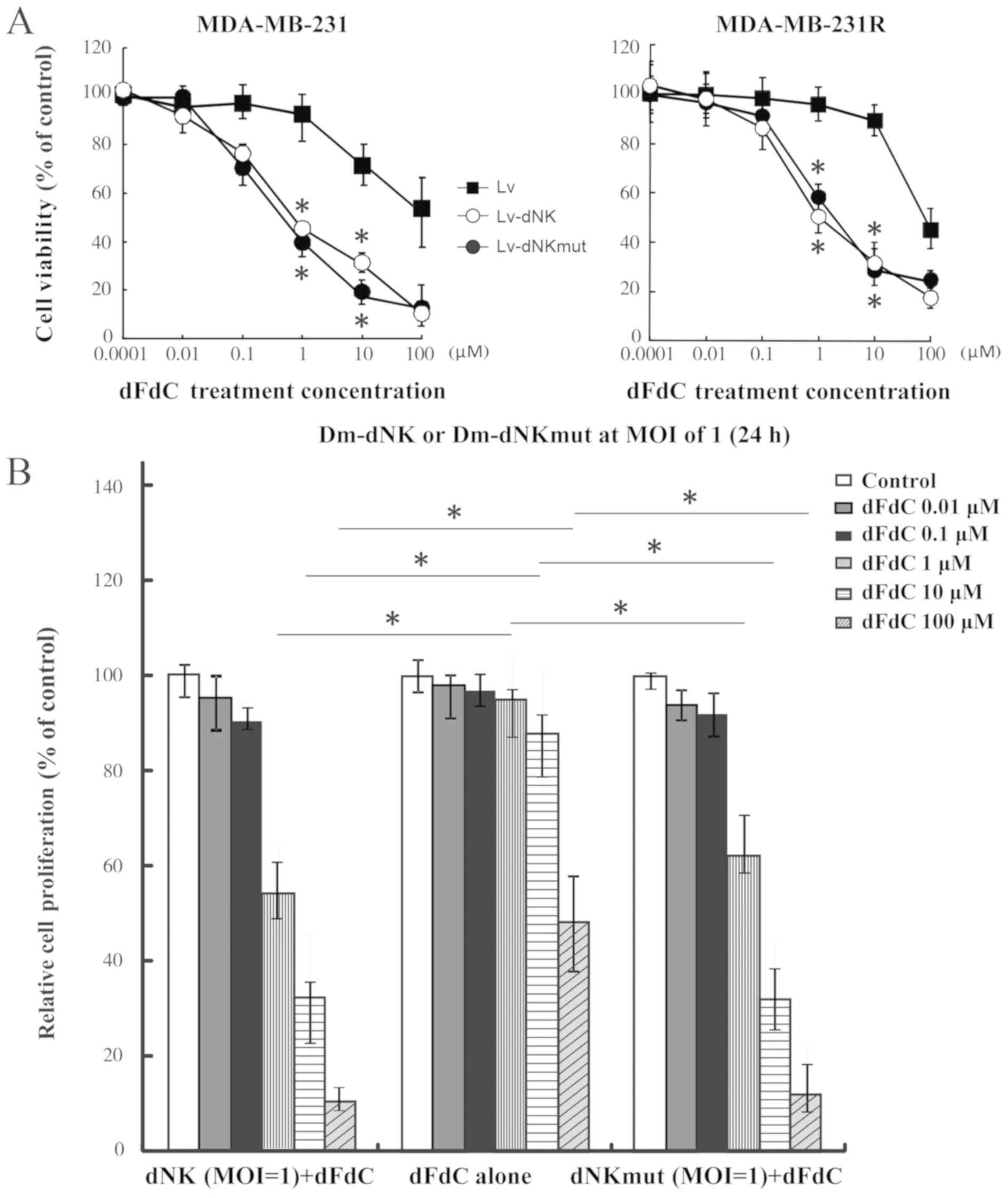
Dm-dNK restores TNBC cell sensitivity
to dFdC
The dFdC resistance of MDA-MB-231R cells was
observed to be reversed following transfection with Lv-dNK or
Lv-dNKmut at the MOI of 1 (Fig. 3A and
B). The IC50 values for dFdC in MDA-MB-231R cells
expressing Dm-dNK or dNKmut were 1.14 and 1.62 µM, respectively;
The IC50 values for dFdC in MDA-MB-231 cells expressing
dNK or dNKmut were 0.81 and 0.78 µM, respectively. These values
were ~30-fold lower compared with those in the Lv group,
irrespective of the protein localization (Fig. 3A and B), indicating that no
differences in sensitivity to nucleoside analogs were observed
between cells expressing nuclear and cytosolic Dm-dNK.
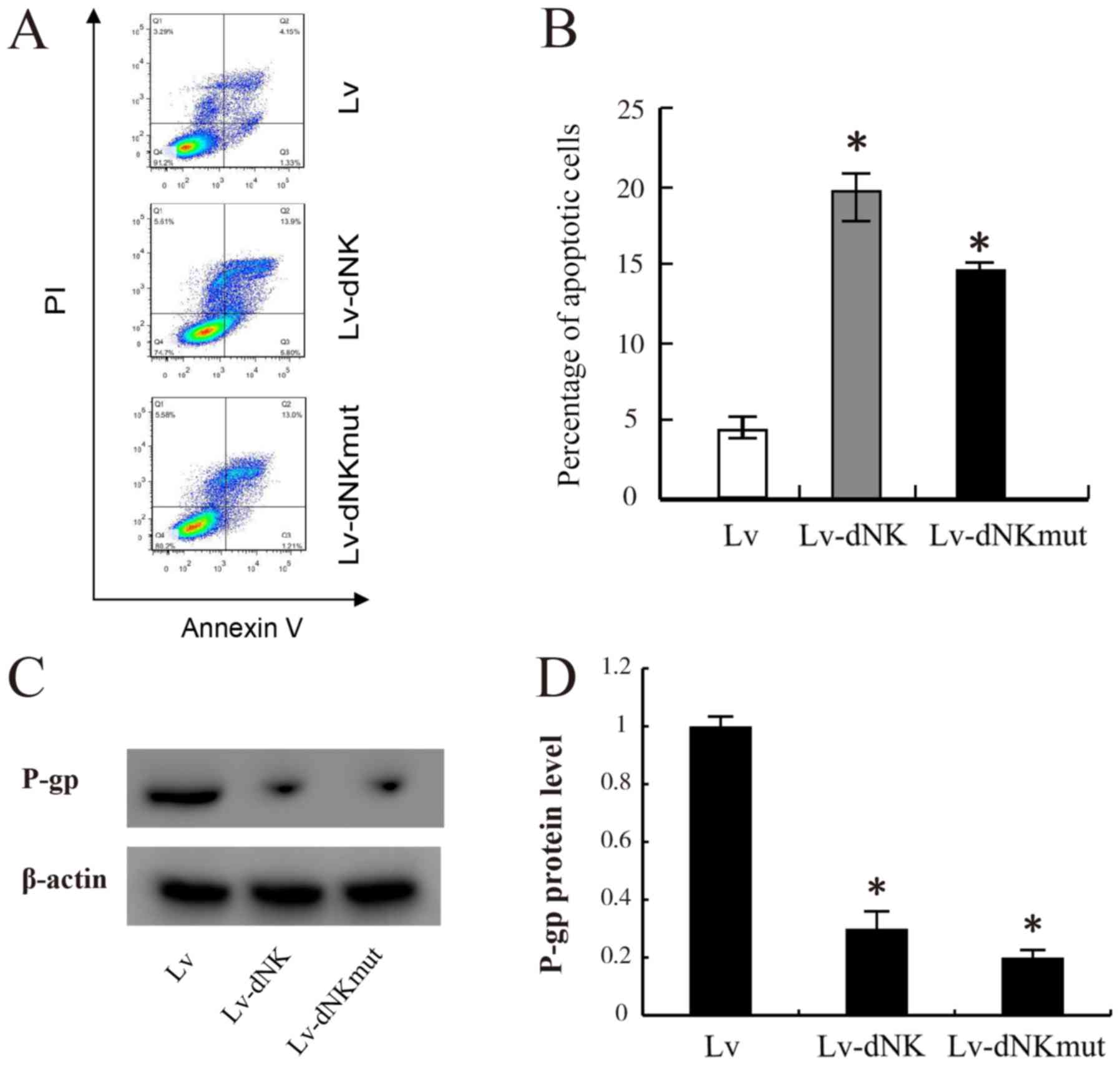
To gain further insights into the anticancer
potential of Lv-dNK and Lv-dNKmut, apoptosis assay was performed in
chemo-resistant MDA-MB-231R cells. As presented in Fig. 4A and B, following 24-h exposure to 1
µM dFdC, a significant increase in the apoptotic rate was observed
in the MDA-MB-231R cells transfected with Lv-dNK and Lv-dNKmut
compared with that in the control cells transduced with lentiviral
vectors. The apoptotic rate in the control cells was 2–3%, whereas
those in the Lv-dNK- and Lv-dNKmut-transfected cells were 19.7 and
14.2%, respectively. In addition, P-gp expression levels were
partly decreased following transfection with Lv-dNK or Lv-dNKmut
compared with the empty lentivirus group in MDA-MB-231R cells
(Fig. 4C and D). These results
revealed that the cell apoptotic rate of MDA-MB-231R cells was
significantly increased due to active drug conversion from the
prodrug.
Discussion
Resistance to anticancer drugs is a crucial problem
that limits the effectiveness of chemotherapy regimens (43). Resistance generally develops with
long-term exposure to the drug (44). In the present study, dFdC resistance
was established in MDA-MB-231 cells through long-term treatment
with dFdC, which was verified by MTT assay. Although the emergence
of drug resistance has been associated with multiple molecular
mechanisms, MDR1, which actively transports toxins out of the
cells, has been strongly associated with the development of
resistance to various chemotherapeutic agents (45). In the dFdC-resistant MDA-MB-231R
cells established in the present study, expression of the
MDR1-encoded P-gp was observed, but this was not the case for the
MDA-MB-231 cells.
dCK is essential for the phosphorylation of dFdC,
and CDA catalyzes the degradation of dFdC (18,19). dCK
and CDA levels have been demonstrated to be significantly
associated with dFdC sensitivity (46). It was reported that a high CDA-to-dCK
ratio may be a marker of resistance to decitabine (an analog of
cytidine) (47). Hosokawa et
al (42) identified high protein
expression levels of CDA and low levels of dCK in their
successfully established HCT116 cells resistant to decitabine and
dFdC. The protein expression levels of CDA and dCK were also
examined in the present study, and similar results were obtained
compared with those of a previous study (40), suggesting that the dFdC-resistant
TNBC cells, which were termed MDA-MB-231R cells, were successfully
established.
The expression of wild-type Dm-dNK in cell lines
leads to nuclear localization of the enzyme, which can be
attributed to the presence of a nuclear localization signal at the
C-terminal region of the protein (40). The nuclear import of the protein is
abolished by the site-directed mutation of arginine-247 to serine,
leading to a predominant cytosolic localization of the enzyme
(37,40). Our research team has previously
investigated Dm-dNK for its potential application as a suicide
gene; the results have revealed that wild-type Dm-dNK retains its
activity when it is expressed in human cells, and it is localized
to the nucleus, resulting in high cell sensitivity to several
cytotoxic nucleoside analogs, including araT, araC, BVDU and dFdC
(25,27,37,40). In
the present study, either the wild-type nuclear Dm-dNK (dNK-GFP) or
the cytosolic arginine-247 Dm-dNK mutant (dNKmut-GFP) was expressed
using lentiviral vectors and the mutant cytosolic Dm-dNK was also
demonstrated to possess highly similar levels of enzymatic activity
and cytotoxicity compared with the wild-type dNK (33,37),
consistent with the findings of the present study. As one of the
most effective substrates for Dm-dNK, dFdC exerts strong effects on
Dm-dNK-expressing MDA-MB-231R cells, which exhibit a 50-fold
decrease in IC50 value for dFdC compared with that of
untransfected cells (40),
suggesting that the nucleoside analogs (prodrug)/Dm-dNK system may
overcome drug resistance by lowering the IC50 values for
these chemotherapeutic agents. As for the underlying mechanism, the
apoptosis assay revealed that such an increase in sensitivity may
be attributed to an enhanced rate of apoptosis. To the best of the
authors' knowledge, the present study was the first study to have
examined the effects of the suicide gene Dm-dNK and its mutant on
reversing drug resistance in TNBC cells.
The present study had certain limitations. Cancer
cells that acquire resistance to one anticancer drug may also
become simultaneously resistant to different drugs, which has been
referred to as multidrug-resistance or cross-resistance (42,48).
Dm-dNK or Dm-dNKmut may also be able to reverse the drug-resistance
that develops in cancer treatments with other chemotherapeutic
agents, such as BVDU and araT, which was not investigated in the
present study. In addition, the underlying mechanism of Dm-dNK
reversing the drug resistance is still unclear and will be
investigated in future studies.
In conclusion, Dm-dNK and Dm-dNKmut may be used to
reverse the drug resistance encountered in cancer chemotherapy.
This may form the basis for novel strategies in the treatment of
patients with TNBC who have developed drug resistance.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Natural
Science Foundation of Liaoning Province (grant no. 20180551215),
Key R & D Plan Guidance Project of Liaoning Province (grant no.
2019JH8/10300020), and the Key Project of China Health Promotion
Foundation (grant no. CHPF-RXO180301).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and XZ designed the study. YZ, MG and CZ
performed the experiments. YZ and HJ collected and interpreted the
data and performed the statistical analysis. YZ, HJ and XZ wrote
and revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Peddi PF, Ellis MJ and Ma C: Molecular
basis of triple negative breast cancer and implications for
therapy. Int J Breast Cancer. 2012:2171852012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas H and Coley HM: Overcoming
multidrug resistance in cancer: An update on the clinical strategy
of inhibiting p-glycoprotein. Cancer Control. 10:159–165. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cleator S, Heller W and Coombes RC:
Triple-negative breast cancer: Therapeutic options. Lancet Oncol.
8:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garrido-Castro AC, Lin NU and Polyak K:
Insights into molecular classifications of triple-negative breast
cancer: Improving patient selection for treatment. Cancer Discov.
9:176–198. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li X, Lewis MT, Huang J, Gutierrez C,
Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC,
et al: Intrinsic resistance of tumorigenic breast cancer cells to
chemotherapy. J Natl Cancer Inst. 100:672–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rong D, Wang C, Zhang X, Wei Y, Zhang M,
Liu D, Farhan H, Ali SA, Liu Y, Taouil A, et al: A novel taxane,
difluorovinyl-ortataxel, effectively overcomes
paclitaxel-resistance in breast cancer cells. Cancer Lett.
491:36–39. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan D, Zhou H, Sun H, Tian R, Xia M, Sun
L and Liu Y: Identification of key genes for guiding
chemotherapeutic management in ovarian cancer using translational
bioinformatics. Oncol Lett. 20:1345–1359. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niero EL, Rocha-Sales B, Lauand C, Cortez
BA, de Souza MM, Rezende-Teixeira P, Urabayashi MS, Martens AA,
Neves JH and Machado-Santelli GM: The multiple facets of drug
resistance: One history, different approaches. J Exp Clin Cancer
Res. 33:372014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou M, Li L, Li L, Lin X, Wang F, Li Q
and Huang Y: Overcoming chemotherapy resistance via simultaneous
drug-efflux circumvention and mitochondrial targeting. Acta Pharm
Sin B. 9:615–625. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Agarwal R and Kaye SB: Ovarian cancer:
Strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Asleh K, Lyck Carstensen S, Tykjaer
Jørgensen CL, Burugu S, Gao D, Won JR, Jensen MB, Balslev E,
Laenkholm AV, Nielsen DL, et al: Basal biomarkers nestin and INPP4B
predict gemcitabine benefit in metastatic breast cancer: Samples
from the phase III SBG0102 clinical trial. Int J Cancer.
144:2578–2586. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diéras V, Bonnefoi H, Alba E, Awada A,
Coudert B, Pivot X, Gligorov J, Jager A, Zambelli S, Lindeman GJ,
et al: Iniparib administered weekly or twice-weekly in combination
with gemcitabine/carboplatin in patients with metastatic
triple-negative breast cancer: A phase II randomized open-label
study with pharmacokinetics. Breast Cancer Res Treat. 177:383–393.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spratlin JL and Mackey JR: Human
equilibrative nucleoside transporter 1 (hENT1) in pancreatic
adenocarcinoma: Towards individualized treatment decisions. Cancers
(Basel). 2:2044–2054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hung SW, Marrache S, Cummins S, Bhutia YD,
Mody H, Hooks SB, Dhar S and Govindarajan R: Defective hCNT1
transport contributes to gemcitabine chemoresistance in ovarian
cancer subtypes: Overcoming transport defects using a nanoparticle
approach. Cancer Lett. 359:233–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Honeywell RJ, Ruiz van Haperen VW, Veerman
G, Smid K and Peters GJ: Inhibition of thymidylate synthase by
2′,2′-difluoro-2′-deoxycytidine (Gemcitabine) and its metabolite
2′,2′-difluoro-2′-deoxyuridine. Int J Biochem Cell Biol. 60:73–81.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitsuno M, Kitajima Y, Ohtaka K, Kai K,
Hashiguchi K, Nakamura J, Hiraki M, Noshiro H and Miyazaki K:
Tranilast strongly sensitizes pancreatic cancer cells to
gemcitabine via decreasing protein expression of ribonucleotide
reductase 1. Int J Oncol. 36:341–349. 2010.PubMed/NCBI
|
|
18
|
Bergman AM, Pinedo HM and Peters GJ:
Determinants of resistance to 2′,2′-difluorodeoxycytidine
(gemcitabine). Drug Resist Updat. 5:19–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baker JA, Wickremsinhe ER, Li CH, Oluyedun
OA, Dantzig AH, Hall SD, Qian YW, Ring BJ, Wrighton SA and Guo Y:
Pharmacogenomics of gemcitabine metabolism: Functional analysis of
genetic variants in cytidine deaminase and deoxycytidine kinase.
Drug Metab Dispos. 41:541–545. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garcia-Diaz M, Murray MS, Kunkel TA and
Chou KM: Interaction between DNA polymerase lambda and anticancer
nucleoside analogs. J Biol Chem. 285:16874–16879. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galmarini CM, Clarke ML, Santos CL,
Jordheim L, Perigaud C, Gosselin G, Cros E, Mackey JR and Dumontet
C: Sensitization of ara-C-resistant lymphoma cells by a
pronucleotide analogue. Int J Cancer. 107:149–154. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Réjiba S, Bigand C, Parmentier C and Hajri
A: Gemcitabine-based chemogene therapy for pancreatic cancer using
Ad-dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia.
11:637–650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saiki Y, Yoshino Y, Fujimura H, Manabe T,
Kudo Y, Shimada M, Mano N, Nakano T, Lee Y, Shimizu S, et al: DCK
is frequently inactivated in acquired gemcitabine-resistant human
cancer cells. Biochem Biophys Res Commun. 421:98–104. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Yang M, Zhang Y, Zhong L and Zheng
X: Novel combination oncolytic adenoviral gene therapy armed with
Dm-dNK and CD40L for breast cancer. Curr Gene Ther. 19:54–65. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dong X, Qu W, Ma S, Zhu Z, Zheng C, He A,
Karlsson A, Xu K and Zheng X: Potent antitumoral effects of
targeted promoter-driven oncolytic adenovirus armed with Dm-dNK for
breast cancer in vitro and in vivo. Cancer Lett. 328:95–103. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang H, Zhao L, Dong X, He A, Zheng C,
Johansson M, Karlsson A and Zheng X: Tanshinone IIA enhances
bystander cell killing of cancer cells expressing Drosophila
melanogaster deoxyribonucleoside kinase in nuclei and
mitochondria. Oncol Rep. 34:1487–1493. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma S, Qu W, Mao L, Zhu Z, Jia L, Zhao L
and Zheng X: Antitumor effects of oncolytic adenovirus armed with
Drosophila melanogaster deoxyribonucleoside kinase in
colorectal cancer. Oncol Rep. 27:1443–1450. 2012.PubMed/NCBI
|
|
28
|
Qu W, Zhu Z, Zhao L, He A and Zheng X:
Conditionally replicating adenovirus SG500-expressed mutant Dm-dNK
gene for breast cancer therapy. Int J Oncol. 41:2175–2183. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang M, Zu C, He A, Wang W, Chen B and
Zheng X: Synergistic antitumor effect of adenovirus armed with
Drosophila melanogaster deoxyribonucleoside kinase and
nucleoside analogs for human breast carcinoma in vitro and in vivo.
Drug Des Devel Ther. 9:3301–3312. 2015.PubMed/NCBI
|
|
30
|
Zhang N, Dong X, Sun Y, Cai X, Zheng C, He
A, Xu K and Zheng X: Cytotoxic effects of adenovirus- and
lentivirus-mediated expression of Drosophila melanogaster
deoxyribonucleoside kinase on Bcap37 breast cancer cells. Oncol
Rep. 29:960–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang N, Zhao L, Ma S, Gu M and Zheng X:
Lentivirus-mediated expression of Drosophila melanogaster
deoxyribonucleoside kinase driven by the hTERT promoter combined
with gemcitabine: A potential strategy for cancer therapy. Int J
Mol Med. 30:659–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng X, Lundberg M, Karlsson A and
Johansson M: Lipid-mediated protein delivery of suicide nucleoside
kinases. Cancer Res. 63:6909–6913. 2003.PubMed/NCBI
|
|
33
|
Zhu Z, Ma S, Zhao L, Sun Z, He A, Xu H and
Zheng X: Adenovirus-mediated Drosophila melanogaster
deoxyribonucleoside kinase mutants combined with gemcitabine harbor
a safe cancer treatment profile. Int J Oncol. 38:745–753.
2011.PubMed/NCBI
|
|
34
|
Ma S, Zhao L, Zhu Z, Liu Q, Xu H,
Johansson M, Karlsson A and Zheng X: The multisubstrate
deoxyribonucleoside kinase of Drosophila melanogaster as a
therapeutic suicide gene of breast cancer cells. J Gene Med.
13:305–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun X, Xu X, Chen Y, Guan R, Cheng T, Wang
Y, Jin R, Song M and Hang T: Danggui buxue decoction sensitizes the
response of non-small-cell lung cancer to gemcitabine via
regulating deoxycytidine kinase and P-glycoprotein. Molecules.
24:20112019. View Article : Google Scholar
|
|
36
|
Sierzega M, Pach R, Kulig P, Legutko J and
Kulig J: Prognostic implications of expression profiling for
gemcitabine-related genes (hENT1, dCK, RRM1, RRM2) in patients with
resectable pancreatic adenocarcinoma receiving adjuvant
chemotherapy. Pancreas. 46:684–689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng X, Johansson M and Karlsson A:
Nucleoside analog cytotoxicity and bystander cell killing of cancer
cells expressing Drosophila melanogaster deoxyribonucleoside
kinase in the nucleus or cytosol. Biochem Biophys Res Commun.
289:229–233. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang J, Wu Y, Wang X, Xu L, Zhao X and
Yang Y: Chemoresistance is associated with overexpression of HAX-1,
inhibition of which resensitizes drug-resistant breast cancer cells
to chemotherapy. Tumour Biol. 39:10104283176922282017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zheng X, Johansson M and Karlsson A:
Retroviral transduction of cancer cell lines with the gene encoding
Drosophila melanogaster multisubstrate deoxyribonucleoside
kinase. J Biol Chem. 275:39125–39129. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fraser SP, Salvador V, Manning EA, Mizal
J, Altun S, Raza M, Berridge RJ and Djamgoz MBA: Contribution of
functional voltage-gated Na+ channel expression to cell
behaviors involved in the metastatic cascade in rat prostate
cancer: I. Lateral motility. J Cell Physiol. 195:479–487. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hosokawa M, Saito M, Nakano A, Iwashita S,
Ishizaka A, Ueda K and Iwakawa S: Acquired resistance to decitabine
and cross-resistance to gemcitabine during the long-term treatment
of human HCT116 colorectal cancer cells with decitabine. Oncol
Lett. 10:761–767. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Erin N, Grahovac J, Brozovic A and Efferth
T: Tumor microenvironment and epithelial mesenchymal transition as
targets to overcome tumor multidrug resistance. Drug Resist Updat.
53:1007152020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang XL, Lin FJ, Guo YJ, Shao ZM and Ou
ZL: Gemcitabine resistance in breast cancer cells regulated by
PI3K/AKT-mediated cellular proliferation exerts negative feedback
via the MEK/MAPK and mTOR pathways. Onco Targets Ther. 7:1033–1042.
2014.PubMed/NCBI
|
|
45
|
Triller N, Korosec P, Kern I, Kosnik M and
Debeljak A: Multidrug resistance in small cell lung cancer:
Expression of P-glycoprotein, multidrug resistance protein 1 and
lung resistance protein in chemo-naive patients and in relapsed
disease. Lung Cancer. 54:235–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kroep JR, Loves WJ, van der Wilt CL,
Alvarez E, Talianidis I, Boven E, Braakhuis BJM, van Groeningen CJ,
Pinedo HM and Peters GJ: Pretreatment deoxycytidine kinase levels
predict in vivo gemcitabine sensitivity. Mol Cancer Ther.
1:371–376. 2002.PubMed/NCBI
|
|
47
|
Qin T, Castoro R, El Ahdab S, Jelinek J,
Wang X, Si J, Shu J, He R, Zhang N, Chung W, et al: Mechanisms of
resistance to decitabine in the myelodysplastic syndrome. PLoS One.
6:e233722011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Stordal B, Pavlakis N and Davey R:
Oxaliplatin for the treatment of cisplatin-resistant cancer: A
systematic review. Cancer Treat Rev. 33:347–357. 2007. View Article : Google Scholar : PubMed/NCBI
|