Introduction
Globally, the incidence of gastroesophageal
adenocarcinomas, primarily including gastric cardia adenocarcinoma
(GCA) and esophageal adenocarcinoma (EA), has increased over the
last 3 decades (1–4). In the Chinese population, EA is
extremely rare (the age standardized rate: Approximately 0.4%) with
a 77-fold lower incidence of esophageal squamous cell carcinoma in
Chinese males, according to data from the GLOBOCAN 2012, and the
majority of adenocarcinomas at the gastroesophageal junction are
defined as GCA (5). GCA exhibits
similar biological behavior to EA (6); however, there are significant
differences with distal gastric adenocarcinoma (DGA). In high-risk
areas of China, which had an annual gastric cancer mortality rate
of 77.67/100,000/year in the 1990s, and were 3.29 and 3.43 times
that of the national rate in 2004 and 2005, respectively (7,8), GCA
shows different immunophenotypical patterns of cytokeratins, mucins
and signaling molecular pathways (9–11). A
genome-wide association study (GWAS) in Asia identified an
association between single nucleotide polymorphisms (SNPs) in
prostate stem cell antigen at rs2294693 and the risk of non-cardia
cancer, but found no evidence for an association with cardia tumors
(12). These findings indicated that
GCA may be an independent entity of gastric cancer (GC).
It is well known that Helicobacter pylori
(H. pylori) is associated with non-cardia adenocarcinomas
(13,14), while gastroesophageal adenocarcinomas
are primarily associated with gastroesophageal reflux disease
(GERD), obesity and Caucasian ethnicity (15,16).
However, the roles of H. pylori in gastroesophageal
carcinogenesis remain controversial. Previous studies have
demonstrated that H. pylori is a potential protective factor
against gastroesophageal adenocarcinomas. In addition, it has also
been shown that H. pylori-mediated chronic gastritis was
inversely associated with GERD and Barrett's esophagus (17). However, in Chinese areas with an
increased incidence rate of upper gastrointestinal cancers and a
high prevalence rate of H. pylori infection, for example the
Chinese Cixian and Zanhuang counties, an increased proportion of
GCA cases has been reported (1). In
addition, a large cohort study in the Chaoshan region revealed that
H. pylori infection was associated with an increased risk of
GCA (18,19). Notably, a GWAS investigating genetic
predisposition factors identified that SNP susceptibility loci and
H. pylori infection were associated with an increased risk
of GCA in a Chinese population (20). However, to the best of our knowledge,
there is no evidence for a causal role of H. pylori in
GCA.
Trefoil factor family (TFF) proteins comprise three
small molecule polypeptides with a clover leaf-like disulfide
structure (21). TFF1 is primarily
secreted in gastric foveolar cells, while TFF2 is expressed in the
neck cells and deep pyloric glands of the stomach, and TFF3 is
highly expressed in intestinal goblet cells and gastrointestinal
metaplasia (21). These peptides are
physiologically resistant to proteolysis and the acidic
environment, and develop to maintain mucosal integrity in stomach
(21–23). In TFF1-knockout mice, the loss of
TFF1 exhibit carcinogenic histological changes from gastritis to
hyperplasia, ultimately leading to malignant adenocarcinoma in the
gastric mucosa (22). Therefore,
H. pylori has been widely considered to exhibit tumor
suppressor effects in gastric carcinogenesis (23,24). It
has been reported that TFF1 and gastrokine 2 (GKN2) are
co-expressed by gastric mucus-secreting cells, and act by forming a
heterodimer (25,26). These findings indicate that GKN2 may
be involved in regulating the important antitumor effects of TFF1,
but not TFF2 and TFF3, by forming a GKN2/TFF1 protein complex,
(27). A recent study revealed that
GKN2 exists in monomeric form in the corpus, and as a TFF1-GKN2
heterodimer in the antrum (28).
However, comparative studies on both genes in different subtypes of
GC are still at a preliminary stage. Therefore, the potential
underlying mechanism of TFF1/GKN2 in H. pylori-mediated
gastroesophageal adenocarcinomas remains elusive.
The present study aimed to determine the roles of
H. pylori in the development of GCA and its effects on
TFF1/GKN2 expression in high-risk areas in China. In addition, the
effects of TFF1 and GKN2 overexpression on H. pylori-induced
cell proliferation and inflammatory responses were also
investigated in vitro.
Materials and methods
Patients
A total of 113 paraffin-embedded GC resection
samples and non-cancerous adjacent tissues were obtained from the
Department of Pathology, The Second Hospital of Hebei Medical
University (Hebei, China), between January 2011 and December 2014.
The center of the GCA was located 1 cm above and 2 cm below the
anatomical location of the gastroesophageal junction, according to
the Siewert classification system (29), while tumors located in the antrum and
angle of the stomach were defined as DGA (9,10). The
clinicopathological parameters are listed in Table SI. None of the patients received
chemotherapy or radiotherapy prior to surgery.
The present study was approved by the local Ethics
Committee, and written informed consent was obtained from all
subjects prior to participation in the study.
Immunohistochemical (IHC)
analysis
All the specimens are fixed with 4% formalin at room
temperature for 4h, and embedded in paraffin. IHC analysis of H.
pylori, TFF1 and GKN2 was conducted on the paraffin-embedded
specimens (4–6 µm). Briefly, following deparaffinization with
xylene and rehydration in a descending alcohol series at room
temperature, high-pressure antigen retrieval was performed in
citrate buffer (pH 6.0) for 5 min, according to the retrieval
protocol. Endogenous peroxidase activity was blocked in 3% hydrogen
peroxidase/methanol for 10 min. Subsequently, three serial sections
were incubated overnight at 4°C in a humidified chamber with
antibodies against H. pylori (dilution, 1:100; cat. no.
ab140128; Abcam), GKN2 (dilution, 1:100; cat. no. ARP65440-p050;
Aviva Systems Biology Corp.) and TFF1 (dilution, 1:100; cat. no.
EPR15377; Epitomics; Abcam). The assay was performed using a rabbit
IHC kit (cat. no. FXP020; 4A Biotech Co., Ltd.) according to the
manufacturer's recommendations. PBS was used as a negative control
instead of the primary antibody. A total of 10 randomly selected
high power fields (10X objective; ×40 magnification) under light
microscope were observed on each slide and samples with >5%
positive cells were categorized as positive. All slides were judged
by an experienced pathologist from the Department of Pathology, The
Second Hospital of Hebei Medical University.
The odds ratio with 95% confidence intervals were
used as measures of the association between H. pylori
infection and the risk of GCA or DGA; and the data was
statistically analyzed using the Mantel-Haenszel χ2
test.
Cell culture and treatment
The normal gastric mucosa (NGM) epithelium cell line
GES-1 was purchased from the Beijing Institute for Cancer Research.
The SKGT-4 distal esophageal adenocarcinoma cell line (cat. no.
CBP60462) was bought from Cobioer Biosciences Co., Ltd. The GES-1
cells were cultured in DMEM (Gibco; Thermo Fisher Scientific,
Inc.), supplemented with 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.), penicillin (100 U/ml) and streptomycin (100 µg/ml), at 37°C
in a humidified atmosphere containing 5% CO2. The SKGT-4
cells were cultured in RPMI-1640 (Gibco; Thermo Fisher Scientific,
Inc.) under the same conditions as GES-1 cells. The cells were then
treated with different concentrations of H. pylori proteins
(2.5, 5, 10, 20, 40, 80 and 160 µg/ml), and the solvent control
group was treated with PBS.
MTT assay
The cells were treated with highly purified H.
pylori protein (cat. no. 30-AH78; Fitzgerald Inc.). The GES-1
and SKGT-4 cells were cultured in the presence of increasing
concentrations of H. pylori protein (2.5, 5, 10, 20, 40, 80
and 160 µg/ml) for 24 h, at 37°C, following which the cells were
incubated with MTT stock solution for 4 h. The inhibitory effect of
H. pylori protein on cell proliferation was measured using a
spectrophotometric microplate reader (BioTek Instruments Inc.) at
450 nm. Cell proliferation was determined and compared with the
control group, using optical density (OD), and the following
equation: Cell proliferation =
OD(experimental)-OD(blank)OD(control)-OD(blank) x 100%.
Transfection
The recombinant plasmids pEZ-M02-GKN2 and
pEZ-Lv105-TFF1, were synthesized by GeneCopoeia Inc.. A total of
1.0×105 cells/ml were seeded into 6-well plates and then
transfected using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
recommendations. Transfection efficiency was determined using
western blot analysis, 48 h after cell transfection with the
targeted expression plasmids of TFF1 and GKN2. Cells transfected
with empty plasmids were regarded as the control group. Finally,
the cells were treated with H. pylori protein (20 µg/ml) for
24 h and harvested for subsequent experimentation.
Reverse transcription-quantitative
(RT-qPCR)
Total RNA was extracted from cells following the
aforementioned treatment using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. Subsequently, cDNA was synthesized using GoScript™
Reverse Transcription System (Promega Corporation) according to the
manufacturer instructions, and PCR amplification was performed with
the appropriate set of primers (Invitrogen; Thermo Fisher
Scientific, Inc.; Table SII) using
the SYBR PrimeScript RT-PCR kit (Takara Bio, Inc.) in an Mx3005p
Real-Time PCR system (Agilent Technologies, Inc.). RT was performed
under the following conditions: 25°C for 5 min, 42°C for 60 min and
70°C for 15 min. The thermocycling conditions of PCR amplification
consisted of an initial denaturation at 95°C for 10 min, followed
by 40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C
for 30 sec and elongation at 60°C for 30 sec. The relative mRNA
expression levels of the targeted genes were calculated according
to the equation 2−ΔΔCq (30), where ΔCq=Cq (targeted gene)-Ct
(internal control gene; β-actin). All assays were performed in
triplicate.
Furthermore, genomic DNA used for qPCR was directly
extracted from the paraffin-embedded tissues, including GC and
non-cancerous mucosa of adjacent tissues, using a Genomic DNA kit
(Tiangen Biotech Co., Ltd.), to detect H. pylori 16S rRNA,
cytotoxin-associated gene A (CagA) and vacuolating toxin A (VacA).
The optimal cut-off points of H. pylori 16S rRNA, CagA and
VacA detection levels were calculated by accepting Ct ≤35 and ΔCt
≤15 as a positive result. It is reported that H. pylori
26695 protein could affect cell proliferation and apoptosis, and
then was involved in GC. Therefore, the primers were designed based
on the complete genome sequence of H. pylori 26695 (GenBank,
AE000511.1).
Western blot analysis
Total protein was extracted from cells using lysis
buffer (1% Triton X-100; 150 mM NaCl; 2 mM EDTA; 50 mM Tris-HCl)
supplemented with a phosphatase inhibitor cocktail. Following
protein quantification by BCA Protein Assay kit (Beijing Solarbio
Science & Technology Co., Ltd.) using Gen5 1.0 software (BioTek
Instruments, Inc.) on a spectrophotometer microplate reader (BioTek
Instruments, Inc.), a total of 20–50 µg protein extract was
analyzed using SDS-PAGE (10–15% gels) and then electrophoretically
transferred onto a polyvinylidene fluoride membrane. Non-specific
sites on the membranes were blocked at room temperature for 60 min
with 5% skimmed milk in TTBS, supplemented with Tween-20. Following
incubation with the primary antibodies overnight at 4°C, the
membranes were then probed with a HRP-conjugated secondary antibody
(dilution, 1:5,000) at room temperature for 2 h. Finally, the
immunoreactive bands were visualized using Pierce™
Electrochemiluminescent Western Blotting Substrate (cat. no.
UC280185; Thermo Fisher Scientific, Inc.) on ImageQuant™ LAS 4,000
(serial no. 2639042; GE Healthcare Life Sciences) and their
densities was quantified and normalized to β-actin.
The primary antibodies used in the present study
included: Rabbit anti-human β-actin (1:10,000; cat. no. AC026), a
monoclonal antibody purchased from ABclonal Biotech Co., Ltd..
Rabbit anti-human cyclin dependent kinase 4 (CDK4; 1:5,000; cat.
no. ab108357), cyclin B1 (1:10,000; cat. no. ab32053), cyclin D
(1:200; cat. no. ab16663), proliferating cell nuclear antigen
(PCNA; 1:2,000; cat. no. ab92552), NF-κB (1:1,000; cat. no.
ab207297) and phosphorylated (p)-NF-κB (1:1,000; cat. no. ab239882)
purchased from Abcam. Goat anti-mouse IgG (H&L), HRP-conjugated
secondary antibody (1:5,000; cat. no. S0002) and goat anti-rabbit
IgG (H&L), HRP-conjugated secondary antibody (1:5,000; cat. no.
S0001) obtained from Affinity Biosciences.
Statistical analysis
SPSS v21.0 software (IBM Corp.) for Windows was used
for statistical analysis, and clinical variables were analyzed
using χ2 or Fisher's exact test, where appropriate. The
qPCR data on H. pylori between GCA and DGA was performed
using χ2 or Fisher's exact test. Quantitative data are
presented as the mean ± SD, and the data were analyzed using an
unpaired t-test. Differences among >2 groups were analyzed using
one-way ANOVA followed by Tukey's post hoc test. All the
statistical tests were two-tailed. P<0.05 was considered to
indicate a statistically significant difference.
Results
H. pylori infection in patients with
GCA and DGA
The expression of 16S rRNA, CagA and VacA was
quantified using qPCR. Among 113 patients with GC, 75.2, 30.1 and
38.9% were positive for H. pylori 16S rRNA, CagA and VacA
expression, respectively.
The comparative analysis of 16S rRNA expression
level revealed no statistically significant difference between
patients with GCA and DGA (73.8 vs. 76.9%; P>0.05; Table I). However, the mRNA expression level
of VacA was significantly higher in patients with GCA (49.2%)
compared with that in patients with DGA (26.9%; P<0.05).
Notably, in samples from male patients >60 years of age, with
lymph node metastasis or stage III GCA, increased VacA levels were
detected compared with those in patients with DGA and matched
pathological features (P<0.05; Table
II). In DGA, but not in GCA (P>0.05), a significant
association between CagA and age (P<0.05), and VacA and lymph
node metastasis (P<0.05) was observed. These findings suggest
that H. pylori may serve an important role in GCA and DGA
pathogenesis. Furthermore, subjects with VacA-positive H.
pylori infection may exhibit an increased risk of GCA in high
incidence areas of China.
 | Table I.H. pylori infection in patients with
GCA or DGA. |
Table I.
H. pylori infection in patients with
GCA or DGA.
| Group | Cases, n | Positive, n
(%) | Negative, n
(%) | OR (95%
CI)a | P-value |
|---|
| H. pylori
16S rRNA |
|
GCA | 61 | 45 (73.8) | 16 (26.2) |
|
|
|
DGA | 52 | 40 (76.9) | 12 (23.1) | 0.844
(0.357–1.996) | 0.699 |
| H. pylori
CagA |
|
GCA | 61 | 19 (31.1) | 42 (68.9) |
|
|
|
DGA | 52 | 15 (28.8) | 37 (71.2) | 1.116
(0.497–2.504) | 0.790 |
| H. pylori
VacA |
|
GCA | 61 | 30 (49.2) | 31 (50.8) |
|
|
|
DGA | 52 | 14 (26.9) | 38 (73.1) | 2.627
(1.190–5.800) | 0.016 |
| Table II.Expression of TFF1 and GKN2 in GCA
and DGA. |
Table II.
Expression of TFF1 and GKN2 in GCA
and DGA.
| Group | N | TFF1 (%) | GKN2 (%) |
|---|
| NGM | 23 | 20 (87.0) | 18 (77.3) |
| GCA | 61 | 26
(42.6)a | 18
(29.5)a |
| DGA | 52 | 22
(42.3)a | 15
(28.8)a |
TFF1 and GKN2 expression levels in GCA
and DGA
It has been reported that TFF peptides are primarily
expressed in gastric mucous cells and are resistant to proteolysis
and acidic environments under physiological conditions; thus,
resulting in maintenance of mucosal integrity in the stomach
(21–23). In addition, GKN2, which acts by
interacting with TFF1 to form a heterodimer, also serves an
important role in maintaining mucosa integrity (25,26).
Therefore, TFF1 and GKN2 protein expression levels were detected in
GC tissues using IHC staining, to further investigate their
association with H. pylori infection. The positive staining
of TFF1 and GKN2 in IHC analysis was indicated by brown granules in
the cytoplasm (Fig. 1A). The
positive rates of TFF1 and GKN2 in 113 GC samples were 42.5 and
29.2%, respectively, and were significantly lower compared with
those in NGM (P<0.05; Table
II).
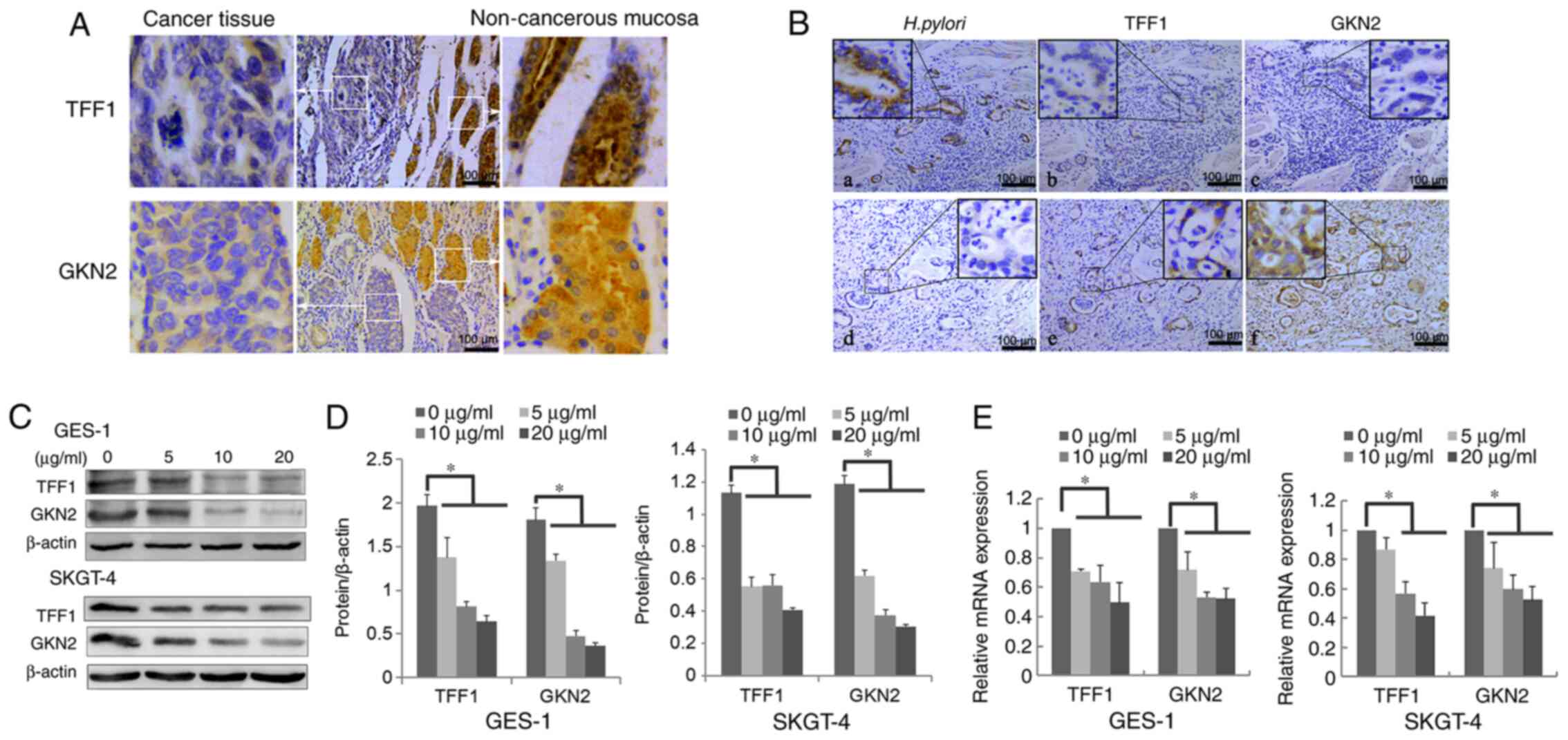 | Figure 1.H. pylori downregulates TFF1
and GKN2 mRNA and protein expression in vivo and in
vitro. (A) Protein expression level of TFF1 and GKN2 in
non-cancerous mucosa (right) and cancer tissue (left) using IHC.
(B) Protein expression of (a) H. pylori+, (b) TFF1-, (c)
GKN2-, (d) H. pylori-, (e) TFF1+ and (f) GKN2+ in continuous
slices of GCA and DGA using IHC. (C-E) H. pylori treatment
for 24 h significantly downregulated the (C and D) protein and (E)
mRNA expression level of TFF1 and GKN2 in GES-1 and SKGT-4 cells
(*P<0.05). Data are presented as the mean ± SD of triplicate
experiments, and the data were analyzed using ANOVA and Tukey's
post hoc test. IHC, immunohistochemistry; GCA, gastric cardia
adenocarcinoma; DGA, distal gastric adenocarcinoma; H.
pylori/Hp, Helicobacter pylori; NGM, normal gastric
mucosa; -, negative; +, positive. |
No statistically significant differences were
detected in TFF1 and GKN2 expression levels between GCA and DGA.
However, the expression levels of both proteins were significantly
lower in both types of gastric adenocarcinoma compared with NGM
(P<0.05; Table II). Furthermore,
TFF1 and GKN2 expression was associated with lymph node metastasis
and invasion depth in DGA (P<0.05; Table III).
| Table III.Association between Helicobacter
pylori, TFF1 and GKN2 and the pathological characteristics in
patients with GCA (n=61) and DGA (n=52). |
Table III.
Association between Helicobacter
pylori, TFF1 and GKN2 and the pathological characteristics in
patients with GCA (n=61) and DGA (n=52).
|
| CGA, n (%) | DGA, n (%) |
|---|
|
|
|
|
|---|
| Pathological
characteristics | Total, n | 16S rRNA | CagA | VacA | TFF1 | GKN2 | Total, n | 16S rRNA | CagA | VacA | TFF1 | GKN2 |
|---|
| Sex |
|
Male | 54 | 39 (72.2) | 16 (29.6) | 27 (50.0) | 24 (44.4) | 17 (31.5) | 42 | 34 (81.0) | 12 (28.6) | 11
(26.2)a | 18 (42.9) | 12 (28.6) |
|
Female | 7 | 6 (85.7) | 3 (42.9) | 3 (42.9) | 2 (28.6) | 1 (14.3) | 10 | 6 (60.0) | 3 (30.0) | 3 (30.0) | 4 (40.0) | 3 (30.0) |
| Age, years |
|
≤60 | 20 | 17 (85.0) | 10 (50.0) | 12 (60.0) | 5 (25.0) | 4 (20.0) | 27 | 23 (85.2) | 11 (40.7) | 9 (33.3) | 11 (40.7) | 7 (25.9) |
|
>60 | 41 | 28 (68.3) | 9
(22.0)b | 18 (43.9) | 21 (51.2) | 14 (34.1) | 25 | 17 (68.0) | 4
(16.0)b | 5
(20.0)a | 11 (44.0) | 8 (32.0) |
| Lymph node
metastasis |
|
Positive | 34 | 25 (73.5) | 10 (29.4) | 16 (47.1) | 12 (35.3) | 8 (23.5) | 30 | 21 (70.0) | 7 (23.3) | 3
(10.0)a | 8 (26.7) | 4 (13.3) |
|
Negative | 27 | 21 (77.8) | 9 (33.3) | 14 (51.9) | 14 (51.9) | 10 (37.0) | 22 | 19 (86.4) | 8 (36.4) | 11
(50.0)c | 14
(63.6)c | 11
(50.0)c |
| Invasion depth |
|
T1+T2 | 10 | 8 (80.0) | 4 (40.0) | 5 (50.0) | 5 (50.0) | 4 (40.0) | 13 | 9 (69.2) | 3 (23.1) | 4 (30.8) | 10 (76.9) | 6 (46.2) |
|
T3+T4 | 51 | 37 (72.5) | 15 (29.4) | 25 (49.0) | 21 (41.2) | 14 (27.5) | 39 | 31 (79.5) | 12 (30.8) | 10 (25.6) | 12
(30.8)d | 9 (23.1) |
| Stage |
| I | 7 | 5 (71.4) | 2 (28.6) | 2 (28.6) | 3 (42.9) | 3 (42.9) | 6 | 5 (83.3) | 0 | 1 (16.7) | 5 (83.3) | 3 (50.0) |
| II | 20 | 15 (75.0) | 7 (35.0) | 12 (60.0) | 12 (60.0) | 6 (30.0) | 19 | 15 (78.9) | 8 (42.1) | 10 (52.6) | 11 (57.9) | 9 (47.4) |
|
III | 31 | 23 (74.2) | 10 (32.3) | 16 (51.6) | 9 (29.0) | 7 (22.6) | 24 | 19 (79.2) | 7 (29.2) | 3
(12.5)a | 6 (25.0) | 3 (12.5) |
| IV | 3 | 2 (66.7) | 0 | 0 | 2 (66.7) | 2 (66.7) | 3 | 1 (33.3) | 0 | 0 | 0 | 0 |
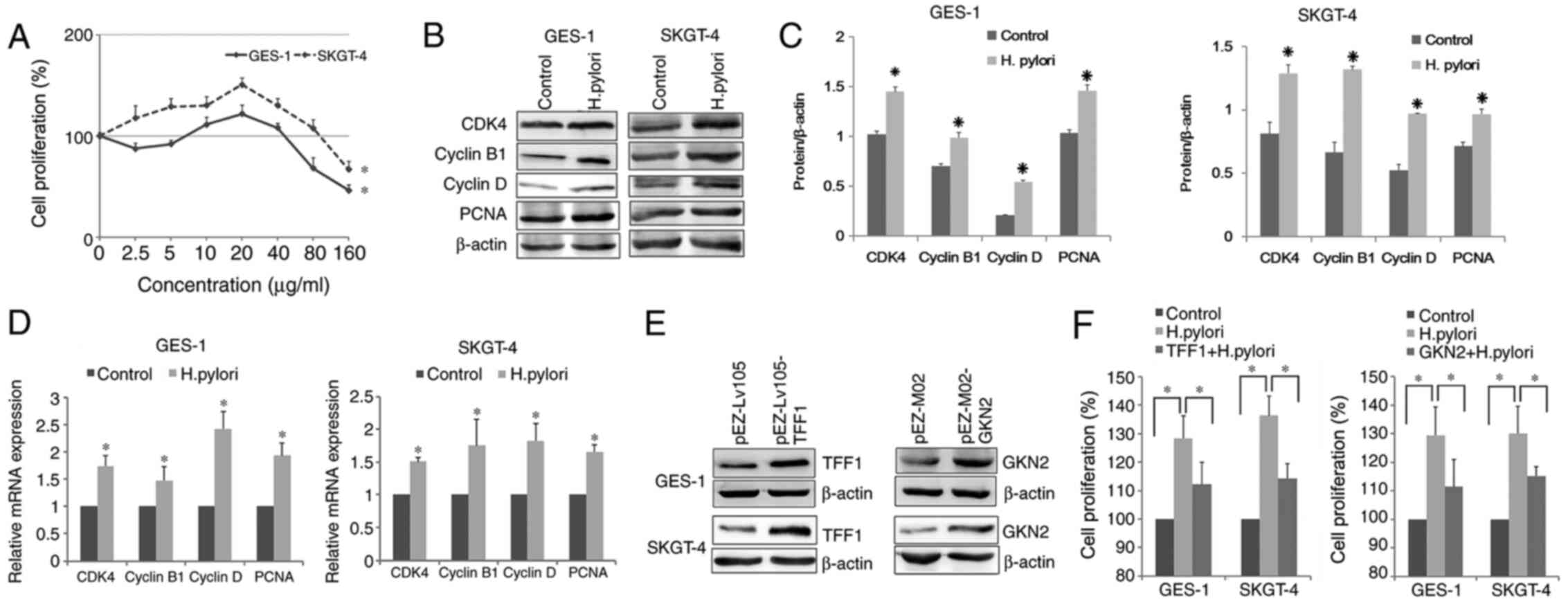
H. pylori downregulates TFF1 and GKN2
expression in vivo and in vitro
The incidence of positive TFF1 and GKN2 expression
was significantly decreased in H. pylori-positive GCA (45
cases) and DGA (40 cases) compared with that in GCA and DGA H.
pylori-negative samples (P<0.05; Fig. 1B and Table IV). Furthermore, to verify the
effects of H. pylori on TFF1 and GKN2 expression, GES-1 and
SKGT-4 cells were treated with different concentrations of H.
pylori protein for 24 h and the results showed that H.
pylori treatment significantly reduced TFF1 and GKN2 mRNA and
protein expression levels (P<0.05; Fig. 1C-E). Taken together, the results
demonstrate that H. pylori infection decreases the
expression of both protective factors in vitro and in
vivo.
| Table IV.Helicobacter pylori infection
decreased the expression of TFF1 and GKN2 in GCA and DGA. |
Table IV.
Helicobacter pylori infection
decreased the expression of TFF1 and GKN2 in GCA and DGA.
|
| GCA | DGA |
|---|
|
|
|
|
|---|
| Groups | n (%) | P-value | n (%) | P-value |
|---|
| TFF1+/H.
pylori+ | 15/45 (33.3) |
| 13/40 (32.5) |
|
| TFF1-/H.
pylori+ | 30/45 (66.7) |
| 27/40 (67.5) |
|
| TFF1+/H.
pylori- | 11/16 (68.8) |
| 9/12 (75.0) |
|
| TFF1-/H.
pylori- | 5/16 (31.3) | 0.014a | 3/12 (25.0) | 0.009a |
| GKN2+/H.
pylori+ | 10/45 (22.3) |
| 8/40 (20.0) |
|
| GKN2-/H.
pylori+ | 35/45 (77.8) |
| 32/40 (80.0) |
|
| GKN2+/H.
pylori- | 8/16 (50.0) |
| 7/12 (58.3) |
|
| GKN2-/H.
pylori- | 8/16 (50.0) | 0.036a | 5/12 (41.7) | 0.010a |
TFF1/GKN2 expression inhibits H.
pylori-induced cell proliferation
It has been reported that H. pylori protein
induces cell proliferation and apoptosis in human gastric
epithelial cell lines (31–33). To further investigate the tumorigenic
potential of H. pylori, its effects on GES-1 and SKGT-4 cell
proliferation were determined. The results showed that cell
survival rate in the H. pylori-treated groups varied with
the different doses (Fig. 2A).
Notably, treatment with 20 µg/ml H. pylori total proteins
for 24 h resulted in the highest cell proliferation rate
(P<0.05; Fig. 2A). Therefore, 20
µg/ml H. pylori total proteins were used for the following
experiment. H. pylori treatment significantly increased the
mRNA and protein expression levels of CDK4, PCNA, cyclin B1 and
cyclin D (P<0.05; Fig. 2B-D).
To investigate whether TFF1/GKN2 regulated H.
pylori-induced proliferation, cells were transfected with TFF1
and GKN2 overexpression plasmids and their protein expression
levels were determined. Western blot analysis demonstrated that
both targeted genes were successfully overexpressed (Fig. 2E). In addition, the overexpression of
TFF1 and GKN2 significantly decreased cell proliferation, compared
with that in the H. pylori-treatment group (P<0.05;
Fig. 2F).
TFF1/GKN2 expression decreases the H.
pylori-induced inflammatory response
To investigate the role of TFF1 and GKN2 in the
regulation of the H. pylori-induced inflammatory response,
the activation status of NF-κB was evaluated using western blot
analysis. The analysis showed that NF-κB p65 were significantly
increased following treatment with H. pylori proteins
compared with those in the control group (P<0.05; Fig. 3A and B). Although there were not
significant differences of the phosphor/total NF-κB p65 ratio
between before and after treatment (Fig.
3C), overexpression of TFF1 and GKN2 inhibited H.
pylori-mediated upregulation of NF-κB p65, compared with H.
pylori-treated group (P<0.05; Fig. 3A and B).
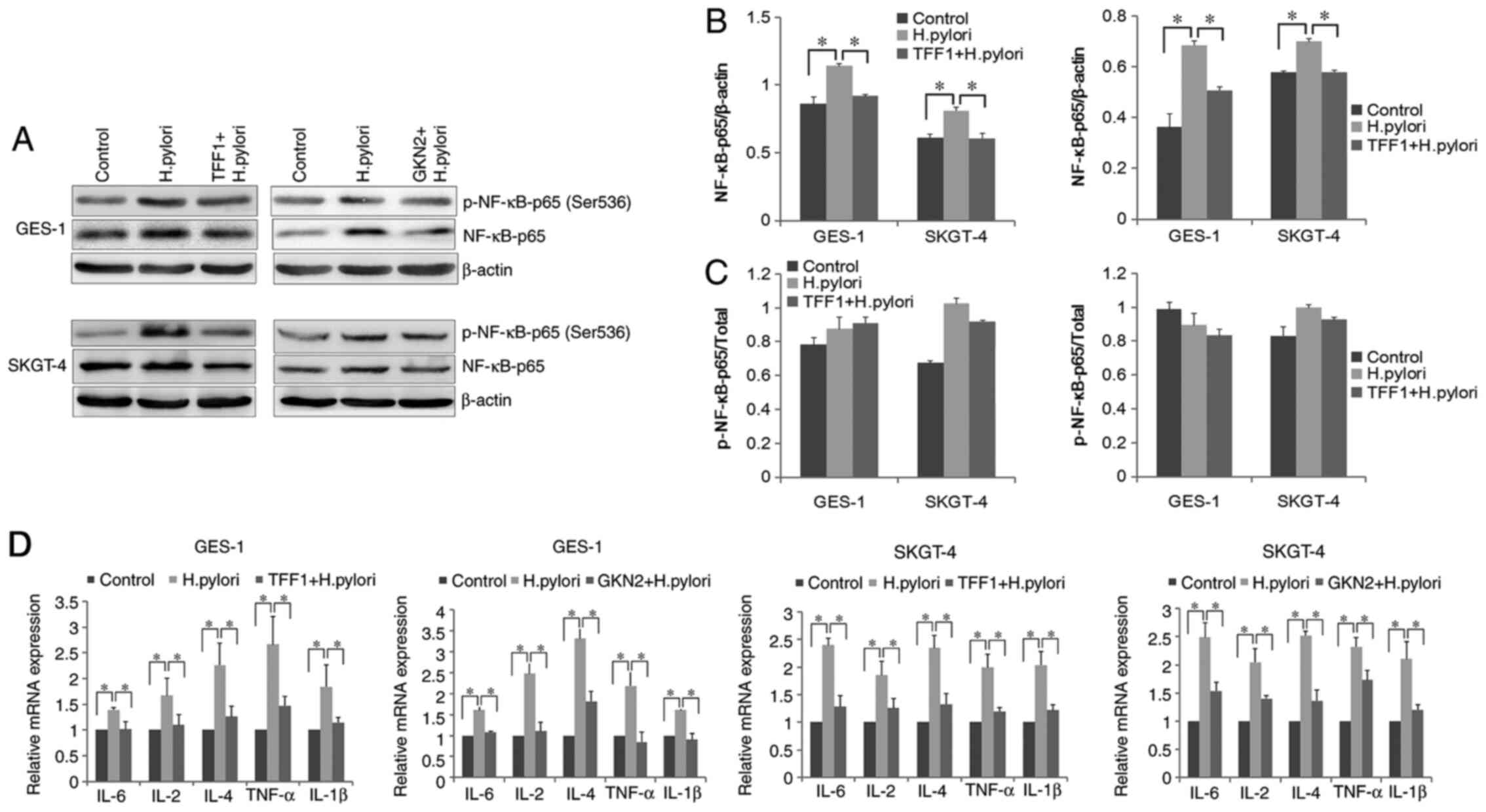 | Figure 3.Overexpression of TFF1 and GKN2
inhibits the H. pylori-induced inflammatory response. (A)
NF-κB-p65 and p-NF-κB-p65 (Ser536) were detected using western blot
analysis. (B and C) NF-κB-p65 and its phosphorylated form (Ser536)
were significantly increased following treatment with H.
pylori proteins, and overexpression of TFF1 and GKN2
significantly inhibited H. pylori-mediated regulation of
NF-κB p65 (*P<0.05). (D) H. pylori (20 µg/ml) significantly
increased the mRNA expression level of IL-6, IL-2, IL-4, IL-1β and
TNF-α (*P<0.05), whereas the overexpression of TFF1 and GKN2
decreased these inflammatory cytokines (*P<0.05). Data are
presented as the mean ± SD of triplicate experiments. H. pylori,
Helicobacter pylori; TFF1, trefoil factor 1; GKN2, gastrokine
2; TNF, tumor necrosis factor; p, phosphorylated. |
It has been reported that bacterial-induced
activation of NF-κB induces the secretion of proinflammatory
cytokines, which in turn leads to inflammation (34–36).
Therefore, the mRNA expression levels of the inflammatory cytokines
IL-6, IL-2, IL-4, IL-1β and tumor necrosis factor α (TNF-α), were
evaluated. RT-qPCR analysis demonstrated that the mRNA expression
levels of all the inflammatory cytokines tested were significantly
increased in the H. pylori-treated group, compared with the
control group (P<0.05; Fig. 3D).
However, overexpression of TFF1 or GKN2 in GES-1 and SKGT-4 cells
significantly downregulated the expression of the aforementioned
cytokines, compared with H. pylori-treated group (P<0.05;
Fig. 3D). These results indicate
that to some extent, TFF1 and GKN2 overexpression suppresses the
H. pylori-induced inflammatory response.
Discussion
The present study aimed to investigate the
association between H. pylori infection and the expression
of TFF1 and GKN2 in GCA and DGA. The results demonstrated that
there was no statistically significant difference in H.
pylori infection rate between GCA and DGA. Furthermore, H.
pylori infection significantly decreased TFF1 and GKN2 in
vitro, and H. pylori protein treatment also inhibited
the mRNA and protein expression level of both genes in vivo.
These findings indicate that H. pylori-mediated loss of TFF1
and GKN2 expression may contribute to the development of GCA and
DGA.
Unlike the clear association between H.
pylori and DGA pathogenesis, its association with GCA remains
controversial. Several studies have reported that there was no
association between H. pylori infection and gastroesophageal
cancers (37,38), while others have demonstrated a
positive association (18,39). It is reported that gastroesophageal
cancers are inversely associated with H. pylori infection,
particularly the CagA strain (40).
In addition, declining prevalence of H. pylori infection has
been reported to coincide with an increased incidence rate of GCA
(14,41). These findings support a potential
protective effect of H. pylori infection against
gastroesophageal cancers. However, a recent meta-analysis revealed
an association between H. pylori infection, particularly
CagA-positive strains, and the relative risk of Barrett's esophagus
(42). In regions, for example
Cixian County and Zanhuang County with a high incidence rate of
gastroesophageal cancers and an increased prevalence of H.
pylori infection in China, GCA accounts for a large proportion
of cases (1). Notably, in a Chinese
population, the studies showed that H. pylori infection was
associated with an increased risk of GCA (18,20). In
the present study, no statistically significant difference was
observed in 16S rRNA positive rate between GCA and DGA. Notably,
GCA samples displayed significantly higher VacA rates compared with
DGA samples. Research in the last few decades has revealed that
VacA is a key toxin for H. pylori pathogenesis and has a
variety of effects on gastric epithelial cells. All identified
H. pylori strains possess the VacA gene; however, there was
significant sequence diversity in VacA genes among numerous
isolated strains (43,44). In the present study, the H.
pylori 26695 strain (GenBank, AE000511.1), including 16S rRNA,
CagA and VacA was detected. Our results showed that there are no
differences in H. pylori infection between GCA and DGA. This
was consistent with several other studies in the Chinese population
(18,20), and further confirms that H.
pylori may be an important factor in GCA pathogenesis and
patients who are VacA-positive may have a high-risk to develop GCA,
in high incidence regions in China.
To further investigate the effects of H.
pylori on host cell self-protection mechanisms, the protein
expression levels of two protective molecules were detected in GC
and normal gastric epithelial tissue specimens. Both GCA and DGA
exhibited significantly lower expression of TFF1 and GKN2 compared
with that in NGM. Notably, H. pylori-positive GCA and DGA
tissues displayed significantly decreased TFF1 and GKN2 expression
compared with that in their respective H. pylori-negative
tumor tissues. It has been reported that TFF1 and GKN2 expression
is downregulated in GC (45,46). The protein and mRNA expression of
GKN1, the other member of GKN family, was decreased in the mucosa
of patients with GC and with H. pylori-positive chronic
gastritis (34), and exosomal GKN1
protein could inhibit gastric carcinogenesis (35). In particular, decreased expression of
GKN2 has been associated with shorter overall survival in the
intestinal subtype of GC (36). A
previous study demonstrated that TFF1 acts by interacting with GKN2
to form a heterodimer, which is stabilized with an intermolecular
disulfide bond (25). GKN2 and TFF1
co-expression induces G1/S cell cycle arrest in MKN1,
MKN28 and MKN45 cells, thus inhibiting the proliferation of gastric
cancer cells (47). Therefore, TFF1
and GKN2 are tumor suppressive factors and their absence may
contribute to carcinogenesis in GC (22). In the present study, H. pylori
significantly downregulated the mRNA and protein expression levels
of these aforementioned protective factors, in two different cell
lines. These findings suggest that H. pylori-mediated TFF1
or GKN2 downregulation may be associated with the development of
GCA and DGA.
It is widely recognized that H. pylori
induces chronic gastritis, which is considered to be the first step
in gastric carcinogenesis (48–50). In
addition, H. pylori serves an important role in the
activation of NF-κB (48).
Therefore, the bacterial-mediated activation of NF-κB induces the
secretion of proinflammatory cytokines such as TNF-α and IL-1β,
which in turn leads to inflammation (48–50).
Furthermore, JHP0290, a functional protein from H. pylori
was found to bind to several cell types, including gastric
epithelial cell lines, macrophage and neutrophils and induce TNF-α
release, which was partly dependent on the activation of NF-κB
(31,32). HP1286, a conserved secreted protein
from strain 26695, could induce apoptosis in adenocarcinoma gastric
cells (33). Previous research in a
TFF1 knock-out mouse model showed that the majority of
inflammation-related genes, such as NF-κB, TNF-α and IL-1β and the
downregulation of TFF1 were involved in the induction of
inflammation, which in turn resulted in gastric tumorigenesis
(49,50). With respect to GKN2, it was found to
recruit neutrophils and induce the release of inflammatory factors,
for example NF-κB and IL-1β, contributing to inflammation in
stress-induced gastric lesions (51). To investigate H. pylori
infection in host cell self-protection mechanisms in the present
study, H. pylori total protein, primarily containing urease,
CagA and cytotoxin A antigens, was selected to treat GES-1 and
SKGT-4 cells in vitro. NF-κB p65 was significantly increased
in both cells following H. pylori protein treatment.
Notably, overexpression of TFF1 and GKN2 inhibited H.
pylori-induced increase of NF-κB p65 and reversed the mRNA
expression levels of inflammatory cytokines (TNF-α, IL-1β, IL-2,
IL-4, and IL-6) following H. pylori-mediated upregulation.
The aforementioned results indicate that TFF1 and GKN2 may protect
gastric epithelial cells from damage mediated by inflammatory
cytokines.
It is well known that inflamed tissues express
several growth factors, including epidermal growth factor and
platelet-derived growth factor, which directly promote cell
proliferation (52). The results of
the current study further demonstrate that H. pylori
regulates cell proliferation differently for distinct
concentrations. In particular, treatment of cells with 20 µg/ml
H. pylori total protein resulted in the highest
proliferation rate. Furthermore, TFF1 and GKN2 overexpression
inhibited H. pylori-mediated cell proliferation. It has been
reported that the proliferation rate of gastric epithelial cells in
patients with CagA-positive H. pylori was significantly
higher compared with that in patients who were CagA-negative
(53). However, in vitro,
CagA acts as a potent inhibitor of cell proliferation, which is
inconsistent with its oncogenic role (54). Therefore, CagA may be involved in a
mechanism that converts the response of the host cells between
growth inhibition and stimulation, depending on varying infective
dosage (53,54). In addition, several H. pylori
virulence factors, for example JHP0290 and SlyD have been
identified to regulate the proliferation of gastric epithelial
cells (32,55). The aforementioned results indicated
that H. pylori, to some extent, induces cell proliferation
and the inflammatory response in host cells. Furthermore,
overexpression of TFF1 and GKN2 may reduce this response.
In conclusion, the present study demonstrated that
there was no statistically significant difference in H.
pylori infection rate between GCA and DGA. This finding
indicates that H. pylori, particularly the VacA+ strain, may
play an important role in GCA, in high risk-areas of China. The
results indicated that H. pylori decreased the mRNA and
protein expression levels of TFF1 and GKN2 in GCA and DGA, while
TFF1/GKN2 overexpression significantly reduced H.
pylori-induced cell proliferation and inflammation in GCA and
DGA; however there was a lack of a suitable negative control in
H. pylori protein treatment. In addition, the mechanism
between H. pylori infection and self-protection of the host
cell remains unclear, and further investigation is required in the
future. Notably, it might be important to select an appropriate
negative control, for example a denatured version of the H.
pylori proteins or proteins derived from harmless gut flora,
for the H. pylori protein experiments.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81301696) and the project of
Educational Commission of Hebei Province (grant no. ZD2016008).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
WL, DZ, BC, XZ and XW performed the experiments and
prepared the figures. JL and LX prepared the figures and analyzed
the data. WL, JL and LX performed critical revision of the
manuscript and supervised the study. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the local Ethics
Committee of Hebei Medical University and written informed consent
was provided by all the participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhao CY, Zhang XH, Xue LY, Xing LX, Wang
JL, Li XM, Mi JM and Jin GL: Analysis of the changing trends of
frequency and localization of gastric cancers arising from
different sites of the stomach in population of the high incidence
area of esophageal and gastric cancers in Hebei province. Zhonghua
Zhong Liu Za Zhi. 30:817–820. 2008.(In Chinese). PubMed/NCBI
|
|
2
|
Dent J: Pathogenesis and classification of
cancer around the gastroesophageal junction-not so different in
Japan. Am J Gastroenterol. 101:934–936. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mattioli S, Ruffato A, Di Simone MP, Corti
B, D'Errico A, Lugaresi ML, Mattioli B and D'Ovidio F:
Immunopathological patterns of the stomach in adenocarcinoma of the
esophagus, cardia, and gastric antrum: Gastric profiles in Siewert
type I and II tumors. Ann Thorac Surg. 83:1814–1819. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou Y, Zhang Z, Zhang Z, Wu J, Ren D, Yan
X, Wang Q, Wang Y, Wang H, Zhang J, et al: A rising trend of
gastric cardia cancer in Gansu Province of China. Cancer Lett.
269:18–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malhotra GK, Yanala U, Ravipati A, Follet
M, Vijayakumar M and Are C: Global trends in esophageal cancer. J
Surg Oncol. 115:564–579. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petrick JL, Hyland PL, Caron P, Falk RT,
Pfeiffer RM, Dawsey SM, Abnet CC, Taylor PR, Weinstein SJ, Albanes
D, et al: Associations between prediagnostic concentrations of
circulating sex steroid hormones and esophageal/gastric cardia
adenocarcinoma among men. J Natl Cancer Inst. 111:34–41. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He Y, Hou J, Liu T and Zhang G: The
association between cancer mortality and socioeconomy status from
1990–1992 in Hebei province, China. China Tumor Bull. 5:111996.(In
Chinese).
|
|
8
|
Zhao P and Kong LZ: Mortality Rate of
Stomach Cancer: Cancer Mortality in China. People's Health Press;
Beijing: 307. 2009
|
|
9
|
Xue L, Zhang X, Li Y, Yang H, Li X, Mi J,
Wang H, Wang J and Yan X: Differences of immunophenotypic markers
and signaling molecules between adenocarcinomas of gastric cardia
and distal stomach. Hum Pathol. 42:594–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xue L, Ouyang Q, Li J, Meng X, Li Y, Xing
L, Wang J, Yan X and Zhang X: Different roles for p16(INK) (4a)-Rb
pathway and INK4a/ARF methylation between adenocarcinomas of
gastric cardia and distal stomach. J Gastroenterol Hepatol.
29:1418–1426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yao D, Wang Y, Xue L, Wang H, Zhang J and
Zhang X: Different expression pattern and significance of
p14ARF-Mdm2-p53 pathway and Bmi-1 exist between gastric cardia and
distal gastric adenocarcinoma. Hum Pathol. 44:844–851. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu N, Wang Z, Song X, Wei L, Kim BS,
Freedman ND, Baek J, Burdette L, Chang J, Chung C, et al:
Genome-wide association study of gastric adenocarcinoma in Asia: A
comparison of associations between cardia and non-cardia tumours.
Gut. 65:1611–1618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Xue L, Xing L, Wang J, Cui J, Mi
J, Xing X, Wang J, Du Z, Misumi J, et al: Low serum pepsinogen I
and pepsinogen I/II ratio and Helicobacter pylori infection
are associated with increased risk of gastric cancer: 14-year
follow up result in a rural Chinese community. Int J Cancer.
130:1614–1619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Backert S and Blaser MJ: The role of CagA
in the gastric biology of Helicobacter pylori. Cancer Res.
76:4028–4031. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Derakhshan MH, Malekzadeh R, Watabe H,
Yazdanbod A, Fyfe V, Kazemi A, Rakhshani N, Didevar R, Sotoudeh M,
Zolfeghari AA and McColl KE: Combination of gastric atrophy, reflux
symptoms and histological subtype indicates two distinct
aetiologies of gastric cardia cancer. Gut. 57:298–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanikini H, Muller DC, Sophiea M, Rinaldi
S, Agudo A, Duell EJ, Weiderpass E, Overvad K, Tjønneland A,
Halkjaer J, et al: Anthropometric and reproductive factors and risk
of esophageal and gastric cancer by subtype and subsite: Results
from the European prospective investigation into cancer and
nutrition (EPIC) cohort. Int J Cancer. 146:929–942. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chow WH, Blaser MJ, Blot WJ, Gammon MD,
Vaughan TL, Risch HA, Perez-Perez GI, Schoenberg JB, Stanford JL,
Rotterdam H, et al: An inverse relation between cagA+ strains of
Helicobacter pylori infection and risk of esophageal and
gastric cardia adenocarcinoma. Cancer Res. 58:588–590.
1998.PubMed/NCBI
|
|
18
|
Wang Y, Liu S, Zhang Y, Bi C, Xiao Y, Lin
R, Huang B, Tian D, Ying S and Su M: Helicobacter pylori
infection and gastric cardia cancer in Chaoshan region. Microbes
Infect. 16:840–844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Wang H, Bi C, Xiao Y and Liu Z:
Expression of CDX2 in gastric cardia adenocarcinoma and its
correlation with H. pylori and cell proliferation.
Oncotarget. 7:54973–54982. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cai M, Dai S, Chen W, Xia C, Lu L, Dai S,
Qi J, Wang M, Wang M, Zhou L, et al: Environmental factors, seven
GWAS-identified susceptibility loci, and risk of gastric cancer and
its precursors in a Chinese population. Cancer Med. 6:708–720.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aihara E, Engevik KA and Montrose MH:
Trefoil factor peptides and gastrointestinal function. Annu Rev
Physiol. 79:357–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soutto M, Belkhiri A, Piazuelo MB,
Schneider BG, Peng D, Jiang A, Washington MK, Kokoye Y, Crowe SE,
Zaika A, et al: Loss of TFF1 is associated with activation of
NF-κB-mediated inflammation and gastric neoplasia in mice and
humans. J Clin Invest. 121:1753–1767. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Omar OM, Soutto M, Bhat NS, Bhat AA, Lu H,
Chen Z and El-Rifai W: TFF1 antagonizes TIMP-1 mediated
proliferative functions in gastric cancer. Mol Carcinog.
57:1577–1587. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soutto M, Peng D, Katsha A, Chen Z,
Piazuelo MB, Washington MK, Belkhiri A, Correa P and El-Rifai W:
Activation of β-catenin signalling by TFF1 loss promotes cell
proliferation and gastric tumorigenesis. Gut. 64:1028–1039. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kouznetsova I, Laubinger W, Kalbacher H,
Kalinski T, Meyer F, Roessner A and Hoffmann W: Biosynthesis of
gastrokine-2 in the human gastric mucosa: Restricted spatial
expression along the antral gland axis and differential interaction
with TFF1, TFF2 and mucins. Cell Physiol Biochem. 20:899–908. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Menheniott TR, O'Connor L, Chionh YT,
Däbritz J, Scurr M, Rollo BN, Ng GZ, Jacobs S, Catubig A, Kurklu B,
et al: Loss of gastrokine-2 drives premalignant gastric
inflammation and tumor progression. J Clin Invest. 126:1383–1400.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
May FE, Griffin SM and Westley BR: The
trefoil factor interacting protein TFIZ1 binds the trefoil protein
TFF1 preferentially in normal gastric mucosal cells but the
co-expression of these proteins is deregulated in gastric cancer.
Int J Biochem Cell Biol. 41:632–640. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heuer J, Heuer F, Stürmer R, Harder S,
Schlüter H, Braga Emidio N, Muttenthaler M, Jechorek D, Meyer F and
Hoffmann W: The tumor suppressor TFF1 occurs in different forms and
interacts with multiple partners in the human gastric mucus
barrier: Indications for diverse protective functions. Int J Mol
Sci. 21:25082020. View Article : Google Scholar
|
|
29
|
Siewert JR, Stein HJ and Feith M:
Adenocarcinoma of the esophago-gastric junction. Scand J Surg.
95:260–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pathak SK, Tavares R, de Klerk N, Spetz AL
and Jonsson AB: Helicobacter pylori protein JHP0290 binds to
multiple cell types and induces macrophage apoptosis via tumor
necrosis factor (TNF)-dependent and independent pathways. PLoS One.
8:e778722013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tavares R and Pathak SK: Helicobacter
pylori protein JHP0290 exhibits proliferative and
anti-apoptotic effects in gastric epithelial cells. PLoS One.
10:e01244072015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Meng FL, He LH and Zhang JZ:
Secreted protein HP1286 of Helicobacter pylori strain 26695
induces apoptosis of AGS cells. Biomed Environ Sci. 25:614–619.
2012.PubMed/NCBI
|
|
34
|
Alarcón-Millán J, Lorenzo-Nazario SI,
Jiménez-Wences H, Campos-Viguri GE, Ortiz-Ortiz J, Mendoza-Catalán
MÁ, Cortés-Malagón EM, Reyes-Navarrete S, Jiménez-López MA,
Castañón-Sánchez CA, et al: Women with chronic follicular gastritis
positive for Helicobacter pylori express lower levels of
GKN1. Gastric Cancer. 23:754–759. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoon J, Ashktorab H, Smoot D, Nam S, Hur H
and Park W: Uptake and tumor-suppressive pathways of
exosome-associated GKN1 protein in gastric epithelial cells.
Gastric Cancer. 23:848–862. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moss SF, Lee JW, Sabo E, Rubin AK, Rommel
J, Westley BR, May FE, Gao J, Meitner PA, Tavares R and Resnick MB:
Decreased expression of gastrokine 1 and the trefoil factor
interacting protein TFIZ1/GKN2 in gastric cancer: Influence of
tumor histology and relationship to prognosis. Clin Cancer Res.
14:4161–4167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kamada T, Kurose H, Yamanaka Y, Manabe N,
Kusunoki H, Shiotani A, Inoue K, Hata J, Matsumoto H, Akiyama T, et
al: Relationship between gastroesophageal junction adenocarcinoma
and Helicobacter pylori infection in Japan. Digestion.
85:256–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang F, Meng W, Wang B and Qiao L:
Helicobacter pylori- induced gastric inflammation and
gastric cancer. Cancer Lett. 345:196–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shakeri R, Malekzadeh R, Nasrollahzadeh D,
Pawlita M, Murphy G, Islami F, Sotoudeh M, Michel A, Etemadi A,
Waterboer T, et al: Multiplex H. pylori serology and risk of
gastric cardia and noncardia adenocarcinomas. Cancer Res.
75:4876–4883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Malfertheiner P, Megraud F, O'Morain CA,
Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY,
Rokkas T, et al: Management of Helicobacter pylori
infection-the maastricht IV/florence consensus report. Gut.
61:646–664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
el Serag HB and Sonnenberg A: Opposing
time trends of peptic ulcer and reflux disease. Gut. 43:327–333.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Erőss B, Farkas N, Vincze Á, Tinusz B,
Szapáry L, Garami A, Balaskó M, Sarlós P, Czopf L, Alizadeh H, et
al: Helicobacter pylori infection reduces the risk of
Barrett's esophagus: A meta-analysis and systematic review.
Helicobacter. 23:e125042018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rassow J: Helicobacter pylori
vacuolating toxin A and apoptosis. Cell Commun Signal. 9:262011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chauhan N, Tay ACY, Marshall BJ and Jain
U: Helicobacter pylori VacA, a distinct toxin exerts diverse
functionalities in numerous cells: An overview. Helicobacter.
24:e125442019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tomita H, Takaishi S, Menheniott TR, Yang
X, Shibata W, Jin G, Betz KS, Kawakami K, Minamoto T, Tomasetto C,
et al: Inhibition of gastric carcinogenesis by the hormone gastrin
is mediated by suppression of TFF1 epigenetic silencing.
Gastroenterology. 140:879–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang W, Li Z, Wang J, Du M, Li B, Zhang L,
Li Q, Xu J, Wang L, Li F, et al: A functional polymorphism in TFF1
promoter is associated with the risk and prognosis of gastric
cancer. Int J Cancer. 142:1805–1816. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim O, Yoon JH, Choi WS, Ashktorab H,
Smoot DT, Nam SW, Lee JY and Park WS: Heterodimeric interaction
between GKN2 and TFF1 entails synergistic antiproliferative and
pro-apoptotic effects on gastric cancer cells. Gastric Cancer.
20:772–783. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wroblewski LE, Peek RM Jr and Wilson KT:
Helicobacter pylori and gastric cancer: Factors that
modulate disease risk. Clin Microbiol Rev. 23:713–739. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Soutto M, Saleh M, Arredouani MS, Piazuelo
B, Belkhiri A and El-Rifai W: Loss of Tff1 promotes
pro-inflammatory phenotype with increase in the levels of RORγt+ T
lymphocytes and Il-17 in mouse gastric neoplasia. J Cancer.
8:2424–2435. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Soutto M, Chen Z, Katsha AM, Romero-Gallo
J, Krishna US, Piazuelo MB, Washington MK, Peek RM Jr, Belkhiri A
and El-Rifai WM: Trefoil factor 1 expression suppresses
Helicobacter pylori-induced inflammation in gastric
carcinogenesis. Cancer. 121:4348–4358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang Z, Xue H, Dong Y, Hu J, Jiang T, Shi
L and Du J: Inhibition of GKN2 attenuates acute gastric lesions
through the NLRP3 inflammasome. Adv Wound Care (New Rochelle).
9:219–232. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wilhelm A, Aldridge V, Haldar D, Naylor
AJ, Weston CJ, Hedegaard D, Garg A, Fear J, Reynolds GM, Croft AP,
et al: CD248/endosialin critically regulates hepatic stellate cell
proliferation during chronic liver injury via a PDGF-regulated
mechanism. Gut. 65:1175–1185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cabral MM, Oliveira CA, Mendes CM, Guerra
J, Queiroz DM, Rocha GA, Rocha AM and Nogueira AM: Gastric
epithelial cell proliferation and cagA status in Helicobacter
pylori gastritis at different gastric sites. Scand J
Gastroenterol. 42:545–554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Murata-Kamiya N, Kurashima Y, Teishikata
Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM
Jr, Azuma T and Hatakeyama M: Helicobacter pylori CagA
interacts with E-cadherin and deregulates the beta-catenin signal
that promotes intestinal transdifferentiation in gastric epithelial
cells. Oncogene. 26:4617–4626. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kang D, Gong Y, Zhu Y, Li A, Dong N, Piao
Y and Yuan Y: The biological activity of H. pylori SlyD in
vitro. Helicobacter. 18:347–355. 2013. View Article : Google Scholar : PubMed/NCBI
|