Introduction
Colorectal cancer ranks as the second most deadly
cancer among all cancer types, and colorectal cancers are commonly
treated with 5-fluorouracil (5-FU), anticancer drug. 5-FU is known
to interfere with DNA synthesis in cancer cells by inhibiting
thymidylate synthase, as a mechanism that inhibits cell
proliferation and leads to cell death (1–3).
Although 5-FU plays a central role in the treatment of colorectal
cancer, it elicits any effect in only 10–15% of colorectal cancer
patients, and approximately 40–50% of treatments are successful
upon combination therapy with other antitumor drugs, including
oxaliplatin, leucovorin, and others. Therefore, outlining the
mechanisms underlying sensitivity to 5-FU among colorectal cancer
cells would be helpful.
Cancer research has shown p53 to be a tumor
suppressor, and p53 dysfunction has been found to lead to the
development of colorectal cancer. Moreover, p53 has been
characterized a key factor influencing drug sensitivity: p53
overexpression has been shown to increase the effect of
chemotherapy in patients with stage III colorectal cancer with
mutant p53 (4–6). The major biological function of p53 is
to initiate cell apoptosis by regulating the cell cycle at the G1/S
and the G2/M check points. Interestingly, p53 studies have
indicated that active p53 exhibits both transcriptional and
non-transcriptional activity and that both can affect apoptosis.
Under various cell death conditions, the non-transcriptional
activity of p53 induces mitochondrial outer membrane
permeabilization (MOMP), and this triggers the release of
pro-apoptotic factors from the mitochondrial inter-membrane space
(7–9).
Chromatin structure and organization influence the
identity of cells by regulating cell-specific signaling networks
(10,11), and recently, various epigenetic
studies have reported a link between alterations in chromatin
structure and cancer progression (12–15).
Measurement of chromatin accessibility during prostate cancer
progression revealed that chromatin relaxation increases the
binding of oncogenic transcription factors, resulting in increased
expression of tumor growth-related genes (16,17).
Another report showed that TGF-β induces chromatin accessibility to
cis-regulatory elements of epithelial-to-mesenchymal
transition-related genes, including CDH1, CDH2, and
VIM. Additionally, research has shown that TGF-β induced
chromatin accessibility increases the transcriptional activity of
RUNX, AP-1, and Etv4/5, and results in epithelial-to-mesenchymal
transition in mammary gland epithelial cells (18). Altogether, these studies indicate
that external stimuli can affect chromatin accessibility and that
such effects are strongly related with stimulus-mediated cellular
phenotypes.
While it is known that p53 plays a key role in
successful chemotherapy, the mechanisms of how p53 affects 5-FU
sensitivity remain unknown. Rubbi and Milner (19) showed that ultraviolet radiation
induces p53-mediated global chromatin relaxation. However, the
authors did not demonstrate the effects of global chromatin
relaxation on DNA damage-induced cell death (19). Therefore, in this study, we
hypothesized that since 5-FU is also a DNA-damaging reagent, like
ultraviolet radiation, TP53-wild-type (TP53-WT) and TP53-knockout
(TP53-KO) colorectal cancer cells would show differences in gene
expression at chromatin accessible regions under 5-FU treatment and
that such differences would affect 5-FU sensitivity depending on
p53 status. To prove our hypothesis, we performed Assay for
Transposase-Accessible Chromatin sequencing (ATAC-seq) to assess
chromatin accessibility and RNA sequencing (RNA-seq) to measure
associated gene expression in both TP53-WT and TP53-KO colorectal
cancer cells exposed to 5-FU treatment.
Materials and methods
Cell culture
The human colorectal carcinoma cell line HCT116
TP53-WT and HCT116 TP53-KO, initially described by Bunz et
al (20), were obtained as a
kind gift from Dr Jae-Hoon Jeong (Korea Institute of Radiological
and Medical Sciences, Korea). The genetic background of TP53-WT and
TP53-KO were verified by performing genotyping using PCR primers
(Table SI), which amplify a 220-bp
band for TP53 exon 2 and a 519-bp band for CTCF exon 12 as the
control (Fig. S1). Target DNA for
genotyping was amplified using Dr. Taq DNA Polymerase (Allforlab)
with initial denaturation at 95°C for 3 min, followed by 35 cycles
of denaturation at 95°C for 10 sec, annealing at 63°C for 30 sec,
extension at 72°C for 1 min, and a final elongation step at 72°C
for 5 min. Cells were cultured in Roswell Park Memorial Institute
1640 medium (Hyclone) supplemented with 10% fetal bovine serum
(Hyclone) and 1% penicillin/streptomycin (100 U/ml:100 µg/ml;
Hyclone) and were grown at 37°C, 5% CO2, and 98%
humidity.
Cell viability assay
Cell viability was analyzed using CellTiter Glo
(Promega, G7570) according to the manufacturer's instructions.
Luminescence was measured using a microplate luminometer (Berthold
Centro XS3 LB 960) and MikroWin2000 software.
ChIPmentation library preparation
Chromatin immunoprecipitation followed by sequencing
(ChIP-seq) assays were performed using p53 antibodies (Santa Cruz
Biotechnology, Inc.; cat. no. sc-126) as described previously
(21) with minor modifications.
ChIP-seq libraries were prepared using a Nextera DNA Sample Prep
kit (Illumina, Inc.) according to the manufacturer's instructions.
Libraries were sequenced on an Illumina HiSeq 2500 system,
generating 100 bp paired-end reads.
RNA-seq library preparation
Total RNA was isolated using Hybrid-R Total RNA kits
(GeneAll Biotechnology Co. Ltd.), and rRNAs were depleted by using
the NEBNext rRNA Depletion kit (NEB; cat. no. E6350). RNA
sequencing (RNA-seq) libraries was prepared using NEBNext Ultra RNA
library prep kits (NEB; cat. no. 7770) according to the
manufacturer's instructions. Libraries were sequenced on an
Illumina HiSeq 2500 system, generating 100 bp paired-end reads.
ATAC-seq library preparation
Assay for Transposase-Accessible Chromatin
sequencing (ATAC-seq) libraries were prepared as described
previously (22) and sequenced on an
Illumina HiSeq 2500 system, generating 100 bp paired-end reads.
ChIP-seq data processing
Raw reads were trimmed using trim galore with
default parameters to remove low quality and adapter sequences.
Trimmed reads were mapped to the human genome reference hg19 using
BWA (23). The reference genome was
downloaded from GENCODE V19. Reads with low mapping quality were
filtered using SAMtools (24). MAPQ
<30 were used to obtain uniquely mapped reads. Duplicated reads
were filtered using Picard (Picard version 2.23.9; http://broadinstitute.github.io/picard).
Reads mapped to mitochondrial chromosomes were filtered. Peaks were
called using MACS2 (25) with input
DNA as a control.
ATAC-seq data processing
Raw reads were trimmed using trim galore with the
same default parameters as in ChIP-seq analysis. Trimmed reads were
mapped to the hg19 reference genome using BWA (23). Fragments below 2,000 bp were allowed.
Reads with low mapping quality were filtered using SAMtools
(24). Duplicated reads and
mitochondrial chromosomes were filtered as described above. Since
Tn5 transposase binds as a dimer and inserts two adaptors separated
by 9 bp, all aligned reads were shifted by +4 bp on the positive
strand and −5 bp on the negative strand using the deepTools
(26) function alignmentSieve. To
obtain nucleosome free regions, fragments over 140 bp in length
were filtered out. ATAC-seq peak calling was performed using MACS2
(25). ATAC-seq peaks across samples
were merged using mergePeaks, and differential peaks were called
using Homer getDifferentialPeaks.pl (Homer version 4.11; http://homer.ucsd.edu). Overlapping of open chromatin
regions with enrichment of p53 was analyzed using bedtools
(27) intersect: Overlap of at least
1-bp intervals was counted. Homer annotatePeaks.pl was used to
annotate differential ATAC-seq peaks to their nearest genes. Homer
findMotifsGenome.pl. was used to identify motifs enriched at open
chromatin regions. ATAC-seq and ChIP-seq data were visualized using
the Integrative Genomics Viewer (28).
RNA-seq data processing
Raw reads were trimmed using trim galore. For
transcript abundance quantification analysis, STAR (29) was used to map the reads to the human
genome. RSEM (30) was used to
quantify transcripts. Differentially expressed genes between groups
were calculated using DESeq2 R package (31). Genes with low read counts were
filtered out before differential analysis. DESeq2 provides a
statistical analysis method and assessment of differential
expression using a model based on negative binomial distribution.
Gene ontology and pathway enrichment analyses were performed using
the Enrichr online tool (32,33).
Statistics
All experimental values are presented as mean ± SD.
Cell viability assays were performed in triplicate, and RNA-seq and
ATAC-seq experiments were performed in duplicate. Most of
statistical significance was calculated by two-way analysis of
variance, in which P<0.05 was considered statistically
significant (GraphPad Prism 7 software; GraphPad Software, Inc.).
Post hoc multiple comparisons were followed by Bonferroni
correction. Two groups were compared using unpaired Student's
t-test, in which P<0.001 was considered statistically
significant (R version 4.0.3; The R Foundation).
Results
Chromatin accessibility changes in
response to 5-FU depending on p53 status
Responding to drug stimulation, p53 has been shown
to increase drug sensitivity in various cancer cells (34,35). In
order to investigate the effects of p53 on responses to 5-FU among
colorectal cancer cells (HCT116 cells), we first treated TP53-WT
and TP53-KO HCT116 cells with 5-FU and then measured mRNA
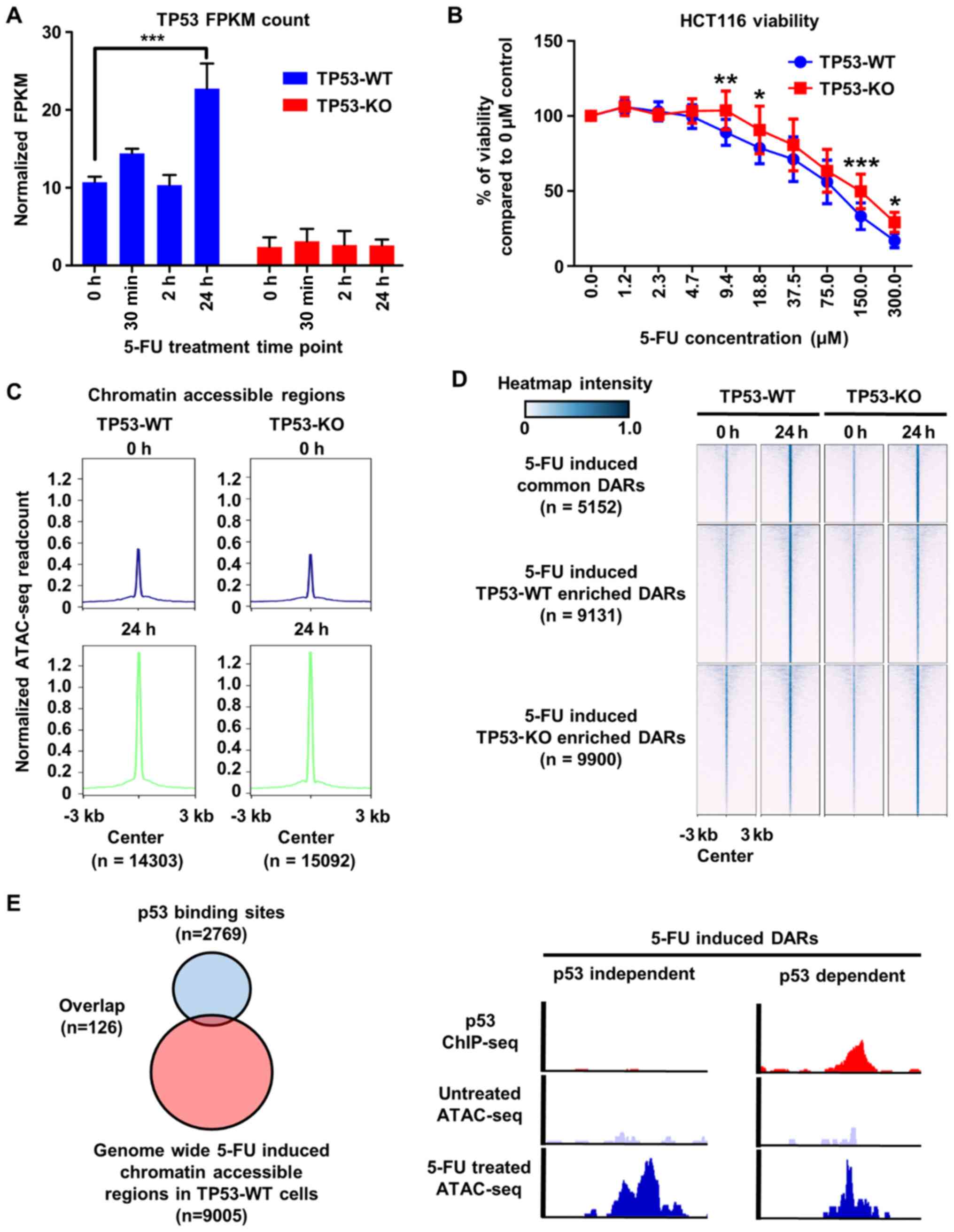
expression levels of p53 using RNA-seq analysis. As expected, we
noted that p53 expression increased under 5-FU treatment at 24 h in
TP53-WT cells, but not TP53-KO cells (Fig. 1A). Next, we performed cell viability
assay and found that the presence of p53 conferred sensitivity to
5-FU among HCT116 cells (Fig. 1B),
clearly confirming the role of p53 in 5-FU sensitivity in HCT116
cells.
Physical DNA accessibility is important in
establishing and maintaining cellular identity through the
regulation of the expression of various genes (36,37).
Moreover, recent research has shown that drug-mediated cellular
replication stress induces chromatin remodeling and that such
modification is highly associated with drug responses in cells
(38,39). To ascertain the effects of 5-FU on
chromatin accessibility, we performed ATAC-seq before and after
5-FU treatment, and found that global chromatin accessibility
increased around 3-fold under 5-FU treatment for 24 h in both
TP53-WT and TP53-KO cells (Fig. 1C).
Next, we analyzed regions showing differences in chromatin
accessibility between TP53-WT and TP53-KO cells after 5-FU
treatment to evaluate the role of p53 in 5-FU-mediated chromatin
remodeling. When we merged and compared all ATAC-seq peaks called
from 5-FU-treated TP53-WT and TP53-KO cells, we found that
chromatin accessibility was equal at 5152 regions in the cells
under 5-FU treatment regardless of the presence of TP53 (Fig. 1D). Respectively, however, 5-FU
elicited higher chromatin accessibility at 9,131 regions in TP53-WT
cells and at 9,900 regions in TP-53-KO cells (Fig. 1D). These data indicated that the
expression status of p53 influences chromatin accessibility in
response to 5-FU.
It is well known that p53 functions as a
transcription factor that binds to specific DNA sequences to
trans-activate a number of genes (40,41). To
determine whether p53 impacts 5-FU-mediated changes in chromatin
accessibility by directly binding to regulatory regions of DNA, we
performed chromatin immunoprecipitation followed by sequencing
(ChIP-seq) to analyze the distribution of binding sites for p53 in
TP53-WT cells. Consistent with a previous report (42), most TP53 binding regions were located
in inaccessible chromatin regions, and only 1.4% of 5-FU-induced
accessible regions overlapped with TP53 binding sites (Fig. 1E). These results demonstrated that
p53 influences 5-FU-mediated changes in chromatin accessibility in
a transcription-independent manner.
Changes in chromatin accessibility are
associated with distinct gene expression profiles in response to
5-FU
Next, we explored genome-wide changes in gene
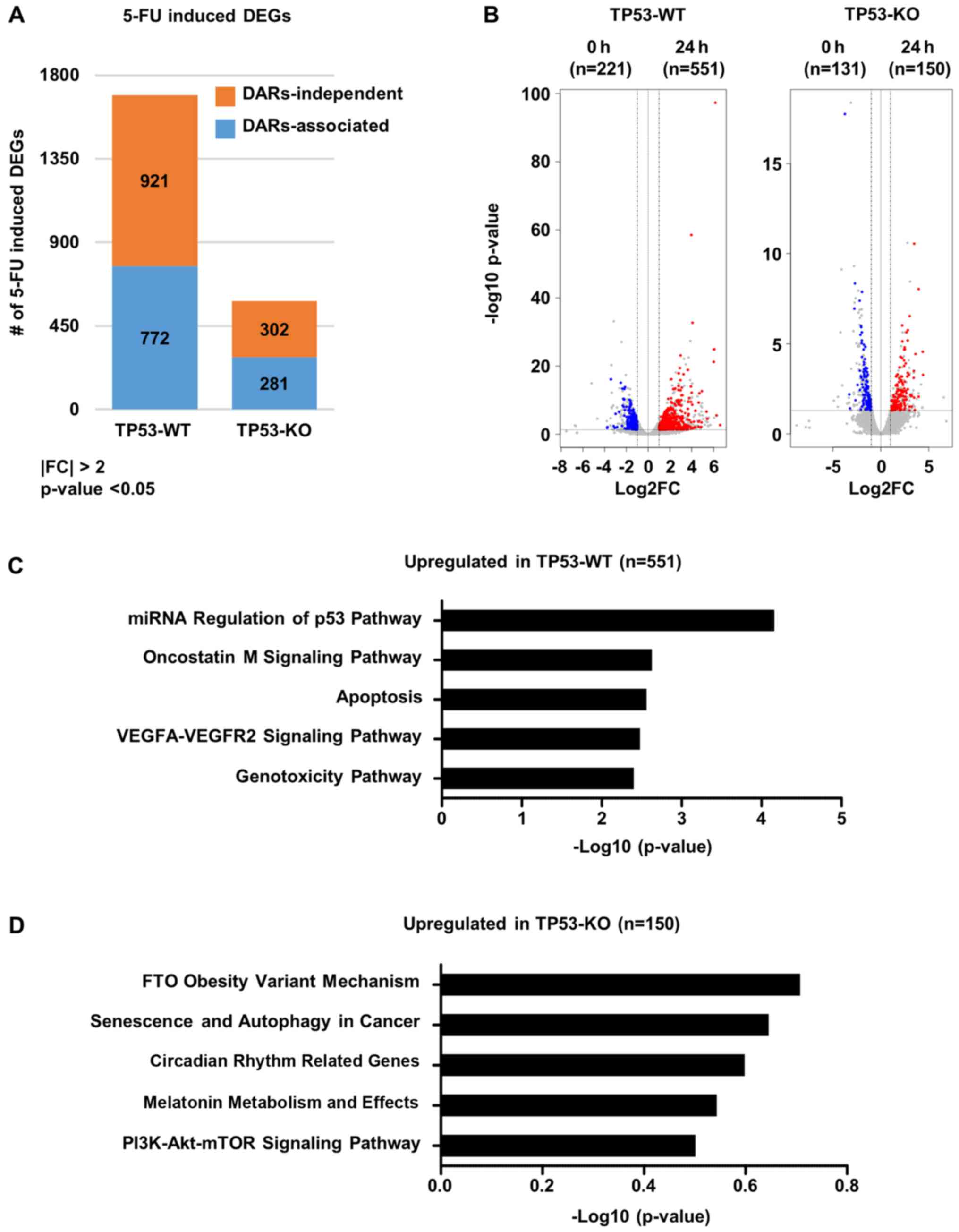
expression profiles following 5-FU treatment by performing RNA-seq.
In response to 5-FU treatment, a larger number of differentially
expressed genes (DEGs, fold change >2, P-value <0.05) in
TP53-WT cells than in TP53-KO cells (1693 genes in TP53-WT versus
583 genes in TP53-KO) (Fig. 2A).
Interestingly, 772 and 281 DEGs from TP53-WT and TP53-KO cells,
respectively, were directly associated with 5-FU-induced changes in
chromatin accessibility (Fig. 2A).
Moreover, expression of 551 and 150 genes were significantly
increased following 5-FU treatment in TP53-WT and TP53-KO cells,
respectively (Fig. 2B). Gene
ontology analysis for 5-FU-mediated upregulated genes harboring
differentially accessible regions (DARs) in TP53-WT cells revealed
significant enrichment of genes known to be associated with p53
pathways, apoptosis, and genotoxicity (Fig. 2C), while those in TP53-KO cells
showed enrichment of genes associated with cancer progression,
including senescence and autophagy, circadian rhythm-related genes,
melatonin metabolism, and PI3K-AKT-mTOR signaling (Fig. 2D). Moreover, we found that 135 genes
were overlapped among the genes whose expressions and chromatin
accessibilities were changed after 5-FU treatment in both TP53-WT
and TP53-KO cells. Gene ontology analysis of these overlapped genes
revealed significant enrichment of genes associated with
transcription regulation, cell differentiation regulation and
apoptosis (Fig. S2). These results
indicated that changes in chromatin accessibility in response to
5-FU elicit distinct gene expression profiles in colorectal cancer
cells depending on p53 status.
Specific chromatin accessible regions
in TP53-WT cells are highly associated with expression of cell
death-related genes
Among the regions showing higher 5-FU-mediated
chromatin accessibility in TP53-WT cells, compared to TP53-KO cells
(Figs. 1D and 3A), we selected DARs that exhibited a
greater than two-fold increase in TP53-WT cells, with an adjusted
P-value of less than 0.05. The annotation of these TP53-WT-specific
DARs to their nearest genes allowed us to identify 95 genes with
significantly higher expression in TP53-WT cells than TP53-KO cells
(Fig. 3B; Table SII). Gene ontology analysis
demonstrated that these TP53-WT-specific upregulated genes were
significantly enriched in cell death pathways (Table I). As a representative example,
Fig. 3C shows that chromatin
accessibility in the promoter regions of two tumor suppressor
genes, SEMA3B and PHLDA3, as well as their mRNA
levels, significantly increased in response to 5-FU treatment in
TP53-WT cells only. De novo transcription factor binding motif
analysis revealed that AP-1 complex motifs were enriched at the
TP53-WT specific chromatin accessible regions
(P=1×10−31) (Fig. 3D).
AP-1 complex is a well characterized transcription factor family
known to be activated in response to various cellular stress
signals, and research has shown that it regulates various genes
related with proliferation, immune response, and cell death
(43). One of the AP-1 family
transcription factors in particular, c-Jun, has been found to
inhibit cell proliferation and to induce apoptosis in response to
genotoxic stress by increasing caspase activity (44–46).
These results suggest that, unlike in TP53-KO cells, 5-FU elicits
increases in chromatin accessibility at specific regions in TP53-WT
cells that promote the binding of AP-1 transcription factor to
genes related to cell death, which may contribute to the higher
sensitivity of TP53-WT cells to 5-FU.
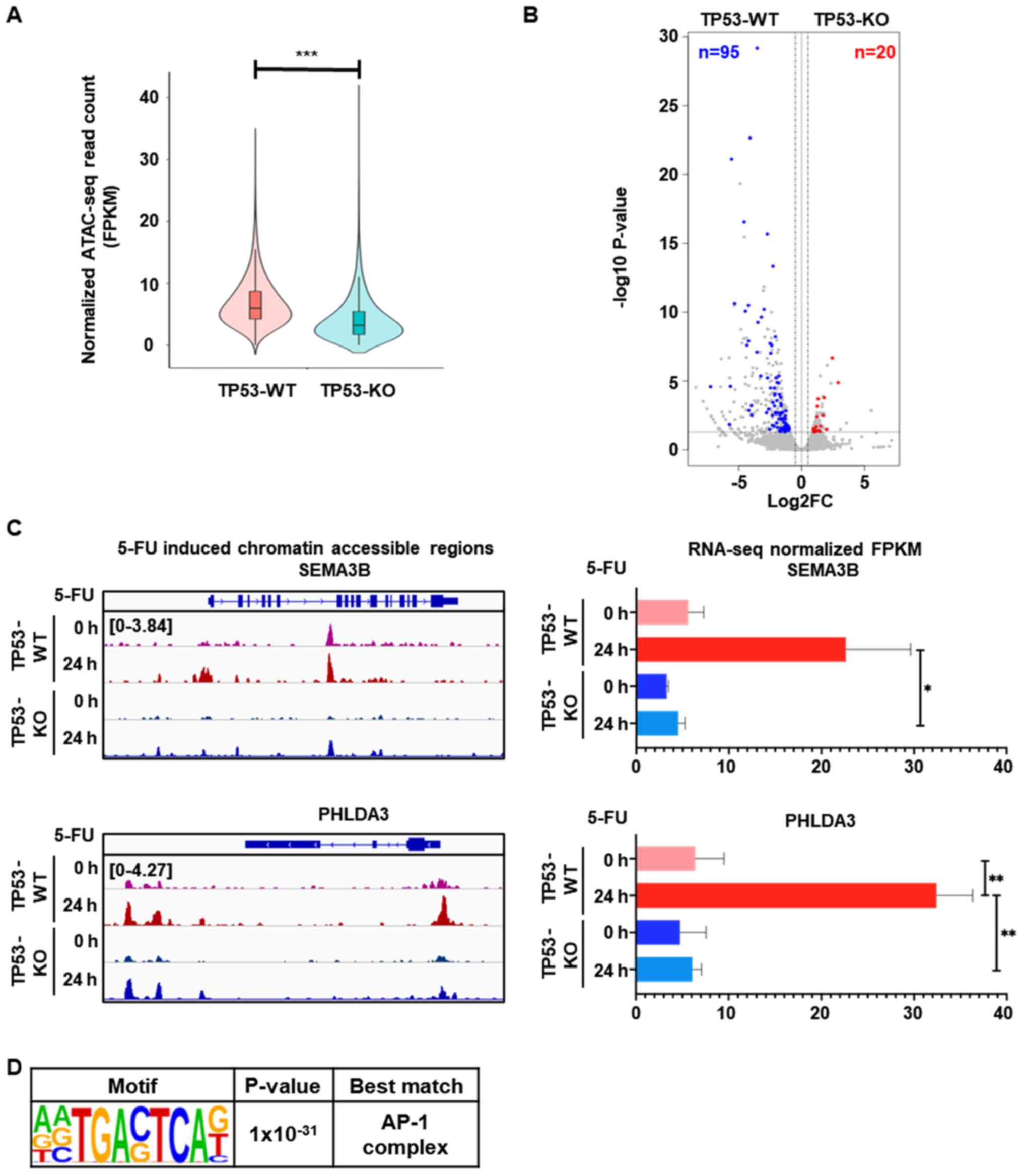 | Figure 3.Distinct 5-FU-induced gene expression
is associated with higher chromatin accessibility in TP53-WT cells
than TP53-KO cells. (A) Violin plot showing 9,131 DARs with higher
5-FU-induced chromatin accessibility in TP53-WT cells, compared to
TP53-KO cells (n=2 per group, ***P<0.001). (B) Volcano plot
showing DEGs associated with 5-FU-induced DARs that exhibited a
greater than two-fold increase in TP53-WT cells, with an adjusted
P-value of less than 0.05. The number of genes that exhibited
greater than two-fold increases in TP53-WT (blue) and TP53-KO (red)
cells with a P-value of less than 0.05 are indicated. (C) Genomic
snapshot of the SEMA3B (upper left) and PHLDA3 (lower
left) genes showing the densities of ATAC-seq reads in TP53-WT and
TP53-KO cells before and after 5-FU treatment for 24 h. Bar graph
shows the mRNA expression levels of SEMA3B (upper right) and
PHLDA3 (lower right) genes measured by RNA-seq from TP53-WT
and TP53-KO cells before and after 5-FU treatment for 24 h (n=2 per
group, *P<0.05; **P<0.01). (D) Enriched de novo motif
analysis for the DARs that showed both higher mRNA expression and
greater chromatin accessibility in TP53-WT cells, compared to
TP53-KO cells, after 5-FU treatment for 24 h. DARs, differentially
accessible regions; TP53-WT, TP53-wild-type; TP53-KO,
TP53-knockout. |
 | Table I.GO analysis for the 95
TP53-WT-specific upregulated genes (related with Fig. 3B). |
Table I.
GO analysis for the 95
TP53-WT-specific upregulated genes (related with Fig. 3B).
| GO biological
process | Fold
enrichment | P-value |
|---|
| Metal ion transport
(GO:0030001) | 4.84 |
1.17×10−2 |
| Regulation of cell
adhesion (GO:0030155) | 4.33 |
4.25×10−2 |
| Regulation of cell
death (GO:0010941) | 2.99 |
1.51×10−2 |
| Cellular response
to chemical stimulus (GO:0070887) | 2.55 |
9.35×10−4 |
| Regulation of cell
communication (GO:0010646) | 2.25 |
6.93×10−3 |
| Regulation of
signaling (GO:0023051) | 2.23 |
7.96×10−3 |
| Response to
chemical (GO:0042221) | 2.16 |
8.63×10−4 |
| Negative regulation
of cellular process (GO:0048523) | 2 |
8.91×10−3 |
| Cell communication
(GO:0007154) | 1.9 |
8.53×10−3 |
| Negative regulation
of biological process (GO:0048519) | 1.88 |
1.49×10−2 |
| Cellular response
to stimulus (GO:0051716) | 1.86 |
2.72×10−4 |
| Signaling
(GO:0023052) | 1.85 |
2.90×10−2 |
| Response to
stimulus (GO:0050896) | 1.76 |
1.13×10−5 |
| Biological
regulation (GO:0065007) | 1.4 |
6.52×10−3 |
Discussion
Modification of the structure of chromatin can
affect the expression of various genes, and it results in changing
molecular signaling networks influencing cellular phenotypes.
According to the research about the chromatin accessibility and
architecture, extracellular stimulation remodels chromatin
structure, and it results in modification of cellular signaling
networks toward unique cellular phenotypes (18,47). In
this study, we found through RNA- and ATAC-seq that 5-FU increases
chromatin accessibility in colorectal cancer cells (HCT116 cells)
and that the regions of increased chromatin accessibility induced
by 5-FU differed depending on p53 status: In cells expressing p53,
5-FU elicited increased chromatin accessibility in genes important
to p53 pathways, apoptosis, and genotoxicity, whereas in cells not
expressing p53, 5-FU elicited increased chromatin accessibility in
genes important to cancer progression.
ATAC-seq is one of the best experimental techniques
with which to measure chromatin accessibility, and it allows
researchers to predict main transcription factors in cis-regulatory
regions. In this study, we used ATAC-seq to prove our hypothesis
that the sensitivity among colorectal cancer cells to 5-FU may be
due to chromatin accessibility and p53 status. ATAC-seq clearly
showed that 5-FU increased chromatin accessibility in HCT116 cells
(Fig. 1C and D). Noteworthy, 5-FU
induced chromatin accessibility differed depending on p53 status in
HCT116 cells (Fig. 1D). To identify
p53 activities related with 5-FU induced chromatin accessibility,
we compared p53 ChIP-seq and ATAC-seq results under 5-FU treatment
in TP53-WT cells. From the integration analysis, we found that the
difference in chromatin accessibility was not the direct effect of
p53 binding: 96% of p53 binding regions were located in chromatin
inaccessible regions (Fig. 1E).
Thus, we deemed that the effects of p53 on 5-FU-induced chromatin
accessibility in TP53-WT cells are related with non-transcriptional
activities. Studies of p53 in relation to drug sensitivity have
primarily focused on correlations between signaling networks and
p53 status, such as genetic mutation and gene silencing, and most
have focused solely on the transcriptional activities of p53
(34,48,49).
Nonetheless, a few studies have recently described the effects of
p53 non-transcriptional activity on apoptosis. Although these
studies only reported on MOMP (7–9), their
results, together with ours, indicate that p53 may carry out
several non-transcriptional functions that await discovery. Given
the high incidence of TP53 mutation in many different types of
cancer, it would be interesting to explore the role of p53 in drug
sensitivity and chromatin structure in diverse cancer cells, which
is beyond the scope of the current study but remains to be our next
project to pursue.
In motif analysis of regions showing increased
chromatin accessibility upon treatment with 5-FU in TP53-WT cells,
we detected the AP-1 complex binding motif (Fig. 3D). Various contradictory functions
have been reported for AP-1 complex: It generally induces cell
proliferation (50) and
differentiation (51); however, one
study has indicated that it also shows an apoptotic function
(45). Of note, the contradictory
functions of AP-1, especially c-JUN, appear to depend on the
cellular context and extracellular stimuli (52). Although the function of AP-1 complex
in relation to apoptotic signaling is still controversial and
unclear, we presume that, through non-transcriptional activity, p53
increases the accessibility of AP-1 binding regions in response to
treatment with 5-FU in TP53-WT cells, resulting in activation of
apoptotic signaling pathways, thereby conferring 5-FU sensitivity.
Unfortunately, however, since motif analysis is only useful in
predicting possible binding proteins based on genomic sequences, we
are unable to confirm the direct effects of AP-1 complex on 5-FU
sensitivity based on these results alone. Further studies are
warranted to confirm our results.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by National Research
Foundation of Korea (NRF) grants funded by the Korean government
(MSIP) (grant nos. 2017M3C9A5029978, 2018M3A9D3079290, and
2020R1A2C2013258 to HPK).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Gene Expression Omnibus
repository, under the accession number GSE158021.
Authors' contributions
HPK conceived and coordinated the study. HPK and CMY
made substantial contributions to the design and interpretation of
data. CMY, MKK, JSJ and YC performed experiments including
ATAC-seq, RNA-seq and ChIP-seq. CMY, WJJ and YJK performed
bioinformatics analysis. HPK, WJJ and CMY confirmed the
authenticity of raw data. HPK and CMY wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Diasio RB and Harris BE: Clinical
pharmacology of 5-fluorouracil. Clin Pharmacokinet. 16:215–237.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jordan VC: A Retrospective: On clinical
studies with 5-fluorouracil. Cancer Res. 76:767–768. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang N, Yin Y, Xu SJ and Chen WS:
5-Fluorouracil: Mechanisms of resistance and reversal strategies.
Molecules. 13:1551–1569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Russo A, Bazan V, Iacopetta B, Kerr D,
Soussi T and Gebbia N; TP53-CRC Collaborative Study Group, : The
TP53 colorectal cancer international collaborative study on the
prognostic and predictive significance of p53 mutation: Influence
of tumor site, type of mutation, and adjuvant treatment. J Clin
Oncol. 23:7518–7528. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giovannetti E, Backus HH, Wouters D,
Ferreira CG, van Houten VM, Brakenhoff RH, Poupon MF, Azzarello A,
Pinedo HM and Peters GJ: Changes in the status of p53 affect drug
sensitivity to thymidylate synthase (TS) inhibitors by altering TS
levels. Br J Cancer. 96:769–775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dominijanni A and Gmeiner WH: Improved
potency of F10 relative to 5-fluorouracil in colorectal cancer
cells with p53 mutations. Cancer Drug Resist. 1:48–58. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moll UM, Marchenko N and Zhang XK: p53 and
Nur77/TR3-transcription factors that directly target mitochondria
for cell death induction. Oncogene. 25:4725–4743. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leu JI, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mihara M, Erster S, Zaika A, Petrenko O,
Chittenden T, Pancoska P and Moll UM: p53 has a direct apoptogenic
role at the mitochondria. Mol Cell. 11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dall'Agnese A, Caputo L, Nicoletti C, di
Iulio J, Schmitt A, Gatto S, Diao Y, Ye Z, Forcato M, Perera R, et
al: Transcription factor-directed re-wiring of chromatin
architecture for somatic cell nuclear reprogramming toward
trans-differentiation. Mol Cell. 76:453–472.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paliou C, Guckelberger P, Schöpflin R,
Heinrich V, Esposito A, Chiariello AM, Bianco S, Annunziatella C,
Helmuth J, Haas S, et al: Preformed chromatin topology assists
transcriptional robustness of Shh during limb development. Proc
Natl Acad Sci USA. 116:12390–12399. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sobczak M, Pitt AR, Spickett CM and
Robaszkiewicz A: PARP1 Co-regulates EP300-BRG1-dependent
transcription of genes involved in breast cancer cell proliferation
and DNA repair. Cancers (Basel). 11:15392019. View Article : Google Scholar
|
|
13
|
Zhou ZH, Wang QL, Mao LH, Li XQ, Liu P,
Song JW, Liu X, Xu F, Lei J and He S: Chromatin accessibility
changes are associated with enhanced growth and liver metastasis
capacity of acid-adapted colorectal cancer cells. Cell Cycle.
18:511–522. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vymetalkova V, Vodicka P, Vodenkova S,
Alonso S and Schneider-Stock R: DNA methylation and chromatin
modifiers in colorectal cancer. Mol Aspects Med. 69:73–92. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uusi-Mäkelä J, Afyounian E, Tabaro F,
Häkkinen T, Lussana A, Shcherban A, Annala N, Nurminen R, Kivinummi
K, Tammela TLJ, et al: Chromatin accessibility analysis uncovers
regulatory element landscape in prostate cancer progression.
bioRxiv. 2020.
|
|
16
|
Hankey W, Chen Z and Wang Q: Shaping
chromatin states in prostate cancer by pioneer transcription
factors. Cancer Res. 80:2427–2436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braadland PR and Urbanucci A: Chromatin
reprogramming as an adaptation mechanism in advanced prostate
cancer. Endocr Relat Cancer. 26:R211–R235. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arase M, Tamura Y, Kawasaki N, Isogaya K,
Nakaki R, Mizutani A, Tsutsumi S, Aburatani H, Miyazono K and
Koinuma D: Dynamics of chromatin accessibility during TGF-β-induced
EMT of Ras-transformed mammary gland epithelial cells. Sci Rep.
7:11662017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rubbi CP and Milner J: p53 is a chromatin
accessibility factor for nucleotide excision repair of DNA damage.
EMBO J. 22:975–986. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park JH, Choi Y, Song MJ, Park K, Lee JJ
and Kim HP: Dynamic long-range chromatin interaction controls
expression of IL-21 in CD4+ T Cells. J Immunol.
196:4378–4389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buenrostro JD, Wu B, Chang HY and
Greenleaf WJ: ATAC-seq: A method for assaying chromatin
accessibility genome-wide. Curr Protoc Mol Biol.
109:21.29.1–21.29.9. 2015. View Article : Google Scholar
|
|
23
|
Li H and Durbin R: Fast and accurate
long-read alignment with burrows-wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence alignment/map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gaspar JM: Improved peak-calling with
MACS2. bioRxiv. 2018.
|
|
26
|
Ramirez F, Ryan DP, Grüning B, Bhardwaj V,
Kilpert F, Richter AS, Heyne S, Dündar F and Manke T: deepTools2: A
next generation web server for deep-sequencing data analysis.
Nucleic Acids Res. 44((W1)): W160–W165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Quinlan AR: BEDTools: The Swiss-Army tool
for genome feature analysis. Curr Protoc Bioinformatics.
47:11.12.1–34. 2014. View Article : Google Scholar
|
|
28
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li B and Dewey CN: RSEM: Accurate
transcript quantification from RNA-Seq data with or without a
reference genome. BMC Bioinformatics. 12:3232011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z,
Meirelles GV, Clark NR and Ma'ayan A: Enrichr: Interactive and
collaborative HTML5 gene list enrichment analysis tool. BMC
Bioinformatics. 14:1282013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44((W1)):
W90–W97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hientz K, Mohr A, Bhakta-Guha D and
Efferth T: The role of p53 in cancer drug resistance and targeted
chemotherapy. Oncotarget. 8:8921–8946. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Munawar U, Roth M, Barrio S, Wajant H,
Siegmund D, Bargou RC, Kortüm KM and Stühmer T: Assessment of TP53
lesions for p53 system functionality and drug resistance in
multiple myeloma using an isogenic cell line model. Sci Rep.
9:180622019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miyamoto K, Nguyen KT, Allen GE, Jullien
J, Kumar D, Otani T, Bradshaw CR, Livesey FJ, Kellis M and Gurdon
JB: Chromatin accessibility impacts transcriptional reprogramming
in oocytes. Cell Rep. 24:304–311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Penalosa-Ruiz G, Bright AR, Mulder KW and
Veenstra GJC: The interplay of chromatin and transcription factors
during cell fate transitions in development and reprogramming.
Biochim Biophys Acta Gene Regul Mech. 1862:1944072019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matilainen O, Sleiman MSB, Quiros PM,
Garcia SMDA and Auwerx J: The chromatin remodeling factor ISW-1
integrates organismal responses against nuclear and mitochondrial
stress. Nat Commun. 8:18182017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weaver IC, Korgan AC, Lee K, Wheeler RV,
Hundert AS and Goguen D: Stress and the emerging roles of chromatin
remodeling in signal integration and stable transmission of
reversible phenotypes. Front Behav Neurosci. 11:412017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fischer M: Census and evaluation of p53
target genes. Oncogene. 36:3943–3956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Andrysik Z, Galbraith MD, Guarnieri AL,
Zaccara S, Sullivan KD, Pandey A, MacBeth M, Inga A and Espinosa
JM: Identification of a core TP53 transcriptional program with
highly distributed tumor suppressive activity. Genome Res.
27:1645–1657. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Younger ST and Rinn JL: p53 regulates
enhancer accessibility and activity in response to DNA damage.
Nucleic Acids Res. 45:9889–9900. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Angel P and Karin M: The role of Jun, Fos
and the AP-1 complex in cell-proliferation and transformation.
Biochim Biophys Acta. 1072:129–157. 1991.PubMed/NCBI
|
|
44
|
Eferl R and Wagner EF: AP-1: A
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Podar K, Raab MS, Tonon G, Sattler M,
Barilà D, Zhang J, Tai YT, Yasui H, Raje N, DePinho RA, et al:
Up-regulation of c-Jun inhibits proliferation and induces apoptosis
via caspase-triggered c-Abl cleavage in human multiple myeloma.
Cancer Res. 67:1680–1688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bossy-Wetzel E, Bakiri L and Yaniv M:
Induction of apoptosis by the transcription factor c-Jun. EMBO J.
16:1695–1709. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Stik G, Vidal E, Barrero M, Cuartero S,
Vila-Casadesús M, Mendieta-Esteban J, Tian TV, Choi J, Berenguer C,
Abad A, et al: CTCF is dispensable for immune cell
transdifferentiation but facilitates an acute inflammatory
response. Nat Genet. 52:655–661. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Parikh N, Hilsenbeck S, Creighton CJ,
Dayaram T, Shuck R, Shinbrot E, Xi L, Gibbs RA, Wheeler DA and
Donehower LA: Effects of TP53 mutational status on gene expression
patterns across 10 human cancer types. J Pathol. 232:522–533. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Herrero AB, Rojas EA, Misiewicz-Krzeminska
I, Krzeminski P and Gutierrez NC: Molecular mechanisms of p53
deregulation in cancer: An overview in multiple myeloma. Int J Mol
Sci. 17:20032016. View Article : Google Scholar
|
|
50
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Obier N, Cauchy P, Assi SA, Gilmour J,
Lie-A-Ling M, Lichtinger M, Hoogenkamp M, Noailles L, Cockerill PN,
Lacaud G, et al: Cooperative binding of AP-1 and TEAD4 modulates
the balance between vascular smooth muscle and hemogenic cell fate.
Development. 143:4324–4340. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gazon H, Barbeau B, Mesnard JM and
Peloponese JM Jr: Hijacking of the AP-1 signaling pathway during
development of ATL. Front Microbiol. 8:26862018. View Article : Google Scholar : PubMed/NCBI
|