Introduction
Ovarian cancer is the eighth most commonly diagnosed
cancer in women after breast, colorectal, lung and uterine cancer
(1). Ovarian cancer is a disease has
poor survival and generally diagnosed at advanced stage. According
to the SEER database, the 5-year relative survival rate for
invasive epithelial ovarian cancer between 2009 and 2015 was 47%
(2). Ovaries consist of different
types of cells, including germ cells, specified gonadal stromal
cells and epithelial cells. Epithelial ovarian cancers constitute
the majority of ovarian cancers, and are responsible for the most
ovarian cancer-associated deaths (3,4). Early
detection of ovarian cancer is difficult due to the lack of ovarian
cancer-specific non-invasive molecular biomarkers. Early diagnosis
is highly important for treatment and survival in ovarian cancer
(5).
Genetic predisposition is known to have a role in
breast and gynecological cancer. The cancer susceptibility risk of
an individual is associated with genetic predisposition in addition
to factors such as reproduction history, the use of oral
contraceptives and hormone replacement, radiation exposure in the
early period of life, alcohol consumption and physical activity
(6). Among a number of risk
evaluation models, mutations in breast-ovarian cancer
syndrome-associated BRCA1 or BRCA2 genes, and
mutations in Lynch II syndrome-associated DNA repair genes have
been identified as risk factors (7–10).
Mutations in BRCA1 and BRCA2 genes and mutations in
mismatch repair genes (Lynch syndrome) are among the most common
causes of hereditary ovarian cancer syndromes (11). DNA methylation is an epigenetic
mechanism that is important in the regulation of gene expression.
Epigenetic changes that affect gene expression without causing a
structural alteration in the DNA sequence have been shown to play a
role in cancer development (12).
Monozygotic (MZ) twins with ~100% identical genetic structure are
known to be good research models for identifying the association
between environmental factors and epigenetic changes in the
occurrence of diseases (13). MZ
twins share the same genotype, but their phenotypic features may
differ. Discordance has been detected in some multifactorial
diseases in MZ twin siblings (14,15). The
mechanism underlying this discordance between MZ twins has been
suggested to involve epigenetic modifications (16).
Various studies have been conducted using twins to
investigate the links between complex diseases and genetic
structure, and the effects of environmental factors on those
associations. In the first large-scale study on DNA methylation in
twins, 20 MZ and 20 dizygotic twin couples were compared. Similar
epigenetic profiles and high epigenetic inheritance were observed
in the MZ twins in the study; however, epigenetic variation was
found to increase with advanced age (17). High-resolution DNA methylation
analyses have detected tissue-specific variations and characterized
the epigenetic meta-stability of ~6,000 unique genomic regions in
MZ twins (18,19). Phenotypic differences have been shown
to develop via epigenetic mechanisms in MZ twin siblings with the
same genotype (13).
In the present study, differences in methylation
were investigated in the whole genome of MZ twins with a pathogenic
BRCA1 mutation, one of whom was healthy while the other was
diagnosed with ovarian cancer. The genomic methylation levels of
the twins were compared with those of their three
BRCA1-mutated sisters and one healthy brother. The findings
suggest that epigenetic differences based on methylation status
could be used as non-invasive biological markers for the early
diagnosis and follow-up of ovarian cancer.
Materials and methods
BRCA1 and BRCA2 mutation
screening
Six siblings who presented to the cancer genetics
clinic at the Institute of Oncology, Istanbul University in 2012
for BRCA1 and BRCA2 mutation testing were included in
the study. Each individual signed an informed consent form. The
study was approved by the Ethics Board of Istanbul University
(approval no. 1552, dated May 18, 2015) in accordance with The
Declaration of Helsinki (20).
DNA samples isolated from the peripheral blood
lymphocytes of the six siblings were used in the study. Genomic DNA
was isolated with a QIAamp DNA Mini QIAcube kit (cat. no./ID:
51326) using the QIAcube automated nucleic acid extraction system
(both Qiagen N.V.). The integrity of the isolated DNA was measured
with the Invitrogen Qubit 4 Fluorometer (Thermo Fisher Scientific,
Inc). The BRCA MASTR Plus Dx (cat. no. MR-2015.024; Multiplicom
N.V., Agilent Technologies GmbH) kit was used for sequencing on the
Illumina MiSeq next generation sequencing (NGS) platform (Illumina,
Inc.) with paired end libraries. Each reaction was conducted using
10 ng DNA. All BRCA1 and BRCA2 coding regions,
including 50-bp intron-exon junctions, were covered with the BRCA
MASTR Plus Dx kit. Sequencing was performed with 200× coverage.
During the run, the presence of small indel mutations and large
deletions and duplications was also investigated and evaluated. The
sequencing run was performed using MiSeq Reagent kit v2 (cat. no.
MS-102-2003; Illumina, Inc.). Sequencing data were analyzed with
the SOPHiA™ DDM clinical NGS data analysis platform (v4; Sophia
Genetics).
Methylation differences were evaluated among the six
siblings, who comprised MZ twins both with BRCA1 pathogenic
mutations but discordant for ovarian cancer, three sisters with
BRCA1 mutations, and one healthy brother with non-mutated
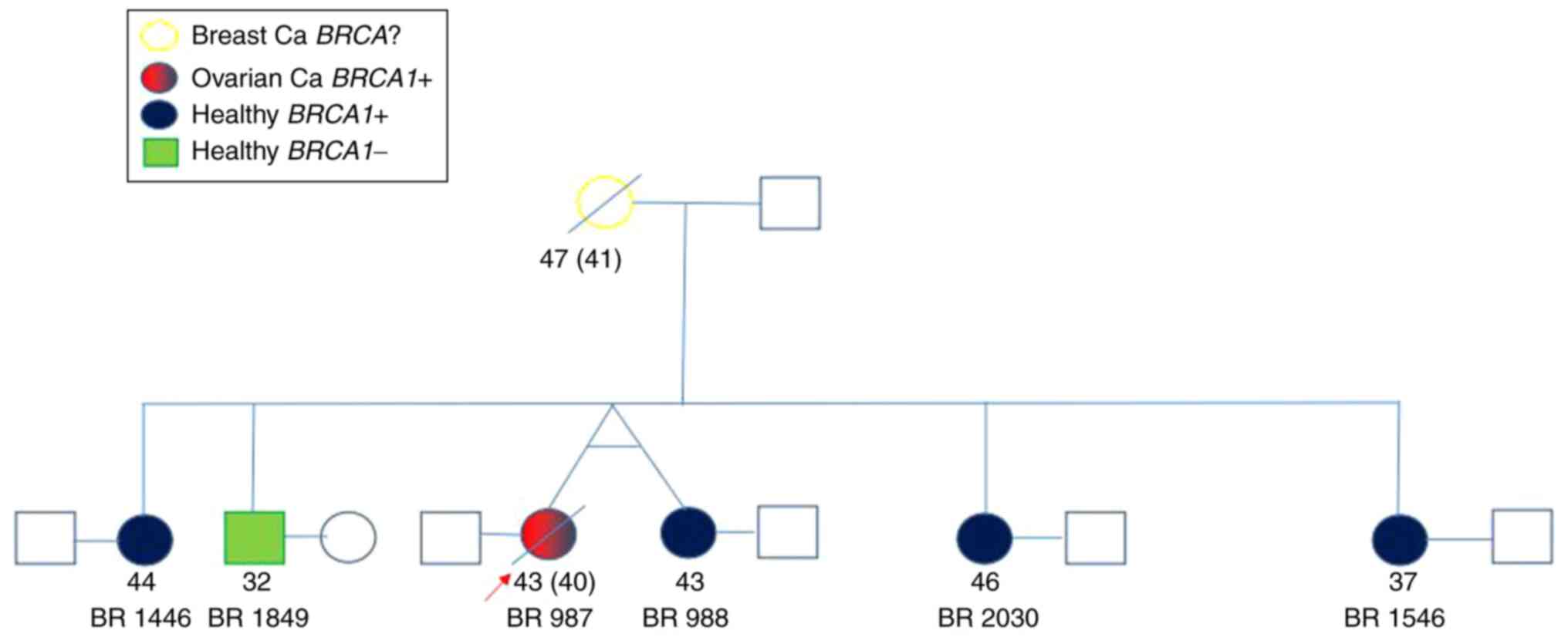
BRCA1. The data and codes of the individuals are presented
in Table I, and their family tree is
shown in Fig. 1.
 | Table I.Baseline information of the study
participants. |
Table I.
Baseline information of the study
participants.
| Sample code | Age (years) | Sex | Diagnosis | Genotype |
|---|
| BR 987 | 43 | Female | Ovarian cancer | HET c.5266dupC
p.Gln1756Profs*74 rs397507247 |
| BR988 | 43 | Female | Healthy | HET c.5266dupC
p.Gln1756Profs*74 rs397507247 |
| BR1446 | 44 | Female | Healthy | HET c.5266dupC
p.Gln1756Profs*74 rs397507247 |
| BR1546 | 37 | Female | Healthy | HET c.5266dupC
p.Gln1756Profs*74 rs397507247 |
| BR1849 | 32 | Male | Healthy | BRCA1 wild
type |
| BR2030 | 46 | Female | Healthy | HET c.5266dupC
p.Gln1756Profs*74 rs397507247 |
Four different comparison groups were generated,
each containing a case and control group, in order to evaluate the
differences in methylation levels according to diagnosis and
BRCA mutation conditions. These groups were as follows:
Group 1, MZ twin siblings with and without ovarian cancer; Group 2,
the MZ twin with ovarian cancer and healthy non-twin siblings;
Group 3, all siblings with the BRCA1 mutation and the
sibling with no BRCA1 mutation; and Group 4, the healthy MZ
twin, and all other healthy siblings. The groups and the codes of
individuals in the groups are presented in Table II.
| Table II.Sample codes of participants in the
comparison groups. |
Table II.
Sample codes of participants in the
comparison groups.
| Group | Case | Control |
|---|
| 1 | BR987 | BR988 |
| 2 | BR987 | BR2030, BR1446,
BR1546, BR1849 |
| 3 | BR987, BR988,
BR2030, BR1446, BR1546 | BR1849 |
| 4 | BR988 | BR2030, BR1446,
BR1546, BR1849 |
The CpG islands at which differences in methylation
level were detected between the case and control groups according
to ovarian cancer etiology and BRCA1 mutation-carrying
status were compared and evaluated. The methylation differences
between the groups were evaluated as 10-fold and in some groups as
25-fold or more to obtain more specific regions.
Preparation of the samples and data
analysis
Following isolation of the DNA, bisulfite
modification was performed for a 500-ng DNA sample in each case.
The bisulfite conversions of DNA samples were conducted using the
EZ DNA Methylation Kit (cat. no. #D5001; Zymo Research Corp.).
The genome-level methylation profiles of the
modified DNA samples were investigated using the Infinium
MethylationEPIC BeadChip Array on an iScan device (Illumina, Inc).
The Infinium MethylationEPIC Array is a genome-wide DNA methylation
analysis system based on bisulfite conversion and Infinium HD
sequencing technology that queries differentiated loci using
region-specific probes designed for methylated and non-methylated
regions. The total methylation level for a queried locus is
determined by calculating the ratio of fluorescent signals from the
methylated and unmethylated regions (21). Data analyses of the experimental
results were conducted using the Lumi libraries within the Illumina
GenomeStudio v2011.1 Methylation Module v1.9.0 (https://www.illumina.com/techniques/microarrays/array-data-analysis-experimental-design/genomestudio.html)
and R 3.0.2 (http://www.r-project.org). The
differences in methylation for >850,000 regions on a point basis
were investigated using this chip system. All six cases in the
study group were evaluated at >850,000 different CpG points.
Pre-processing and quality control of
the samples
Background corrections and dye bias equalization
filtering, transformation and normalization of the data were
conducted using library(lumi) in R 3.0.2 and were performed to
minimize the rate of possible systematic statistical error. After
filtering all samples by P-value, a mean of 866,309.3 CpG regions
were identified by P<0.01 and 866,518.5 CpG regions were
identified by P<0.05. Probes were regarded as erroneous and
excluded from the CpG analysis when no detection could be taken
from the same probe in >25% of all samples (P≥0.05).
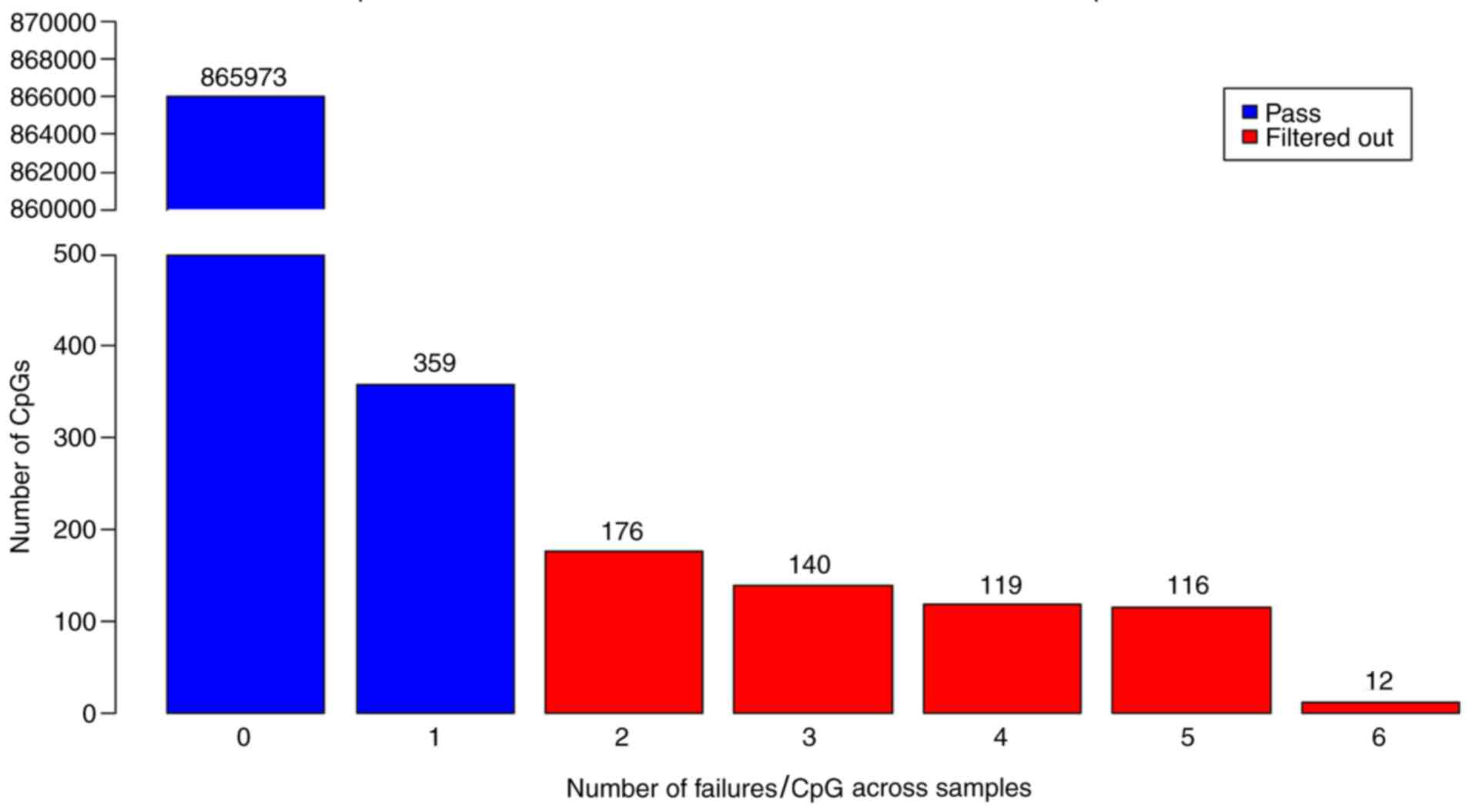
Probes readable in all samples or having only 1
sample with no readable value were included in the analysis, and
the other probes were filtered out (Fig.
2). Accordingly, a total of 563 CpG regions that were found to
have insignificant results according to their detection P-value
were excluded from the analysis. Thus, 866,332 CpG regions were
analyzed in accordance with the Beta Mixture Quantile (BMIQ)
normalization procedure using the BMIQ function in R 3.0.2
(22).
Data quality control
Boxplot and density diagrams were drawn for
comparison of the distributions before and after BMIQ normalization
and data conversion to avoid false results and reduce systematic
bias.
A rating diagram was prepared to observe the degree
of repetitiveness between the samples using the M-value with
Pearson's correlation. The M-value is calculated as the log2 ratio
of the densities of the methylated probe and the unmethylated probe
(23). The interval of this rating
diagram was established to provide a correlation coefficient (r) of
−1≤ r ≤1. The samples were identified to have a strong positive
correlation if r was close to +1. Our samples were identified as
r=0.99 with strong positive correlation.
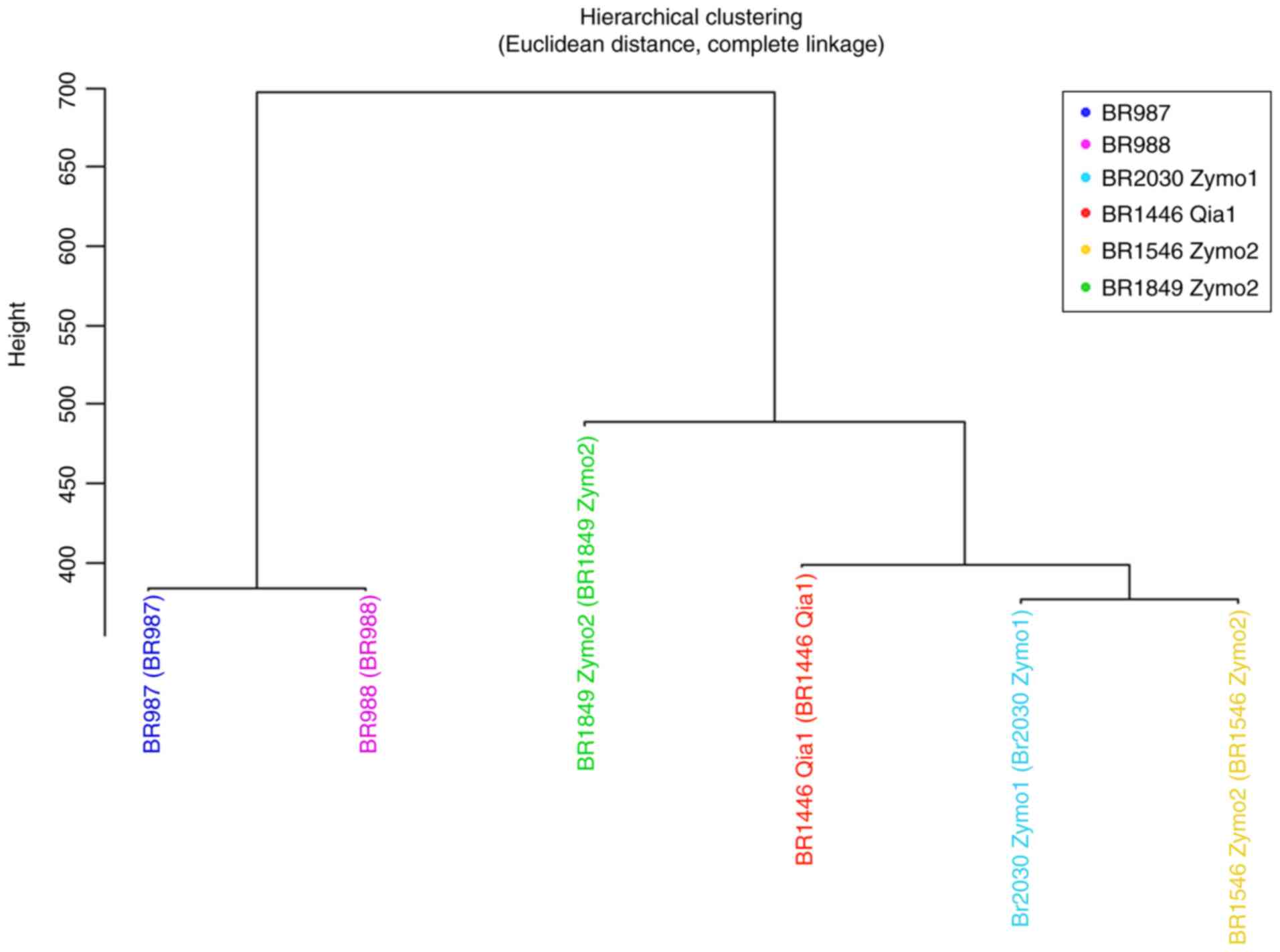
A dendrogram was drawn using M-values for the
samples grouped using hierarchical clustering with Euclidean
distance and complete linkage methods (Fig. 3). The diseased and healthy MZ twins
were classified together into one group in accordance with the
Euclidean distance clustering approach, and the other 4 healthy
siblings were classified into a separate group. Investigation of
the healthy siblings showed that the BRCA negative brother
was classified into a different group from the BRCA
mutation-carrying sisters. After clustering with the Euclidean
distance method, methylation expression levels were analyzed in the
aforementioned case and control groups. The differences between
these groups were investigated with regard to two different
aspects, namely association with disease and the presence of
BRCA1 mutation.
Functional association analysis
between genes and proteins
Protein-protein interaction (PPI) analysis provides
new data on protein functions and the general organizational
principles of functional cellular networks (24). The STRING scores are indicators of
confidence and rank from 0 to 1, with 1 being the highest possible
confidence (25).
Biological function analyses
The biological functions of the genes were evaluated
using the Protein Analysis Through Evolutionary Relationships
(PANTHER) classification system, which is designed to classify
proteins and genes according to their functions (26).
Results
DNA samples from six individuals from the same
family with ovarian cancer risk were analyzed. BRCA1 and
BRCA2 gene analyses were performed, and the presence of
mutations in certain members of the family was demonstrated. The
mutation was detected to be the HET c.5266dupC p.Gln1756Profs*74
rs397507247 mutation in exon 20 of the BRCA1 gene.
Differences in methylation levels in
the comparison groups
The sites with variations in methylation levels were
identified in four different comparison groups for analysis
according to diagnosis and BRCA1 mutation conditions.
The regions that were identified to have >10-fold
differences in methylation levels by comparison of the MZ twins
that were discordant for the presence of ovarian cancer in Group 1
are presented in Table III. In the
MZ twin with ovarian cancer, hypermethylation was detected in the
promoter region of the PR/SET domain 6 (PRDM6)
(NM_001136239), RB-binding protein 7, chromatin repair factor
(RBBP7) (NM_002893), ankyrin repeat domain 23
(ANKRD23) (NM_144994), RIB43A domain with coiled-coils 1
(RIBC1) (NM_001031745), C6orf227 (NR_027908) and
clustered mitochondria homolog (CLUH) (NM_015229) genes, and
hypomethylation was detected in the promoter region of the
cytochrome B5 reductase 4 (CYB5R4) (NM_016230) gene. The
sites of hypermethylation were located in CpG islets of the
RBBP7, ANKRD23, RIBC1 and C6orf227 genes, and the
northern (N) shore region of the CLUH gene, while the
CYB5R4 gene was hypomethylated in the southern (S) shore
region.
| Table III.Genes with methylation differences
between the discordant monozygotic twins. |
Table III.
Genes with methylation differences
between the discordant monozygotic twins.
| CpG no. | FC value | Chromosome | UCSC RefGene | UCSC RefGene
group | Regulatory
characteristic | Methylation |
|---|
| cg07490070 | 11.71 | 2 | ANKRD23 | Body | Promoter
associated |
Hypermethylated |
| cg10632209 | 17.60 | 5 | PRDM6 | Body | Unclassified |
Hypermethylated |
| cg04329454 | 11.30 | 6 |
C6orf227 | Body | Unclassified |
Hypermethylated |
| cg22356173 | 11.02 | 17 | CLUH | 5′UTR | – |
Hypermethylated |
| cg20246257 | 10.45 | X | RBBP7 | TSS1500,
TSS200 | Promoter
associated |
Hypermethylated |
| cg16978043 | 11.49 | X | RBBP7 | TSS200 | Promoter
associated |
Hypermethylated |
| cg17880859 | 11.81 | X | RBBP7 | 1stExon, 5′UTR | Promoter
associated |
Hypermethylated |
| cg11449070 | 11.42 | X | RIBC1 | TSS200,
TSS1500 | Promoter
associated |
Hypermethylated |
| cg26130726 | −11.39 | 6 | CYB5R4 | Body | Promoter
associated | Hypomethylated |
The regions with >10-fold difference in
methylation levels when the MZ twin with ovarian cancer was
compared with the other healthy siblings in Group 2 are presented
in Table IV. The differentially
methylated sites of the genes were as follows: RPL9, LIAS
and CYP2U1 in CpG islets; ACTN3 and ZFA in S
shelf regions; MYCBP, GJA9 and SEMA4D in S shore
regions; and ZNF714, OR4D and CLUH in N shore
regions.
| Table IV.Genes demonstrating methylation
differences between the monozygotic twin diagnosed with ovarian
cancer and healthy siblings. |
Table IV.
Genes demonstrating methylation
differences between the monozygotic twin diagnosed with ovarian
cancer and healthy siblings.
| CpG no. | FC value | Chromosome | UCSC RefGene | UCSC RefGene
group | Regulatory
characteristic | Methylation |
|---|
| cg01802772 |
14.69 | 1 | ACOT11 | Body | Unclassified |
Hypermethylated |
| cg10767615 |
10.36 | 1 | CAPZB | Body, 5′UTR | – |
Hypermethylated |
| cg06279067 |
12.44 | 1 | CASQ2 | Body | – |
Hypermethylated |
| cg13324406 |
10.18 | 1 | CHD1L | Body | – |
Hypermethylated |
| cg03544800 |
11.11 | 1 | DNTTIP2 | Body | – |
Hypermethylated |
| cg10195365 |
12.94 | 3 | FLNB | Body | – |
Hypermethylated |
| cg05393861 |
16.12 | 3 | ITIH3 | TSS200 | – |
Hypermethylated |
| cg17004290 |
15.06 | 4 | CYP2U1 | Body | Promoter
associated |
Hypermethylated |
| cg16104636 |
12.30 | 5 | CTNND2 | Body, 5′UTR | – |
Hypermethylated |
| cg07611121 |
10.12 | 5 | TRIO | Body | – |
Hypermethylated |
| cg10613215 |
11.44 | 6 | HIVEP2 | 5′UTR | Promoter
associated |
Hypermethylated |
| cg12134602 |
13.53 | 7 | C7orf45 | 3′UTR | – |
Hypermethylated |
| cg21499289 |
12.72 | 9 |
C9orf171 | Body | – |
Hypermethylated |
| cg22356173 |
15.03 | 17 | CLUH | 5′UTR | – |
Hypermethylated |
| cg09255886 | −14.88 | 1 | LUZP1 | 5′UTR | – | Hypomethylated |
| cg24051749 | −17.15 | 1 |
MYCBP;GJA9 | TSS1500, body | – | Hypomethylated |
| cg03967651 | −17.55 | 1 |
SLC2A1-AS1 | Body | – | Hypomethylated |
| cg00409995 | −14.51 | 2 | HDAC4 | Body | – | Hypomethylated |
| cg03192919 | −12.31 | 3 | FBXW12 | TSS1500 | – | Hypomethylated |
| cg19311470 | −26.28 | 4 |
RPL9,LIAS | TSS150, 5′UTR,
TSS200, TSS200 | Promoter
associated | Hypomethylated |
| cg15421137 | −11.74 | 5 |
FAM114A2 | 3′UTR | – | Hypomethylated |
| cg17386240 | −21.99 | 5 | TGFBI | Body | – | Hypomethylated |
| cg16792234 | −10.42 | 7 |
SLC25A13 | Body | – | Hypomethylated |
| cg10584449 | −12.97 | 8 | TG | Body | – | Hypomethylated |
| cg21927991 | −10.03 | 8 | ZFAT | Body | – | Hypomethylated |
| cg21203249 | −10.19 | 9 | SEMA4D | Body | Gene associated
cell type specific | Hypomethylated |
| cg12208638 | −18.40 | 11 | ACTN3 | Body | – | Hypomethylated |
| cg27079096 | −14.59 | 11 | OR52B4 | TSS200 | – | Hypomethylated |
| cg17040924 | −10.63 | 11 | OR52M1 | TSS1500 | – | Hypomethylated |
| cg14167033 | −13.45 | 11 | SHANK2 | Body | – | Hypomethylated |
| cg09581911 | −11.28 | 12 | DYRK4 | TSS200 | Promoter
associated | Hypomethylated |
| cg00645020 | −16.70 | 16 | KLHL36 | Body | – | Hypomethylated |
| cg11189272 | −14,04 | 17 | OR4D1 | 1stExon | – | Hypomethylated |
| cg01462799 | −13,84 | 19 | UPF1 | Body | – | Hypomethylated |
| cg19882830 | −11,24 | 19 | ZNF714 | TSS200 | – | Hypomethylated |
| cg01483656 | −14,22 | 19 | ZNF714 | TSS200 | Promoter
associated | Hypomethylated |
Regions with >10-fold hypomethylation and with
>25-fold hypermethylation (there were too many regions over
10-fold in this group, therefore 25-fold was used, for which more
specific regions should be given) when the BRCA1 positive
cases were compared with the BRCA1 negative case in Group 3
are presented in Table V. The
hypermethylation sites of genes PQBP1, TIMM17B, FMR1 and
AIFM1 were located in CpG islets, while those of the
ARHGEF9, AR and RPL36A genes were in N shore regions.
The TSC22D3 and DOCK11 genes were found to be
hypermethylated at S shore sites.
| Table V.Genes demonstrating methylation
differences between the BRCA1 positive and negative
cases. |
Table V.
Genes demonstrating methylation
differences between the BRCA1 positive and negative
cases.
| CpG no. | FC value | Chromosome | UCSC RefGene | UCSC RefGene
group | Regulatory
characteristic | Methylation |
|---|
| cg27519679 | 27.84 | X | AIFM1 | 1stExon | Promoter
associated |
Hypermethylated |
| cg19493242 | 27.70 | X | AR | 1stExon | – |
Hypermethylated |
| cg06316979 | 25.69 | X | ARHGEF9 | 1stExon, 5′UTR,
body | – |
Hypermethylated |
| cg00723034 | 25.00 | X | CXorf26 | Body | – |
Hypermethylated |
| cg18785414 | 32.78 | X | DOCK11 | Body | Promoter
associated |
Hypermethylated |
| cg14972002 | 25.23 | X | FAM122C | TSS200, body | Promoter
associated |
Hypermethylated |
| cg17430903 | 26.33 | X | FMR1 | TSS200 | Promoter
associated |
Hypermethylated |
| cg14332086 | 26.22 | X | PQBP1;
TIMM17B | TSS1500, TSS200,
5′UTR | Promoter
associated |
Hypermethylated |
| cg00029931 | 30.19 | X | RPL36A | TSS1500 | Promoter
associated |
Hypermethylated |
| cg13801593 | 29.57 | X | TSC22D3 | Body, 1stExon, | Promoter
associated |
Hypermethylated |
|
|
|
|
| TSS1500, 5′UTR |
|
|
| cg17018422 | −10.25 | 2 |
TRAPPC12 | Body | – | Hypomethylated |
| cg09819502 | −11.41 | 5 | C5orf33 | TSS1500 | − | Hypomethylated |
| cg03966322 | −15.68 | 5 | NADK2 | TSS1500,
TSS200 | − | Hypomethylated |
| cg14022523 | −11.83 | 6 | SFT2D1 | TSS200 | Promoter
associated | Hypomethylated |
| cg10637509 | −13.34 | 6 | FNDC1 | Body | – | Hypomethylated |
| cg18847598 | −13.65 | 11 | ASAM | Body | − | Hypomethylated |
| cg07541959 | −11.05 | 14 | TXNDC16 | Body | − | Hypomethylated |
| cg05522042 | −13.87 | 16 |
KIAA0513 | 3′UTR | Unclassified
cell | Hypomethylated |
|
|
|
|
|
| type specific |
|
| cg06589596 | −12.87 | 17 | GSDMA | 5′UTR | − | Hypomethylated |
| cg25929399 | −14.06 | 17 | KRT38 | TSS200 | − | Hypomethylated |
| cg13176022 | −10.47 | X | XG | Body | Unclassified | Hypomethylated |
| cg02351050 | −10.89 | Y | CD24; | 1stExon, body,
5′UTR, | Unclassified
cell | Hypomethylated |
|
|
|
| TTTY14 | TSS1500,
TSS200 | type specific |
|
| cg23654549 | −10.88 | Y | CD24; | 1stExon, body, | − | Hypomethylated |
|
|
|
| TTTY14 | TSS200, 5′UTR |
|
|
| cg16227841 | −10.68 | Y | CD24; | 1stExon, body, | − | Hypomethylated |
|
|
|
| TTTY14 | TSS200, 5′UTR |
|
|
| cg01150227 | −10.46 | Y | CD24; | 1stExon, body,
5′UTR, | Unclassified
cell | Hypomethylated |
|
|
|
| TTTY14 | TSS1500,
TSS200 | type specific |
|
| cg14683071 | −11.20 | Y | CD24; | 1stExon, body, | – | Hypomethylated |
|
|
|
| TTTY14 | TSS200, 5′UTR |
|
|
The regions with >10-fold differences in
methylation levels when the healthy MZ twin was compared with the
other healthy siblings in Group 4 are presented in Table VI. With regard to hypomethylated
sites, those of RPL9, LIAS and PRDM6 were located in
CpG islets, ACTN3 and ZFAT were in S shelf regions,
MYCBP, GJA9 and KLHL36 were in S shore regions, and
OR4D1 was in the N shore region. The CYP2U1 gene was
hypermethylated on CpG islets, the LOC25 3724 gene was
hypermethylated in the S shelf region, and the CYB5R4 gene
was hypermethylated in the S shore region.
| Table VI.Genes demonstrating methylation
differences between the healthy monozygotic twin, and the other
healthy siblings. |
Table VI.
Genes demonstrating methylation
differences between the healthy monozygotic twin, and the other
healthy siblings.
| CpG no | FC value | Chromosome | UCSC RefGene | UCSC RefGene
group | Regulatory
characteristics | Methylation |
|---|
| cg01802772 | 14.00 | 1 | ACOT11 | Body | Unclassified |
Hypermethylated |
| cg06279067 | 11.58 | 1 | CASQ2 | Body | – |
Hypermethylated |
| cg03544800 | 11.26 | 1 | DNTTIP2 | Body | – |
Hypermethylated |
| cg10195365 | 13.63 | 3 | FLNB | Body | – |
Hypermethylated |
| cg09371091 | 10.17 | 3 | HRH1 | 5′UTR | – |
Hypermethylated |
| cg05393861 | 15.47 | 3 | ITIH3 | TSS200 | – |
Hypermethylated |
| cg17004290 | 16.01 | 4 | CYP2U1 | Body | Promoter
associated |
Hypermethylated |
| cg16104636 | 12.17 | 5 | CTNND2 | Body, 5′UTR | – |
Hypermethylated |
| cg07611121 | 10.00 | 5 | TRIO | Body | – |
Hypermethylated |
| cg26130726 | 14.50 | 6 | CYB5R4 | Body | Promoter
associated |
Hypermethylated |
| cg10613215 | 13.24 | 6 | HIVEP2 | 5′UTR | Promoter
associated |
Hypermethylated |
| cg12134602 | 13.58 | 7 | C7orf45 | 3′UTR | – |
Hypermethylated |
| cg21499289 | 12.73 | 9 |
C9orf171 | Body | – |
Hypermethylated |
| cg13815695 | 10.72 | 12 |
LOC253724 | Body | – |
Hypermethylated |
| cg09255886 | −14.51 | 1 | LUZP1 | 5′UTR | – | Hypomethylated |
| cg24051749 | −14.41 | 1 |
MYCBP;GJA9 | TSS1500, body | – | Hypomethylated |
| cg03967651 | −18.79 | 1 |
SLC2A1-AS1 | Body | – | Hypomethylated |
| cg00409995 | −14.53 | 2 | HDAC4 | Body | – | Hypomethylated |
| cg01427108 | −11.99 | 3 | LTF | Body | – | Hypomethylated |
| cg03192919 | −12.55 | 3 | FBXW12 | TSS1500 | – | Hypomethylated |
| cg16570885 | −11.45 | 3 | IGF2BP2 | Body | – | Hypomethylated |
| cg19311470 | −20.13 | 4 |
RPL9;LIAS | TSS1500, 5′UTR,
TSS200 | Promoter
associated | Hypomethylated |
| cg15421137 | −11.62 | 5 |
FAM114A2 | 3′UTR | – | Hypomethylated |
| cg10632209 | −17.03 | 5 | PRDM6 | Body | Unclassified cell
type specific | Hypomethylated |
| cg17386240 | −17.46 | 5 | TGFBI | Body | – | Hypomethylated |
| cg16792234 | −11.71 | 7 |
SLC25A13 | Body | – | Hypomethylated |
| cg10584449 | −13.63 | 8 | TG | Body | – | Hypomethylated |
| cg21927991 | −10.92 | 8 | ZFAT | Body | – | Hypomethylated |
| cg12208638 | −18.53 | 11 | ACTN3 | Body | – | Hypomethylated |
| cg14167033 | −16.07 | 11 | SHANK2 | Body | – | Hypomethylated |
| cg27079096 | −14.72 | 11 | OR52B4 | TSS200 | – | Hypomethylated |
| cg00474091 | −11.18 | 12 | AEBP2 | Body | – | Hypomethylated |
| cg09581911 | −11.28 | 12 | DYRK4 | TSS200 | – | Hypomethylated |
| cg00645020 | −13.81 | 16 | KLHL36 | Body | – | Hypomethylated |
| cg11189272 | −13.02 | 17 | OR4D1 | 1stExon | – | Hypomethylated |
| cg01462799 | −11.99 | 19 | UPF1 | Body | – | Hypomethylated |
Evaluation of the comparisons between
groups
Comparison of the MZ twin with ovarian cancer and
the healthy MZ twin (Group 1) showed that the PRDM6 gene was
hypermethylated in the MZ twin with ovarian cancer. However,
PRDM6 was found to be hypomethylated in the healthy MZ twin
compared with the other healthy siblings (Group 4). The
CYB5R4 gene was demonstrated to be hypomethylated in the MZ
twin with ovarian cancer compared with the healthy MZ twin (Group
1), but hypermethylated in the healthy MZ twin compared with the
other healthy siblings (Group 4). Furthermore, the CYB5R4
gene was found to be hypermethylated in all healthy individuals,
regardless of whether they were negative or positive for the
BRCA1 mutation (Group 3). The genes RPL9, LIAS, TGFBI,
ACTN3, SLC2A1-AS1, MYCBP, GJA9, KLHL36, LUZP1, HDAC4, OR4D1, UPF1,
SHANK2, TG, FBXW12, FAM114A2, DYRK4, SLC25A13 and ZFAT
were found to be hypomethylated in the MZ twin with ovarian cancer
compared with the non-twin healthy siblings (Group 2) and the
healthy MZ twin compared with the other healthy siblings (Group 4),
while the genes TRIO, DNTTIP2, HIVEP2, CTNND2, CASQ2, C9orf171,
FLNB, C7orf45, ACOT11, CYP2U, and ITIH3 were found to be
hypermethylated in these comparison groups.
The genes RBBP7, ANKRD23, RIBC1, C6orf227 and
CLUH were hypermethylated in the MZ twin with ovarian cancer
compared with the healthy MZ twin (Group 1), while the
CYB5R4 gene was hypomethylated. The ZNF714, OR52M1
and SEMA4D genes were observed to be hypomethylated, while
the CHD1L, CAPZB and CLUH genes were hypermethylated
in the MZ twin with ovarian cancer compared with non-twin healthy
siblings (Group 2). Evaluation of the results obtained from all the
comparison groups in the present study suggests that the
methylation conditions of the PRDM6, CYB5R4, ZNF714, OR52M1,
SEMA4D, CHD1L, CAPZB, CLUH, RBBP7, ANKRD23, RIBC and
C6orf227 genes may be effective for the differentiation of
ovarian cancer.
The comparison of BRCA1 positive and
BRCA1 negative cases (Group 3) showed that the genes
NADK2, KRT38, KIAA0513, ASAM, FNDC1, GSDMA, SFT2D1, C5orf33,
CD24, TTTY14, TXNDC16, XG and TRAPPC12 were hypomethylated, and
the genes CXorf26, FAM122C, ARHGEF9, PQBP1, TIMM17B, FMR1, AR,
AIFM1, TSC22D3, RPL36A and DOCK11 were hypermethylated
between the comparison groups. These methylation changes may be
associated with the presence or absence of the BRCA1 gene
mutation.
Functional association analysis
between genes and proteins
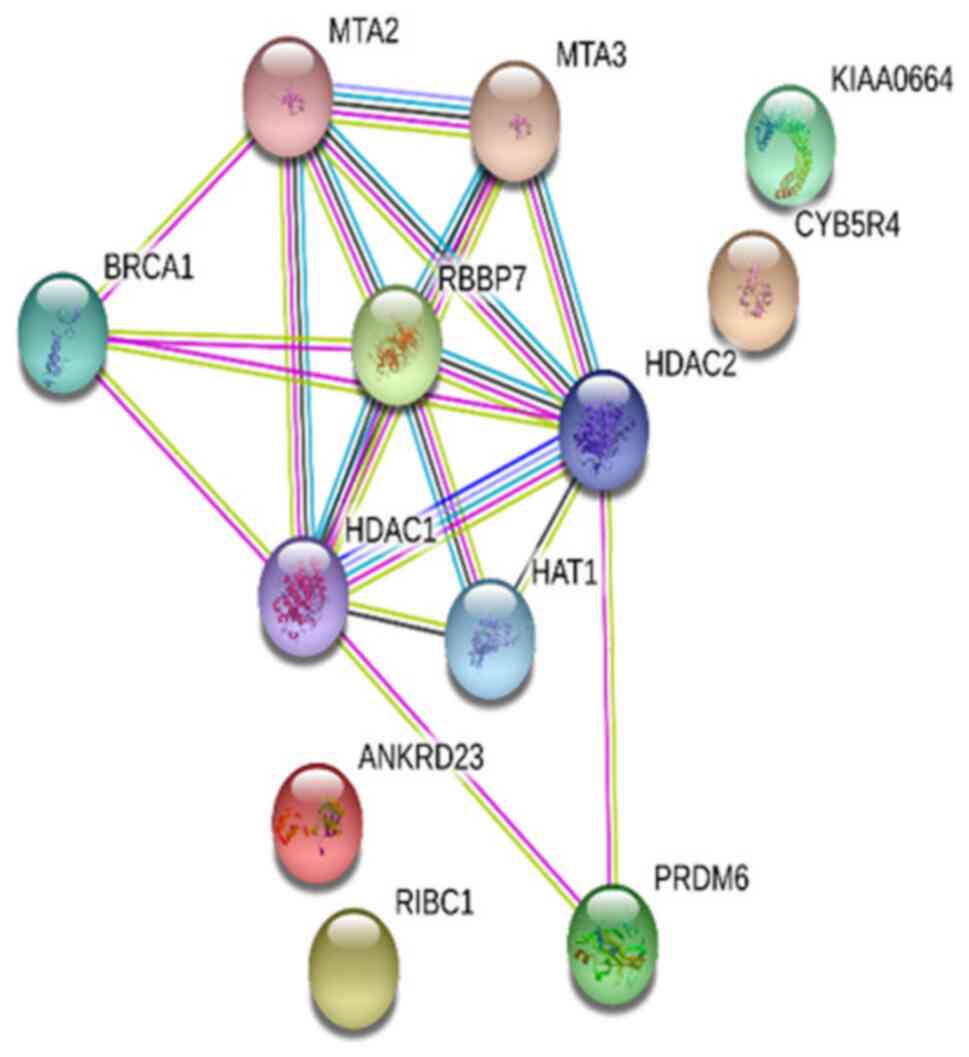
A PPI network was established in the present study
to analyze the functions of the proteins encoded by the
differentially methylated genes. The experimental data and the
STRING functional protein association network v.10.5, which
provides data on estimated interactions, was used in the analysis
of the interactions. The protein network obtained was shown to have
significantly higher protein interaction than expected (P=0.000331;
Fig. 4). Two more STRING analyses
were performed, which reported connections between CAPZB and
FLNB, and BRCA1 and AR (data not shown).
Biological function analyses
The results of the PANTHER analysis revealed that
the genes identified in the study had roles in biological processes
including ‘biologic adhesion’, ‘regulation’, ‘cellular components
organization’ and ‘biogenesis’, ‘immune system functioning’,
‘metabolic functioning’ and ‘localization’. Also, these genes were
found to have roles in molecular functions including ‘attachment’,
‘catalytic activity’, ‘receptor activity’, ‘signal transmission’
and ‘translational regulation’.
Discussion
Although there have been promising developments in
cancer studies in recent years, data on the biological basis of
ovarian cancer are limited. All cancers develop as a consequence of
the accumulation of genetic changes or other molecular disorders
such as epigenetic changes. Epigenetic and genetic changes are
known to contribute to the development of ovarian cancer.
Epigenetic studies on ovarian cancer have demonstrated the role of
epigenetics in ovarian cancer development, and its association with
various signaling pathways (27–29). DNA
methylation detected in the CpG islands of the promoter regions of
genes associated with the development of cancer has been found to
be frequently associated with the reduced expression or silencing
of those genes (27).
Researchers investigating methylation in MZ twins
demonstrated that age and other non-genetic factors triggered
epigenetic variations and provided strong evidence for epigenetic
inheritance (18,19). Therefore, the present study
investigated genome level methylation differences among MZ twins
with BRCA1 gene mutations, one with ovarian cancer and one
without, and their healthy siblings in the present study. The
potential effects of the differentially methylated genes and their
association with ovarian cancer were investigated. Some of the
significantly differentially hypermethylated and hypomethylated
genes that were identified in the methylation analyses conducted in
the present study were consistent with those in previous
studies.
In the present study, promoter hypermethylation of
the genes PRDM6, RBBP7, ANKRD23, RIBC1, C6orf227 and
CLUH was identified in the MZ twin with ovarian cancer
compared with the healthy MZ twin. PRDM6 encodes a protein
that binds to nucleic acid and has a histone-lysine
N-methyltransferase activity. PRDM6 is a transcription factor
associated with enzymes that have roles in chromatin remodeling and
gene expression, including heterochromatin protein-1, histone
deacetylase (HDAC)1, HDAC2 and HDAC3, histone acetyltransferase
p300 and histone methyltransferase G9a (30). The STRING analysis in the present
study showed that PRDM6 was associated with HDAC1 and HDAC2, which
themselves bound to BRCA1 and RBBP7 proteins. We suggest that the
PRDM6 gene is specific to ovarian cancer because the
PRDM6 gene was hypermethylated in the MZ twin with cancer
compared with the healthy MZ twin, but hypomethylated in the
healthy MZ twin compared with other healthy siblings. The STRING
analysis indicates that the effect of this molecule in the
development of ovarian cancer may be mediated through HDAC and
BRCA1 proteins. The carriers of the BRCA1 mutation in the
present study, and the methylation changes between the MZ twins and
other siblings who were discordant for ovarian cancer supports
STRING analysis. The PRDM6 gene was hypermethylated in the
MZ twin with cancer compared with the healthy MZ twin, but
hypomethylated in the healthy MZ twin compared with other healthy
siblings.
The RBBP7 gene encodes a highly expressed
protected nuclear protein that directly binds to retinoblastoma
protein and thereby regulates cell proliferation. Retinoblastoma
and retinal cancers are associated with RBBP7 (31). BRCA1 has been shown to
interact in vivo and in vitro with the Rb-binding
proteins RBBP7 (also known as RbAp4) and RbAp48 (31). The effect of the BRCA1 gene on
various processes, including transcription, DNA repair and
recombination, has been explained by the association of BRCA1 with
HDAC1 and HDAC2 (31). A >10-fold
higher hypermethylation of the RBBP7 gene was detected in
the MZ twin with cancer compared with the healthy twin. The STRING
analysis performed in this group suggests that RBBP7 interacts with
the BRCA1 tumor suppressor protein, thus resulting in the
development of ovarian cancer via roles in the regulation of
cellular proliferation and differentiation. In addition,
RBBP7 has previously been shown to have NF-kB modulating,
NOTCH1-associated pathway and HDAC activities by Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathway analysis. These activities have
been reported to serve a role in different cancers (32).
Detection of hypomethylation of the CYB5R4
gene in the MZ twin with ovarian cancer compared with the healthy
MZ twin, and hypermethylation in all healthy siblings regardless of
BRCA1 mutation status suggests that hypomethylation of
CYB5R4 gene might be involved in the development of ovarian
cancer. Although the effect of the CYB5R4 gene is unclear in
cancer, this gene has been reported to be mutated in patients with
breast cancer (33).
VEGF has been shown to suppress the expression of
Semaphorin 4D (SEMA4D) in epithelial ovarian cancer tissues
(34). In the present study,
ZNF714, OR52M1 and SEMA4D genes were found to be
hypomethylated in the MZ twin with ovarian cancer compared with the
non-twin healthy siblings. This suggests that the hypomethylation
of SEMA4D may be associated with ovarian cancer. The
overexpression of CHD1L protein has been reported to be associated
with metastasis in ovarian cancer, and CHD1L protein expression
evaluated using immunohistochemistry has been suggested be a new
prognostic biomarker for patients with ovarian cancer (35). The observation of CHD1L
hypermethylation in the twin with ovarian cancer compared with the
non-twin healthy siblings, in contrast with the literature,
suggests that the development of ovarian cancer might occur with
the suppression of CHD1L expression.
In the STRING analysis conducted in the present
study, a connection was identified between CAPZB and
FLNB. In a previous study, the CpG regions with different
FLNB DNA methylation levels between men and women were
identified. The genes with higher methylation in either sex were
subjected to KEGG pathway analysis. The ‘cell adhesion molecules’
pathway was enriched with genes having a higher methylation level
in women, and the ‘adipocytokine signaling pathway’ was enriched
with genes having a higher methylation level in men (36). In another study, FLNB was
shown to be inhibited by mir-223, let-7d and mir-130a (37). The miRNA molecules shown to inhibit
the FLNB gene in the previous study were found to have high
expression levels in MZ twin siblings with ovarian cancer in
another study conducted by our group (unpublished data).
NADK2, KRT38, KIAA0513, ASAM, FNDC1, GSDMA,
SFT2D1, C5orf33, CD24, TTTY14, TXNDC16, XG and TRAPPC12
genes were hypomethylated, and CXorf26, FAM122C, ARHGEF9, PQBP1,
TIMM17B, FMR1, AR, AIFM1, TSC22D3, RPL36A and DOCK11
genes were hypermethylated in the siblings with the BRCA1
mutation compared with the sibling with wild-type BRCA1. It
has been suggested that the determined genes are completely related
to wild-type BRCA1 because they have different gene profiles
according to the comparisons. These genes are completely different
from the genes detected in the other comparison groups. An
association was detected between BRCA1 and AR
proteins in the STRING analysis. Previous studies reported that
AR promoter hypermethylation was associated with decreased
AR expression in breast cancer cell lines (38), and that AR gene promoter
methylation was higher in patients with wild-type BRCA1/2
compared with mutated BRCA1/2 in men diagnosed with breast
cancer (39). In general, the data
in the literature indicate that AR is associated with male
breast cancer (39). The detection
of hypermethylated AR in the brother with wild-type
BRCA1 in the present study is consistent with the
literature.
The present study was performed using the peripheral
blood lymphocytes of BRCA1 mutation carrying discordant MZ
twins and other healthy siblings with and without BRCA1
mutation. Therefore, the study has a limitation that no methylation
analysis was performed in the tissues of the MZ twins or the
siblings who underwent preventive surgery. However, we plan to
investigate the expression and methylation of these genes in these
tissues and in larger ovarian cancer patient cohorts in future
studies.
The results obtained in the present study suggest
that the differential methylation of 12 different genes, namely
PRDM6, CYB5R4, ZNF714, OR52M1, SEMA4D, CHD1L, CAPZB, CLUH,
RBBP7, ANKRD23, RIBC1 and C6orf227 might be associated
with the development of ovarian cancer. Also, we suggest that the
differential methylation levels of 24 genes, namely NADK2,
KRT38, KIAA0513, ASAM, FNDC1, GSDMA, SFT2D1, C5orf33, CD24; TTTY14,
TXNDC16, XG, TRAPPC12, CXorf26, FAM122C, ARHGEF9, PQBP1, TIMM17B,
FMR1, AR, AIFM1, TSC22D3, RPL36A and DOCK11 are
associated with the BRCA1 mutation. To the best of our
knowledge, the present study is the first to report the effect of
methylation differences in the full genome in ovarian cancer. The
comparison of the identified genes in larger ovarian cancer patient
cohorts, in benign ovarian disease, and a population-based healthy
cohort, and an investigation of the mRNA and protein expression
levels of these genes would be appropriate in the future.
Acknowledgements
Not applicable.
Funding
This study was supported by the Scientific Research
Projects Unit of Istanbul University (project no. 59477). In the
study, the funding body has no role in the preparation, data
collection, analysis and interpretation of the manuscript.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to restrictions of the
Local and Clinical Research Ethics Committee of Istanbul University
to protect patient privacy.
Authors' contributions
Conceptualization and methodology, OSE and HY;
formal analysis of the data, OSE, SK, DAO, SBT, GKT, BC and MA;
writing the original draft of the manuscript, OSE, SK, DAO, SBT,
GKT, BC and MA; writing, reviewing and editing the manuscript, OSE
and HY; visualization, supervision, scientific contribution and
criticism, HY. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study complies with ethical standards WMA
Declaration of Helsinki-Ethical Principles for Medical Research
Involving human subjects) (20). The
Ethics Board of Istanbul University granted approval for the study
to be conducted in the Department of Basic Oncology, Istanbul
University, Institute of Oncology (approval date May 18, 2015;
approval no. 1552). All patients were informed about the study and
granted consent in the scope of the cancer genetics polyclinic.
Patient consent for publication
Individual consent was signed by each sibling.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MZ
|
monozygotic
|
|
NGS
|
next generation sequencing
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Noone AM, Krapcho M, Miller D, Brest A, Yu
M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, et al: SEER
Cancer statistics review. National Cancer Institute Bethesda; MD:
1975-2015
|
|
3
|
Yancik R: Ovarian cancer. Age contrasts in
incidence, histology, disease stage at diagnosis, and mortality.
Cancer. 71:517–523. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kurman RJ: Origin and molecular
pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol. 24
(Suppl 10):x16–x21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
US Preventive Services Task Force, ;
Bibbins-Domingo K, Grossman DC, Curry SJ, Barry MJ, Davidson KW,
Doubeni CA, Epling JW Jr, García FA, Kemper AR, et al: Screening
for gynecologic conditions with pelvic examination: US preventive
services task force recommendation statement. JAMA. 317:947–953.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
La Vecchia C: Ovarian cancer: Epidemiology
and risk factors. Eur J Cancer Prev. 26:55–62. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Whittemore AS, Harris R and Itnyre J:
Characteristics relating to ovarian cancer risk: Collaborative
analysis of 12 US case-control studies. II. Invasive epithelial
ovarian cancers in white women. Collaborative Ovarian Cancer Group.
Am J Epidemiol. 136:1184–1203. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pharoah PD, Day NE, Duffy S, Easton DF and
Ponder BA: Family history and the risk of breast cancer: A
systematic review and meta-analysis. Int J Cancer. 71:800–809.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Crum CP, Drapkin R, Miron A, Ince TA, Muto
M, Kindelberger DW and Lee Y: The distal fallopian tube: A new
model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol.
19:3–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kurman RJ and McConnell TG:
Characterization and comparison of precursors of ovarian and
endometrial carcinoma: Parts I and II. Int J Surg Pathol. 18 (Suppl
3):S181–S189. 2010. View Article : Google Scholar
|
|
11
|
Mutch D, Denny L and Quinn M; FIGO
Committee on Gynecologic Oncology, : Hereditary gynecologic
cancers. Int J Gynaecol Obstet. 124:189–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wolffe AP and Matzke MA: Epigenetics:
Regulation through repression. Science. 286:481–486. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chatterjee A and Morison IM: Monozygotic
twins: Genes are not the destiny? Bioinformation. 7:369–370. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ready K, Litton JK and Arun BK: Clinical
application of breast cancer risk assessment models. Future Oncol.
6:355–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Petronis A, Gottesman II, Kan P, Kennedy
JL, Basile VS, Paterson AD and Popendikyte V: Monozygotic twins
exhibit numerous epigenetic differences: Clues to twin discordance?
Schizophr Bull. 29:169–178. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong AH, Gottesman II and Petronis A:
Phenotypic differences in genetically identical organisms: The
epigenetic perspective. Hum Mol Genet 14 Spec No. 1:R11–R18. 2005.
View Article : Google Scholar
|
|
17
|
Fraga MF, Ballestar E, Paz MF, Ropero S,
Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M,
Benitez J, et al: Epigenetic differences arise during the lifetime
of monozygotic twins. Proc Natl Acad Sci USA. 102:10604–10609.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heijmans BT, Kremer D, Tobi EW, Boomsma DI
and Slagboom PE: Heritable rather than age-related environmental
and stochastic factors dominate variation in DNA methylation of the
human IGF2/H19 locus. Hum Mol Genet. 16:547–554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh
GH, Wong AH, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, et al:
DNA methylation profiles in monozygotic and dizygotic twins. Nat
Genet. 41:240–245. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
The Helsinki Declaration of the World
Medical Association (WMA): Ethical principles of medical research
involving human subjects. Pol Merkur Lekarski. 36:298–301. 2014.(In
Polish). PubMed/NCBI
|
|
21
|
Pidsley R, Zotenko E, Peters TJ, Lawrence
MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C
and Clark SJ: Critical evaluation of the Illumina MethylationEPIC
BeadChip microarray for whole-genome DNA methylation profiling.
Genome Biol. 17:2082016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Du P, Zhang X, Huang CC, Jafari N, Kibbe
WA, Hou L and Lin SM: Comparison of Beta-value and M-value methods
for quantifying methylation levels by microarray analysis. BMC
Bioinformatics. 11:5872010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stelzl U, Worm U, Lalowski M, Haenig C,
Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A,
Koeppen S, et al: A human protein-protein interaction network: A
resource for annotating the proteome. Cell. 122:957–968. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thomas PD, Kejariwal A, Campbell MJ, Mi H,
Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A,
Rabkin S, et al: PANTHER: A browsable database of gene products
organized by biological function, using curated protein family and
subfamily classification. Nucleic Acids Res. 31:334–341. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng M, Hu Y, Gou R, Wang J, Nie X, Li X,
Liu Q, Liu J and Lin B: Integrated multi-omics analysis of
genomics, epigenomics, and transcriptomics in ovarian carcinoma.
Aging (Albany NY). 11:4198–4215. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maldonado L and Hoque MO: Epigenomics and
ovarian carcinoma. Biomark Med. 4:543–570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davis CA, Haberland M, Arnold MA,
Sutherland LB, McDonald OG, Richardson JA, Childs G, Harris S,
Owens GK and Olson EN: PRISM/PRDM6, a transcriptional repressor
that promotes the proliferative gene program in smooth muscle
cells. Mol Cell Biol. 26:2626–2636. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yarden RI and Brody LC: BRCA1 interacts
with components of the histone deacetylase complex. Proc Natl Acad
Sci USA. 96:4983–4988. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu N, Zhang P, Wang L, He XJ, Yang SS and
Lu HJ: RBBP7 is a prognostic biomarker in patients with esophageal
squamous cell carcinoma. Oncol Lett. 16:7204–7211. 2018.PubMed/NCBI
|
|
33
|
Xu M, Wang W, Frontera JR, Neely MC, Lu J,
Aires D, Hsu FF, Turk J, Swerdlow RH, Carlson SE and Zhu H: Ncb5or
deficiency increases fatty acid catabolism and oxidative stress. J
Biol Chem. 286:11141–11154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Y, Zhang L, Liu WX and Wang K: VEGF
and SEMA4D have synergistic effects on the promotion of
angiogenesis in epithelial ovarian cancer. Cell Mol Biol Lett.
23:22018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
He WP, Zhou J, Cai MY, Xiao XS, Liao YJ,
Kung HF, Guan XY, Xie D and Yang GF: CHD1L Protein is overexpressed
in human ovarian carcinomas and is a novel predictive biomarker for
patients survival. BMC Cancer. 12:4372012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hall E, Volkov P, Dayeh T, Esguerra JL,
Salö S, Eliasson L, Rönn T, Bacos K and Ling C: Sex differences in
the genome-wide DNA methylation pattern and impact on gene
expression, microRNA levels and insulin secretion in human
pancreatic islets. Genome Biol. 15:5222014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen B and Li CW: Big Mechanisms of aging
via system ıdentification and big database mining. Big Mechanisms
in Systems Biology. Elsevıer; pp. 671–735. 2017, View Article : Google Scholar
|
|
38
|
Peters KM, Edwards SL, Nair SS, French JD,
Bailey PJ, Salkield K, Stein S, Wagner S, Francis GD, Clark SJ and
Brown MA: Androgen receptor expression predicts breast cancer
survival: The role of genetic and epigenetic events. BMC Cancer.
12:1322012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rizzolo P, Silvestri V, Valentini V, Zelli
V, Zanna I, Masala G, Bianchi S, Palli D and Ottini L:
Gene-specific methylation profiles in BRCA-mutation positive and
BRCA-mutation negative male breast cancers. Oncotarget.
9:19783–19792. 2018. View Article : Google Scholar : PubMed/NCBI
|