Introduction
Breast cancer is the third most common cancer
worldwide in 2016 and is considered a paramount public health issue
that seriously endangers the lives of millions of women (1). Globally, 1 in 20 women develop breast
cancer in their lifetime, and the incidence continues to increase
(1). Triple-negative breast cancer
(TNBC), whose expression levels of progesterone receptor (PR),
estrogen receptor (ER) and human epidermal growth factor receptor 2
(HER2) are negative, comprises ~15% of all breast cancers, with the
worst prognosis compared with other subtypes irrespective of race,
age, or stage (2). TNBCs are
characterized by a poor prognosis and high rates of proliferation
and metastases, and occur frequently in younger patients, where
tumors generally present unfavorable clinical features, such as
larger size, higher histologic grade and lymph node involvement
(3–5). Due to the defect of promising molecular
markers, conventional chemotherapy and radiation are the primary
systemic therapeutic strategies (5).
Thus, it remains critical to discover novel biomarkers for therapy
patients with TNBC.
With the rapid development of genomic and proteomic
technologies, bioinformatics have facilitated the discovery of
reliable biomarkers for diagnosis, survival and prognosis of
diseases (6). Recent studies have
focused on the therapeutic targets of TNBC by microarray analysis
of gene expression profiles, including CCNA2, CDC20 and BUB1, which
are upregulated in TNBC tissues compared with normal tissues
(7,8). However, lack of direct experimental
validation of the upregulated genes decreases the reliability of
these conclusions.
To identify differentially expressed genes (DEGs) in
TNBC tissues compared with adjacent normal breast tissues, the
present study analyzed two microarray expression datasets, GSE38959
(9) and GSE65212 (10), from the Gene Expression Omnibus (GEO)
database and RNA sequencing (RNA-seq) data of TNBC tissues and
adjacent normal breast tissues from The Cancer Genome Atlas (TCGA)
database. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analyses were performed to determine the
significant functional terms of overlapping DEGs across the three
datasets. Centrality and survival analyses were performed to
determine the pivotal genes with higher importance and prognostic
values. Reverse transcription-quantitative (RT-q)PCR analysis was
performed to detect the expression levels of the hub genes in
clinical TNBC tissues and adjacent normal tissues. Gene set
enrichment analysis was performed to investigate the potential
biological functions associated with the hub genes.
Materials and methods
Data source
A total of two microarray expression datasets
(GSE38959 and GSE65212) were downloaded from the GEO database
(https://www.ncbi.nlm.nih.gov/geo). The
GSE38959 dataset had 30 TNBC tissues and 13 adjacent normal breast
tissues, while the GSE65212 dataset had 41 TNBC tissues and 11
adjacent normal breast tissues. All samples included both TNBC
tissues and normal breast tissues, and each microarray contained
>40 samples. In addition, gene expression profiles together with
corresponding clinical data of 1,109 breast cancer tissues and 113
adjacent normal tissues were obtained from TCGA database
(http://gdac.broadinstitute.org/runs/analyses_2016_01_28/data/BRCA).
Following filtration via immunohistochemistry (IHC) information of
ER/PR/HER2 in clinical data of TCGA-BRCA, 88 TNBC tissues and 6
normal tissues, with detailed clinical information and without
history of neoadjuvant chemotherapy, were enrolled in the present
study. The aforementioned information was freely available
online.
Tissue samples
A total of 25 TNBC tissues and matched adjacent
normal tissues were collected from patients diagnosed with TNBC via
biopsy IHC staining at the First Affiliated Hospital of Chongqing
Medical University (Chongqing, China) between January 2019 and
October 2019. The age of patients ranged from 37–75 years (median
age, 50 years). The extracted normal tissues were 3 cm away from
the tumor border, all tissue samples were snap-frozen in liquid
nitrogen after surgery, and subsequently stored in liquid nitrogen
until subsequent experimentation. Patients' initial treatment was
surgery without receiving prior treatment with radiation and/or
chemotherapy. The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Chongqing Medical
University (Chongqing, China, approval no. 2020-124) and written
informed consent was provided by all patients prior to the study
start.
Identification of DEGs
The gene expression datasets from the GEO database
(GSE38959 and GSE65212) were analyzed using GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r),
an online tool that can compare gene expression levels between two
sample groups (11), to identify
DEGs between TNBC tissues and adjacent normal tissues. The RNA-seq
level 3 normalized data from TCGA database was performed using R
package of edgeR (v3.28.1, http://bioinf.wehi.edu.au/edgeR). Genes with |log2
fold change|>1.5 and P<0.05 were differentially expressed.
Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny), a Venn
diagram web tool, was used to identify the overlapping DEGs across
the three datasets.
Functional enrichment analysis of
DEGs
GO functions were analyzed based on overlapped
genes, whose functions were classified into biological process
(BP), molecular function (MF) and cellular component (CC) terms.
The Search Tool for the Retrieval of Interacting Genes (STRING)
database (version 11.0; http://string-db.org) was used to export results of GO
enrichment analysis (12). The
Database for Annotation, Visualization and Integrated Discovery
(DAVID) (version 6.8, http://david.ncifcrf.gov) (13) was used to perform KEGG pathway
enrichment analysis of the upregulated and downregulated DEGs,
respectively. P<0.05 was considered to indicate statistically
significant GO terms and KEGG pathways. Gene count thresholds for
the GO terms and KEGG pathways were set to ≥20 and ≥4,
respectively.
Protein-protein interaction (PPI)
network construction
To assess the potential associations among DEGs, the
STRING database (12) was used to
construct a PPI network. The results were visualized using
Cytoscape software v3.7.1 (14). A
combined score of >0.4 was considered to indicate a
statistically significant result.
Centrality analysis based on the PPI
network
Based on the PPI network, two significant
topological parameters, degree and betweenness centrality, were
used to identify potential pivotal genes in this network. The two
centrality scores of each node were exported using NetworkAnalyzer
(v2.7) in Cytoscape software (15).
The Venn diagram was applied to demonstrate the intersections of
top 50% DEGs sorted by the degree value and the betweenness
value.
Module analysis of the PPI
network
MCODE Cytoscape plugin (v1.6.1) was applied to
screen the modules considered essential parts of the network
(16). For each significant module,
the default criteria were as follows: Degree cut-off, 2; node score
cut-off, 0.2; k-core, 2 and max depth, 100. The genes in the 1st
ranked module with high degree and betweenness values were selected
as candidate genes for further analysis.
Survival analysis
To assess the clinical outcome, the candidate genes
were subjected to the Kaplan-Meier plotter (http://kmplot.com/analysis), which assessed the
effects of 22,277 genes on breast cancer prognosis, using
microarray data (17). A total of
255 patients with TNBC were selected from 3,955 patients in the
Kaplan-Meier plotter breast cancer database by restricting the IHC
negative status of ER, PR and HER2. In the present study,
relapse-free survival (RFS) curves were drawn and exported using
the online survival analysis tool, Kaplan-Meier plotter. According
to the median expression of each gene, the cohorts were divided
into two groups, high expression group (127 patients) and low
expression group (128 patients). Log-rank P<0.05 was considered
to indicate a statistically significant difference.
RT-qPCR
Total RNA was extracted from TNBC tissues and
adjacent normal tissues using TRIzol® reagent (Takara
Bio, Inc.), according to the manufacturer's instructions. Briefly,
TRIzol® reagent was added to each tissue sample and
homogenized. Subsequently, chloroform was used to separate the
components and isopropanol was added to precipitate RNA. The eluted
RNA precipitation was assessed using NanoDrop 2000 (Thermo Fisher
Scientific, Inc.). Total RNA was reverse transcribed into cDNA
using the PrimeScript RT reagent kit (Takara Bio, Inc., cat. no.
RR047A). Temperature protocol for RT was as follows: 37°C for 15
min, 85°C for 5 sec and 4°C for 15 min. qPCR was subsequently
performed using SYBR Premix Ex Taq™ (Takara Bio, Inc.) to determine
the amplification of mRNAs in the CFX96 Real Time system (Bio-Rad
Laboratories, Inc.). The primer sequences used for qPCR are listed
in Table I. Relative expression
levels were calculated using the 2−ΔΔCq method (18) and normalized to the internal
reference gene GAPDH. All experiments were performed in
triplicate.
 | Table I.Primer sequences used for
quantitative PCR. |
Table I.
Primer sequences used for
quantitative PCR.
| Primer | Sequence
(5′-3′) |
|---|
| GAPDH-F |
GTCTTCCTGGGCAAGCAGTA |
| GAPDH-R |
CTGGACAGAAACCCCACTTC |
| CCNB1-F |
AACTTTCGCCTGAGCCTATTTT |
| CCNB1-R |
TTGGTCTGACTGCTTGCTCTT |
| GINS2-F |
AGGGTCTCGTTCTGTCATCC |
| GINS2-R |
TCTTTTGGTCCCAGTCTTCC |
| NCAPG-F |
TTTGTATTGGTGTGCCCTTT |
| NCAPG-R |
AGCCAGCAGTTTTTTTCTTC |
| MCM4-F |
CTCATCCACAACCGCTCC |
| MCM4-R |
TTCACTCTGTCCCCAGGC |
| RRM2-F |
CTCCAAGGACATTCAGCAC |
| RRM2-R |
GGAAGCCATAGAAACAGCG |
Gene set enrichment analysis
(GSEA)
GSEA was performed on TCGA RNA-seq data using R
package ‘clusterprofiler’ (19).
Based on Spearman's correlation coefficients between the expression
levels of the hub genes and other genes in TNBC samples of TCGA
cohort, GSEA was implemented on a set of 50 hallmark signatures
(20). Gene signatures with adjusted
P<0.05 were significantly enriched. The reference gene set
‘h.all.v7.0.symbols.gmt.txt’ was downloaded from the Molecular
Signatures Database (MSigDB, http://broadinstitute.org/gsea/msigdb/index.jsp).
Statistical analysis
Statistical analysis was performed using the ggpubr
package (v 0.4.0, http://CRAN.R-project.org/package=ggpubr) in R version
3.6.2 (https://www.R-project.org). Paired
Student's t-test was used to compare differences between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Filtration of DEGs
A total of 1,800, 2,347 and 2,244 DEGs were obtained
from the GSE38959 and GSE65212 datasets and TCGA TNBC cohort,
respectively. A total of 377 overlapping DEGs were identified
between TNBC tissue samples and non-tumor breast tissue samples via
Venn analysis. Among these genes, 260 genes were upregulated
(Fig. 1A) and 117 genes were
downregulated (Fig. 1B).
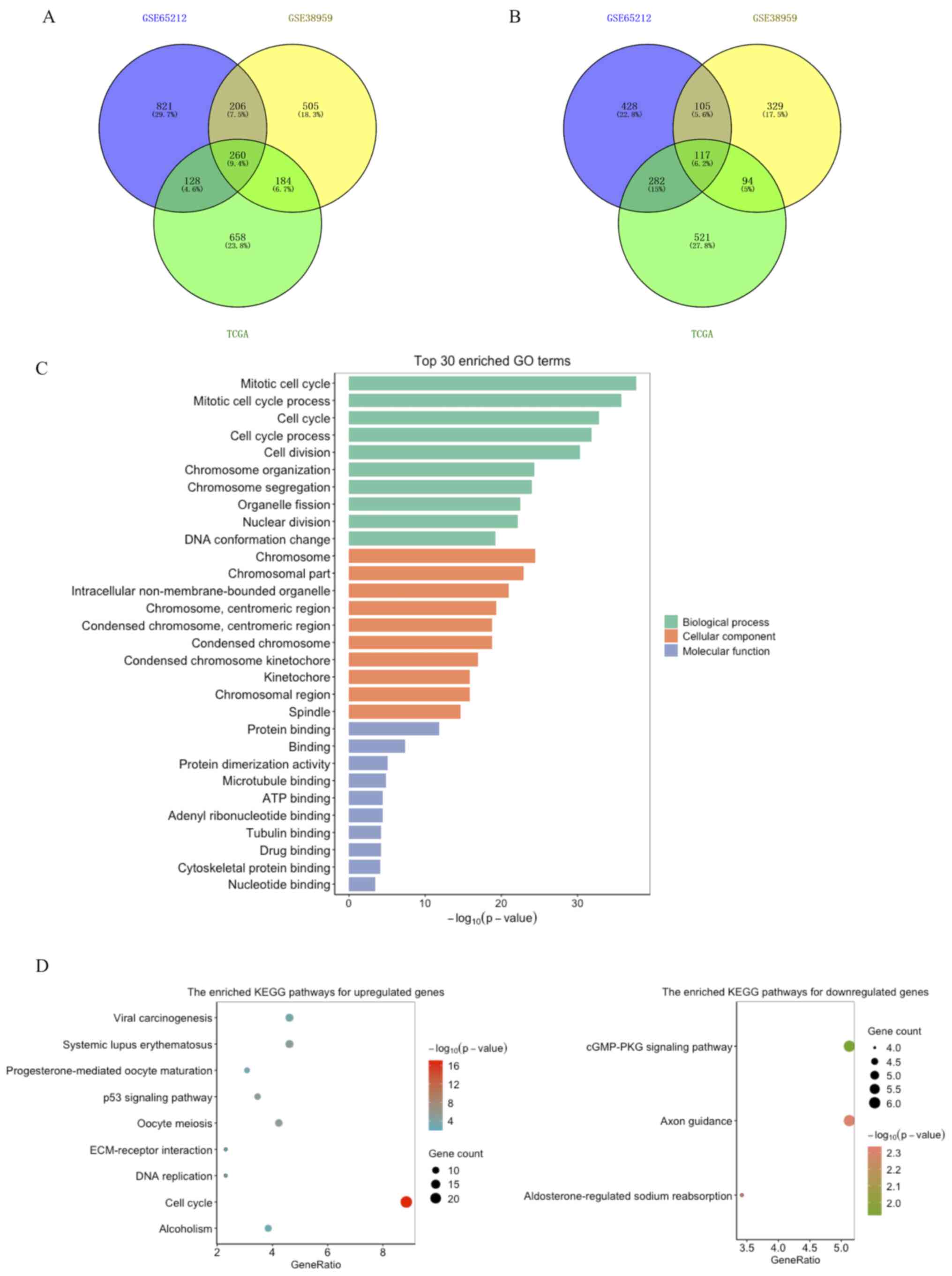 | Figure 1.Common DEGs among the GSE65212,
GSE38959 and TCGA datasets, and GO and KEGG functional enrichment
analyses of 377 DEGs. (A) Upregulated and (B) downregulated DEGs.
(C) Significantly enriched GO terms of DEGs, including top 10
cellular components, molecular functions and biological processes,
according to the -log10 (P-value). (D) Significantly
enriched KEGG pathway terms for upregulated (left) and
downregulated (right) DEGs. The size of each node represents the
gene number in the corresponding pathway, whereas the color change
from blue to red or from green to salmon indicates the P-values
from the big to the small for the corresponding pathway. DEGs,
differentially expressed genes; TCGA, The Cancer Genome Atlas; GO,
gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
Functional enrichment analysis of
DEGs
The functions of the 377 filtrated DEGs were
assessed via GO function and KEGG enrichment analyses (Table SI). GO analysis demonstrated that
the DEGs were associated with ‘mitotic cell cycle process’,
‘mitotic nuclear division’, ‘DNA conformation change’, ‘chromosome
segregation’, ‘centromeric region’, ‘condensed chromosome’ and
‘binding of protein, ATP and microtubule’ (Fig. 1C). KEGG analysis for the upregulated
DEGs demonstrated that the genes were markedly enriched in the ‘p53
signaling pathway’, ‘cell cycle’, ‘DNA replication’, ‘alcoholism’,
‘extracellular matrix (ECM)-receptor interaction’ and
‘progesterone-mediated oocyte maturation’. For the downregulated
DEGs, the most enriched pathways were ‘axon guidance’, ‘cGMP-PKG
signaling pathway’ and ‘aldosterone-regulated sodium reabsorption’
(Fig. 1D).
Construction of the PPI network
To determine the interactions of the 377 identified
DEGs, a PPI network was constructed, which comprised of 335 nodes
and 6,026 edges (Fig. S1). These
DEGs were regarded as potential crucial genes in TNBC
pathogenesis.
Centrality analysis of the PPI
network
Centrality analysis of the PPI network was performed
based on two significant parameters, degree and betweenness
centrality. Degree centrality refers to the sum of edges connected
to other vertexes, which symbolizes importance of each node in the
network. While betweenness centrality refers to the sum of times
each vertex is included in all-pairs shortest paths, indicating the
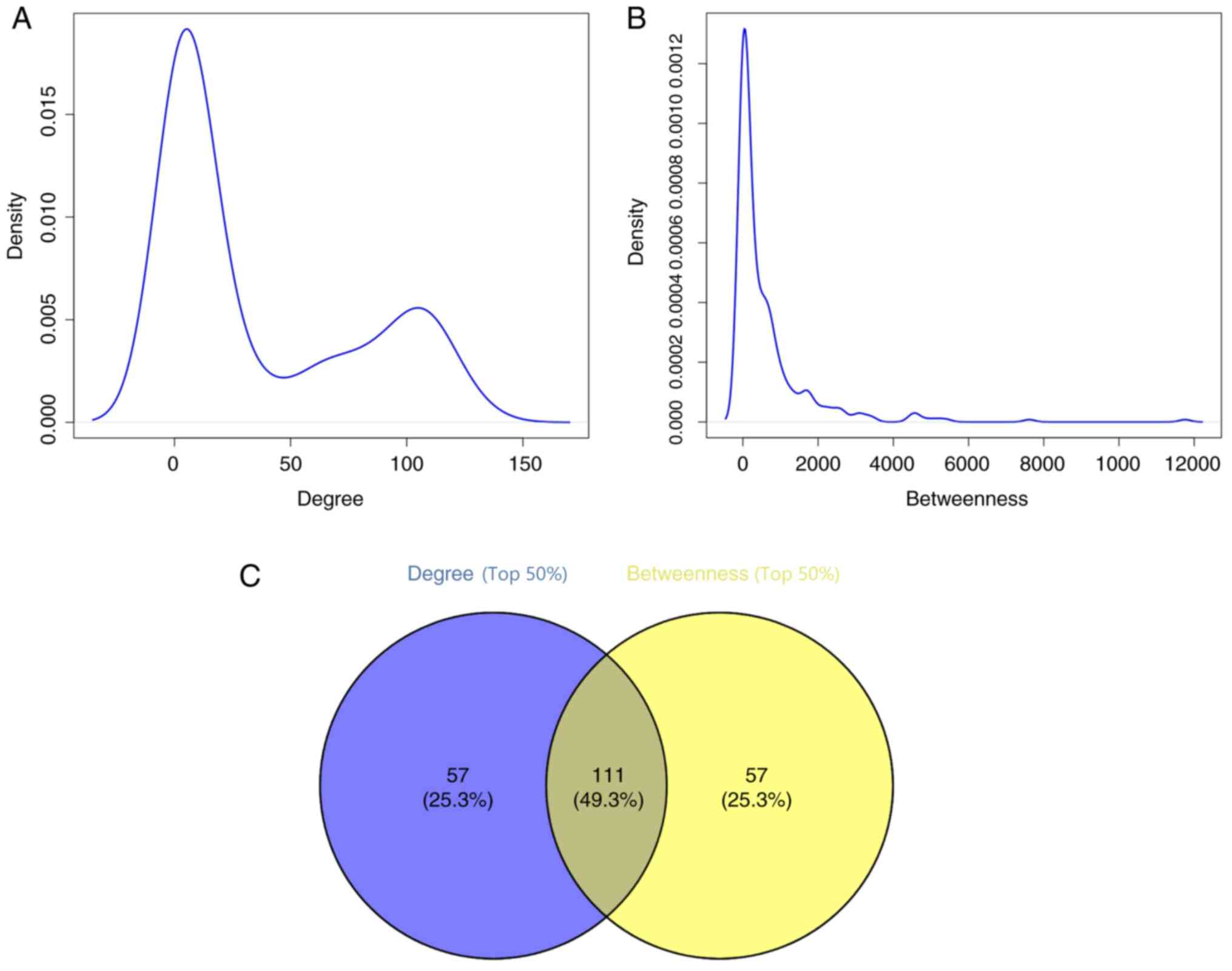
intermediate influence of each node (21). The results demonstrated that the
degree and betweenness values displayed power-law distributions
(Fig. 2A and B). Subsequently, the
top 50% of each parameter was chosen for further investigations,
and 111 DEGs were obtained based on the results of the Venn
analysis (Fig. 2C).
Modules analysis of the PPI
network
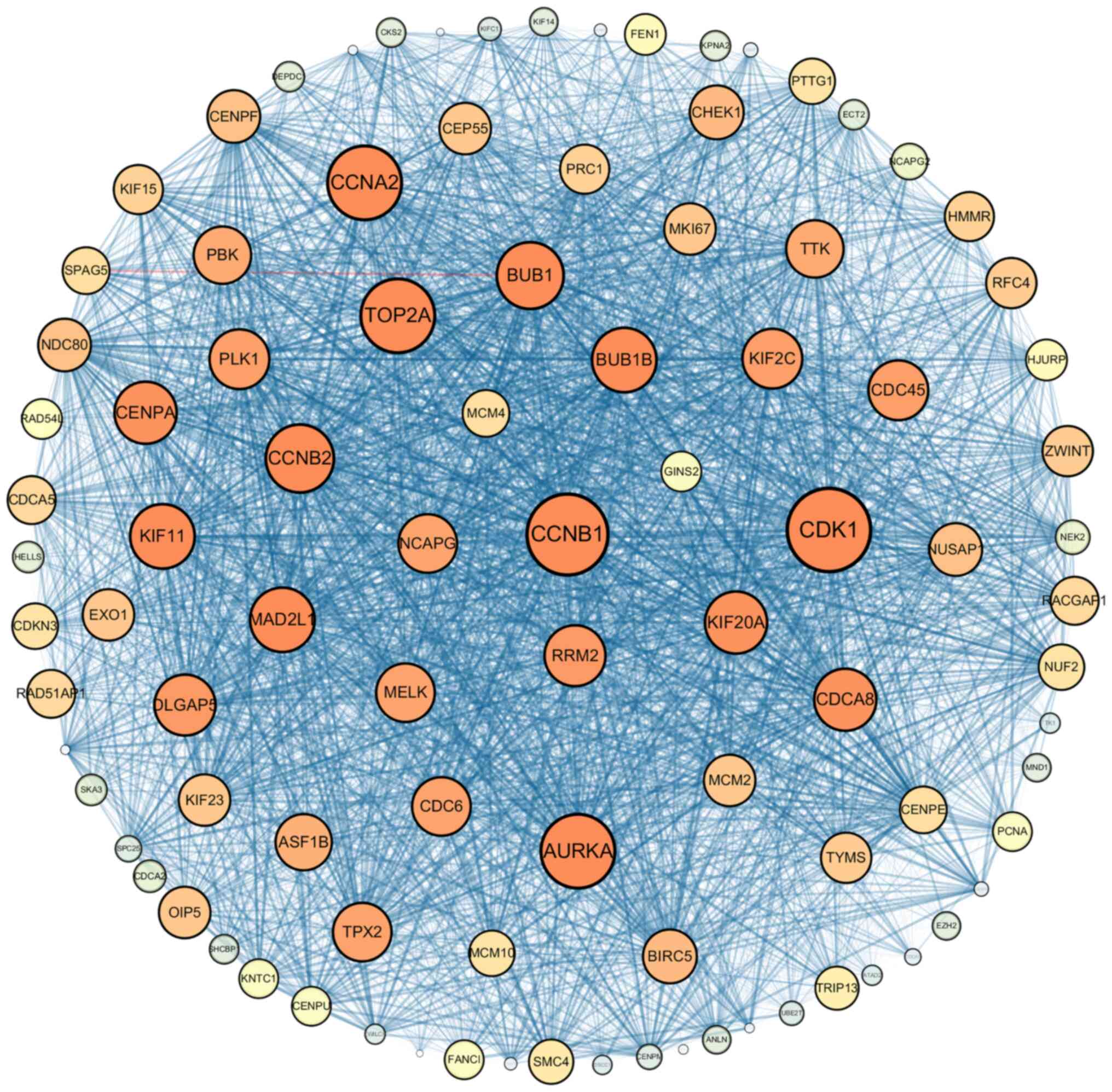
Modules analysis was performed using Cytoscape
software, and the module with the highest score, module 1 (Fig. 3), was further screened from the PPI
network. And the results demonstrated that module 1 contained 96
nodes and 4,064 edges, with a 85.56 MCODE score. Among the 96 nodes
in module 1, 66 nodes had high degree and betweenness values,
suggesting that these nodes may act as potential key genes with
essential physiological or pathological regulatory functions. Thus,
these 66 nodes were selected as candidate genes for further
analyses.
Survival analysis to identity the hub
genes
RFS analysis in the Kaplan-Meier plotter platform
was performed to determine the prognostic value of the 66 potential
candidate genes. The results demonstrated that upregulated CCNB1,
GINS complex subunit 2 (GINS2), NCAPG, MCM4 and RRM2 expression
levels were associated with unfavorable RFS of patients with TNBC
(log-rank P<0.05; Fig. 4).
Further details of the five hub genes are presented in Table II.
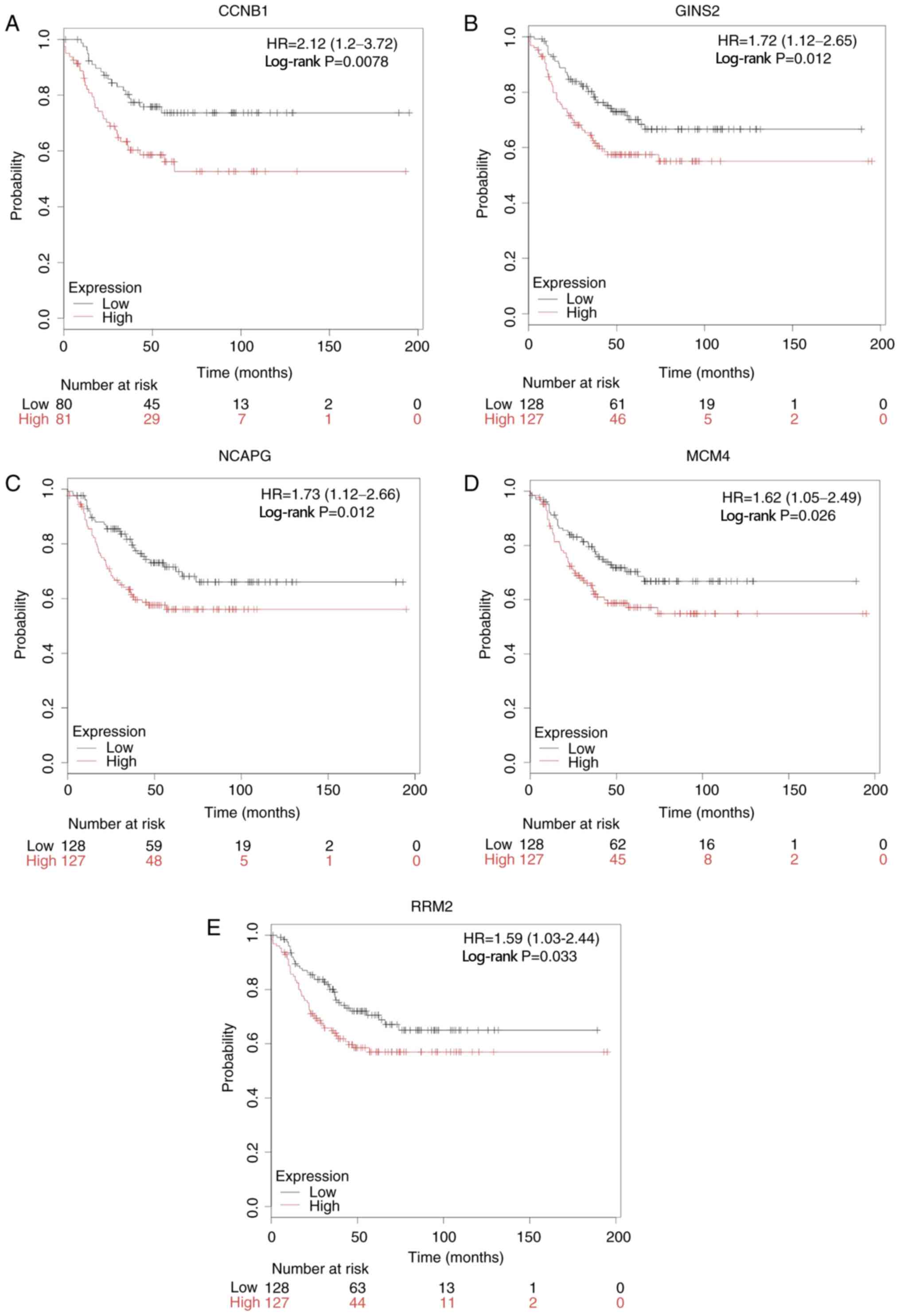 | Figure 4.RFS analysis of hub genes in patients
with TNBC using Kaplan-Meier plotter. High expression levels of (A)
CCNB1, (B) GINS2, (C) NCAPG, (D) MCM4, (E) RRM2 were significantly
associated with unfavorable prognosis of patients with TNBC. RFS,
relapse-free survival; TNBC, triple-negative breast cancer; CCNB1,
cyclin B1; GINS2, GINS complex subunit 2; NCAPG, non-SMC condensin
I complex subunit G; MCM4, minichromosome maintenance 4; RRM2,
ribonucleotide reductase regulatory subunit M2; HR, hazard
ratio. |
| Table II.Information on the five hub genes
from the protein-protein interaction network. |
Table II.
Information on the five hub genes
from the protein-protein interaction network.
| Gene symbol | Gene
description | Expression in
TNBC | Degree value | Betweenness
value | P-value |
|---|
| CCNB1 | Cyclin B1 | Up | 132 | 4,591.6 | 0.0078 |
| GINS2 | GINS complex
subunit 2 | Up | 97 | 712.8 | 0.0120 |
| NCAPG | Non-SMC condensin I
complex subunit G | Up | 113 | 199.9 | 0.0120 |
| MCM4 | Minichromosome
maintenance 4 | Up | 103 | 204.0 | 0.0260 |
| RRM2 | Ribonucleotide
reductase regulatory subunit M2 | Up | 115 | 1,317.9 | 0.0330 |
Validation of hub genes in clinical
samples via RT-qPCR analysis
RT-qPCR analysis was performed to detect the
expression levels of the five hub genes in 25 clinical TNBC tissues
and adjacent normal breast tissues. The results demonstrated that
the expression levels of CCNB1, NCAPG, MCM4 and RRM2 were elevated
in TNBC tissues compared with adjacent normal tissues (Fig. 5A-D). These experimental results were
in accordance with the bioinformatics-predicted results. However,
no significant difference in GINS2 expression was observed between
the TNBC tissues and adjacent normal tissues (Fig. 5E).
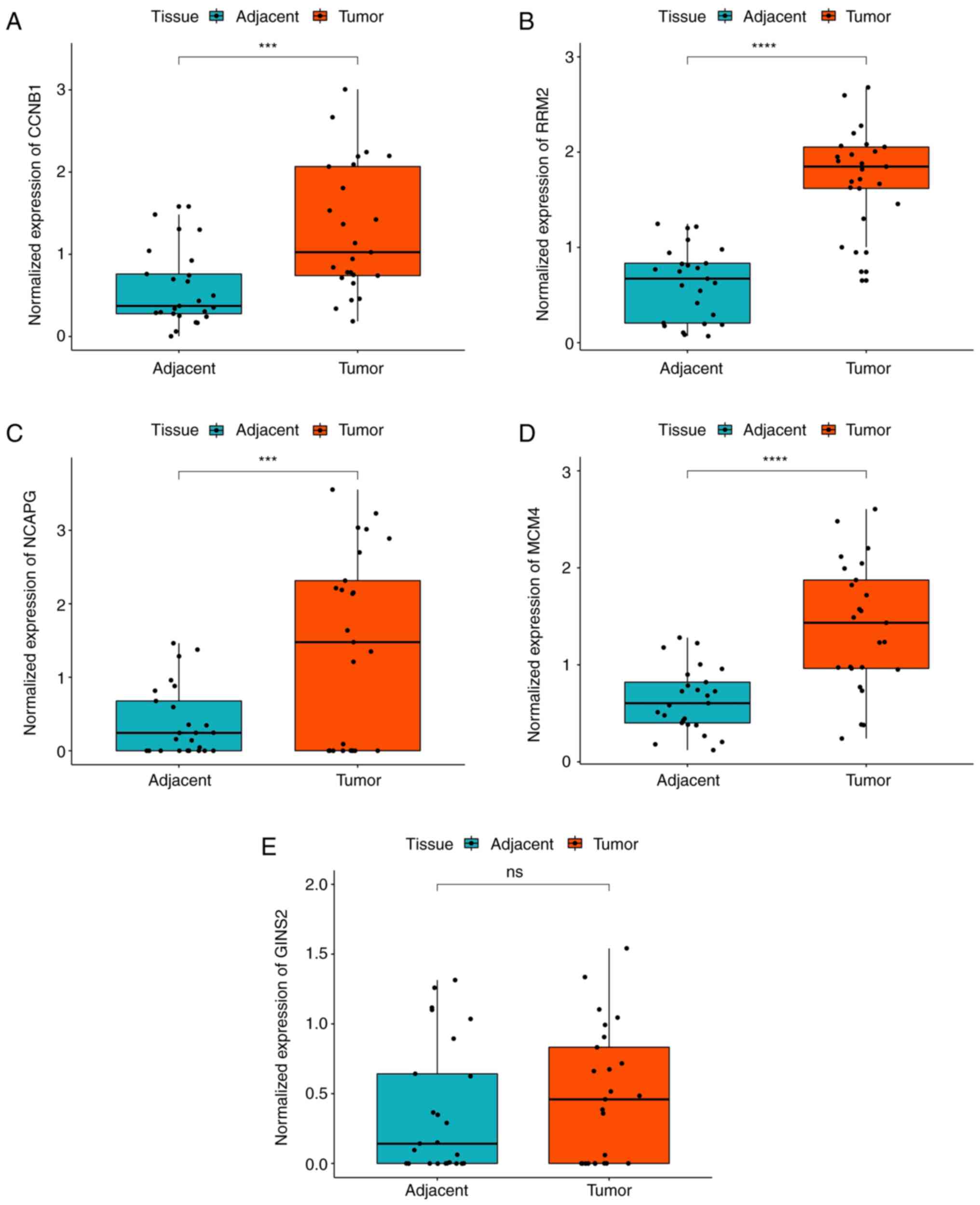 | Figure 5.Validation of the gene expression
levels of CCNB1, RRM2, NCAPG, MCM4, GINS2 between TNBC tissues and
adjacent normal breast tissues via reverse
transcription-quantitative PCR analysis. (A-D) CCNB1, RRM2, NCAPG
and MCM4 expression levels were significantly upregulated in TNBC
tissues compared with adjacent normal breast tissues. (E) No
significant difference in GINS2 expression was observed between the
TNBC tissues and adjacent normal breast tissues. ***P<0.001,
****P<0.0001. TNBC, triple-negative breast cancer; ns, no
significance; CCNB1, cyclin B1; GINS2, GINS complex subunit 2;
NCAPG, non-SMC condensin I complex subunit G; MCM4, minichromosome
maintenance 4; RRM2, ribonucleotide reductase regulatory subunit
M2. |
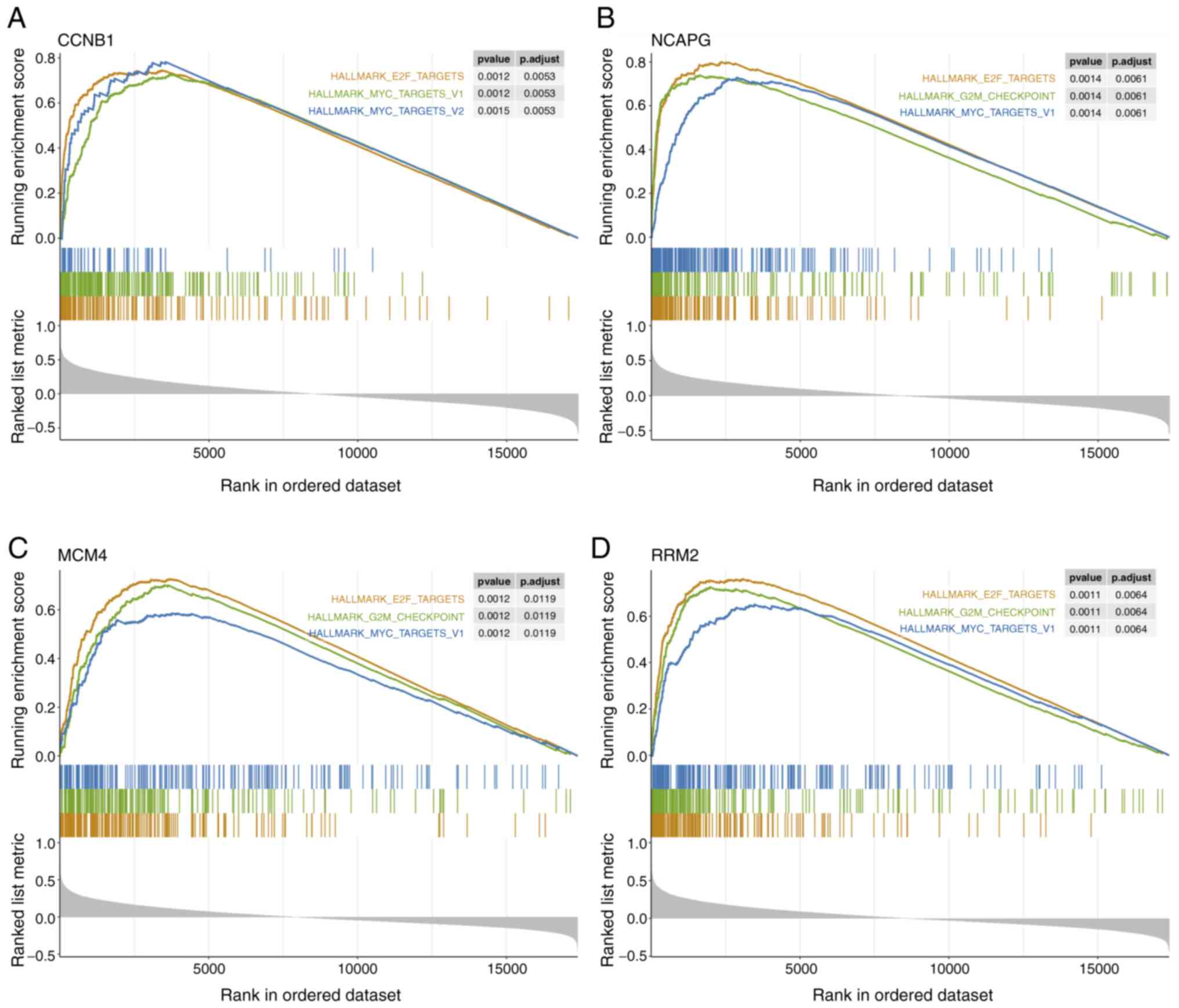
Four hub oncogenes significantly
associated with tumor proliferation
To further investigate the potential biological
functions associated with hub genes, GSEA was performed on mRNA
expression data of TCGA TNBC samples. The results demonstrated
prominent enrichments of hallmark proliferation gene sets for genes
associated with high expression levels of the four hub oncogenes
(CCNB1, NCAPG, MCM4 and RRM2), including ‘E2F_TARGETS’,
‘G2M_CHECKPOINT’, ‘MYC_TARGETS_V1’ and ‘MYC_TARGETS_V2’ (Fig. 6A-D). Taken together, these results
suggest that the four identified hub genes are significantly
associated with cell proliferative processes.
Discussion
In 2016, breast cancer was the third most common
cancer worldwide and the leading cause of cancer-associated
mortality (535,000 deaths) in women (1). TNBC is a unique subtype of breast
cancer characterized by poor prognosis and limited effective
treatments (2). Due to the absence
of targeted therapies, conventional chemotherapy and radiation are
the primary systemic therapeutic strategies (5). Recently, the rapid development of next
generation sequencing in GEO and integrated multi-omics
measurements in TCGA database has revealed significant molecular
heterogeneity of breast cancer (22). Thus, bioinformatics analyses are
performed to identify specific molecular targets for TNBC.
In the initial stages of the study, three
microarrays were assessed (GSE65212, GSE38959 and GSE76250).
However, GSE76250 was excluded due to the difference in its design
from the other two datasets. Based on the GSE65212 and GSE38959
datasets from the GEO database and a breast cancer cohort from TCGA
database, 377 DEGs between TNBC tissues and adjacent normal human
breast tissues were screened, including 260 upregulated genes and
117 downregulated genes. GO and KEGG functional enrichment analyses
demonstrated that the most enriched GO terms of the DEGs were
‘mitotic cell cycle process’, ‘chromosome segregation’ and ‘mitotic
nuclear division’. KEGG pathways, such as the ‘p53 signaling
pathway’, ‘progesterone-mediated oocyte maturation’, ‘DNA
replication’, ‘alcoholism’ and ‘ECM-receptor interaction’ were
predominantly associated with the upregulated genes, while a few
pathways, such as ‘axon guidance’, ‘cGMP-PKG signaling pathway’
were enriched in the downregulated genes.
It is well-known that defects in cell cycle
regulation, such as sustaining proliferation and unlimited
replication, are fundamental characteristics of cancer pathogenesis
(23), and some newly discovered
TNBC-associated small molecule inhibitors have been demonstrated to
induce cell cycle arrest (5).
Similarly, chromosome segregation with nuclear division in M phase
and DNA replication in S phase are essential processes during
mitotic cell division (24). In
tumorigenesis, driven by oncogene activation, DNA replication
stress and its adverse impact on chromosome segregation are
associated with genome instability (25). In addition, the p53 pathway is a
classic signaling pathway involved in the occurrence and
progression of cancer, which plays essential roles in tumor
suppression, regulating cell migration and invasion (26). The frequency of TP53 gene mutation in
basal-like breast tumors/TNBCs is ~80% (22), and based on molecular mechanisms of
the p53 pathway, a few chemicals indicate potential therapeutic
intervention in breast cancer (27,28). In
addition, alcohol has a deleterious effect on women by increasing
the risk of breast cancer (29), and
in vitro experiments have demonstrated that alcohol promotes
TNBC cell proliferation, migration and invasion (30). However, to the best of our knowledge,
alcoholism in TNBC has not been reported by other datasets
enrichment analyses. Cell-cell adhesion alterations and attachment
to the ECM are common events in diverse epithelial malignancies,
which are associated with cellular invasion and metastasis
(22). Excessiveness of ECM
deposition may enhance tumor cell invasion in the breast cancer
(31). Dysregulated microRNAs
(miRNAs) associated with progesterone-mediated oocyte maturation
may have an impact on follicular growth arrest and metabolic
disorders (32). Furthermore, oocyte
meiosis and progesterone-mediated oocyte maturation pathways are
enriched in survival associated miRNAs of ovarian carcinomas
(33). Taken together, these results
suggest that these DEGs may be associated with the pathogenesis and
development of TNBC.
To investigate the interactions between these DEGs,
the PPI network complex was constructed. Following centrality
analysis, the results demonstrated that the degree and betweenness
parameters displayed power-law distributions. It is well-known that
power-law distributions frequently appear in several disease or
metabolic biological networks (34),
suggesting that the PPI network in the present study has similar
scale-free characteristics with other biological networks. Modules
analysis identified four sub modules, including the first-ranked
module, which contained 96 nodes. Increasing evidence suggest that
modules analysis has been extensively applied for identifying hub
genes in diverse cancers, such as colorectal cancer (35), oral cancer (36) and renal carcinoma (37). Thus, these DEGs and interactions in
the first-ranked module may be the core of the network. A total of
66 candidate DEGs with high degree and betweenness values among the
96 nodes in the first-ranked module were selected. Collectively,
the results of the present study suggest that the 66 candidate
genes may be pivotal in regulating the occurrence and progression
of TNBC.
Survival analysis demonstrated that high expression
levels of the five hub genes (CCNB1, GINS2, NCAPG, MCM4 and RRM2)
among the 66 candidate genes were significantly associated with
shorter RFS times in patients with TNBC (P<0.05). This suggests
that the five hub genes may be indispensable to tumorigenesis and
progression in TNBC. Reverse transcription quantitative PCR
analysis was performed to validate the expression levels of the
five hub genes in TNBC clinical samples and their matched adjacent
normal controls. The results demonstrated that CCNB1, NCAPG, MCM4
and RRM2 expression levels were significantly upregulated in TNBC
samples compared with the controls, and no significant difference
in GINS2 expression was observed between the two groups. GSEA was
performed to investigate the potential biological functions of the
four oncogenes, which revealed significant enrichment of cell
proliferation markers for high expression levels of CCNB1, NCAPG,
MCM4 and RRM2.
CCNB1 is a checkpoint protein in the G2-M
transition phase during cell cycle (38). CCNB1 protein is upregulated in TNBCs
compared with other subtypes, which is closely associated with
adverse clinical prognosis in patients with breast cancer (7,39). In
clinical practice, CCNB1 has been applied as a cell proliferation
biomarker to evaluate breast cancer recurrence risk in a genetic
test called the 21-gene expression assay (40). Recent studies have reported that some
drugs, such as Dipalmitoylphosphatidic acid (41) and F1012-2 (a material isolated from
Eupatorium lindleyanum DC) (42), inhibited TNBC tumor growth by
suppressing CCNB1 expression. NCAPG is a constituent of the
condensin complex, which serves as a major molecular effector of
chromosome condensation and segregation during mitosis (43). Upregulated NCAPG expression is
significantly associated with adverse prognosis in various
malignant tumors, particularly in hepatocellular cancer (44). In TNBC, upregulated NCAPG expression
is associated with Ki67 index, a biomarker of mitosis and
proliferation of tumor cells (45).
MCM4 is part of the MCM2-7 heterohexameric complex, which is
important for DNA replication initiation, elongation and
replication licensing (46). It has
been reported that overexpression of MCM4 is associated with tumor
progression, high histological grade and poor survival outcomes in
patients with breast cancer (47).
Both elevated mRNA and protein expression levels of MCM4 have been
observed in TNBC tissues (48).
Overexpressed mutant p53 shows a protein-protein interaction with
MCM4, and after inhibiting this interaction with the poly
ADP-ribose polymerase, TNBC cells with mutant p53 undergo apoptosis
(49). RRM2 is an important
component of ribonucleotide reductase, which catalyzes the
rate-limiting step for DNA synthesis and repair (50). RRM2 expression is elevated in the
TNBC subtype, with respect to non-TNBC subtypes (51). Notably, RRM2 expression is
upregulated in tamoxifen-resistant breast cancer cells, the effects
of which are reversed following inhibition of RRM2 (52), which suggests that RRM2 promotes the
conversion of ER-positive to ER-negative subtype. Several studies
have demonstrated that upregulated RRM2 expression is associated
with oncogenic cellular activities, such as anti-apoptotic, cell
proliferation and invasiveness, as well as angiogenesis in breast
cancer (53,54). GINS2, a subunit of the DNA
replication complex GINS, is crucial to initiation of DNA
replication (55). Zheng et
al (56) reported that
upregulated GINS2 expression is associated with histological grade,
metastasis and endocrine therapy resistance in patients with breast
cancer. Peng et al (57)
confirmed that GINS2 mediates cell cycle progression and
proliferation, and that GINS2 knockdown inhibits the migratory and
invasive abilities of TNBC cells. The results of the present study
demonstrated that GINS2 expression was not significantly elevated
in TNBC tissues compared with adjacent normal tissues. This may be
attributed to limited samples and imprecise primer extension
reaction temperature, which require confirmation with large sample
size and perfect reaction conditions.
Taken together, the results of the present study
suggest that CCNB1, NCAPG, MCM4 and RRM2 may be potential
prognostic factors and therapeutic targets for TNBC. However,
further studies, including in vivo and in vitro
experiments are required to determine the molecular mechanisms of
these genes.
In conclusion, based on bioinformatics analysis of
three independent datasets, the present study filtered 377 DEGs of
TNBC primarily, which were significantly enriched in the cell cycle
process, p53 pathway and DNA replication. Furthermore, the TNBC
related PPI network was constructed, consisting of 335 nodes and
6,026 edges. A total of 66 candidate genes with high centrality
values in a significant module were identified. Reverse
transcription-quantitative PCR analysis revealed that CCNB1, NCAPG,
MCM4 and RRM2 were upregulated in TNBC tissue samples, and high
expression levels of these oncogenes were associated with
unfavorable survival outcomes. In addition, the four oncogenes were
significantly associated with tumor cell proliferation.
Collectively, the results of the present study provide theoretical
guidance for TNBC prognosis evaluation and prospective molecular
targeted therapy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81700639).
Availability of data and materials
The gene expression profiles of TNBC included in the
present study are accessible through GEO accession numbers GSE38959
and GSE65212. Gene expression data and clinical information of TCGA
breast cancer cohort can be accessed at http://gdac.broadinstitute.org/runs/analyses_2016_01_28/data/BRCA.
Authors' contributions
XX and XC contributed toward study conception and
design. XX drafted the initial manuscript and acquired the data. ZZ
improved the study design and revised the manuscript for important
intellectual content. RL and YS performed statistical analysis. RP
and JW helped analyze and interpret the data. XX and XC confirmed
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved and supervised by the
Ethics Committee of the First Affiliated Hospital of Chongqing
Medical University (Chongqing, China; approval no. 2020-124).
Written informed consent was provided by all patients prior to the
study start.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T,
Alizadeh-Navaei R, Allen C, Alsharif U, Alvis-Guzman N, Amini E, et
al: Global, regional, and national cancer incidence, mortality,
years of life lost, years lived with disability, and
disability-adjusted life-years for 29 cancer groups, 1990 to 2016:
A systematic analysis for the global burden of disease study. JAMA
Oncol. 4:1553–1568. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hwang KT, Kim J, Jung J, Chang JH, Chai
YJ, Oh SW, Oh S, Kim YA, Park SB and Hwang KR: Impact of breast
cancer subtypes on prognosis of women with operable invasive breast
cancer: A population-based study using SEER database. Clin Cancer
Res. 25:1970–1979. 2019.PubMed/NCBI
|
|
3
|
Bauer KR, Brown M, Cress RD, Parise CA and
Caggiano V: Descriptive analysis of estrogen receptor
(ER)-negative, progesterone receptor (PR)-negative, and
HER2-negative invasive breast cancer, the so-called triple-negative
phenotype: A population-based study from the California cancer
registry. Cancer. 109:1721–1728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: Disease entity or title of
convenience? Nat Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hwang SY, Park S and Kwon Y: Recent
therapeutic trends and promising targets in triple negative breast
cancer. Pharmacol Ther. 199:30–57. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kulasingam V and Diamandis EP: Strategies
for discovering novel cancer biomarkers through utilization of
emerging technologies. Nat Clin Pract Oncol. 5:588–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li MX, Jin LT, Wang TJ, Feng YJ, Pan CP,
Zhao DM and Shao J: Identification of potential core genes in
triple negative breast cancer using bioinformatics analysis. Onco
Targets Ther. 11:4105–4112. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lv X, He M, Zhao Y, Zhang L, Zhu W, Jiang
L, Yan Y, Fan Y, Zhao H, Zhou S, et al: Identification of potential
key genes and pathways predicting pathogenesis and prognosis for
triple-negative breast cancer. Cancer Cell Int. 19:1722019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Komatsu M, Yoshimaru T, Matsuo T, Kiyotani
K, Miyoshi Y, Tanahashi T, Rokutan K, Yamaguchi R, Saito A, Imoto
S, et al: Molecular features of triple negative breast cancer cells
by genome-wide gene expression profiling analysis. Int J Oncol.
42:478–506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maire V, Némati F, Richardson M,
Vincent-Salomon A, Tesson B, Rigaill G, Gravier E, Marty-Prouvost
B, De Koning L, Lang G, et al: Polo-like kinase 1: A potential
therapeutic option in combination with conventional chemotherapy
for the management of patients with triple-negative breast cancer.
Cancer Res. 73:813–823. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41((Database Issue)):
D991–D995. 2013.PubMed/NCBI
|
|
12
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47D:D607–D613. 2019.
View Article : Google Scholar
|
|
13
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Doncheva NT, Assenov Y, Domingues FS and
Albrecht M: Topological analysis and interactive visualization of
biological networks and protein structures. Nat Protoc. 7:670–685.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bader GD and Hogue CWV: An automated
method for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Györffy B, Lanczky A, Eklund AC, Denkert
C, Budczies J, Li Q and Szallasi Z: An online survival analysis
tool to rapidly assess the effect of 22,277 genes on breast cancer
prognosis using microarray data of 1,809 patients. Breast Cancer
Res Treat. 123:725–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liberzon A, Birger C, Thorvaldsdóttir H,
Ghandi M, Mesirov JP and Tamayo P: The molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Freeman LC: Centrality in social networks
conceptual clarification. Soc Netw. 1:215–239. 1978. View Article : Google Scholar
|
|
22
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hartwell LH and Weinert TA: Checkpoints:
Controls that ensure the order of cell cycle events. Science.
246:629–634. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang BN, Bueno Venegas A, Hickson ID and
Chu WK: DNA replication stress and its impact on chromosome
segregation and tumorigenesis. Semin Cancer Biol. 55:61–69. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Joerger AC and Fersht AR: The p53 pathway:
Origins, inactivation in cancer, and emerging therapeutic
approaches. Annu Rev Biochem. 85:375–404. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beberok A, Wrześniok D, Rok J, Rzepka Z,
Respondek M and Buszman E: Ciprofloxacin triggers the apoptosis of
human triple-negative breast cancer MDA-MB-231 cells via the
p53/Bax/Bcl-2 signaling pathway. Int J Oncol. 52:1727–1737.
2018.PubMed/NCBI
|
|
28
|
Zhu X, Wang K, Zhang K, Zhang T, Yin Y and
Xu F: Ziyuglycoside I inhibits the proliferation of MDA-MB-231
breast carcinoma cells through inducing p53-mediated G2/M cell
cycle arrest and intrinsic/extrinsic apoptosis. Int J Mol Sci.
17:19032016. View Article : Google Scholar
|
|
29
|
Singletary KW and Gapstur SM: Alcohol and
breast cancer: Review of epidemiologic and experimental evidence
and potential mechanisms. JAMA. 286:2143–2151. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao M, Howard EW, Parris AB, Guo Z, Zhao
Q and Yang X: Alcohol promotes migration and invasion of
triple-negative breast cancer cells through activation of p38 MAPK
and JNK. Mol Carcinog. 56:849–862. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shekhar MP, Pauley R and Heppner G: Host
microenvironment in breast cancer development: Extracellular
matrix-stromal cell contribution to neoplastic phenotype of
epithelial cells in the breast. Breast Cancer Res. 5:130–135. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu S, Zhang X, Shi C, Lin J, Chen G, Wu
B, Wu L, Shi H, Yuan Y, Zhou W, et al: Altered microRNAs expression
profiling in cumulus cells from patients with polycystic ovary
syndrome. J Transl Med. 13:2382015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuznetsov VA, Tang Z and Ivshina AV:
Identification of common oncogenic and early developmental pathways
in the ovarian carcinomas controlling by distinct prognostically
significant microRNA subsets. BMC Genomics. 18 (Suppl 6):S6922017.
View Article : Google Scholar
|
|
34
|
Cheng L, Yang H, Zhao H, Pei X, Shi H, Sun
J, Zhang Y, Wang Z and Zhou M: MetSigDis: A manually curated
resource for the metabolic signatures of diseases. Brief Bioinform.
20:203–209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo Y, Bao Y, Ma M and Yang W:
Identification of key candidate genes and pathways in colorectal
cancer by integrated bioinformatical analysis. Int J Mol Sci.
18:7222017. View Article : Google Scholar
|
|
36
|
Zhao X, Sun S, Zeng X and Cui L:
Expression profiles analysis identifies a novel three-mRNA
signature to predict overall survival in oral squamous cell
carcinoma. Am J Cancer Res. 8:450–461. 2018.PubMed/NCBI
|
|
37
|
Luo Y, Shen D, Chen L, Wang G, Liu X, Qian
K, Xiao Y, Wang X and Ju L: Identification of 9 key genes and small
molecule drugs in clear cell renal cell carcinoma. Aging (Albany
NY). 11:6029–6052. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gavet O and Pines J: Progressive
activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell.
18:533–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Agarwal R, Gonzalez-Angulo AM, Myhre S,
Carey M, Lee JS, Overgaard J, Alsner J, Stemke-Hale K, Lluch A,
Neve RM, et al: Integrative analysis of cyclin protein levels
identifies cyclin b1 as a classifier and predictor of outcomes in
breast cancer. Clin Cancer Res. 15:3654–3662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sparano JA, Gray RJ, Makower DF, Pritchard
KI, Albain KS, Hayes DF, Geyer CE Jr, Dees EC, Perez EA, Olson JA
Jr, et al: Prospective validation of a 21-gene expression assay in
breast cancer. N Engl J Med. 373:2005–2014. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang QQ, Chen J, Zhou DL, Duan YF, Qi CL,
Li JC, He XD, Zhang M, Yang YX and Wang L: Dipalmitoylphosphatidic
acid inhibits tumor growth in triple-negative breast cancer. Int J
Biol Sci. 13:471–479. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tian S, Chen Y, Yang B, Lou C, Zhu R, Zhao
Y and Zhao H: F1012-2 inhibits the growth of triple negative breast
cancer through induction of cell cycle arrest, apoptosis, and
autophagy. Phytother Res. 32:908–922. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thadani R, Uhlmann F and Heeger S:
Condensin, chromatin crossbarring and chromosome condensation. Curr
Biol. 22:R1012–R1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Q, Su R, Shan C, Gao C and Wu P:
Non-SMC condensin I complex, subunit G (NCAPG) is a novel mitotic
gene required for hepatocellular cancer cell proliferation and
migration. Oncol Res. 26:269–276. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen J, Qian X, He Y, Han X and Pan Y:
Novel key genes in triple-negative breast cancer identified by
weighted gene co-expression network analysis. J Cell Biochem.
120:16900–16912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tye BK: MCM proteins in DNA replication.
Annu Rev Biochem. 68:649–686. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kwok HF, Zhang SD, McCrudden CM, Yuen HF,
Ting KP, Wen Q, Khoo US and Chan KY: Prognostic significance of
minichromosome maintenance proteins in breast cancer. Am J Cancer
Res. 5:52–71. 2014.PubMed/NCBI
|
|
48
|
Issac MSM, Yousef E, Tahir MR and Gaboury
LA: MCM2, MCM4, and MCM6 in breast cancer: Clinical utility in
diagnosis and prognosis. Neoplasia. 21:1015–1035. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qiu WG, Polotskaia A, Xiao G, Di L, Zhao
Y, Hu W, Philip J, Hendrickson RC and Bargonetti J: Identification,
validation, and targeting of the mutant p53-PARP-MCM chromatin axis
in triple negative breast cancer. NPJ Breast Cancer. 3:12017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Aye Y, Li M, Long MJ and Weiss RS:
Ribonucleotide reductase and cancer: Biological mechanisms and
targeted therapies. Oncogene. 34:2011–2021. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen WX, Yang LG, Xu LY, Cheng L, Qian Q,
Sun L and Zhu YL: Bioinformatics analysis revealing prognostic
significance of RRM2 gene in breast cancer. Biosci Rep.
39:BSR201820622019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shah KN, Wilson EA, Malla R, Elford HL and
Faridi JS: Targeting ribonucleotide reductase M2 and NF-κB
activation with didox to circumvent tamoxifen resistance in breast
cancer. Mol Cancer Ther. 14:2411–2421. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang WH, Li N, Yuan ZQ, Qian XL and Wang
ZH: DSCAM-AS1 promotes tumor growth of breast cancer by reducing
miR-204-5p and up-regulating RRM2. Mol Carcinog. 58:461–473. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jones DT, Lechertier T, Mitter R, Herbert
JM, Bicknell R, Jones JL, Li JL, Buffa F, Harris AL and
Hodivala-Dilke K: Gene expression analysis in human breast cancer
associated blood vessels. PLoS One. 7:e442942012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Labib K and Gambus A: A key role for the
GINS complex at DNA replication forks. Trends Cell Biol.
17:271–278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zheng M, Zhou Y, Yang X, Tang J, Wei D,
Zhang Y, Jiang J, Chen Z and Zhu P: High GINS2 transcript level
predicts poor prognosis and correlates with high histological grade
and endocrine therapy resistance through mammary cancer stem cells
in breast cancer patients. Breast Cancer Res Treat. 148:423–436.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Peng L, Song Z, Chen D, Linghu R, Wang Y,
Zhang X, Kou X, Yang J and Jiao S: GINS2 regulates matrix
metallopeptidase 9 expression and cancer stem cell property in
human triple negative Breast cancer. Biomed Pharmacother.
84:1568–1574. 2016. View Article : Google Scholar : PubMed/NCBI
|