Introduction
One of the most difficult challenges in developing
anticancer treatment strategies is overcoming the phenotypic
plasticity of cancer cells. Oxygen deprivation (also known as
hypoxia) to a level lower than the physiological concentration is
frequently accompanied by the rapid proliferation of cancer cells
in vivo, and functions as a strong extracellular factor to
stimulate cancer cell plasticity (1). In hypoxia, cancer cells induce various
response pathways. To provide the energy necessary for cellular
activities when the oxygen supply is scarce, cells upregulate
anaerobic glycolysis-associated pathways (2). Angiogenesis is also stimulated to
generate blood vessels for tumour growth (3). Furthermore, hypoxia enables the
induction of the epithelial-to-mesenchymal transition (EMT) and
subsequent cell motility, which reflect the phenotypical transition
of cancer cells to aggressive metastasis (1).
The reprogramming of gene expression and/or
signalling pathways is involved in hypoxia-induced changes in the
state of cancer cells. Hypoxia-inducible factor-1 (HIF-1) is a key
transcription factor involved in hypoxia-induced transcriptional
alteration (4). Accumulation of
HIF-1 in hypoxia alters the expression of numerous target genes,
which mediates phenotypical changes in cancer cells. Chromatin
alteration is also closely associated with hypoxia-induced
transcriptional reprogramming. Among various chromatin
modifications, histone lysine demethylation as a response to
hypoxia is of particular interest, since Jumonji C (JmjC)
domain-containing lysine demethylases (KDMs) require oxygen as a
substrate for catalysis. While a lack of oxygen attenuates the
activity of KDMs, the expression of multiple KDMs, some of which
are HIF-1 dependent, is upregulated in hypoxic cells (5). Increased cellular levels of KDMs may
compensate attenuated KDM catalytic activity. In addition, the
direct molecular function of KDMs in hypoxia-induced responses have
been reported: KDMs alter the chromatin structure of numerous
target genes (6). These findings
suggest that the mechanisms by which KDMs are involved in
hypoxia-induced responses are important to understand the
progression of aggressive forms of cancer.
Among the KDMs upregulated directly by HIF-1, KDM4C,
which demethylates histone H3 at lysine 9 (H3K9), functions as a
coactivator of HIF-1 and mediates breast cancer progression
(7). KDM3A, another H3K9
demethylase, also enhances HIF-1 transcriptional activity. In
endothelial cells, KDM3A associates with and demethylates H3K9 at
the SLC2A3 locus in a HIF-dependent manner (8). KDM3A cooperates with HIF-1 to
upregulate the expression of androgen receptor target genes in
prostate cancer cells (9). KDM3A
accumulation is also required for the proliferation of different
cancer cell lines under hypoxic conditions (10). Furthermore, gene expression profiling
of mouse embryonic stem cells under hypoxia revealed that KDM3A
knockout affected the expression of numerous hypoxia-responsive
genes (11). These findings
collectively suggest that KDM3A is an important chromatin regulator
for hypoxia-induced transcriptional reprogramming to mediate
aggressive cancer progression. Accordingly, to gain insight into
developing a means to overcome cancer cell plasticity in hypoxic
tumour microenvironments, the present study investigated the
molecular mechanisms by which KDM3A functions in cancer cell
phenotypic transitions in hypoxia.
Materials and methods
Cell culture
The MCF7 cell line was purchased from the American
Type Culture Collection and was maintained in high-glucose
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% foetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) and 1X antibiotic-antimycotic
(Gibco; Thermo Fisher Scientific, Inc.). To provide hypoxic culture
conditions where oxygen levels were lower than physiological
normoxic levels (21% O2), cells were cultured in 1% (for
cell death, invasion, reporter assay and ChIP assay) or 5%
O2 (for cell proliferation assay) with 5% CO2
(MCO-5M; Sanyo Electric Co., Ltd.), while normoxic culture
conditions were maintained under 21% O2 with 5%
CO2 (Heraeus BB15 CO2 Incubator; Thermo
Fisher Scientific, Inc.).
Reagents, transfection and
antibodies
Lipofectamine® Plus (Invitrogen; Thermo
Fisher Scientific, Inc.) and RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) reagents were employed for DNA plasmid and small
interfering (si)RNA transfection, respectively. siRNA against KDM3A
(si-KDM3A; 5′-GUCUAUGUGGGAAUUCCCA-3′) and a negative control
(si-CTL; 5′-CCUACGCCACCAAUUUCGU-3′) were purchased from Bioneer
Corporation. The primary and secondary antibodies used in the
present study were as follows: antiKDM3A (Santa Cruz Biotechnology,
Inc.; sc-376608; 1:1,000), anti-E-cadherin (Santa Cruz
Biotechnology, Inc.; sc-21791; 1:1,000), anti-Slug (Santa Cruz
Biotechnology, Inc.; sc-1666477; 1:1,000), α-Snail (Santa Cruz
Biotechnology, Inc.; sc-271977; 1:1,000), anti-Twist (Santa Cruz
Biotechnology, Inc.; sc-81417; 1:1,000), anti-β-actin (Santa Cruz
Biotechnology, Inc.; sc-47778; 1:2,000), anti-Flag (Sigma-Aldrich;
Merck KGaA; F3165; 1:1,000), anti-IgG chromatin immunoprecipitation
(ChIP) grade (Abcam; ab2410; 1 µg/ml) and horseradish
peroxidase-conjugated anti-rabbit IgG (EMD Millipore; AP132P;
1:5,000). A plasmid construct encoding Flag-KDM3A derived from
pcDNA3.1 was provided by Dr Hyun-Soo Cho (Korea Research Institute
of Bioscience and Biotechnology). SNAI2 promoter activity reporter
constructs were constructed by subcloning DNA fragments
encompassing the transcriptional start site (TSS) of the SNAI2 gene
into the Dual-Luciferase Reporter Assay System (Promega
Corporation). The primers used for subcloning were as follows:
Reporter (Rep)-A-forward (F), 5′-AAGGATACCGTTCCAAATGACAGTTAC-3′;
Rep-B-F, 5′-AAGGATACCCGCCTTTGTCTTCCCGCTTCC-3′; and Rep-R,
5′-TTCTCGAGTTTTTGGAGGCGTTGAAATG-3′. To generate different 5′
regions of reporter constructs with the shared 3′ region, a common
Rep-R primer was used for the subcloning of both Rep-A and Rep-B
constructs.
Cell proliferation assay
Proliferation of cells seeded at the same number
(1×105/well) in 6-well plates under normoxic or hypoxic
conditions was examined by counting the cell numbers 0, 2 and 4
days after seeding. The number of viable cells with an automated
cell counter (Invitrogen™ Countess™ II FL Automated Cell Counter;
Thermo Fisher Scientific, Inc.).
Apoptotic cell death measurement
Apoptotic cell death was measured using an Apo-ONE™
Homogeneous Caspase-3/7 Assay kit (Promega Corporation). Assays
were performed according to the manufacturer's protocols. Cells
transfected with 0.1 µM si-CTL or si-KDM3A were cultured in
normoxic conditions (21% O2) for 48 h and seeded in
96-well plates (5×103cells/well), followed by incubation
in normoxic (21% O2) or hypoxic conditions (1%
O2) for 24 h. The fluorescence signal (485-nm excitation
wavelength/527-nm emission wavelength) generated from cleaved
proluminescent caspase-3/7 DEVD-aminoluciferin substrate by active
caspase-3/7 in apoptotic cells was recorded with a fluorometer
(BioTek Instruments, Inc.).
Reverse transcription (RT)-PCR
Total RNA was isolated using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and assessed
with an ND-1000 spectrophotometer (NanoDrop Technologies; Thermo
Fisher Scientific, Inc.). First-strand cDNA synthesis was produced
with RevertAid™ M-MuLV Reverse Transcriptase (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. RT-PCR
was performed with AccuPower PCR Master Mix (Bioneer Corporation)
and a GeneAmp PCR System 970 (Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: Pre-denaturation at 94°C
for 5 min, followed by 20–25 cycles of denaturation at 94°C for 20
sec, annealing at 55–60°C for 20 sec and extension at 72°C for 20
sec, with a final extension at 72°C for 5 min. The PCR products
were electrophoresed on 1%-agarose gels and stained with RedSafe™
(Intron, Inc.). PCR products were visualized with GelDoc™ XR+
(Bio-Rad Laboratories, Inc.) and ImageLab™ v6.0.1 software (Bio-Rad
Laboratories, Inc.) and semi-quantified by measuring densitometry
using ImageJ (National Institutes of Health). The primers used for
RT-PCR were as follows: KDM3A-F, 5′-CCAGCCTCAAAGGAAGACCT-3′;
KDM3A-R, 5′-ACTGCACCAAGAGTCGGTTT-3′; E-cadherin (CDH1)-F,
5′-GCTTTGACGCCGAGAGCTACACGTT-3′; CDH1-R,
5′-AGGAGTTGGGAAATGTGAGCAATTC-3′; β-actin (ACTB)-F,
5′-GGAGTCCTGTGGCATCCACG-3′; and ACTB-R,
5′-CTAGAAGCATTTGCGGTGGA-3′.
Luciferase assay
Luciferase assay was performed using a
Dual-Luciferase Reporter Assay System (Promega Corporation)
according to the manufacturer's instructions. Cells
(7.5×104/well) cultured in 12-well plates for 24 h were
co-transfected with luciferase reporter (0.2 µg), Renilla
reporter (0.05 µg) and si-CTL or KDM3A (100 nM). At 48 h
post-transfection, the cells were exposed to hypoxic conditions for
an additional 24 h. Luciferase activities were determined using a
luminometer (Luminoskan Ascent; Thermo Fisher Scientific, Inc.).
Data represent the signals normalized to Renilla luciferase
activity.
Immunoblot analysis
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH
7.5, 150 mM NaCl, 10 mM EDTA and 1% Triton X-100) with protease
inhibitor cocktail (Roche Diagnostics) or 2X sample buffer (120 mM
Tris-HCl pH 6.8, 20% glycerol, 4% SDS and 5% β-mercaptoethanol).
Proteins extracted with RIPA were quantified with the Bradford
assay and equal amounts (30 µg) of proteins were used for each
sample. For resolving by SDS-PAGE, 5X SDS sample buffer [250 mM
Tris-HCl (pH 6.8), 10% SDS, 50% glycerol, 0.5 M dithiothreitol,
0.25% bromophenol] was added to cell lysates and boiled at 100°C
for 10 min. After SDS-PAGE, the proteins were transferred to
nitrocellulose membranes and the membranes were blocked at room
temperature for 1 h using 5% skimmed milk in TBST (0.1% Tween-20)
buffer. Primary antibodies were added and incubated at 4°C for 16
h, and then HRP-conjugated secondary antibody, which was incubated
with the membranes at room temperature for 1 h, was used for
detecting the chemiluminescent signal (Pierce™ ECL Western Blotting
Substrate; Thermo Fisher Scientific, Inc.) from specific target
proteins. The immunoblots were developed with LAS-4000 (Luminescent
Image Analyzer; Fujifilm Wako Pure Chemical Corporation).
Invasion assays
The outsides of Transwell inserts were pre-coated
with human collagen-type IV at room temperature for 1 h. For the
invasion assays, the upper chambers of the Transwell inserts were
coated with 250 µg/ml Matrigel (Becton-Dickinson and Company) at
room temperature for 1 h. MCF7 cells were transfected with 0.1 µM
si-CTL or si-KDM3A. At 24 h post-transfection, the cells were
incubated under either hypoxic (1% O2) or normoxic (21%
O2) conditions. After 24 h of incubation at different
oxygen concentrations, the cells were harvested with 0.025%
trypsin-EDTA and transferred to Transwell inserts (1×105
cells/chamber) in 6-well plates. After 24 h of incubation in
normoxic conditions (21% O2), the inserts were fixed
with 100% methanol and stained with 0.5% crystal violet. The
invaded cells were counted manually under a light microscope (×200
magnifications; 5 fields per replicate).
ChIP assay
ChIP assay was performed using a ChIP assay kit
following the manufacturer's protocol (Upstate Biotechnology, Inc).
Cells (2×106) were preincubated with a dimethyl
3,3′-dithiobispropionimidate-HCl solution (5 mM; Pierce; Thermo
Fisher Scientific, Inc.) for 30 min on ice and then treated with 1%
formaldehyde at room temperature for 15 min. ChIP-enriched DNA
samples were visualized by PCR and electrophoresed on a
1.5%-agarose gel stained with RedSafe™ (Intron, Inc.). Input DNA
(1%) used for ChIP with individual antibody served as the control.
The thermocycling conditions were as follows: Pre-denaturation at
94°C for 5 min, followed by 20 cycles of denaturation at 94°C for
20 sec, annealing at 55–60°C for 20 sec and extension at 72°C for
15 sec, with a final extension at 72°C for 5 min. Densitometry of
visualized DNA was semi-quantitatively measured by ImageJ (National
Institutes of Health). The primer pairs used for the ChIP assays,
which were specific to regions within the SNAI2 promoter and the
downstream adjacent DNA element (from −1,500 to +1,000) separated
by a ~200-bp interval, as labelled in Fig. 4A, were as follows: A-F,
5′-ACCTGACAATGCATTTTCTCT-3′; A-R, 5′-TCACATGAAGATCACCCTACTCT-3′;
d-F, 5′-GGGTTCTTAAAATTTAATCAATCTA-3′; d-R,
5′-AAAGCCATTAAAATCCATCTCA-3′; e-F, 5′-GTGAGAGAATGTCCGGTCCT-3′; e-R,
5′-GGCTGATCGGAAGAACTGGAA-3′; f-F, 5′-GCGGAAGCCCTGAGTAGC-3′; f-R,
5′-TTGAAAAAGGAAGGGGGAAGCGGGAAG-3′; B-F, 5′-CCTTCTCCTGGCGGACACT-3′;
B-R, 5′-ACGCTCTCTGGGAGCTAGG-3′; g-F, 5′-CTGTTCACAGCTGTCCCAGAG-3′;
g-R, 5′-TTACGAACTGAGCCCGTTTT-3′; h-F, 5′-AGACCCGCTGGCAAGATG-3′;
h-R, 5′-TCTTTTTACCTGTATGTGTCCA-3′; i-F, 5′-TTCGTTTGCTAAAGCCTGT-3′;
i-R, 5′-GCCGCCTTCTAAAGGA-3′; C-F, 5′- AGCCCATACGTAACATGGAAAA-3′;
C-R, 5′-TGCACGGAGCTATAGGTGCTC-3′; j-F, 5′-CCATGCCTGTCATACCACA-3′;
and j-R, 5′-GAGAGGCCATTGGGTAGCTG-3′.
Statistical analysis
All data are presented as means of three independent
experiments ± standard deviation. GraphPad Prism 7 software
(GraphPad Software, Inc.) was used for statistical analyses.
Student's t-tests (two-tailed, unpaired) were used to determine
significant differences between si-CTL versus si-KDM3A and Mock
versus Flag-KDM3A in Fig. 1. In
Fig. 1, data from normoxia (21%
O2) or hypoxia (1% O2) groups were compared
independently. Differences in KDM3A ChIP signals between normoxia
(21% O2) and hypoxia (1% O2) in Fig. 4C was also analysed with Student's
t-test (two-tailed, unpaired). Data in Figs. 3B, C and 4E were analysed with one-way ANOVA followed
by Tukey's post hoc test to determine significant differences.
P<0.05 was considered to indicate a statistically significant
difference.
Results
KDM3A is involved in breast cancer
cell invasion in hypoxia
To examine the role of KDM3A in aggressive cancer
cell behaviour in the hypoxic microenvironment, the involvement of
KDM3A in hypoxia-induced cancer cell phenotypes was determined in
the present study. Given the prominent clinical association between
hypoxia-induced response and prognosis of patients with breast
cancer (12), the function of KDM3A
in MCF7 breast cancer cells was examined. First, the requirement of
KDM3A for cell proliferation in hypoxia conditions was observed. It
was notable that MCF7 cells incubated in 1% O2 for
longer than 24 h showed a higher death rate. Thus, cellular
proliferation was determined in milder hypoxic conditions, 5%
O2, rather than 1% O2. Under hypoxic
conditions (5% O2), MCF7 cells proliferated at a ~5-fold
decreased rate compared with that of MCF7 cells cultured in
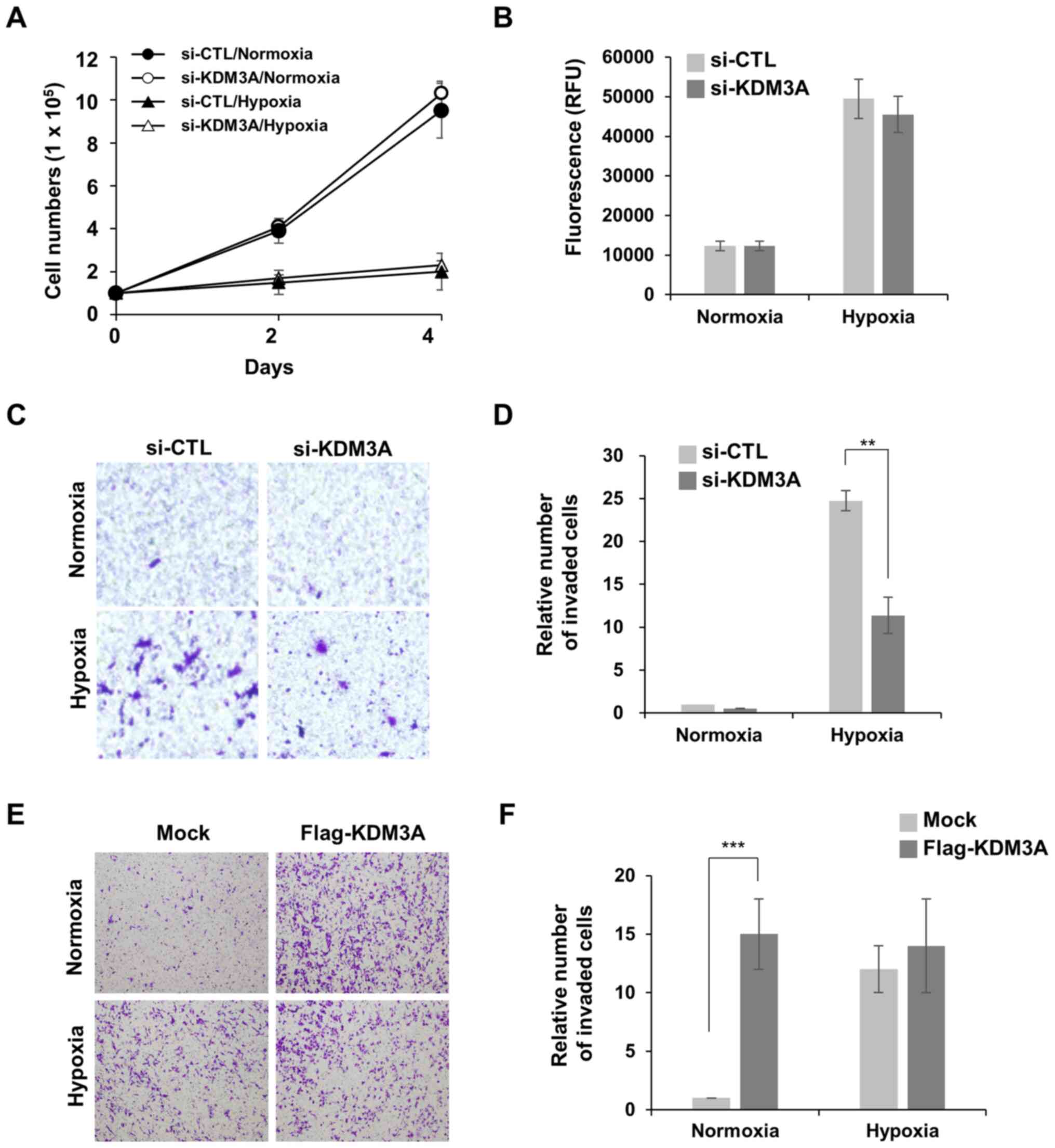
normoxic conditions (21% O2; Fig. 1A). KDM3A depletion did not further
aggravate MCF7 cell proliferation in hypoxic conditions. Notably,
KDM3A knockdown minimally affected MCF7 cell proliferation in
normoxic conditions. In addition, the loss of KDM3A did not trigger
cell death in normoxic or hypoxic conditions (1% O2),
which was determined by the fluorescent signals generated from
cleaved substrate by active caspases in apoptotic cells (Fig. 1B). These results indicate that KDM3A
is dispensable for MCF7 cell survival and proliferation under
hypoxic stress conditions.
Next, the present study investigated whether KDM3A
is associated with hypoxia-induced cell motility, another
hypoxia-responsive cell phenotype that promotes the spread of
aggressive cancer cells. MCF7 cells poorly invaded through
Matrigel-coated Transwell chambers in normoxic conditions, whereas
preincubation in hypoxia (1% O2) for 24 h induced cell
invasion (Fig. 1C and D). Notably,
KDM3A depletion decreased hypoxia-induced invasion of MCF7 cells by
>2-fold compared with that of si-CTL cells. This finding implies
that KDM3A is involved in hypoxia-induced invasive cell motility.
To assess whether the upregulation of KDM3A expression in hypoxia
is sufficient to induce cancer cell invasion, and whether other
hypoxia-induced signalling pathways are necessary, the effect of
KDM3A overexpression on MCF7 cell invasion in normoxic conditions
was examined (Fig. 1E and F). The
invasive capacity of cells transfected with ectopic Flag-KDM3A
increased by ~15-fold compared with that of Mock-transfected cells,
even in normoxia. This finding supports the notion that the
enhanced expression of endogenous KDM3A in hypoxic conditions is
sufficient to promote hypoxia-induced breast cancer cell invasion.
It is notable that ectopic expression of KDM3A had no effect on
MCF7 cell invasion in hypoxia. It can be speculated that the robust
increase in endogenous KDM3A expression in hypoxia (Fig. 2) may be sufficient to induce cell
invasion to the level that is not further enhanced by additional
exogenous KDM3A.
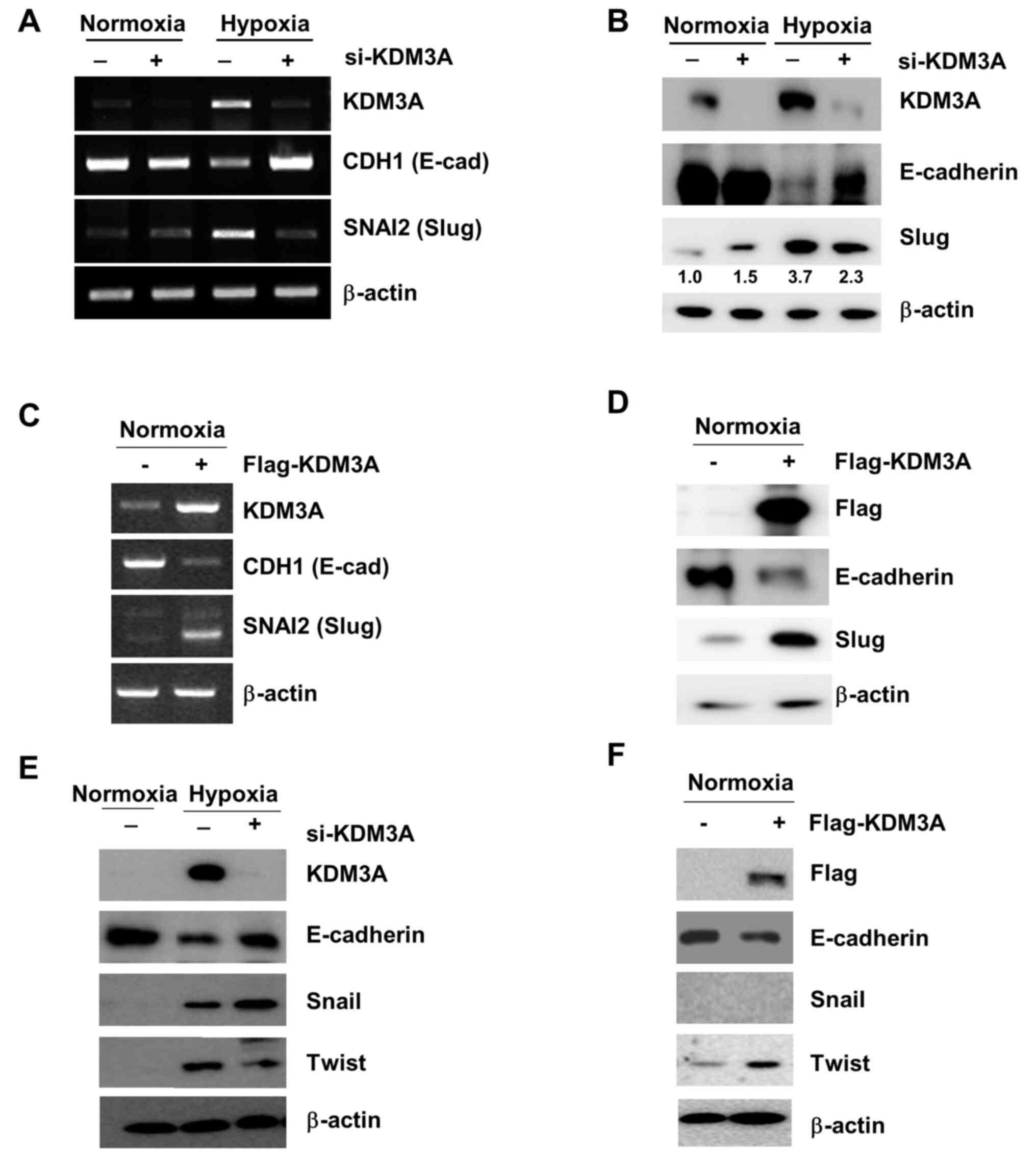 | Figure 2.Elevated KDM3A expression in hypoxia
upregulates Slug, which suppresses CDH1 expression. (A and B) KDM3A
knockdown impairs CDH1 (E-cadherin) downregulation as well as SNAI2
(Slug) upregulation in hypoxia. MCF7 cells transfected with either
si-CTL (−) or si-KDM3A (+) were cultured for 48 h in normoxia.
Next, the cells were divided for culture under normoxic (21%
O2) or hypoxic conditions (1% O2) for 24 h.
The expression of mRNA specific to each target gene, as determined
by RT-PCR, is displayed in panel A, while the protein expression
levels measured by IB using specific antibodies against the
proteins indicated in the figure are shown in panel B. (C and D)
KDM3A overexpression decreases CDH1 expression while increasing
SNAI2 expression in normoxia. MCF7 cells transfected with either
empty vector (−) or Flag-tagged KDM3A-encoding vector (+) were
cultured for 48 h, and CDH1 and SNAI2 expression at the (C) mRNA
and (D) protein level was examined using RT-PCR and IB,
respectively. (E) KDM3A knockdown in MCF7 cells decreases Twist
upregulation in hypoxia while increasing the hypoxic induction of
Snail expression. (F) KDM3A overexpression increases Twist
expression in MCF7 cells cultured under normoxic conditions. The
experiments were performed as described in (D). si, small
interfering; RT-PCR, reverse transcription-PCR; KDM, lysine
demethylase; IB, immunoblotting. |
KDM3A promotes hypoxia-induced Slug
expression
Transcriptional alteration is critical for cancer
cells to switch phenotypic states in response to microenvironmental
conditions. It was hypothesised that KDM3A-mediated chromatin
modification is involved in regulating the transcriptional
expression of key players promoting cancer cell invasion.
E-cadherin, which is encoded by CDH1, is a well-known epithelial
cell marker. CDH1 downregulation is a characteristic of EMT, which
induces cell motility (13). Under
hypoxic conditions (1% O2 for 24 h), CDH1 mRNA
expression in MCF7 cells was largely decreased compared with that
in cells cultured under normoxic conditions (Fig. 2A). Compromised CDH1 expression in
hypoxic conditions was also observed at the protein level (Fig. 2B). By contrast, KDM3A depletion
restored CDH1 expression under hypoxic conditions to levels
comparable to those observed in control cells under normoxic
conditions (Fig. 2A and B). This
finding implies that KDM3A is necessary for hypoxia-induced
downregulation of E-cadherin, which is associated with the
induction of cell invasion.
Given that KDM3A removes H3K9 methylation, which
represses transcription, KDM3A is expected to function as a
transcriptional activator. However, enhanced CDH1 expression in
KDM3A-depleted hypoxic cells suggests that KDM3A negatively
regulates CDH1 expression. To explain this discordant observation,
it was hypothesised that decreased CDH1 expression in hypoxia may
be a consequence of KDM3A-mediated activation of a transcription
factor involved in CDH1 expression. Multiple transcription factors
are involved in EMT-associated changes in CDH1 expression (14). Among them, Slug, which directly
represses CDH1 expression, is induced in hypoxia (15). The present study assessed whether
Slug expression was affected by the loss of KDM3A. Consistent with
a previous report (15), hypoxic
stress (1% O2 for 24 h) increased Slug expression in
MCF7 cells compared with that of control cells. By contrast, KDM3A
knockdown led to a prominent decrease in Slug expression levels
under hypoxic conditions at both the mRNA and protein levels
(Fig. 2A and B). This result
suggests that KDM3A upregulates the transcriptional expression of
Slug. Consistent with this finding, ectopic overexpression of KDM3A
in MCF7 cells increased the expression of Slug, even under normoxic
conditions (Fig. 2C and D). In
addition, the expression of CDH1 (E-cadherin) at both the mRNA and
protein levels was largely decreased by KDM3A overexpression
(Fig. 2C and D). Together, these
results support that enhanced KDM3A expression upregulates the
transcription of Slug, subsequently downregulating CDH1 expression,
which is associated with EMT-associated cell invasion. Furthermore,
KDM3A depletion decreased the hypoxia-induced expression of Twist,
another EMT-associated transcription factor that represses
E-cadherin expression (Fig. 2E).
Consistently, the ectopic overexpression of KDM3A increased Twist
expression under normoxic conditions (Fig. 2F). By contrast, KDM3A knockdown
increased hypoxia-induced Snail expression (Fig. 2E). In normoxic cells transfected with
KDM3A-overexpressing constructs, Snail protein expression was not
detected (Fig. 2E and F). It is
likely that an increase in KDM3A alone is not sufficient to induce
Snail expression in normoxia but requires additional factor(s)
activated in hypoxia. It was not determined as to whether
KDM3A-knockdown affected Snail expression in normoxia, as Snail
expression appeared not to be associated with KDM3A-dependent
E-cadherin repression. To summarise, these results imply that KDM3A
mediates the upregulation of Twist in addition to Slug, both of
which are involved in the repression of E-cadherin expression.
KDM3A positively regulates the
transcriptional activity of the SNAI2 promoter
To understand the molecular mechanism by which KDM3A
upregulates the transcription of Slug, the effect of KDM3A
depletion on the promoter of the SNAI2 gene, which encodes Slug,
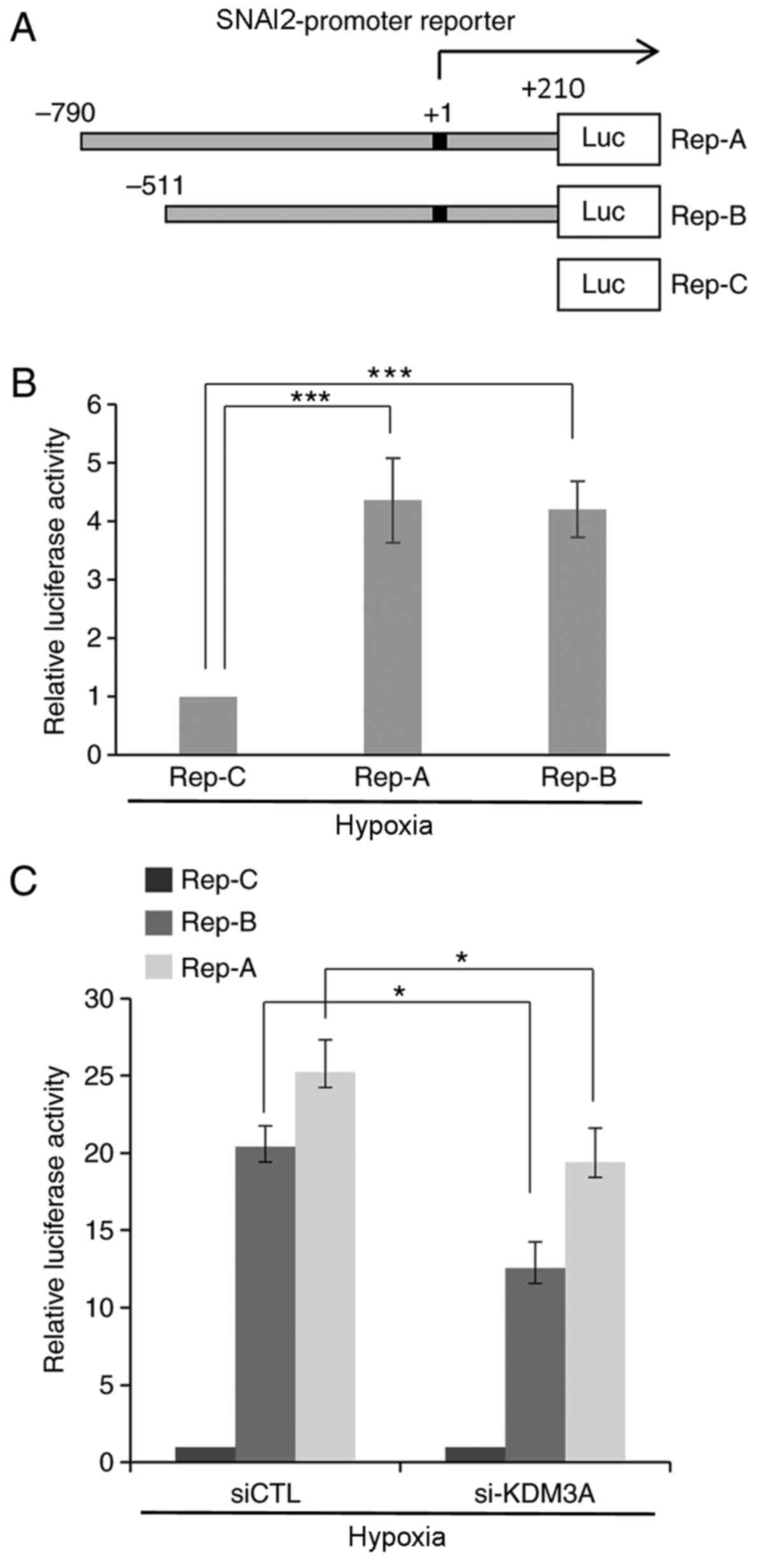
was examined. First, a reporter system carrying an ~1-kb fragment
encompassing 790 bp upstream to 210 bp downstream of the SNAI2 TSS
(+1) fused to the luciferase coding region (Rep-A) was constructed
(Fig. 3A). To further define the
promoter of SNAI2, an additional reporter system lacking the DNA
region spanning −790 bp to −511 bp from the TSS (Rep-B) was
generated. A reporter containing only the luciferase gene without a
promoter element was used as a negative control (Rep-C). MCF7 cells
transfected with each reporter construct were subjected to
luciferase assays to assess SNAI2 promoter activity through the
expression of luciferase. Cells transfected with both the Rep-A and
Rep-B reporters expressed prominent levels of luciferase under
hypoxic conditions (1% O2), as detected by luminescence
(Fig. 3B). This finding indicates
that the cloned DNA fragments 5′ upstream of the SNAI2 TSS function
as competent promoter elements under hypoxic conditions. Next,
KDM3A-specific siRNA along with reporter constructs were
simultaneously introduced into MCF7 cells, and reporter activity
was measured under the same hypoxic conditions using a quantitative
luminescence assay (Fig. 3C).
Compared with their activities in si-CTL-transfected cells, cells
transfected with si-KDM3A exhibited a statistically significant
decrease in the reporter activities of both Rep-A and Rep-B. This
finding demonstrates that KDM3A depletion leads to a defect in the
transcriptional activity of the SNAI2 promoter under hypoxic
conditions.
Dimethylation of histone H3 at lysine
9 (H3K9me2) at the SNAI2 promoter is regulated by the recruitment
of KDM3A
Next, to determine whether KDM3A functions as a
histone demethylase to alter the chromatin structure of the SNAI2
promoter, whether histone methylation at the SNAI2 promoter region
was altered by the loss of KDM3A was examined. In total, 10 primer
pairs specific to regions within the SNAI2 promoter and the
downstream adjacent DNA element (from −1,500 to +1,000) separated
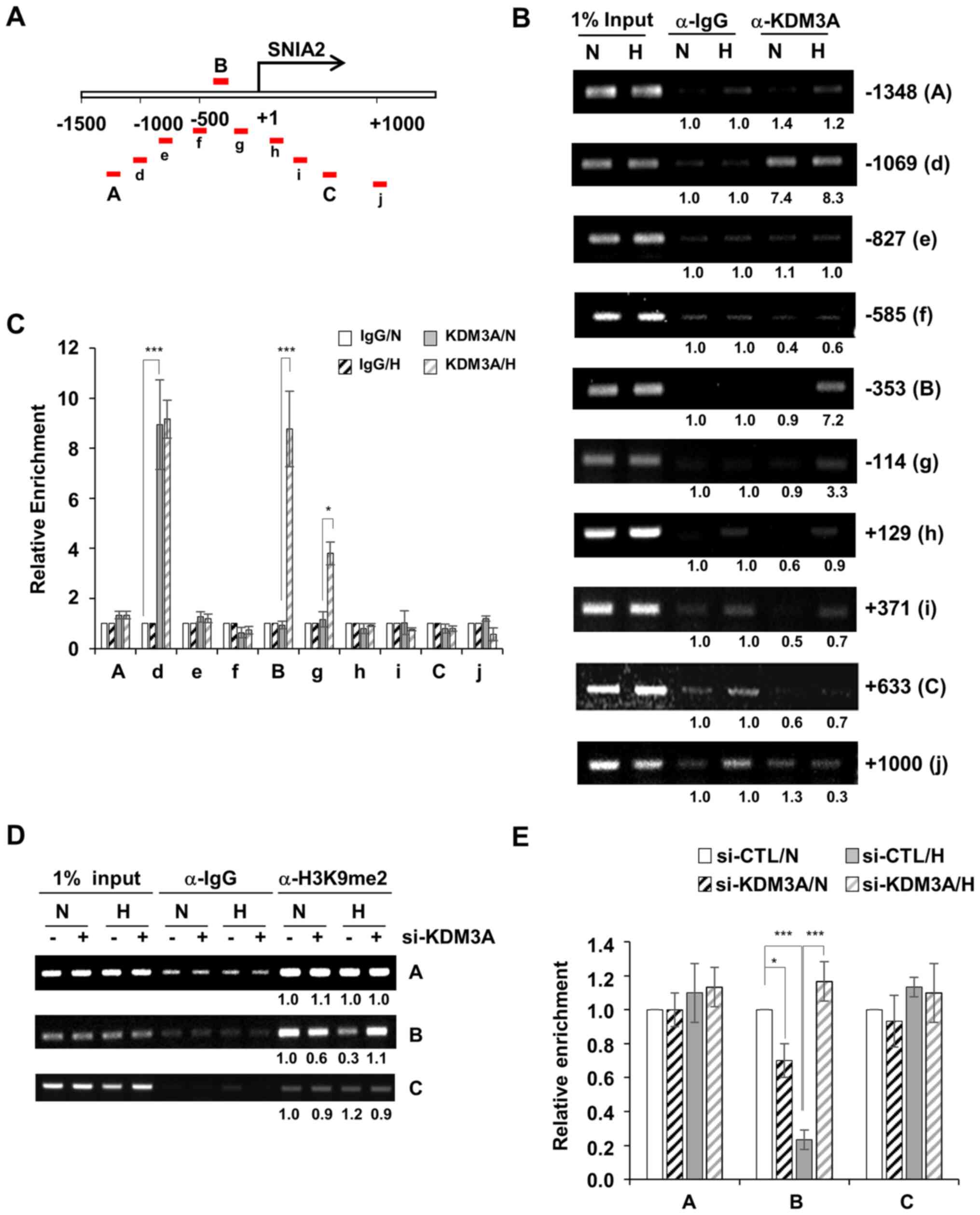
by a ~200-bp interval were designed (Fig. 4A). First, to determine the
recruitment of KDM3A to the SNAI2 promoter region, MCF7 cells
cultured under normoxic or hypoxic conditions (1% O2)
for 24 h were subjected ChIP using an anti-KDM3A-specific antibody,
followed by PCR-mediated amplification of the antibody-associated
DNA regions. As a non-specific enrichment control, anti-IgG was
used as a control antibody. The KDM3A-specific ChIP signals were
semi-quantitatively measured by normalization to the IgG ChIP
signals. The enrichment of KDM3A was observed in three regions,
namely regions d, B and g, which are located approximately −1,100,
−350 and −110 bp away from the TSS (+1), respectively. In region d,
KDM3A accumulation was detected under both normoxic and hypoxic
conditions (Fig. 4B and C). By
contrast, KDM3A enrichment at regions B and g was specific to
hypoxia. All other regions (A, e, f, h, j, C and j) exhibited
minimal KDM3A ChIP signalling; implying that, while KDM3A
associates with the SNAI2 promoter region ~1,000 bp upstream of the
TSS regardless of the oxygen concentration, additional KDM3A is
recruited specifically to the SNAI2 promoter region spanning ~350
bp to 110 bp upstream of the TSS (encompassing region B and g)
under hypoxic conditions.
Next, chromatin modification at the SNAI2 promoter
in MCF7 cells was assessed by ChIP against H3K9me2. In hypoxia,
H3K9me2 levels at region B, where hypoxia-induced KDM3A
accumulation was detected (Fig. 4B and
C), were largely decreased compared with the levels in the
other KDM3A-accumulated regions (region A and C; Fig. 4D and E). By contrast, when KDM3A was
depleted with siRNA, H3K9me2 levels at region B were not decreased
in hypoxic conditions compared with that in normoxic conditions.
Therefore, H3K9me2 at region B of the SNAI2 promoter is likely to
be demethylated by KDM3A, which is specifically recruited under
hypoxic conditions (Fig. 4B and C).
Thus, this result suggests H3K9me2-mediated transcriptional
repression of SNAI2 is relieved under low-oxygen conditions. KDM3A
depletion mediated a decrease in H3K9me2 at region B under normoxic
conditions (Fig. 4D and E). It is
possible that the functional involvement of KDM3A in demethylating
the SNAI2 promoter may be distinctive between normoxic and hypoxic
conditions, which was not further characterized in the present
study.
Discussion
The phenotypic change of cancer cells in the hypoxic
tumour microenvironment promotes aggressive cancer progression
(1). Understanding the molecular
mechanisms underlying hypoxia-mediated cancer cell state transition
will pave the way to develop treatment strategies. Epigenetic
modifications to mediate transcriptional alteration are crucial for
cellular state transitions (16).
Accordingly, attempts have been made to characterize the functional
implication of epigenetic modifications, including lysine
demethylase-mediated chromatin alterations in hypoxia-mediated
transcriptional modulation (17). In
this context, the present study demonstrated the functional
involvement of KDM3A in the hypoxia-induced metastatic potential of
breast cancer cells through increased invasion. Mechanistically,
KDM3A, the expression of which is increased by HIF-1 under hypoxic
conditions, is recruited to remove the transcriptionally repressive
histone modification marker H3K9me2 from the promoter of SNAI2,
which encodes Slug, an EMT transcription factor. Consequently, the
increased expression of Slug decreases the expression of
CDH1-encoded E-cadherin, which is a distinctive characteristic of
EMT that confers cancer cell metastatic potential. These results
collectively suggest that KDM3A is a transcriptional coactivator of
Slug expression that induces cancer cell invasion in hypoxia. In
addition, KDM3A is involved in the induction of Twist expression in
hypoxia, which also promotes invasive cancer cell motility. Of
note, the changes in Slug or Twist expression in the cells with or
without KDM3A knockdown were modest compared with those in
E-cadherin expression. It is likely that the dysregulation of
E-cadherin expression in cells with KDM3A depletion is
cooperatively mediated by a simultaneous decrease in Slug and Twist
expression following KDM3A knockdown. Future studies may reveal
whether KDM3A would be also recruited to the Twist promoter in
hypoxia to regulate its chromatin structure.
Consistent with the results of the present study,
the transcriptional coactivator activity of KDM3A has previously
been observed in rat trophoblast stem cell differentiation under
hypoxic conditions by Chakraborty et al (18). By systemic analyses of the
upregulated genes in rats exposed to low oxygen, the study found
that matrix metallopeptidase 12 (MMP12) is a crucial gene
associated with hypoxia-induced cell invasion. In addition, the
study showed that, in hypoxia, KDM3A-mediated H3K9 demethylation is
required for the expression of MMP12, which is not a direct target
of HIF-1 (18). This finding implies
that KDM3A, the expression of which is upregulated by HIF-1,
functions as a transcriptional coactivator to facilitate the
expression of genes involved in hypoxia-induced cellular state
transitions such as MMP12. Additionally, in urinary bladder cancer,
KDM3A cooperates with HIF-1 to enhance glycolysis and thereby
promote cancer cell proliferation (19). By demonstrating that the
catalytically inactive KDM3A mutant failed to act as a
transcriptional coactivator of HIF-1, the bladder cancer study
suggests the importance of KDM3A-mediated chromatin modification in
the transcriptional regulation associated with aggressive cancer
progression in the hypoxic tumour microenvironment. The
KDM3A-mediated decrease in H3K9me2 at the SNAI2 promoter in hypoxic
breast cancer cells observed in the present study also strongly
supports the involvement of KDM3A-dependent chromatin modifications
in hypoxic cancer progression.
The involvement of KDM3A in aggressive cancer cell
phenotypes was also reported in an aggressive form of breast cancer
in a previous study (20). In
contrast to the results of the present study demonstrating the
necessity of KDM3A in the induction of cellular invasion under
hypoxic conditions, the aforementioned study showed that, in the
metastatic breast cancer cell lines MDA-MB-231 and Hs578T, KDM3A
knockdown inhibited cell invasion under normoxic conditions by
demethylating H3K9 at pro-invasive genes such as MMP9 and S100A4.
This result, along with our finding that KDM3A is required for the
hypoxic induction of breast cancer cell invasion likely through
promoting the expression of Slug, an EMT-associated transcription
factor, suggests that KDM3A not only establishes but also maintains
the metastatic potential of breast cancer cells. Notably, while
Wade et al (21) reported the
requirement of KDM3A for estrogen receptor signalling-dependent
proliferation of MCF7 breast cancer cells in
oestradiol-supplemented medium, the present study failed to observe
the detrimental effect of KDM3A on MCF7 cell survival or
proliferation in high-glucose DMEM (Fig.
1A and B). It can be speculated that the requirement of KDM3A
for MCF7 breast cancer cell survival and proliferation may be
largely dependent on growth conditions The mechanism by which KDM3A
is recruited to distinctive target genes in different oxygen
concentrations such as MDA-MB-231 in normoxia vs. MCF7 in hypoxia,
remains unknown. Future studies to identify KDM3A-interacting
transcription factors that promote cancer cell invasion will reveal
detailed information about hypoxia-induced transcriptional
alterations associated with the transition of cancer cells to a
more aggressive state.
Acknowledgements
The authors would like to thank Dr Hyun-Soo Cho
(Korea Research Institute of Bioscience and Biotechnology, Daejeon,
Republic of Korea) for providing the Flag-KDM3A construct.
Funding
The present study was supported by the National
Research Foundation of Korea funded by the Ministry of Science and
Information & Communication Technology (grant nos.
NRF-2013R1A1A1006638 and NRF-2019R1A2C1086151) and by the KRIBB
Research Initiative Program.
Authors' contributions
HJA and JAK conceived and designed the study. HJA,
BM, and MP performed the experiments and analyzed the data. JAK
wrote the manuscript. All authors read and approved the final
manuscript and agreed to be accountable for all aspects of the
research.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rankin EB, Nam JM and Giaccia AJ: Hypoxia:
Signaling the Metastatic Cascade. Trends Cancer. 2:295–304. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eales KL, Hollinshead KE and Tennant DA:
Hypoxia and metabolic adaptation of cancer cells. Oncogenesis.
5:e1902016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chouaib S, Messai Y, Couve S, Escudier B,
Hasmim M and Noman MZ: Hypoxia promotes tumor growth in linking
angiogenesis to immune escape. Front Immunol. 3:212012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xia X, Lemieux ME, Li W, Carroll JS, Brown
M, Liu XS and Kung AL: Integrative analysis of HIF binding and
transactivation reveals its role in maintaining histone methylation
homeostasis. Proc Natl Acad Sci USA. 106:4260–4265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hancock RL, Dunne K, Walport LJ, Flashman
E and Kawamura A: Epigenetic regulation by histone demethylases in
hypoxia. Epigenomics. 7:791–811. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luo W, Chang R, Zhong J, Pandey A and
Semenza GL: Histone demethylase JMJD2C is a coactivator for
hypoxia-inducible factor 1 that is required for breast cancer
progression. Proc Natl Acad Sci USA. 109:E3367–E3376. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mimura I, Nangaku M, Kanki Y, Tsutsumi S,
Inoue T, Kohro T, Yamamoto S, Fujita T, Shimamura T, Suehiro J, et
al: Dynamic change of chromatin conformation in response to hypoxia
enhances the expression of GLUT3 (SLC2A3) by cooperative
interaction of hypoxia-inducible factor 1 and KDM3A. Mol Cell Biol.
32:3018–3032. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee HY, Yang EG and Park H: Hypoxia
enhances the expression of prostate-specific antigen by modifying
the quantity and catalytic activity of Jumonji C domain-containing
histone demethylases. Carcinogenesis. 34:2706–2715. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park SJ, Kim JG, Son TG, Yi JM, Kim ND,
Yang K and Heo K: The histone demethylase JMJD1A regulates
adrenomedullin-mediated cell proliferation in hepatocellular
carcinoma under hypoxia. Biochem Biophys Res Commun. 434:722–727.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ueda J, Ho JC, Lee KL, Kitajima S, Yang H,
Sun W, Fukuhara N, Zaiden N, Chan SL, Tachibana M, et al: The
hypoxia-inducible epigenetic regulators Jmjd1a and G9a provide a
mechanistic link between angiogenesis and tumor growth. Mol Cell
Biol. 34:3702–3720. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Favaro E, Lord S, Harris AL and Buffa FM:
Gene expression and hypoxia in breast cancer. Genome Med. 3:552011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang J, Tang YL and Liang XH: EMT: A new
vision of hypoxia promoting cancer progression. Cancer Biol Ther.
11:714–723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flavahan WA, Gaskell E and Bernstein BE:
Epigenetic plasticity and the hallmarks of cancer. Science.
357:eaal23802017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perez-Perri JI, Acevedo JM and Wappner P:
Epigenetics: New questions on the response to hypoxia. Int J Mol
Sci. 12:4705–4721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chakraborty D, Cui W, Rosario GX, Scott
RL, Dhakal P, Renaud SJ, Tachibana M, Rumi MA, Mason CW, Krieg AJ,
et al: HIF-KDM3A-MMP12 regulatory circuit ensures trophoblast
plasticity and placental adaptations to hypoxia. Proc Natl Acad Sci
USA. 113:E7212–E7221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wan W, Peng K, Li M, Qin L, Tong Z, Yan J,
Shen B and Yu C: Histone demethylase JMJD1A promotes urinary
bladder cancer progression by enhancing glycolysis through
coactivation of hypoxia inducible factor 1α. Oncogene.
36:3868–3877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramadoss S, Guo G and Wang CY: Lysine
demethylase KDM3A regulates breast cancer cell invasion and
apoptosis by targeting histone and the non-histone protein p53.
Oncogene. 36:47–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wade MA, Jones D, Wilson L, Stockley J,
Coffey K, Robson CN and Gaughan L: The histone demethylase enzyme
KDM3A is a key estrogen receptor regulator in breast cancer.
Nucleic Acids Res. 43:196–207. 2015. View Article : Google Scholar : PubMed/NCBI
|