According to the report by the International Agency
for Research on Cancer of 2018 (1),
prostate cancer (PCa) has the second highest estimated
age-standardized incidence rate worldwide (29.3 per 100,000) and is
the sixth cause of cancer-associated death in men (7.6 per
100,000). The problem with the management of PCa is due to the
difficulty in stratifying between indolent and aggressive cases.
Although <5% of patients exhibit advanced disease, up to 40% of
patients will eventually develop metastatic disease despite local
therapy (2,3). Other patients with PCa undergo hormonal
therapy treatment, radical prostatectomy (RP) or radiotherapy, so,
numerous cases of PCa only require expectant management; thus, in
these patients, an overtreatment may result in significant
morbidity.
High Grade Prostatic Intraepithelial Neoplasia
(HGPIN) is a prostate preneoplastic lesion, which may develop
towards an invasive PCa (25 to 30%) during a process, which may
take over 10 years (4), according to
a study conducted at Johns Hopkins University School of Medicine,
in patients of Baltimore (Maryland, United States of America).
Both, HGPIN and PCa are multifocal, and HGPIN foci coexist in
adjacent areas of PCa sharing chromosome deletions and interstitial
translocations that originate the TMPRSS2-ERG fusion gene and
genetic alterations, such as hypermethylation of the π-class
glutathione S-transferase (GSTP1) promoter, which suggests a common
origin (5). The heterogeneous and
multifocality nature of the disease makes it difficult to
understand prostate carcinogenesis (6). Currently, there is no adequate method
to differentiate patients with poor prognosis of PCa from those
with indolent disease, who should only have a controlled follow-up.
The primary method of determining the suitable treatment option for
a patient with PCa is based on the Gleason classification (5), an assessment of its morphological
heterogeneity, which is associated with prognosis. Pathologists can
classify each focus of PCa using Gleason patterns (GP) ranging from
1 to 5, and assigning a Gleason score (GS); or using the updated
Gleason grade group, which includes the two most representative GPs
in the tumor (7,8). Despite the association between Gleason
classification and tumor behavior (the degree of differentiation of
the neoplastic cells) the association between morphological
heterogeneity and molecular heterogeneity has not been elucidated
(9).
Molecular studies of PCa have revealed numerous
recurrent DNA alterations associated with deregulating genes
involved in the development of the prostate, such as the deletions
and interstitial translocations that originate the TMPRSS2-ERG
fusion gene, chromatin modification, cell cycle regulation and
androgen signaling (10,11). Over the last decade (12), the investigation into PCa has focused
on identifying the exclusive molecular events in the development of
PCa, which could represent early and divergent events and could
direct the course of the disease (13); thus, it is crucial to elucidate the
carcinogenesis of PCa and utilize the information in the treatment
of patients.
The present review will describe an updated review
of intratumoral heterogeneity in multifocal PCa, to understand the
carcinogenic process and its implication in the management of the
disease; as the vast majority of molecular studies in PCa performed
are single focused, and do not take into account molecular
heterogeneity, which could contribute to limiting the use of
molecular subtypes in the prognosis and treatment of the
disease.
The origin of PCa could be defined by the occurrence
of chromosomal rearrangements that simultaneously, and in a
coordinated manner, cause the inactivation of tumor suppressor
genes (TSG) and the creation of oncogenic fusions, which would
support a model of punctuated development in PCa (11,14).
This could in turn be associated with the appearance of canonical
alterations, and with the molecular subtypes involved in a broad
genomic and transcriptomic diversity within and among
intraprostatic PCa foci. Several studies have shown potential for
their utilization as prognostic biomarker signatures (15–17).
Recently published data from The Cancer Genome Atlas (TCGA)
(17) supports the division of the
major molecular subclasses of localized PCa into erythroblast
transformation specific (ETS)-rearrangement PCa [PCa with
rearrangements and overexpression of ETS transcription factor ERG
(ERG), ETS variant transcription factor (ETV)-1, −4, or other ETS
family transcription factors], SPOP-mutated and CHD1-deleted
[speckle type BTB/POZ protein (SPOP)/chromodomain helicase DNA
binding protein 1] altered cancers (17), and several smaller categories, such
as FOXA1 or IDH1deletion, which have been described in Table I. The use of molecular classifiers to
personalize treatment shows promise; however, it is still in its
infancy and additional validation and optimization are required to
ensure it can be used in a clinical setting.
There are several molecular alterations in the PCa,
such as copy number changes, gene fusions, single nucleotide
mutations and polymorphisms, methylation, microRNAs and long
non-coding RNAs, one of the most characteristic involves multiple
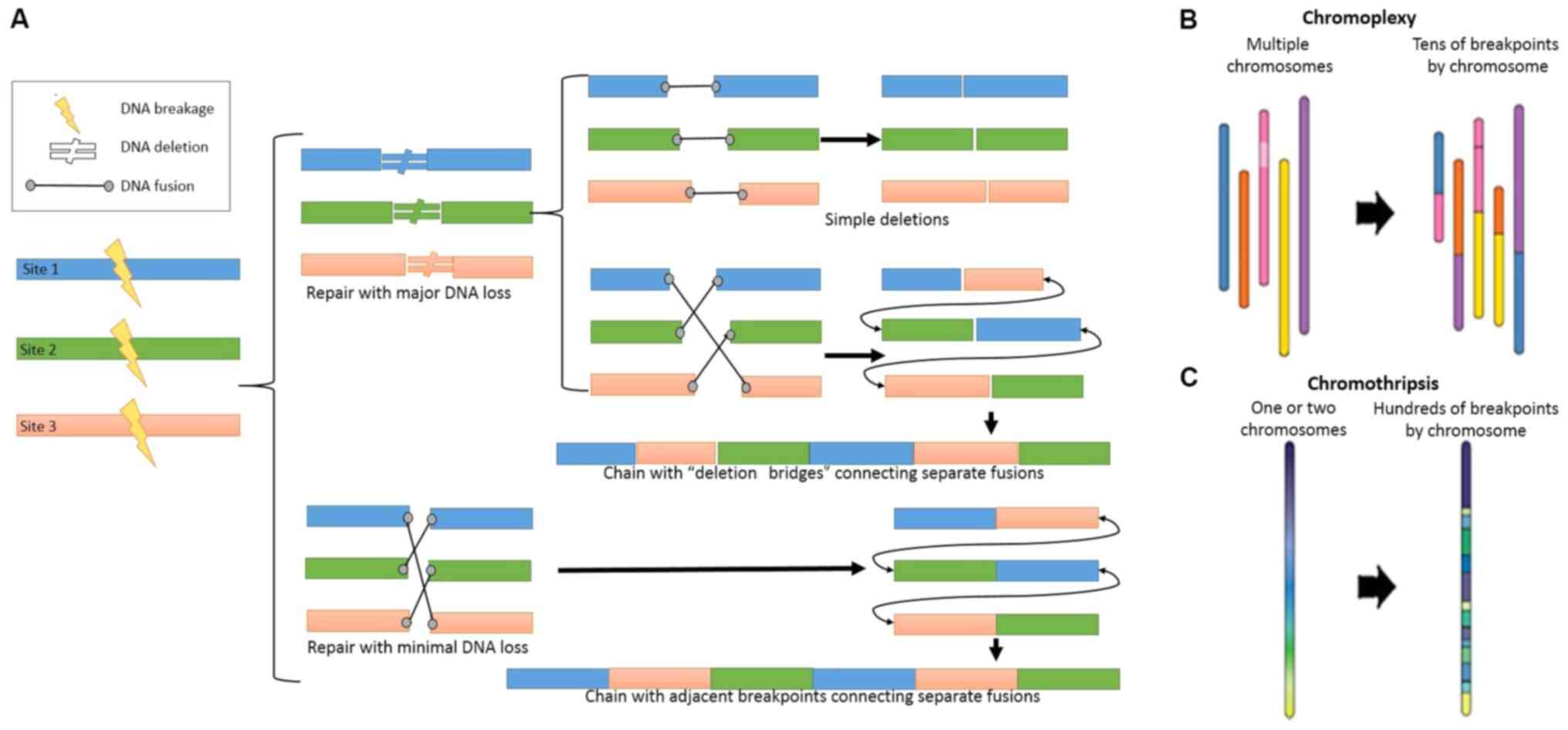
complex chromosomal rearrangement processes (Fig. 1A), which has been reported in 63% of
PCa cases (18). These
rearrangements can be classified as chromothripsis or chromoplexy
(Fig. 1B and C), and some
coordinated structured rearrangements may have intermediate
chromothripsis and chromoplexy properties (11), for instance both chromothripsis and
chromoplexy display random breakage and fusion of genomic segments
with low copy numbers, most likely mediated by non-homologous
end-joining.
It has been found that the breaking points of DNA
rearrangements are more likely to occur near specific DNA
sequences, where the androgen receptor (AR) binds as a
transcription factor, known as androgen response elements (ARE),
compared with that in other randomly predicted locations, that is
anywhere else in the genome (20,21).
This finding suggests that AR-ARE complexes may be predisposed to
genomic rearrangements through transcriptional stress, since
androgen signaling promotes co-recruitment of androgen receptor and
topoisomerase II β (TOP2β) to sites of TMPRSS2-ERG genomic
breakpoints, triggering recombinogenic TOP2β-mediated DNA
double-strand breaks. For example, it has been found that
transmembrane serine protease 2 (TMPRSS2)-ERG fusion is induced by
the interaction of androgens with AR (14,22).
In the development of PCa, chromothripsis is
relatively rare and occurs as one clonal early event; in contrast,
chromoplexy is a common and sequential event, which is detected at
clonal or sub-clonal level (14,18,23). In
a study performed using 57 patients with PCa, Baca et al
(11) identified over 5,000 somatic
rearrangements associated with this type of chromosomal
rearrangements. Several cancer genes were repeatedly deleted or
rearranged by chromoplexy, including PTEN, NK3 homeobox 1, cyclin
dependent kinase inhibitor 1B, tumor protein p53 (TP53), and RB
transcriptional corepressor 1. These multiple complex chromosomal
rearrangements are one of the primary reasons for the high
molecular heterogeneity in PCa.
Over the last decade, several studies have focused
on determining the excluding molecular events in the development of
PCa, which are able to establish different subtypes, and are
associated with the prognosis of the disease, and have the
potential to be developed into targeted therapies (11,16,17,24).
Therefore, the current status of PCa subtypes, will be subsequently
discussed, using TCGA study (17) as
a reference for the majority of the comparisons, the prognostic
involvement, and the therapies of inhibitors of target oncogenes
associated with subtypes, such as EZH2 or ERG (Table I), which have been used or are
currently in the experimental phase, in this way some treatments
that apply to cases that are ETS(+) will not work for those that
are ETS(−), such as blocking function of ERG regulating co-factors,
such as PARP1 (25–27) (Table
I). Table I summarizes the main
clinical-pathological characteristics of the subtypes and
treatments that have been investigated as a development of
personalized medicine (13,17,24–26,28–63).
The ETS family of transcription factors consists of
phosphorylated proteins with DNA-binding domains (ETS domain) that
act as either activators or repressors of transcription. The family
consists of 30 identified genes, 28 of which are found in the human
genome. Previous studies have found that between 50 and 70% of
patients with PCa overexpress ETS, following gene fusion, which
causes ETS to be controlled by ARE (12). Due to the prevalence of these types
of rearrangements, efforts to molecularly characterize PCa begin by
separating the cases which have gene fusion from those that do not,
termed ETS(+) and ETS(−), respectively in numerous studies
(20,38,64).
TCGA study (17) has characterized
four different ETS(+) subtypes, depending on the type of ETS
involved in the fusion: ERG, ETV1, ETV4 and Fli-1 proto-oncogene,
ETS transcription factor ERG is the most frequently overexpressed
ETS. TMPRSS2 the most frequent fusion gene in all ETS fusions;
however, fusions with other androgen-regulated genes have also been
described, including solute carrier family 45 member 3 and N-myc
downstream regulated 1 (65).
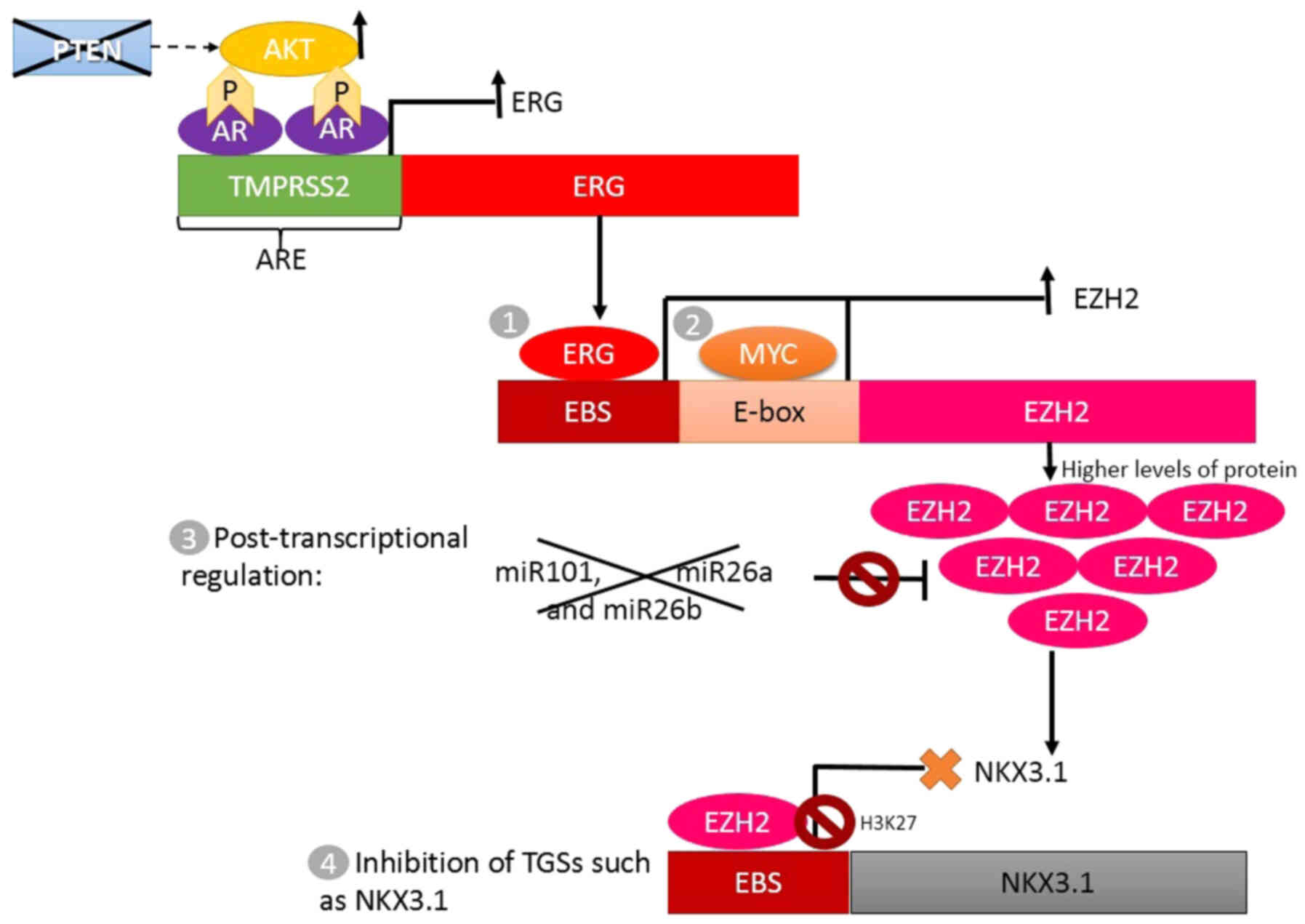
The TMPRSS2-ERG gene fusion causes ERG
overexpression, which initiates a cascade of events that continues
with Enhancer of zeste 2 polycomb repressive complex 2 subunit
(EZH2) overexpression and decreased NKX3-1 expression (Fig. 2) (20,28).
EZH2 is a methyltransferase from the polycomb group, involved in
tissue-specific differentiation by histone methylation (H3K27),
while NKX3-1 is an androgen-regulated, prostate-specific gene,
which encodes a critical transcription factor during prostate
development by downregulating epithelial cell growth, and is
considered a TSG (20,28,66).
Other common molecular alterations in PCa are
deletions and point mutations of PTEN, leading to activation of
Akt, which can subsequently lead to over-activation of AR
signaling; and in the presence of TMPRSS2-ERG gene fusion, it can
increase the activation of a cascade involving ERG, EZH2 and NKX3-1
(Fig. 2) (20,31,33).
ETS(−) are PCa subtypes with a canonic alteration,
which is different to that in the ETS fusion genes. Whereas
rearrangements occurring in ETS(+) tumors display features of
chromoplexy, ETS(−) tumors display a higher frequency of
chromothripsis (11). Among the
ETS(−) subtypes, different studies have found the existence of a
highly-expressed EZH2 subtype, which is independent of ERG
expression (38,39). The overexpression of EZH2 has been
described as well; it is caused by nuclear phosphoprotein MYC,
which is involved in the cell cycle progression, apoptosis and
cellular transformation. MYC oncogene also represses transcription
of microRNA (miR)-26a and miR-26b, which are targets of EZH2, thus
contributing to overexpression of EZH2 (36,38). In
addition, the genomic or functional loss of miR-101, a TSG whose
targets include the EZH2 gene, causes the overexpression of this
gene (37) (Fig. 2).
Since epigenetic abnormalities driven by MYC
overexpression have been associated with a poorer prognosis of PCa
(progression-free survival after radical prostatectomy), therapies
targeting these abnormalities may favor patients with EZH2
overexpression (25–27) (Table
I). Furthermore, TCGA describes 3 ETS(−) subtypes,
characterized by mutations in genes, including SPOP, overexpression
of serine protease inhibitor Kazal type 1 (SPINK1), forkhead box A1
(FOXA1) or isocitrate dehydrogenase (NAPD+)1 (IDH1)
(17) (Table I). Previous studies have reported an
overexpression of SPINK, as an independent subtype (66,67);
however, its overexpression was associated with the mutated SPOP
subtype in TCGA study (17). In
2018, Wu et al (63) reported
a novel subtype, characterized by the inactivation of cyclin
dependent kinase 12, which could benefit from immune checkpoint
inhibition therapy (Table I).
In TCGA study, 26% of PCa cases could not be
classified into any of the identified subtypes (17). There were three major groups of
prostate cancers in the study, one with mostly unaltered genomes
(referred to as quiet), a second group encompassing 50% of all
tumors with an intermediate level of SCNAs, and a third group with
a high burden of arm level genomic gains and losses (17). These PCa were clinically and
genomically heterogeneous; low-pass and high-pass whole-genome
sequencing (WGS) on 100 and 19 tumor/normal pairs, some of had
numerous somatic copy number alterations (SCNA) and a high GS,
which is an indicator of poor prognosis; 33% of them were
genomically similar to the SPOP and FOXA1 subtypes, others were
enriched for mutations of TP53, KDM6A, and KMT2D (lysine
methyltransferase 2D) or specific SCNAs spanning MYC and CCND1
(cyclin D1) and other cases had a low GS (GS 6) with fewer genomic
alterations (38% in the ‘quiet’ class vs. 8% in the class with the
greatest burden of alterations), such as SCNA and DNA methylation
patterns similar to those in normal tissue (17).
The Gleason classification, using histological
methods, that heterogeneity in multifocal PCa can be recognized,
which is higher when analyzed from a molecular point of view
(6,69). This intratumoral heterogeneity makes
it difficult to associate specific molecular alterations, detected
in a single focus, with the clinical behavior of a patient with
multifocal PCa (6,69). In molecular studies, the pattern of
the index tumor is typically obtained to assign molecular
alterations and subtypes; however, other foci are not taken into
account (6). The index tumor is the
largest tumor focus, which in most cases (89%) can be associated
with significant pathological parameters, such as the highest GS,
the largest tumor volume and extraprostatic extension (70); however, this is not always the case
(69). Numerous studies have found
that a single clone is responsible for the dispersal of all
metastatic foci (13,71,72);
which suggests that identifying the ‘deadly’ clone is of utmost
importance, and should not necessarily start from the index
tumor.
Due to the heterogeneity caused by multifocality in
PCa, it is necessary to perform studies, that analyze the molecular
alterations in various foci, which would allow the identification
of the impact of different molecular subtypes in the same patient
(4,64). Some studies using the TMPRSS2-ERG
fusion (73,74), PTEN deletion (75), SPINK1, ERG (67), and whole genome sequencing (6,75–77) have
found a markedly interfocal discordance, which was consistent with
the concept that multiple foci of PCa have a multiclonal origins
(6). Wei et al (6) also found this heterogeneity when using
commercial PCa diagnostic kits (Decipher, Prolaris and Oncotype
DX), which are based on the expression of various genes, including
immune response genes like testis-specific basic protein and PBX
homeobox 1 of Decipher, cell cycle-related genes CDC20 (Cell
Division Cycle 2), CDKN3 (Cyclin Dependent Kinase Inhibitor 3),
CDC2 (cell division control protein 2 homolog) of Prolaris, genes
of androgen signaling AZGP1 (Alpha-2-Glycoprotein 1) and FAM13C
(Family With Sequence Similarity 13 Member C) of Oncotype DX. These
results suggest that applying a single subtype of the molecular
taxonomy of PCa proposed by TCGA to a patient, i.e., studying a
single focus, is an over simplified and incorrect view of the
molecular landscape of PCa.
As aforementioned, the origins of PCa begins as a
pre-neoplastic lesion which progresses to localized cancer, and can
subsequently metastasize. Elimination of androgens using surgical
or chemical castration, in numerous cases, results in control of
PCa (78,79). However, when relapse occurs despite
treatment, PCa has progressed to an androgen-independent form of
cancer or CRPC, which is considered the most aggressive form of PCa
(80).
Most of the molecular alterations in PCa have been
described from HGPIN (or associated with HGPIN). However, there are
studies, which have found associations between some of these
alterations with different stages of PCa progression. ETS fusions
and FOXA1 mutations frequently occur in HGPIN (14,23).
Overexpression of SPINK1 (~10–25%) and SPOP (~11%), TP53 (~25%),
IDH1 (~1%), MAP3K7 and CHD1 (~10–20%) mutations (11,13,17,23,49,81) are
more frequent in localized PCa, while the highest and lowest
expression levels of EZH2 and NKX3-1, respectively, together with
PTEN deletion, have been found in metastatic CRPC (11,23,82).
Monoallelic loss of PTEN is present in up to 60% of localized
prostate cancers and complete loss of PTEN in prostate cancer is
linked to metastasis and androgen-independent progression (86). Alteration of the AR signaling pathway
compared with that in other pathways in CRPC suggest that AR
signaling continues being the ‘master regulator’ for PCa
progression (45,55,84),
including AR copy number gain (24% of CRPCs) or AR point mutation
(20% of CRPCs). These results assist to define the sequence of the
molecular events in the development of PCa, from the origins of the
disease through to its progression into metastasis, resistance to
treatment and death.
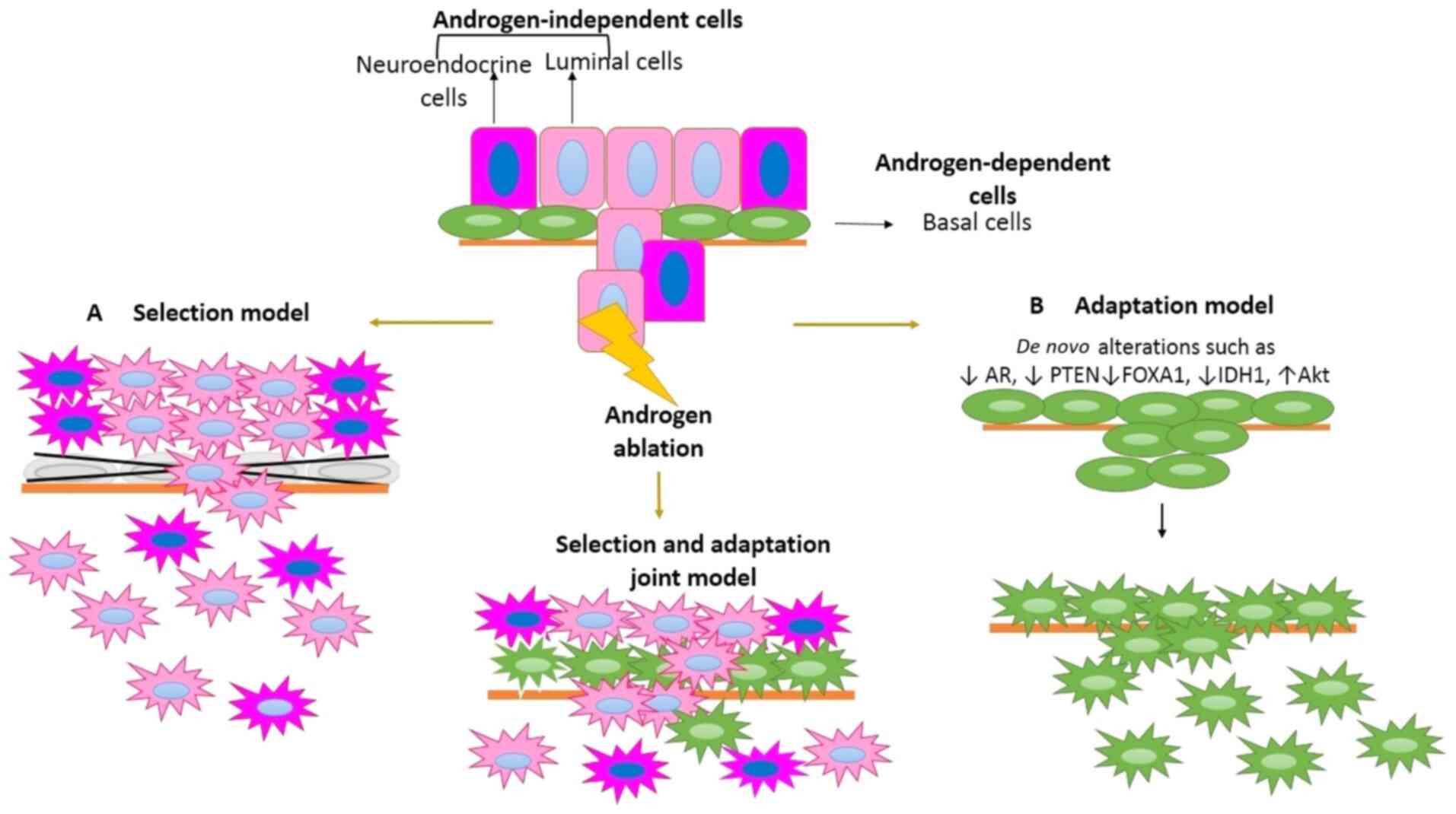
There are 3 types of cells which interact in PCa to
survive androgen ablation treatment: Androgen-dependent, androgen
producing, and androgen-independent cells (85). The interaction among them determines
whether CRPC will develop or not. The pathway which leads from the
development of androgen-dependent to androgen-independent cells is
still unknown. However, there are two models which have been used
to explain this process. Some studies suggest that there is a
collection of androgen-independent preexisting cells following
therapy (selection model) (86,87); in
contrast, other studies postulate that cells acquire novel
alterations which allow them to survive in the absence of androgens
(adaptation model) (88) for
developing CRPC (Fig. 3).
In this model, primary PCa consists of a
heterogeneous mix of luminal, neuroendocrine and stem cells. When
the patient undergoes androgen ablation treatment, most of the
androgen-dependent cells undergo apoptosis, while
androgen-independent cells persist and survive due to their low
androgen requirement (Fig. 3)
(86,87,89).
The vast majority of PCa are luminally
differentiated adenocarcinomas with the presence of neuroendocrine
cells, and respond to hormonal therapy (90); however, there are some tumors, which
consist of aggressive and highly proliferating neuroendocrine cells
only, for example small cell neuroendocrine PCa, which do not
respond to hormone therapy, therefore platinum-based chemotherapy
(phase II trial) is used (90,91).
These malignant neuroendocrine cells, which share their origin with
normal prostatic neuroendocrine cells (93), express epidermal growth factor
receptor (EGFR) and receptor tyrosine-protein kinase erbB-2; for
these reasons, they are classified as androgen-independent cells
(94), and their abundance is
considered a promising prognostic marker for the development of
CRPC (95).
A previous study compared global transcriptomic
profiles of normal basal and luminal epithelial lineages from
samples of patients with PCa and PCa cell lines (87). It was found that PCa cells exhibited
a gene expression profile similar to that found in a luminal cell
and aggressive and neuroendocrine PCa were similar to basal cells
(87).
In addition to cell type, in a few cases, point
mutations in AR can cause cells, which were originally
androgen-dependent, to become androgen-independent cells, and can
be resistant to therapy, that is, those pre-existing mutations in
the localized disease confer a selective advantage with
threapy-resistant cells (84,95–97).
The S646F mutation within AR, in the hinge region, has been
associated with a short response to endocrine therapy, due to a
markedly increased transcriptional activity on ARE-containing
promoters (95). In addition, AR
gene copies (two to four copies), due to polysomy of the
X-chromosome, are present in a subgroup of localized PCa, and these
specimens may have an advantage in low concentrations of androgens
due to therapy (96), since the
additional AR copies may be a factor leading to a poor clinical
outcome of antiandrogen therapy as there is a compensatory
mechanism allowing activation of the AR post-castration.
Furthermore, that study concluded that high stage primary prostate
cancer may be associated with increased frequencies of aneuploidies
of the X chromosome resulting in an increased AR gene copies
number.
The adaptation model suggests that resistance to
castration is the result of the acquisition of genetic and/or
epigenetic alterations in response to therapy, which allows cells
that were previously dependent on androgens to proliferate at low
concentrations of androgens due to therapy (97). Adaptations to androgen ablation
treatment include the occurrence of mutations with a copy number
gain of the AR gene, changes in the expression of AR co-regulation
molecules (48,84,98), and
deregulation of key molecules in proliferation (99), such as Akt overexpression (29).
With the widespread use of novel drugs targeting the
androgenic axis, such as abiraterone acetate and enzalutamide,
there has been a rapid increase in the incidence of small cell
neuroendocrine carcinoma (91,105),
on the basis of autopsy series and other studies this type of PCa
may represent approximately 25% of late stage of PCa (106), which will become a major challenge
in the treatment of these patients. The clarification of the
determining factors that lead to CRPC will be key to understanding
the carcinogenesis process and guiding the clinical management of
each patient.
The use of molecular subtypes in PCa to personalize
treatment is promising; however, it is necessary to consider
multifocality. The lack of an association between subtype and
prognosis in PCa may be due to the fact that only the index tumor
is investigated. It is important to analyze the subtypes in
multiple foci, to elucidate the development of PCa, which could
include different molecular subtypes; during the development of
tumor foci, these would be selected according to their adaptive
advantages, such as resistance to castration and the ability to
metastasize.
The presence or absence of a specific alteration in
any of the foci may be associated with the potential of PCa to
progress to CRPC or to be the target for the development of
targeted therapy, which does not necessarily have to be found in
the index lesion.
Not applicable.
The present study was funded by the National
Institute of Cancerology of Bogota, Colombia (grant no.
C190103001-09).
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
YYSM and MLS were responsible for writing the
manuscript, editing, acquisition, analysis, interpretation of the
data, contributed to data acquisition and edited references. YYSM,
MCSS, RV, JAM and MLS contributed to the conception and design of
the article and were involved in critical revision of the
manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
IARC: Global Cancer Observatory
https://gco.iarc.fr/today/home: GLOBOCAN, . 2018.The Global Cancer
Observatory (GCO) is an interactive web-based platform presenting
global cancer statistics to inform cancer control and research.
|
|
2
|
Beltran H, Yelensky R, Frampton GM, Park
K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus
DM, et al: Targeted next-generation sequencing of advanced prostate
cancer identifies potential therapeutic targets and disease
heterogeneity. Eur Urol. 63:920–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu-Yao G, Albertsen PC, Stanford JL,
Stukel TA, Walker-Corkery E and Barry MJ: Screening, treatment, and
prostate cancer mortality in the Seattle area and Connecticut:
Fifteen-year follow-up. J Gen Intern Med. 23:1809–1814. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Partin AW: High-grade prostatic
intraepithelial neoplasia on a prostate biopsy-what does it mean?
Rev Urol. 4:157–158. 2002.PubMed/NCBI
|
|
5
|
De Marzo AM, Marchi VL, Epstein JI and
Nelson WG: Proliferative inflammatory atrophy of the prostate:
Implications for prostatic carcinogenesis. Am J Pathol.
155:1985–1992. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei L, Wang J, Lampert E, Schlanger S,
DePriest AD, Hu Q, Gomez EC, Murakam M, Glenn ST, Conroy J, et al:
Intratumoral and intertumoral genomic heterogeneity of multifocal
localized prostate cancer impacts molecular classifications and
genomic prognosticators. Eur Urol. 71:183–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Epstein JI, Allsbrook WC, Amin MB and
Egevad LL; ISUP Grading Committee, : The 2005 International society
of urological pathology (ISUP) consensus conference on gleason
grading of prostatic carcinoma. Am J Surg Pathol. 29:1228–1242.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Epstein JI, Egevad L, Amin MB, Delahunt B,
Srigley JR and Humphrey PA; Grading Committee, : The 2014
International Society of Urological Pathology (ISUP) Consensus
Conference on Gleason Grading of Prostatic Carcinoma: Definition of
Grading Patterns and Proposal for a New Grading System. Am J Surg
Pathol. 40:244–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Nunzio C, Pastore AL, Lombardo R,
Simone G, Leonardo C, Mastroianni R, Collura D, Muto G, Gallucci M,
Carbone A, et al: The new Epstein gleason score classification
significantly reduces upgrading in prostate cancer patients. Eur J
Surg Oncol. 44:835–839. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baca SC and Garraway LA: The genomic
landscape of prostate cancer. Front Endocrinol (Lausanne).
3:692012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baca SC, Prandi D, Lawrence MS, Mosquera
JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi
M, et al: Punctuated evolution of prostate cancer genomes. Cell.
153:666–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomlins SA, Rhodes DR, Perner S,
Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J,
Kuefer R, et al: Recurrent fusion of TMPRSS2 and ETS transcription
factor genes in prostate cancer. Science. 310:644–648. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet.
44:685–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berger MF, Lawrence MS, Demichelis F,
Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger
D, Sougnez C, et al: The genomic complexity of primary human
prostate cancer. Nature. 470:214–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tomlins SA, Laxman B, Dhanasekaran SM,
Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et
al: Distinct classes of chromosomal rearrangements create oncogenic
ETS gene fusions in prostate cancer. Nature. 448:595–599. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tomlins SA, Alshalalfa M, Davicioni E,
Erho N, Yousefi K, Zhao S, Haddad Z, Den RB, Dicker AP, Trock BJ,
et al: Characterization of 1577 primary prostate cancers reveals
novel biological and clinicopathologic insights into molecular
subtypes. Eur Urol. 68:555–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cancer Genome Atlas Research Network, .
The Molecular Taxonomy of Primary Prostate Cancer. Cell.
163:1011–1025. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dzamba M, Ramani AK, Buczkowicz P, Jiang
Y, Yu M, Hawkins C and Brudno M: Identification of complex genomic
rearrangements in cancers using CouGaR. Genome Res. 27:107–117.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chun TY: Coincidence of bladder and
prostate cancer. J Urol. 157:65–67. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu J, Mani RS, Cao Q, Brenner CJ, Cao X,
Wang X, Wu L, Li J, Hu M, Gong Y, et al: An integrated network of
androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in
prostate cancer progression. Cancer Cell. 17:443–454. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin C, Yang L, Tanasa B, Hutt K, Ju BG,
Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK and Rosenfeld MG: Nuclear
receptor-induced chromosomal proximity and DNA breaks underlie
specific translocations in cancer. Cell. 139:1069–1083. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mani RS, Tomlins SA, Callahan K, Ghosh A,
Nyati MK, Varambally S, Palanisamy N and Chinnaiyan AM: Induced
chromosomal proximity and gene fusions in prostate cancer. Science.
326:12302009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jung SH, Shin S, Kim MS, Baek IP, Lee JY,
Lee SH, Kim TM, Lee SH and Chung YJ: Genetic progression of high
grade prostatic intraepithelial neoplasia to prostate cancer. Eur
Urol. 69:823–830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lapointe J, Li C, Higgins JP, van de Rijn
M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim
U, et al: Gene expression profiling identifies clinically relevant
subtypes of prostate cancer. Proc Natl Acad Sci USA. 101:811–816.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao
Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al: Mechanistic
rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene
fusion-positive prostate cancer. Cancer Cell. 19:664–678. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Williamson SR and Cheng L: Potential for
targeted therapy in prostate cancers with ERG abnormalities. Asian
J Androl. 13:781–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karpova Y, Wu C, Divan A, McDonnell ME,
Hewlett E, Makhov P, Gordon J, Ye M, Reitz AB, Childers WE, et al:
Non-NAD-like PARP-1 inhibitors in prostate cancer treatment.
Biochem Pharmacol. 167:149–162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kunderfranco P, Mello-Grand M, Cangemi R,
Pellini S, Mensah A, Albertini V, Malek A, Chiorino G, Catapano CV
and Carbone GM: ETS transcription factors control transcription of
EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1
in prostate cancer. PLoS One. 5:e105472010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang S, Gao J, Lei Q, Rozengurt N,
Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et
al: Prostate-specific deletion of the murine Pten tumor suppressor
gene leads to metastatic prostate cancer. Cancer Cell. 4:209–221.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao T, Mei Y, Sun H, Nie Z, Liu X and Wang
S: The association of Phosphatase and tensin homolog (PTEN)
deletion and prostate cancer risk: A meta-analysis. Biomed
Pharmacother. 83:114–121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leinonen KA, Saramäki OR, Furusato B,
Kimura T, Takahashi H, Egawa S, Suzuki H, Keiger K, Ho Hahm S,
Isaacs WB, et al: Loss of PTEN is associated with aggressive
behavior in ERG-positive prostate cancer. Cancer Epidemiol
Biomarkers Prev. 22:2333–2344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ngollo M, Lebert A, Dagdemir A, Judes G,
Karsli-Ceppioglu S, Daures M, Kemeny JL, Penault-Llorca F, Boiteux
JP, Bignon YJ, et al: The association between histone 3 lysine 27
trimethylation (H3K27me3) and prostate cancer: Relationship with
clinicopathological parameters. BMC Cancer. 14:9942014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishigami-Yuasa M, Ekimoto H and Kagechika
H: Class IIb HDAC inhibition enhances the inhibitory effect of
Am80, a synthetic retinoid, in prostate cancer. Biol Pharm Bull.
42:448–452. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bai Y, Zhang Z, Cheng L, Wang R, Chen X,
Kong Y, Feng F, Ahmad N, Li L and Liu X: Inhibition of enhancer of
zeste homolog 2 (EZH2) overcomes enzalutamide resistance in
castration-resistant prostate cancer. J Biol Chem. 294:9911–9923.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Taplin ME, Hussain A, Shah S, Neal D.
Shore, Manish Agrawal, William Clark, et al: ProSTAR: A phase Ib/II
study of CPI-1205, a small molecule inhibitor of EZH2, combined
with enzalutamide (E) or abiraterone/prednisone (A/P) in patients
with metastatic castration-resistant prostate cancer (mCRPC). J
Clin Oncol. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koh CM, Iwata T, Zheng Q, Bethel C,
Yegnasubramanian S and De Marzo AM: Myc enforces overexpression of
EZH2 in early prostatic neoplasia via transcriptional and
post-transcriptional mechanisms. Oncotarget. 2:669–683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Varambally S, Cao Q, Mani RS, Shankar S,
Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Börno ST, Fischer A, Kerick M, Fälth M,
Laible M, Brase JC, Kuner R, Dahl A, Grimm C, Sayanjali B, et al:
Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative
prostate cancers implicate an EZH2-dependent mechanism with miR-26a
hypermethylation. Cancer Discov. 2:1024–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Melling N, Thomsen E, Tsourlakis MC, Kluth
M, Hube-Magg C, Minner S, Koop C, Graefen M, Heinzer H, Wittmer C,
et al: Overexpression of enhancer of zeste homolog 2 (EZH2)
characterizes an aggressive subset of prostate cancers and predicts
patient prognosis independently from pre- and postoperatively
assessed clinicopathological parameters. Carcinogenesis.
36:1333–1340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Uchiyama N, Tanaka Y and Kawamoto T:
Aristeromycin and DZNeP cause growth inhibition of prostate cancer
via induction of mir-26a. Eur J Pharmacol. 812:138–146. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kirschner AN, Wang J, van der Meer R,
Anderson PD, Franco-Coronel OE, Kushner MH, Everett JH, Hameed O,
Keeton EK, Ahdesmaki M, et al: PIM kinase inhibitor AZD1208 for
treatment of MYC-driven prostate cancer. J Natl Cancer Inst.
107:dju4072015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rebello RJ, Kusnadi E, Cameron DP, Pearson
HB, Lesmana A, Devlin JR, Drygin D, Clark AK, Porter L, Pedersen J,
et al: The dual inhibition of RNA Pol I transcription and PIM
kinase as a new therapeutic approach to treat advanced prostate
cancer. Clin Cancer Res. 22:5539–5552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boysen G, Barbieri CE, Prandi D, Blattner
M, Chae SS, Dahija A, Nataraj S, Huang D, Marotz C, Xu L, et al:
SPOP mutation leads to genomic instability in prostate cancer.
Elife. 4:e092072015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rodrigues LU, Rider L, Nieto C, Romero L,
Karimpour-Fard A, Loda M, Lucia MS, Wu M, Shi L, Cimic A, et al:
Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate
cancer. Cancer Res. 75:1021–1034. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Grasso CS, Wu YM, Robinson DR, Cao X,
Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC,
et al: The mutational landscape of lethal castration-resistant
prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shen C, Zhang J, Qi M, Chang YWY and BH:
Roles of serine protease inhibitor kazal type 1 (SPINK1) in
prostate cancer. Med chem. 4:725–728. 2014. View Article : Google Scholar
|
|
47
|
Liu D, Takhar M, Alshalalfa M, Erho N,
Shoag J, Jenkins RB, Karnes RJ, Ross AE, Schaeffer EM, Rubin MA, et
al: Impact of the SPOP mutant subtype on the interpretation of
clinical parameters in prostate cancer. JCO Precis Oncol.
2018:102018.
|
|
48
|
Johnson MH, Ross AE, Alshalalfa M, Erho N,
Yousefi K, Glavaris S, Fedor H, Han M, Faraj SF, Bezerra SM, et al:
SPINK1 Defines a molecular subtype of prostate cancer in men with
more rapid progression in an at risk, natural history radical
prostatectomy cohort. J Urol. 196:1436–1444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yun SJ, Kim SK, Kim J, Cha EJ, Kim JS, Kim
SJ, Ha YS, Kim YH, Jeong P, Kang HW, et al: Transcriptomic features
of primary prostate cancer and their prognostic relevance to
castration-resistant prostate cancer. Oncotarget. 8:114845–114855.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tiwari R, Manzar N, Bhatia V, Yadav A,
Nengroo MA, Datta D, Carskadon S, Gupta N, Sigouros M, Khani F, et
al: Androgen deprivation upregulates SPINK1 expression and
potentiates cellular plasticity in prostate cancer. Nat Commun.
11:3842020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Geng C, Rajapakshe K, Shah SS, Shou J,
Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M,
et al: Androgen receptor is the key transcriptional mediator of the
tumor suppressor SPOP in prostate cancer. Cancer Res. 74:5631–5643.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lu D, Lee J, Lee A and Lee R: Development
of a new approach for the therapy of prostate cancer with SPOP
mutations. J Cancer Therapy. 6:841–848. 2015. View Article : Google Scholar
|
|
53
|
Boysen G, Rodrigues DN, Rescigno P, Seed
G, Dolling D, Riisnaes R, Crespo M, Zafeiriou Z, Sumanasuriya S,
Bianchini D, et al: SPOP-Mutated/CHD1-deleted lethal prostate
cancer and abiraterone sensitivity. Clin Cancer Res. 24:5585–5593.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ateeq B, Tomlins SA, Laxman B, Asangani
IA, Cao Q, Cao X, Li Y, Wang X, Feng FY, Pienta KJ, et al:
Therapeutic targeting of SPINK1-positive prostate cancer. Sci
Transl Med. 3:72ra172011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Stelloo S, Nevedomskaya E, Kim Y,
Schuurman K, Valle-Encinas E, Lobo J, Krijgsman O, Peeper DS, Chang
SL, Feng FY, et al: Integrative epigenetic taxonomy of primary
prostate cancer. Nat Commun. 9:49002018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Imamura Y, Sakamoto S, Endo T, Utsumi T,
Fuse M, Suyama T, Kawamura K, Imamoto T, Yano K, Uzawa K, et al:
FOXA1 promotes tumor progression in prostate cancer via the
insulin-like growth factor binding protein 3 pathway. PLoS One.
7:e424562012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Adams EJ, Karthaus WR, Hoover E, Liu D,
Gruet A, Zhang Z, Cho H, DiLoreto R, Chhangawala S, Liu Y, et al:
FOXA1 mutations alter pioneering activity, differentiation and
prostate cancer phenotypes. Nature. 571:408–412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Song B, Park SH, Zhao JC, Fong KW, Li S,
Lee Y, Yang YA, Sridhar S, Lu X, Abdulkadir SA, et al: Targeting
FOXA1-mediated repression of TGF-β signaling suppresses
castration-resistant prostate cancer progression. J Clin Invest.
129:569–582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gui B, Gui F, Takai T, Feng C, Bai X,
Fazli L, Dong X, Liu S, Zhang X, Zhang W, et al: Selective
targeting of PARP-2 inhibits androgen receptor signaling and
prostate cancer growth through disruption of FOXA1 function. Proc
Natl Acad Sci USA. 116:14573–14582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ghiam AF, Cairns RA, Thoms J, Dal Pra A,
Ahmed O, Meng A, Mak TW and Bristow RG: IDH mutation status in
prostate cancer. Oncogene. 31:38262012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mondesir J, Willekens C, Touat M and de
Botton S: IDH1 and IDH2 mutations as novel therapeutic targets:
Current perspectives. J Blood Med. 7:171–180. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wu YM, Cieślik M, Lonigro RJ, Vats P,
Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al:
Inactivation of CDK12 delineates a distinct immunogenic class of
advanced prostate cancer. Cell. 173:1770–1782.e14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tomlins SA, Rhodes DR, Yu J, Varambally S,
Mehra R, Perner S, Demichelis F, Helgeson BE, Laxman B, Morris DS,
et al: The role of SPINK1 in ETS rearrangement-negative prostate
cancers. Cancer Cell. 13:519–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hermans KG, van Marion R, van Dekken H,
Jenster G, van Weerden WM and Trapman J: TMPRSS2:ERG fusion by
translocation or interstitial deletion is highly relevant in
androgen-dependent prostate cancer, but is bypassed in late-stage
androgen receptor-negative prostate cancer. Cancer Res.
66:10658–10663. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Thangapazham R, Saenz F, Katta S, Mohamed
AA, Tan SH, Petrovics G, Srivastava S and Dobi A: Loss of the
NKX3.1 tumorsuppressor promotes the TMPRSS2-ERG fusion gene
expression in prostate cancer. BMC Cancer. 14:162014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fontugne J, Davis K, Palanisamy N, Udager
A, Mehra R, McDaniel AS, Siddiqui J, Rubin MA, Mosquera JM and
Tomlins SA: Clonal evaluation of prostate cancer foci in biopsies
with discontinuous tumor involvement by dual ERG/SPINK1
immunohistochemistry. Mod Pathol. 29:157–165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Løvf M, Zhao S, Axcrona U, Johannessen B,
Bakken AC, Carm KT, Hoff AM, Myklebost O, Meza-Zepeda LA and Lie
AK: Multifocal primary prostate cancer exhibits high degree of
genomic heterogeneity. Eur Urol. 75:498–505. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Huang CC, Deng FM, Kong MX, Ren Q, Melamed
J and Zhou M: Re-evaluating the concept of ‘dominant/index tumor
nodule’ in multifocal prostate cancer. Virchows Arch. 464:589–594.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
McNeal JE, Price HM, Redwine EA, Freiha FS
and Stamey TA: Stage A versus stage B adenocarcinoma of the
prostate: Morphological comparison and biological significance. J
Urol. 139:61–65. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu W, Laitinen S, Khan S, Vihinen M,
Kowalski J, Yu G, Chen L, Ewing CM, Eisenberger MA, Carducci MA, et
al: Copy number analysis indicates monoclonal origin of lethal
metastatic prostate cancer. Nat Med. 15:559–565. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Haffner MC, Mosbruger T, Esopi DM, Fedor
H, Heaphy CM, Walker DA, Adejola N, Gürel M, Hicks J, Meeker AK, et
al: Tracking the clonal origin of lethal prostate cancer. J Clin
Invest. 123:4918–4922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Barry M, Perner S, Demichelis F and Rubin
MA: TMPRSS2-ERG fusion heterogeneity in multifocal prostate cancer:
clinical and biologic implications. Urology. 70:630–633. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Furusato B, Gao CL, Ravindranath L, Chen
Y, Cullen J, McLeod DG, Dobi A, Srivastava S, Petrovics G and
Sesterhenn IA: Mapping of TMPRSS2-ERG fusions in the context of
multi-focal prostate cancer. Mod Pathol. 21:67–75. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yoshimoto M, Ding K, Sweet JM, Ludkovski
O, Trottier G, Song KS, Joshua AM, Fleshner NE, Squire JA and Evans
AJ: PTEN losses exhibit heterogeneity in multifocal prostatic
adenocarcinoma and are associated with higher Gleason grade. Mod
Pathol. 26:435–447. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Boutros PC, Fraser M, Harding NJ, de Borja
R, Trudel D, Lalonde E, Meng A, Hennings-Yeomans PH, McPherson A,
Sabelnykova VY, et al: Spatial genomic heterogeneity within
localized, multifocal prostate cancer. Nat Genet. 47:736–745. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cooper CS, Eeles R, Wedge DC, Van Loo P,
Gundem G, Alexandrov LB, Kremeyer B, Butler A, Lynch AG, Camacho N,
et al: Analysis of the genetic phylogeny of multifocal prostate
cancer identifies multiple independent clonal expansions in
neoplastic and morphologically normal prostate tissue. Nat Genet.
47:367–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Crawford ED, Heidenreich A, Lawrentschuk
N, Tombal B, Pompeo ACL, Mendoza-Valdes A, Miller K, Debruyne FMJ
and Klotz L: Androgen-targeted therapy in men with prostate cancer:
Evolving practice and future considerations. Prostate Cancer
Prostatic Dis. 22:24–38. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Vickers AJ, Bianco FJ, Serio AM, Eastham
JA, Schrag D, K EA, Reuther AM, Kattan MW, Pontes JE and Scardino
PT: The surgical learning curve for prostate cancer control after
radical prostatectomy. J Natl Cancer Inst. 99:1171–1177. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yu EY, Gulati R, Telesca D, Jiang P, Tam
S, Russell KJ, Nelson PS, Etzioni RD and Higano CS: Duration of
first off-treatment interval is prognostic for time to castration
resistance and death in men with biochemical relapse of prostate
cancer treated on a prospective trial of intermittent androgen
deprivation. J Clin Oncol. 28:2668–2673. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Huang KC, Evans A, Donnelly B and Bismar
TA: SPINK1 Overexpression in localized prostate cancer: A rare
event inversely associated with ERG expression and exclusive of
homozygous PTEN deletion. Pathol Oncol Res. 23:399–407. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Green SM, Mostaghel EA and Nelson PS:
Androgen action and metabolism in prostate cancer. Mol Cell
Endocrinol. 360:3–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Phin S, Moore MW and Cotter PD: Genomic
rearrangements of PTEN in prostate cancer. Front Oncol. 3:2402013.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Taylor BS, Schultz N, Hieronymus H,
Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva
B, et al: Integrative genomic profiling of human prostate cancer.
Cancer Cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang J, Cunningham JJ, Brown JS and
Gatenby RA: Integrating evolutionary dynamics into treatment of
metastatic castrate-resistant prostate cancer. Nat Commun.
8:18162017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lawson DA, Zong Y, Memarzadeh S, Xin L,
Huang J and Witte ON: Basal epithelial stem cells are efficient
targets for prostate cancer initiation. Proc Natl Acad Sci USA.
107:2610–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang D, Park D, Zhong Y, Lu Y, Rycaj K,
Gong S, Chen X, Liu X, Chao HP, Whitney P, et al: Stem cell and
neurogenic gene-expression profiles link prostate basal cells to
aggressive prostate cancer. Nat Commun. 7:107982016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Nouri M, Caradec J, Lubik AA, Li N,
Hollier BG, Takhar M, Altimirano-Dimas M, Chen M, Roshan-Moniri M,
Butler M, et al: Therapy-induced developmental reprogramming of
prostate cancer cells and acquired therapy resistance. Oncotarget.
8:18949–18967. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu C, Wyatt AW, Lapuk AV, McPherson A,
McConeghy BJ, Bell RH, Anderson S, Haegert A, Brahmbhatt S, Shukin
R, et al: Integrated genome and transcriptome sequencing identifies
a novel form of hybrid and aggressive prostate cancer. J Pathol.
227:53–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lipianskaya J, Cohen A, Chen CJ, Hsia E,
Squires J, Li Z, Zhang Y, Li W, Chen X, Xu H and Huang J:
Androgen-deprivation therapy-induced aggressive prostate cancer
with neuroendocrine differentiation. Asian J Androl. 16:541–544.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Aparicio AM, Harzstark AL, Corn PG, Wen S,
Araujo JC, Tu SM, Pagliaro LC, Kim J, Millikan RE, Ryan C, et al:
Platinum-based chemotherapy for variant castrate-resistant prostate
cancer. Clin Cancer Res. 19:3621–3630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bonkhoff H and Remberger K:
Differentiation pathways and histogenetic aspects of normal and
abnormal prostatic growth: A stem cell model. Prostate. 28:98–106.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cortés MA, Cariaga-Martinez AE, Lobo MV,
Martín Orozco RM, Motiño O, Rodríguez-Ubreva FJ, Angulo J,
López-Ruiz P and Colás B: EGF promotes neuroendocrine-like
differentiation of prostate cancer cells in the presence of
LY294002 through increased ErbB2 expression independent of the
phosphatidylinositol 3-kinase-AKT pathway. Carcinogenesis.
33:1169–1177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Abrahamsson PA, Wadström LB, Alumets J,
Falkmer S and Grimelius L: Peptide-hormone- and
serotonin-immunoreactive tumour cells in carcinoma of the prostate.
Pathol Res Pract. 182:298–307. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thompson J, Hyytinen ER, Haapala K,
Rantala I, Helin HJ, Jänne OA, Palvimo JJ and Koivisto PA: Androgen
receptor mutations in high-grade prostate cancer before hormonal
therapy. Lab Invest. 83:1709–1713. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Röpke A, Erbersdobler A, Hammerer P,
Palisaar J, John K, Stumm M and Wieacker P: Gain of androgen
receptor gene copies in primary prostate cancer due to X chromosome
polysomy. Prostate. 59:59–68. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nouri M, Ratther E, Stylianou N, Nelson
CC, Hollier BG and Williams ED: Androgen-targeted therapy-induced
epithelial mesenchymal plasticity and neuroendocrine
transdifferentiation in prostate cancer: An opportunity for
intervention. Front Oncol. 4:3702014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Han G, Buchanan G, Ittmann M, Harris JM,
Yu X, Demayo FJ, Tilley W and Greenberg NM: Mutation of the
androgen receptor causes oncogenic transformation of the prostate.
Proc Natl Acad Sci USA. 102:1151–1156. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kaarbø M, Mikkelsen OL, Malerød L, Qu S,
Lobert VH, Akgul G, Halvorsen T, Maelandsmo GM and Saatcioglu F:
PI3K-AKT-mTOR pathway is dominant over androgen receptor signaling
in prostate cancer cells. Cell Oncol. 32:11–27. 2010.PubMed/NCBI
|
|
100
|
Terry S and Beltran H: The many faces of
neuroendocrine differentiation in prostate cancer progression.
Front Oncol. 4:602014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Choi N, Zhang B, Zhang L, Ittmann M and
Xin L: Adult murine prostate basal and luminal cells are
self-sustained lineages that can both serve as targets for prostate
cancer initiation. Cancer Cell. 21:253–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Germann M, Wetterwald A, Guzmán-Ramirez N,
vander Pluijm G, Culig Z, Cecchini MG, Williams ED and Thalmann GN:
Stem-like cells with luminal progenitor phenotype survive
castration in human prostate cancer. Stem Cells. 30:1076–1086.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Evans AJ, Humphrey PA, Belani J, van der
Kwast TH and Srigley JR: Large cell neuroendocrine carcinoma of
prostate: A clinicopathologic summary of 7 cases of a rare
manifestation of advanced prostate cancer. Am J Surg Pathol.
30:684–693. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen H, Sun Y, Wu C, Magyar CE, Li X,
Cheng L, Yao JL, Shen S, Osunkoya AO, Liang C and Huang J:
Pathogenesis of prostatic small cell carcinoma involves the
inactivation of the P53 pathway. Endocr Relat Cancer. 19:321–331.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Beltran H, Prandi D, Mosquera JM, Benelli
M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV,
Varambally S, et al: Divergent clonal evolution of
castration-resistant neuroendocrine prostate cancer. Nat Med.
22:298–305. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Aparicio A, Logothetis CJ and Maity SN:
Understanding the lethal variant of prostate cancer: Power of
examining extremes. Cancer Discov. 1:466–468. 2011. View Article : Google Scholar : PubMed/NCBI
|