Introduction
Although its incidence is declining, gastric cancer
(GC) is still a leading cause of cancer-associated mortality and a
major socio-economic burden worldwide (1). Helicobacter pylori (H.
pylori) infection is a major risk factor of GC (2). Environmental factors are subordinate to
the effect of H. pylori infection, which has been listed as
a class I carcinogen for GC by the World Health Organization since
1994 (2). H. pylori infection
causes gastric mucosal inflammation, as well as genetic and
epigenetic changes, which lead to genetic instability in host
gastric epithelial cells (2).
However, the exact molecular mechanism of H.
pylori-associated GC is not yet clear (3). At present, several studies have
demonstrated that H. pylori induces the
epithelial-mesenchymal transition (EMT) process in gastric
epithelial cells, which promotes invasion and metastasis of GC via
the TGF-β, NF-κB and Hippo signaling pathways (4–6).
EMT is a process in which epithelial cells lose
their polarity and transform their morphology by developing
fiber-like structures (7).
Simultaneously, the contacts with the surrounding cells and cell
adhesion are reduced and an increased migratory and invasive
activity is noted. Both of these processes promote tumor metastasis
(7). The molecular changes of EMT
are mainly characterized by the downregulation of the expression of
epithelial cell markers and the upregulation of the expression of
mesenchymal phenotypic markers (8).
In particular, the absence of E-cadherin is an important EMT signal
and a prerequisite for tumor cell invasion (9). The functional molecules of mesenchymal
cells are mainly skeletal proteins derived from mesenchyme or
embryos (e.g., N-cadherin) that are normally not expressed or
expressed at low levels in epithelial cells, while the expression
of these molecules is increased during the EMT process (10).
Multiple signaling pathways are involved in the
development of EMT (11). For
example, hepatocyte growth factor/scatter factor and early growth
response gene 1 synergistically activate the MAPK signaling
pathway, which enhances the activity of snail and inhibits the
expression of the epithelial marker E-cadherin, promoting EMT
(12). The AKT/GSK3β signaling
pathway has attracted considerable attention due to its involvement
in EMT (13). AKT, which is also
known as protein kinase B, is a serine/threonine protein kinase
associated with a number of signaling pathways that promote cell
proliferation, survival and inhibition of apoptosis (14). GSK3β is a serine/threonine protein
kinase that acts downstream of AKT (15). Unlike other kinases, GSK3β is
normally dephosphorylated in its active state. The phosphorylation
of GSK3β (Ser9) by phosphorylated (p-)AKT leads to inhibition of
its activity (15). Activation of
AKT results in GSK3β phosphorylation, which reduces β-catenin
degradation, promotes Snail expression and eventually induces EMT
in combination with T cell factor/lymphocyte enhancer factor
(13,16).
Previous studies demonstrated that H. pylori
activated the AKT signaling pathway in human gastric epithelial
cells (GES-1), thereby causing disorders in cell proliferation and
apoptosis (17,18). However, to the best of our knowledge,
the role of the AKT/GSK3β signaling pathway in H.
pylori-associated EMT is not yet clear. The aim of the present
study was to investigate whether H. pylori infection induces
EMT and promotes the development and metastasis of cancer in normal
gastric mucosa. In addition, the present study investigated whether
this process was dependent on AKT activation. The pathogenic
mechanism of H. pylori was thoroughly examined and the data
provided a novel theoretical basis for the development of
prevention strategies for H. pylori infection-associated
diseases.
Materials and methods
Patients and sample collection
Between January 2007 and September 2012, 165
formalin-fixed (10% formalin at room temperature for 24 h) and
paraffin-embedded gastric tissue samples from 165 patients who
underwent gastroduodenoscopy were obtained from the Digestive
Centre of The First Affiliated Hospital of Nanchang University
(Nanchang, China). Patients aged between 18–70 years and without
previous H. pylori treatment history were enrolled in the
present study, and patients who underwent gastric surgery were
excluded. The clinical characteristics of patients were stratified
according to pathological diagnosis and are shown in Table I. The patients had a median age of 55
years (range, 36–69 years) and 86 patients (52%) were male. Among
these patients, 19 out of the 39 who were diagnosed with chronic
non-atrophic gastritis (CNAG), 19 out of the 40 who had intestinal
metaplasia (IM), 19 out of the 39 who had dysplasia (Dys) and 22
out of the 47 who had GC were H. pylori-positive. The age
and gender distribution of these patients were compared and the
data revealed no significant differences (P>0.05; Table I). Gastric tissue samples were
collected from the gastric antrum and from the individual lesions
of each patient. Pathological diagnosis and classification were
based on the criteria of the World Health Organization and the
updated Sydney system (19,20). A non-atrophy gastritis diagnosis was
made when all biopsies indicated a non-atrophy gastritis result
(19,20). The rapid urease test and Giemsa
staining were used for the detection of H. pylori infection,
as previously described (21). The
present study was approved by the Human Ethics Committee of the
First Affiliated Hospital of Nanchang University (Nanchang, China).
All patients signed an informed consent form for their
participation in the study.
 | Table I.Clinical characteristics of patients
with different stages of gastric carcinogenesis. |
Table I.
Clinical characteristics of patients
with different stages of gastric carcinogenesis.
| Characteristic | CNAG | IM | Dys | GC | P-value |
|---|
| No. | 39 | 40 | 39 | 47 |
|
| Age, years (mean ±
SD) | 50.80±14.08 | 54.32±12.18 | 56.68±10.81 | 55.06±14.01 | >0.05 |
| Male, n | 20 | 21 | 20 | 25 | >0.05 |
| Female, n | 19 | 19 | 19 | 22 | >0.05 |
| H. pylori
positive, n | 19 | 19 | 19 | 22 | >0.05 |
| H. pylori
negative, n | 20 | 21 | 20 | 25 | >0.05 |
H. pylori strain and culture
conditions
The wild-type H. pylori CagA+
VaCA+ strain (ATCC43504; American Type Culture
Collection) was used in the present study. Briefly, H.
pylori was cultured on Campylobacter agar (Oxoid; Thermo Fisher
Scientific, Inc.) plates with 5% sheep blood (Gibco; Thermo Fisher
Scientific, Inc.) and incubated at 37°C with 5% CO2 for
subsequent in vitro experiments. Following 24 h of
incubation, H. pylori strains were grown in Brucella broth
(Gibco; Thermo Fisher Scientific, Inc.) with 10% sheep serum
(Gibco; Thermo Fisher Scientific, Inc.) for 16 h under
microaerophilic conditions (5% O2, 10% CO2
and 85% N2). H. pylori co-culture experiments
with GES-1 cells were conducted at an MOI of 50:1.
Animals and infections
Male Mongolian gerbils (specific pathogen-free; age,
5–8 weeks; weight, 30–50 g; obtained from Zhejiang Academy of
Medical Sciences (Zhejiang, China) http://www.zjams.com.cn/) were utilized in the present
study between August 2018 and June 2019. A total of 79 gerbils were
maintained in an isolated clean room with regulated temperature
(20-22°C), humidity (~55%) and a 12/12-h light/dark cycle, with
ad libitum access to rodent diet and water as previously
described, animal health and behavior were monitored twice a day,
and no gerbils were found dead during experiments (22). Following fasting for 12 h, the
animals were orogastrically challenged with 0.2 ml sterile Brucella
broth, which was used as a control group (n=25) or H. pylori
strains (n=54) at 10 time points (every 3 days). The Mongolian
gerbils were humanely euthanized via CO2 inhalation (30%
of the chamber volume per min) at 9 months after the challenge and
gastric tissues were harvested. Euthanasia was confirmed by
observation of each gerbil for lack of respiration and faded eye
color. The First Affiliated Hospital of Nanchang University Ethics
Committee (Nanchang, China) approved all experiments and
procedures.
Cell line and reagents
The immortalized human gastric cell line GES-1
(Beijing Institute for Cancer Research, Beijing, China) was grown
in DMEM (Gibco; Thermo Fisher Scientific, Inc.) with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin at 37°C with 5% CO2. The AKT
inhibitor VIII was purchased from Merch KGaA.
Western blotting
GES-1 cells were co-cultured with H. pylori
strain 43504, and total protein was extracted in RIPA lysis buffer
(Beyotime Institute of Biotechnology). Protein concentration was
determined using a BCA assay kit (Thermo Fisher Scientific, Inc.).
Protein (20 µg/lane) from each sample was separated by 10% SDS-PAGE
and transferred to PVDF membranes, as previously described
(21). Following blocking with 5%
non-fat milk at room temperature for 1 h, the blots were incubated
with the primary antibodies overnight at 4°C and with
HRP-conjugated secondary antibodies (dilution, 1:5,000; cat. no.
ZB-2301; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.)
at 4°C for 4 h. The protein bands were visualized using the
Super-Signal West Femto Maximum Sensitivity Substrate Kit (Thermo
Fisher Scientific, Inc.) and exposed by the Bio-Rad Image Lab
system (Bio-Rad Laboratories, Inc.). The band intensities of the
target proteins were normalized to those of β-actin using Image Lab
software (version 5.2.1; Bio-Rad Laboratories, Inc.). The primary
antibodies and their sources were as follows: E-cadherin
(anti-E-cadherin antibody; dilution, 1:1,000; cat. no. ab1416;
Abcam), N-cadherin (anti-N-cadherin antibody; dilution, 1:1,000;
cat. no. ab18203; Abcam), AKT (anti-AKT antibody; dilution,
1:1,000; cat. no. 4691; Cell Signaling Technology, Inc.), p-AKT
(Ser473) (anti-p-AKT antibody; dilution, 1:1,000; cat. no. 9271;
Cell Signaling Technology, Inc.), GSK3β (anti-GSK3β antibody;
dilution, 1:1,000; cat. no. 9315S; Cell Signaling Technology,
Inc.), p-GSK3β (Ser9) (anti-p-GSK3β antibody; dilution, 1:1,000;
cat. no. 9322; Cell Signaling Technology, Inc.) and β-actin
(β-actin antibody; dilution, 1:1,000; cat. no. sc-1615-R; Santa
Cruz Biotechnology, Inc.).
Immunohistochemical analysis
Biopsy specimens and animal gastric tissues were
processed for immunohistochemical analysis as previously described
(21). Briefly, specimens were
sectioned at 4-µm thickness and baked at 70°C for 1.5 h.
Subsequently, sections were de-paraffinized in xylene and
rehydrated with gradient ethanol (100, 95 and 85% ethanol). The
sections were repaired with citrate buffer solution (TransGen
Biotech Co., Ltd.) at 100°C for 15 min, and then washed in PBS
solution (TransGen Biotech Co., Ltd.) three times. Following
blocking with 3% H2O2 solution at room
temperature for 8 min and incubation with 5% goat serum (Beyotime
Institute of Biotechnology) at room temperature for 30 min,
sections were incubated with the primary antibodies at 4°C
overnight and with secondary antibodies (dilution, 1:100; 100 µl
per section; cat. no. PV-6000; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd.) at 37°C for 30 mins. Sections were stained
with DAB solution (Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.) and evaluated under a fluorescence microscope (Olympus
bx53 biomicroscope; Olympus Corporation). The samples were stained
with anti-E-cadherin antibody (biopsy and gastric tissues;
dilution, 1:300; cat. no. ab1416; Abcam), anti-N-cadherin antibody
(biopsy and gastric tissues; dilution, 1:800; cat. no. ab18203;
Abcam), anti-AKT antibody (gastric tissues; dilution, 1:500; cat.
no. ab8805; Abcam), anti-p-AKT antibody (Ser473) (gastric tissues;
dilution, 1:500; cat. no. ab66138; Abcam), anti-GSK3β antibody
(gastric tissues; dilution, 1:400; cat. no. ab98081; Abcam) and
anti-p-GSK3β antibody (Ser9) (gastric tissues; dilution, 1:200;
cat. no. ab75814; Abcam). Two pathologists (Department of
Pathology, The First Affiliated Hospital of Nanchang University,
Nanchang, China) who were blinded to the clinicopathological data
scored (0–12 points) and assessed (grade 1–4) the stained sections
by evaluating and calculating the intensity (0–3 points) and
frequency (0–4 points) of staining. The relative expression levels
of the proteins were reported as grades as follows: grade 1 (0–2
points), grade 2 (3–5 points), grade 3 (6–8 points) and grade 4
(9–12 points) (18). Grading
discrepancies were re-reviewed and discussed to obtain a final
score.
Statistical analysis
The results are presented as the mean ± standard
deviation of three independent experiments or the percentage
compared with the control samples. The χ2 and
Mann-Whitney tests were used to analyze the statistical difference
between two groups. One-way ANOVA followed by Tukey's post hoc test
or the Kruskal-Wallis test followed by Bonferroni post hoc test
were used to determine the differences among multiple groups based
on the normality of the data (SPSS software; version 18.0; SPSS,
Inc.). P≤0.05 was considered to indicate a statistically
significant difference.
Results
Expression levels of EMT-associated
proteins in gastric carcinogenesis
To evaluate the expression levels of EMT-associated
proteins in patients with gastric carcinogenesis, an
immunohistochemical analysis of human gastric tissues of different
gastric lesions was performed. The expression levels of the
epithelial marker E-cadherin were decreased at the IM stage
compared with CNAG stage and continued to decrease in the GC stage.
E-cadherin expression was significantly reduced in GC tissues
compared with in CNAG tissues (P<0.001). However, the expression
levels of the mesenchymal marker N-cadherin were increased at the
IM stage compared with CNAG stage and continued to increase in the
subsequent stages of gastric carcinogenesis. N-cadherin expression
was significantly increased in GC tissues compared with in CNAG
tissues (P<0.05; Fig. 1A). These
results indicated that EMT may be involved in the initiation of the
early stage of gastric carcinogenesis.
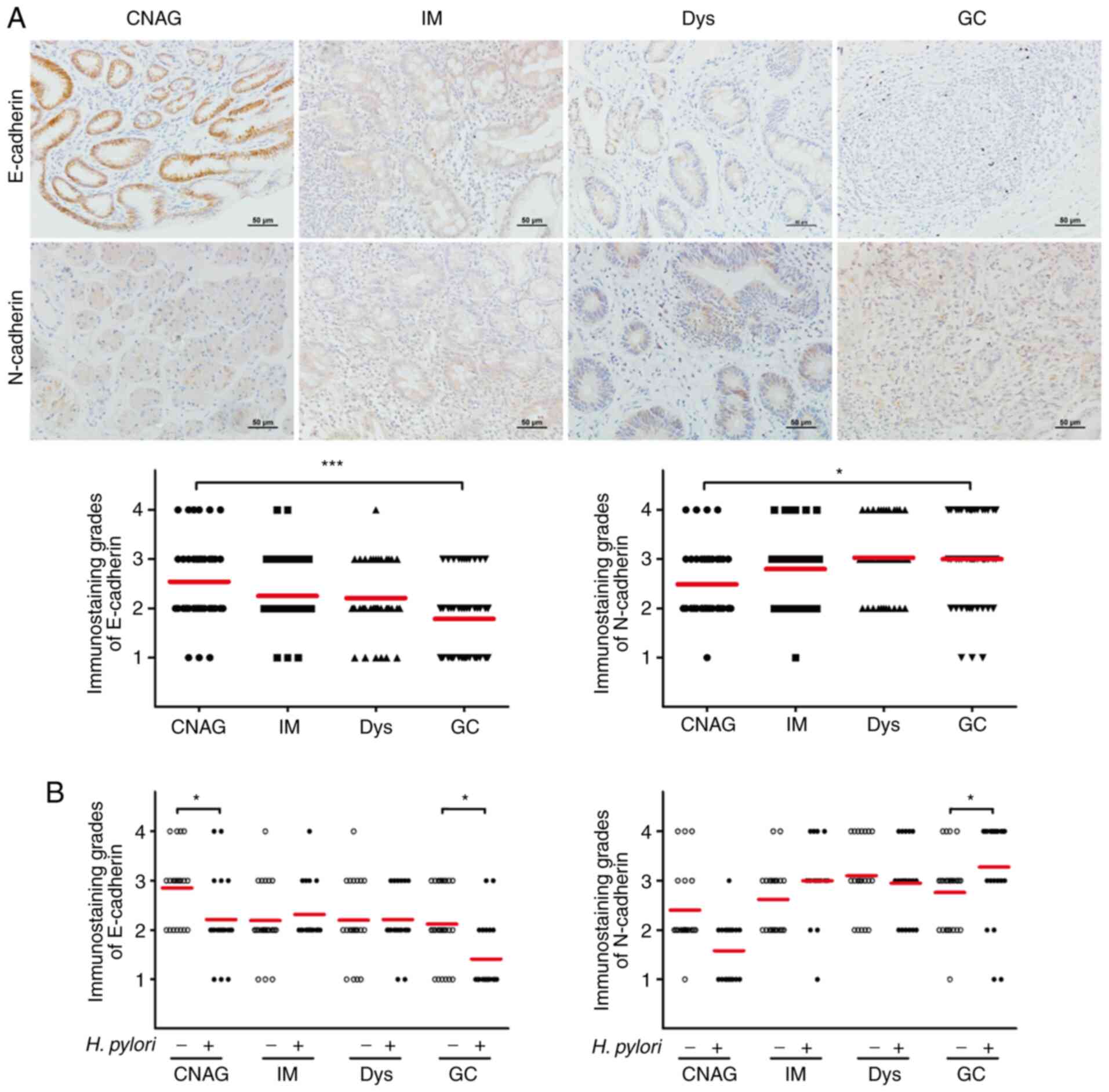 | Figure 1.Detection of the expression levels of
EMT-associated proteins in gastric carcinogenesis. (A) Expression
levels of the EMT-associated markers E-cadherin and N-cadherin in
165 gastric tissue samples at different disease stages (CNAG, IM,
Dys and GC) were detected by immunohistochemical analysis. The
expression grades were assessed by the staining intensity and
frequency. Scale bar, 50 µm. (B) Expression levels of E-cadherin
and N-cadherin were assessed in 165 gastric tissue samples at
different disease stages according to H. pylori infection
status. H. pylori infection was assessed by rapid urease
tests and Giemsa staining. *P<0.05, ***P<0.001. EMT,
epithelial-mesenchymal transition; CNAG, chronic non-atrophic
gastritis; IM, intestinal metaplasia; Dys, dysplasia; GC, gastric
cancer; H. pylori, Helicobacter pylori. |
H. pylori is a known important risk factor
that triggers gastric carcinogenesis (2). The association between H. pylori
infection and the expression levels of E-cadherin and N-cadherin
was examined in the patients. The immunohistochemical results
indicated that the expression levels of E-cadherin were
significantly lower following H. pylori infection compared
with those of non-infected patients (P<0.05) in CNAG and GC
stage, whereas the expression levels of N-cadherin were
significantly higher following H. pylori infection compared
with those of non-infected patients (P<0.05; Fig. 1B) in GC stage. There was no
significant difference in E-cadherin and N-cadherin expression in
IM and Dys stages. Overall, these results suggested that H.
pylori infection may induce EMT.
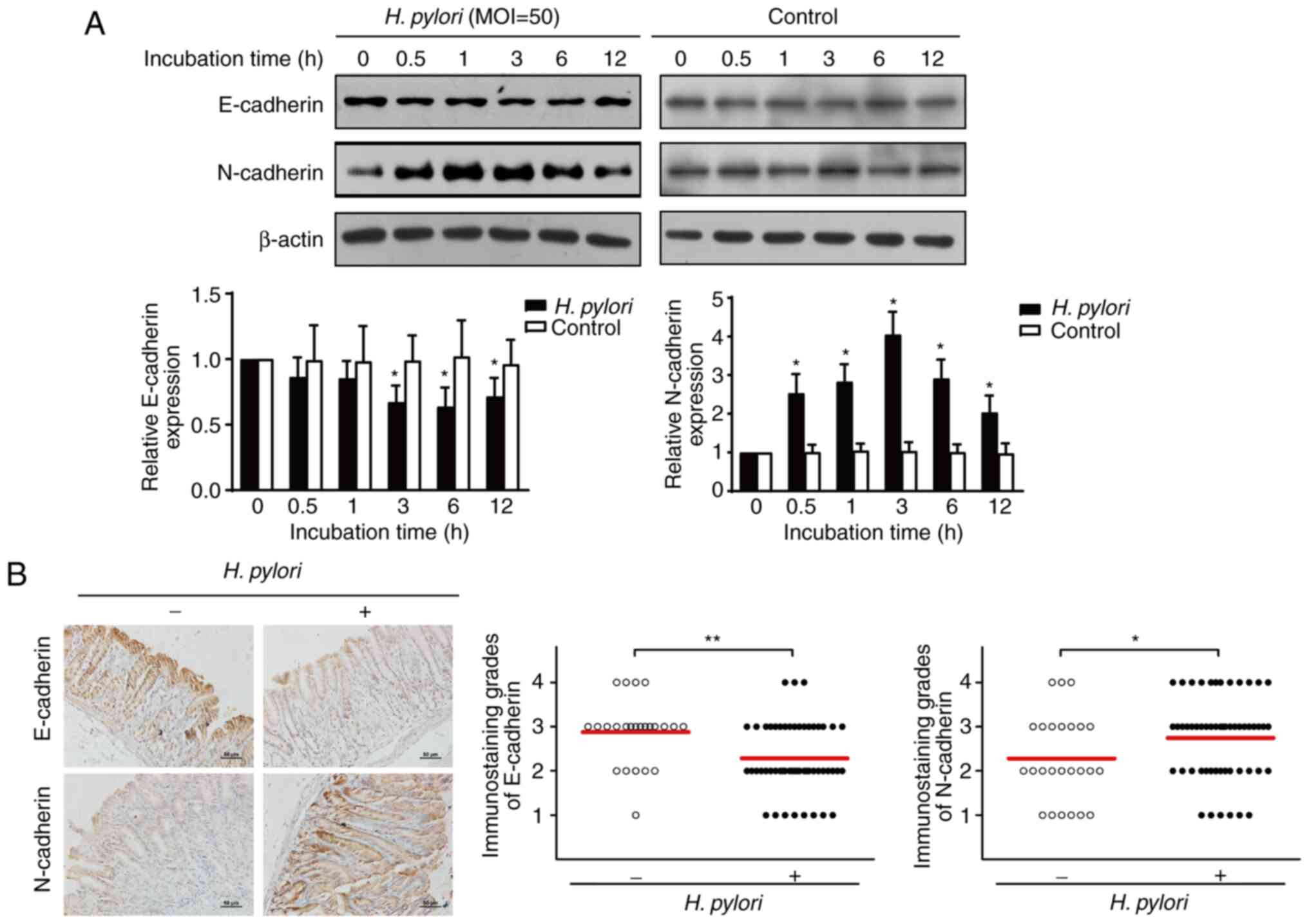
H. pylori infection induces EMT in
GES-1 cells and Mongolian gerbils
The initial observation demonstrated an association
between H. pylori infection and the expression levels of
EMT-associated markers in human gastric tissues. Subsequently, the
effects of H. pylori infection on EMT were investigated in
gastric epithelial GES-1 cells and in an animal model. GES-1 cells
were co-cultured with H. pylori and cellular proteins were
extracted at different time points. Protein detection was performed
by western blotting. The results demonstrated that H. pylori
infection significantly decreased the expression levels of
E-cadherin following 3 h of co-culture, whereas it increased the
expression levels of N-cadherin following 0.5 h of co-culture
(P<0.05; Fig. 2A). Subsequently,
immunohistochemical analysis was performed in order to examine the
expression levels of E-cadherin and N-cadherin in gastric tissues
of Mongolian gerbils following H. pylori infection and in
the corresponding tissues of the control group. H. pylori
infection significantly increased the expression levels of
N-cadherin and decreased the expression levels of E-cadherin
(P<0.05; Fig. 2B). In summary,
these in vivo and in vitro data demonstrated that
H. pylori infection may induce EMT.
H. pylori-induced EMT is regulated via
the AKT/GSK3β signaling pathway
The molecular mechanisms of H. pylori
infection were investigated with regard to EMT induction. The
AKT/GSK3β signaling pathway has previously been demonstrated to
induce EMT (13). To investigate
whether the AKT/GSK3β signaling pathway was involved in H.
pylori-induced EMT, the effects of H. pylori infection
on the AKT/GSK3β signaling pathway were further investigated in
GES-1 cells. Western blot analysis demonstrated that the relative
expression ratio of p-AKT (p-AKT/AKT) was significantly increased
in GES-1 cells following incubation of the cells with H.
pylori for 0.5, 1, 3 and 6 h, and no significant difference was
observed at 12 h compared with in cells in the group without H.
pylori incubation (P<0.05; Fig.
3A). The relative expression ratio of p-GSK3β (p-GSK3β/GSK3β)
was significantly increased in GES-1 cells that were incubated with
H. pylori for 3, 6 and 12 h compared with in cells in the
group without H. pylori incubation (P<0.05; Fig. 3A).
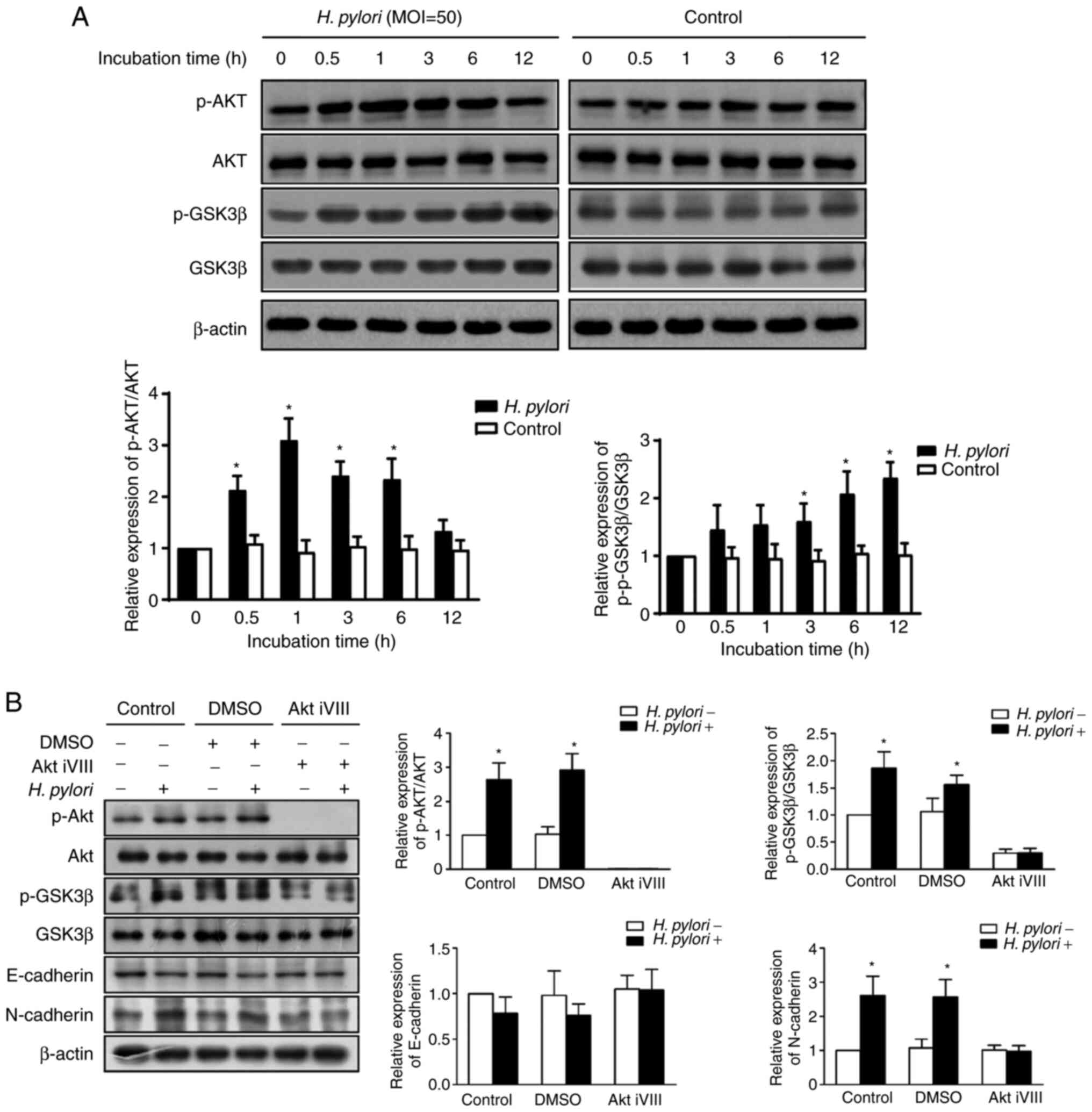 | Figure 3.H. pylori-induced
epithelial-mesenchymal transition is regulated via the AKT-GSK3β
signaling pathway. (A) Expression levels of the AKT-GSK3β signaling
pathway-associated proteins AKT, p-AKT (Ser473), GSK3β and p-GSK3β
(Ser9) were detected by western blotting in GES-1 cells following
0, 0.5, 1, 3, 6 and 12 h co-culture with or without H.
pylori. The statistical analysis of three independent
experiments is shown. (B) GES-1 cells were pretreated with DMSO,
the Akt iVIII or control medium. The expression levels of the
AKT-GSK3β signaling pathway-associated proteins were detected by
western blot analysis in GES-1 cells with or without H.
pylori infection. The statistical analysis of three independent
experiments is shown. *P<0.05 vs. 0 h in (A) and H.
pylori (−) group in (B). Akt iVIII, AKT inhibitor VIII; H.
pylori, Helicobacter pylori; p-, phosphorylated. |
The role of AKT in H. pylori-induced EMT was
further explored. As shown in Figs.
2 and 3, H.
pylori-infected GES-1 cells exhibited a prominent increase in
the levels of N-cadherin, p-AKT and p-GSK3β, whereas a decrease was
noted in the expression levels of E-cadherin compared with the
corresponding expression levels of the control group (P<0.05).
The results were similar in GES-1 cells that were pre-treated with
DMSO. H. pylori infection did not result in a change in the
expression levels of E-cadherin, N-cadherin, p-AKT and p-GSK3β in
GES-1 cells that were pre-treated with the AKT inhibitor VIII
(P>0.05; Fig. 3B). Therefore,
H. pylori-induced EMT may be blocked by AKT inhibition,
suggesting that H. pylori-induced EMT may be regulated by
the AKT/GSK3β signaling pathway.
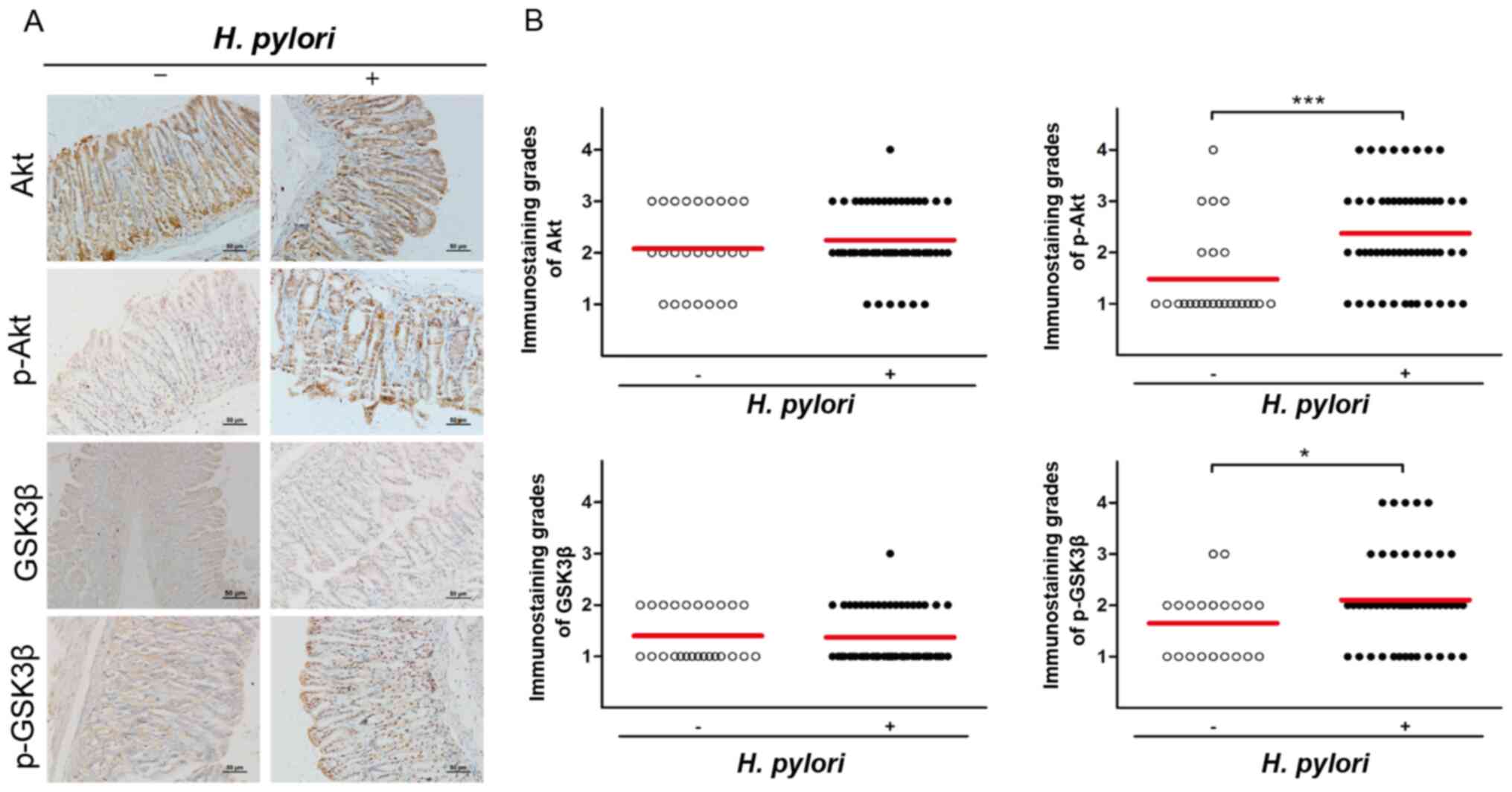
AKT and GSK3β are activated in H.
pylori-infected Mongolian gerbils
To further validate the role of the AKT/GSK3β
signaling pathway in H. pylori-induced EMT,
immunohistochemical analysis was performed to assess the expression
levels of AKT, p-AKT, GSK3β and p-GSK3β in gastric tissues of
Mongolian gerbils following H. pylori infection and they
were compared with those of the control group. Immunohistochemical
assays indicated that although the expression levels of AKT and
GSK3β did not exhibit significant differences between the two
groups (P>0.05), H. pylori infection significantly
increased the expression levels of p-AKT and p-GSK3β (P<0.05;
Fig. 4). Overall, these results
further validated the role of the AKT/GSK3β signaling pathway in
H. pylori-induced EMT.
Discussion
H. pylori infection is the main risk factor
for GC and is responsible for the initiation of gastric
carcinogenesis in different developmental stages, including CNAG,
IM, Dys and eventually GC (23). It
has been reported that various oncogenic signaling pathways are
involved in the pathogenic effects of H. pylori (24).
Sun et al (25) assessed the expression levels of
E-cadherin by immunohistochemical analysis in 58 GC tissues, 40
adjacent tissues and 42 CNAG tissues. Sun et al (25) demonstrated that the expression levels
of E-cadherin in specimens of patients with GC were significantly
lower than those of patients with CNAG, which is consistent with
the present findings. Furthermore, the present study highlighted
that the changes in the expression levels of the EMT markers
E-cadherin and N-cadherin were initially noted at the IM stage of
gastric adenocarcinoma formation, suggesting that EMT may
contribute to the initiation of gastric adenocarcinoma.
Subsequently, the specimens were divided into two groups according
to H. pylori infection status and the data demonstrated that
at the CNAG and GC stages, the expression levels of E-cadherin in
the H. pylori-positive group were significantly lower than
those in the H. pylori-negative group, while at the GC
stage, the expression levels of N-cadherin were significantly
higher in the H. pylori-positive group than in the H.
pylori-negative group. A recent study by Li et al
(6) indicated that H. pylori
infection reduces E-cadherin expression via the YY1 associated
protein 1 signaling pathway in patients with CNAG, which is
consistent with the results of the present study. These results
strongly suggest that H. pylori infection initiates the EMT
process at an early stage of gastric carcinogenesis, which may
subsequently promote growth, metastasis and invasion of GC.
Subsequently, GES-1 cells were infected with H.
pylori in vitro, which induced EMT, as detected by decreased
E-cadherin expression and increased N-cadherin expression in GES-1
cells in the early stage of H. pylori infection. In
addition, the effect of H. pylori infection on EMT was
further demonstrated by experiments in H. pylori-infected
Mongolian gerbils. H. pylori infection induced a decrease in
E-cadherin levels and an increase in N-cadherin expression, which
was consistent with the in vitro results.
Activation of the AKT signaling pathway is involved
in the EMT process by directly affecting the expression of specific
transcription factors, such as Twist, Snail and Slug, the
expression of integrin-linked kinase and extracellular matrix or by
interacting with other signaling pathways, such as the TGF-β,
NF-κB, Ras and Wnt/β-catenin signaling pathways (13). Activation of AKT/GSK3β can increase
the transcriptional activity and intracellular levels of β-catenin,
which exerts a positive regulatory effect on the Wnt signaling
pathway (26,27). The present results indicated that the
levels of p-AKT and p-GSK3β in GES-1 cells began to increase
following 0.5 and 3 h, respectively, of co-culture with H.
pylori and that this trend of p-AKT persisted until the 9 h
time point and the trend for p-GSK3β persisted until the 12 h time
point. Furthermore, GES-1 cells were pre-treated with the AKT
inhibitor VIII or DMSO, which was used as a negative control.
Following 1 h of co-culture with H. pylori, the protein
expression levels of p-AKT, p-GSK3β, N-cadherin and E-cadherin in
GES-1 cells that were pre-treated with the AKT inhibitor VIII did
not change significantly compared with those noted in the cells
that were not infected with H. pylori. These results
indicated that inhibition of AKT activity blocked the development
of EMT, demonstrating that the H. pylori-associated EMT
process was AKT-dependent.
To further validate the role of the AKT/GSK3β
signaling pathway in H. pylori-induced EMT, the levels of
AKT, p-AKT, GSK3β and p-GSK3β were examined in Mongolian gerbils
with or without H. pylori infection. Although no significant
differences were noted in the expression levels of AKT and GSK3β
between the two groups, H. pylori infection activated
phosphorylated AKT and phosphorylated GSK3β. These results were
consistent with the in vitro findings.
There were a few limitations of the present study.
Although the role of the AKT/GSK3β signaling pathway in H.
pylori-induced EMT was thoroughly examined, other signaling
pathways involved in the mechanism underlying this process require
further exploration. Although the present study focused on the role
of the AKT/GSK3β signaling pathway, whether this signaling pathway
interacts with other signaling pathways in H. pylori-induced
EMT remains to be explored. In addition, there is still a gap
between activation of AKT and EMT, and the exact mechanism by which
the AKT/GSK3β signaling pathway regulates EMT is not clear. Future
studies should investigate this gap in further.
In conclusion, the present study indicated that
H. pylori infection activated AKT and resulted in the
phosphorylation and inactivation of GSK3β, which promoted
early-stage EMT development. These effects were AKT-dependent and
were blocked by application of the AKT inhibitor VIII.
Acknowledgements
The authors would like to thank Professor Jianzhong
Zhang (Chinese Center for Disease Control and Prevention, Beijing,
China) for providing the wild-type H. pylori
CagA+ VaCA+ strain (ATCC43504; originally
from American Type Culture Collection).
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81060038,
81460377, 81670507, 81760106, 81870395, 81960112 and 81900500), the
Natural Science Foundation of Jiangxi Province of China (grant nos.
20142BAB215036 and 20171BAB205012), the Jiangxi Provincial
Department of Science & Technology (grant no. 20192BAB215006)
and the Subject of Jiangxi Provincial Department of Health (grant
no. 202110024).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YO, GL, JH and NLu conceived and designed the study.
YO and GL performed the experiments, and NLu analyzed the data. WX
and ZY collected human specimens and analyzed immunohistochemical
data. CX, CZ and JC provided assistance with analyses of the data
and technical issues. YO, NLi and YZ interpreted the data and
drafted the manuscript. NLu supervised the study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The use of human tissues was approved by the Human
Ethics Committee of the First Affiliated Hospital of Nanchang
University. All patients signed an informed consent form for their
participation in the study protocol. The First Affiliated Hospital
of Nanchang University Ethics Committee approved all animal
experiment protocols.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Karimi P, Islami F, Anandasabapathy S,
Freedman ND and Kamangar F: Gastric cancer: Descriptive
epidemiology, risk factors, screening, and prevention. Cancer
Epidemiol Biomarkers Prev. 23:700–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malfertheiner P, Megraud F, O'Morain CA,
Gisbert JP, Kuipers EJ, Axon AT, Bazzoli F, Gasbarrini A, Atherton
J, Graham DY, et al: Management of Helicobacter pylori
infection-the maastricht V/florence consensus report. Gut. 66:6–30.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Graham DY: Helicobacter pylori
update: Gastric cancer, reliable therapy, and possible benefits.
Gastroenterology. 148:719–731.e3. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watanabe T, Takahashi A, Suzuki K,
Kurusu-Kanno M, Yamaguchi K, Fujiki H and Suganuma M:
Epithelial-mesenchymal transition in human gastric cancer cell
lines induced by TNF-α-inducing protein of Helicobacter
pylori. Int J Cancer. 134:2373–2382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choi YJ, Kim N, Chang H, Lee HS, Park SM,
Park JH, Shin CM, Kim JM, Kim JS, Lee DH and Jung HC:
Helicobacter pylori-induced epithelial-mesenchymal
transition, a potential role of gastric cancer initiation and an
emergence of stem cells. Carcinogenesis. 36:553–563. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li N, Feng Y, Hu Y, He C, Xie C, Ouyang Y,
Artim SC, Huang D, Zhu Y, Luo Z, et al: Helicobacter pylori
CagA promotes epithelial mesenchymal transition in gastric
carcinogenesis via triggering oncogenic YAP pathway. J Exp Clin
Cancer Res. 37:2802018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong SHM, Fang CM, Chuah LH, Leong CO and
Ngai SC: E-cadherin: Its dysregulation in carcinogenesis and
clinical implications. Crit Rev Oncol Hematol. 121:11–22. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ye X and Weinberg RA:
Epithelial-mesenchymal plasticity: A central regulator of cancer
progression. Trends Cell Biol. 25:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dongre A and Weinberg RA: New insights
into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. Nat Rev Mol Cell Biol. 20:69–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu W, Yang Z and Lu N: A new role for the
PI3K/Akt signaling pathway in the epithelial-mesenchymal
transition. Cell Adh Migr. 9:317–324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Warfel NA and Kraft AS: PIM kinase (and
Akt) biology and signaling in tumors. Pharmacol Ther. 151:41–49.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Beurel E, Grieco SF and Jope RS: Glycogen
synthase kinase-3 (GSK3): Regulation, actions, and diseases.
Pharmacol Ther. 148:114–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katoh M and Katoh M: Cross-talk of WNT and
FGF signaling pathways at GSK3beta to regulate beta-catenin and
SNAIL signaling cascades. Cancer Biol Ther. 5:1059–1064. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shu X, Yang Z, Li ZH, Chen L, Zhou XD, Xie
Y and Lu NH: Helicobacter pylori Infection activates the
akt-Mdm2-p53 signaling pathway in gastric epithelial cells. Dig Dis
Sci. 60:876–886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang Z, Xie C, Xu W, Liu G, Cao X, Li W,
Chen J, Zhu Y, Luo S, Luo Z and Lu N: Phosphorylation and
inactivation of PTEN at residues Ser380/Thr382/383 induced by
Helicobacter pylori promotes gastric epithelial cell
survival through PI3K/Akt pathway. Oncotarget. 6:31916–31926. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hamilton SR and Aaltonen LA: Pathology and
genetics of tumours of the digestive system. IARC Press; Lyon:
2000
|
|
20
|
Dixon MF, Genta RM, Yardley JH and Correa
P: Classification and grading of gastritis. The updated sydney
system. International workshop on the histopathology of gastritis,
houston 1994. Am J Surg Pathol. 20:1161–1181. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ
and Lu NH: Reduced expression of PTEN and increased PTEN
phosphorylation at residue Ser380 in gastric cancer tissues: A
novel mechanism of PTEN inactivation. Clin Res Hepatol
Gastroenterol. 37:72–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu W, Huang Y, Yang Z, Hu Y, Shu X, Xie C,
He C, Zhu Y and Lu N: Helicobacter pylori promotes gastric
epithelial cell survival through the PLK1/PI3K/Akt pathway. Onco
Targets Ther. 11:5703–5713. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suerbaum S and Michetti P: Helicobacter
pylori infection. N Engl J Med. 347:1175–1186. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naumann M, Sokolova O, Tegtmeyer N and
Backert S: Helicobacter pylori: A paradigm pathogen for
subverting host cell signal transmission. Trends Microbiol.
25:316–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun GY, Wu JX, Wu JS, Pan YT and Jin R:
Caveolin-1, E-cadherin and β-catenin in gastric carcinoma,
precancerous tissues and chronic non-atrophic gastritis. Chin J
Cancer Res. 24:23–28. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sheng S, Qiao M and Pardee AB: Metastasis
and AKT activation. J Cell Physiol. 218:451–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qiao M, Sheng S and Pardee AB: Metastasis
and AKT activation. Cell Cycle. 7:2991–2996. 2008. View Article : Google Scholar : PubMed/NCBI
|