Introduction
Intercellular adhesion is the structural basis for
the growth and development of multicellular organisms. The abnormal
expression of proteins involved in the process of intercellular
adhesion can cause various complications, such as kidney,
cardiovascular and autoimmune diseases, as well as cancer (1–3).
Cadherins are essential for cell adhesion. In vertebrates, it is
generally accepted that there are six main subfamilies of
cadherins: Classical cadherins, desmosomal cadherins,
protocadherins, Flamingo/Celsr, Dachsous and FAT atypical cadherins
(FAT) (4).
In the 1920s, FAT was discovered in
Drosophila due to a lethal mutation (5). Drosophila has two FAT cadherin
members, Ft and Ft2, which are considered tumor suppressors
(6). Most functional mutations of
the Ft gene result in an ‘epithelial overgrowth phenotype’ in
Drosophila larvae, which can affect the wings, legs, eye
antennae, glands and genital imaging disc (6,7). The
overgrowth phenotype observed following Ft inactivation is
hypothesized to be partly associated with the fact that Ft can
affect the localization and expression levels of the Hippo
signaling pathway transcription factors, Yorkie (Yki), Warts (Wts)
and Expanded (8–10). In addition, in the eyes of FAT mutant
Drosophila, the omentum exhibits reversed dorsal-ventral
polarity (11). This is hypothesized
to be caused by the effect of Ft on planar cell polarity (PCP)
(11). The other FAT cadherin member
in Drosophila, Ft2, is necessary for morphogenesis and
maintenance of tubular structures of ectodermal origin (7,12).
Deletion of Ft2 results in abnormal development of renal tubular
structures, such as loss of the trachea, gastric glands and
salivary glands (12). The
established functional roles of Ft and Ft2 in Drosophila
primarily include regulation of morphogenesis, growth control and
PCP (8,11,12),
providing a platform for the study of FAT cadherins in
vertebrates.
From Drosophila to vertebrates, the FAT
cadherin family has expanded from two members to four, but their
structure and function remain conserved across species (13). The FAT cadherin family in vertebrates
consists of FAT1-4 (5), all of which
have unique arrangements and number of laminin G motifs and
EGF-like motifs, which bestows upon them unique functions. FAT1 and
FAT4 contain 34 extracellular cadherin repeats, whereas FAT2
contains 32 repeats and FAT3 contains 33 repeats (Fig. 1) (14). As FAT mRNA transcripts and coding
proteins are large, understanding the function of FAT proteins is
challenging. At present, the majority of research on FAT proteins
have focused on FAT1, as FAT1 was the first member to be identified
in humans, and the effects of FAT1 mutations in tumors, such as
acute lymphoblastic leukemia (ALL) (15) and hepatocellular carcinoma (HCC)
(16), are more prominent (7).
The human FAT1 gene was cloned from a human T-cell
ALL (T-ALL) cell line in 1995 (15).
The gene is located on chromosome 4q34-35 and consists of 27 exons
in humans (15). It is a type I
transmembrane protein, consisting of an extracellular region,
transmembrane region and cytoplasmic tail (17).
It has been reported that FAT1 is involved in
numerous processes, such as cell adhesion, proliferation and
migration via protein-protein interactions of its cytoplasmic tail
(7). Existing evidence has shown
that FAT1 can bind to the actin-regulatory protein Enabled
(Ena)/vasodilator-stimulated phosphoprotein (Vasp) and β-catenin to
regulate cell proliferation and migration, and that it serves a
role in the Wnt/β-catenin, Hippo and MAPK/ERK signaling pathways,
as well affecting epithelial-mesenchymal transition (EMT) (18–22). At
present, knowledge regarding the exact downstream signaling
pathways mediated by FAT1 remains incomplete, but an increasing
body of knowledge is indicating that changes in FAT1 expression are
associated with several diseases, such as facioscapulohumeral
muscular dystrophy (FSHD) (23),
bipolar disorder (BPAD) (24) and
ALL (15). In the present review,
the latest progress in understanding the role of FAT1 is discussed,
with the aim of promoting interest in FAT1 as a biomarker and a
potential therapeutic target for the management of several
diseases.
Signaling pathways and mechanisms
FAT1 and Ena/Vasp
Ena/Vasp is an important regulator of actin
dynamics. The Ena/Vasp protein family regulates the assembly of the
actin cytoskeleton by antagonizing the dissociation of capping
proteins and promoting actin filament branching connections, which
serve a vital role in morphogenesis, axon guidance, cell migration
and other processes (25–28). The role of FAT1 in the pathways
necessary to determine the polarity of cells in the tissue plane
has been demonstrated in Drosophila (29). The FAT1 cytoplasmic domain contains
an EVH1 binding motif (4433DFPPPPEE). EVH1 binding motifs are
predicted to interact with members of the Ena/Vasp protein family
that contain EVH1 domains (30).
Thus, FAT1 may serve a role in regulating cell migration by
participating in the regulation of Ena/Vasp-dependent frontier
cytoskeletal dynamics (29,30). Tanoue and Takeichi (30) found that FAT1 and Ena/Vasp proteins
are co-localized at the edge of the plate fat, the tip of the silk
shaft and at early contact site between cells. Knockdown of FAT1
causes Vasp to no longer accumulate in these locations, and the
cytoskeleton of connecting actin cannot be formed correctly
(30). Additionally, it was shown
that actin filaments were recruited to the junction and formed
radial actin cables during the early stages of cell-cell contact in
mouse cells (30). After knocking
down FAT1, the connecting actin filaments were poorly arranged, and
the connecting actin cytoskeleton could not be formed correctly
(30). Additionally, FAT1 can
promote the formation of actin stress fibers in renal epithelial
cells (30). These observations
indicate that FAT1 serves an important role in actin dynamics
(30). Moeller et al
(29) reported that in cells where
FAT1 expression was knocked out, the amount of Vasp protein that
accumulated at the front of cells was significantly decreased, the
cells did not become polarized and the migratory ability of cells
was decreased. In in vitro wound healing assays,
FAT1-knockout NRK-52E cells (rat kidney epithelioid cells)
exhibited abnormal lamella dynamics at the wound edge, which
included an absence of lamella or abnormal lamella lipid membranes,
decreased lamella size and abnormally structured lamellae, all of
which resulted in delayed wound closure (31). These results indicate that FAT1 is
necessary for physiological lamellipodial dynamics, and
FAT1-knockout disrupts normal lamellipodial dynamics to a certain
extent (29,30). This may provide an explanation for
the weakened cell migration in FAT1-deficient cells.
FAT1 and the Wnt/β-catenin signaling
pathway
The role of the Wnt/β-catenin signaling pathway in
human development and maintaining adult tissue homeostasis is
well-established. Abnormalities in the Wnt signaling pathway are
closely associated with the occurrence of numerous diseases, such
as osteoporosis, Alzheimer's disease and pigmented diseases
(32). Abnormal activation of the
Wnt/β-catenin signaling pathway is widely considered to be a
promoter of tumorigenesis and cancer cell proliferation in several
types of cancer (33), such as HCC
(34) and colon cancer (35). The Wnt/β-catenin signaling pathway
primarily consists of three steps: Wnt signal transduction at the
membrane, stable regulation of β-catenin in the cytoplasm and
activation of Wnt target genes in the nucleus. FAT1 protein may
affect Wnt signaling via modulation of the latter two steps
(36–38).
Just as classical cadherin can bind to β-catenin and
regulate its transcriptional activity, FAT1 can also bind to
β-catenin and limit its translocation to the nucleus (39). This links FAT1 to the typical Wnt
signaling pathway. A study by Hou et al (36) confirmed this interaction by
demonstrating increased FAT1 expression following carotid artery
injury in rats. The rat FAT1 cytoplasmic tail was shown to bind to
β-catenin, preventing its translocation to the nucleus and thus
preventing transcription of its target genes, including the known
transcriptional target cyclin D1, which is a target of the typical
Wnt signaling pathway (36). In
addition, the results of FAT1-knockout revealed that increased FAT1
expression limited cyclin D1 expression in mouse vascular smooth
muscle cells (VSMCs) (40). Cyclin
D1 is a known Tcf/β-catenin target gene, which serves a key role in
the regulation of G1 phase progression and
G1max/S cell cycle transition (36). Increased FAT1 expression after injury
may slow down the proliferation of VSMCs by decreasing cyclin D1
expression (36). The association
between FAT1 and the Wnt signaling pathway serves an important role
in the overall regulation of VSMC proliferation.
Morris et al (19) revealed that endogenous FAT1 bound to
β-catenin in human cells. In experiments using glioma cells and
immortalized human brain astrocytes, FAT1-knockout resulted in a
decrease in plasma membrane β-catenin staining, and a significant
increase in nuclear β-catenin staining (19). Inactivated FAT1 expression could not
sequester β-catenin at the cell membrane, thereby allowing for
canonical Wnt signal transduction and tumor growth (19). Overexpression of β-catenin promoted
cell proliferation and cell cycle progression, and increased the
incorporation of BrdU (indicative of increased proliferation), but
these effects were inhibited by FAT1 (19). Knockout of CTNNB1 (encoding
β-catenin) in glioblastoma multiforme cells decreased cell
proliferation, cell cycle progression and BrdU incorporation. These
effects were largely reversed by FAT1-knockout (19). After knocking down FAT1 in glioma
cells and immortalized human astrocytes using small interfering
RNAs, β-catenin-mediated transcription was significantly increased
(19). Another means by which FAT1
affects the Wnt/β-catenin signaling pathway is that the deletion of
FAT1 alters gene expression and affects the expression of
components of the Wnt/β-catenin signaling pathway (19). Knockout of FAT1 mRNA resulted in
consistent upregulation of multiple Wnt/β-catenin target genes,
including myc, cyclin D1, inhibitor of DNA binding 2, transcription
factor 4, claudin and zinc finger E-box binding homeobox 1 (ZEB1)
(41–43). Similar results were obtained based on
analysis of data obtained from an ovarian cancer dataset from The
Cancer Genome Atlas (19,44). Based on the identification of 189
significantly differentially expressed genes, it was revealed that
tumors with low FAT1 expression exhibited enrichment of genes
associated with the Wnt/β-catenin signaling pathway (44). These results suggest that FAT1
mutations may be a prominent cause of the dysregulation of the Wnt
signaling pathway in cancer (44).
FAT1 and the Hippo signaling
pathway
The Hippo signaling pathway, also known as the
Salvador (Sav)-Wts-Hippo signaling pathway, includes three primary
components: Upstream regulators, core kinase cassettes and
downstream transcriptional activators (45,46). The
Drosophila kinase cassette includes Wts and the
serine/threonine kinase Hippo, as well as its two cofactors Sav and
Mob as tumor suppressors (referred to as Mats) (45). The downstream transcriptional
co-activator, Yki, controls the activity of several transcription
factors through subcellular localization (45,47).
When the core kinase cassette is activated, Yki is phosphorylated
and retained in the cytoplasm, where it is eventually degraded
(45). In the absence of upstream
activation signals, Yki exists in a non-phosphorylated state and is
transferred to the nucleus, where its transcription program is
formulated, thereby promoting cell proliferation (45). The Ft mutation in the overgrowth
phenotype of Drosophila is considered to exert its effects
upstream of the Hippo signaling pathway, and is mediated by
affecting the subcellular localization of Yki (48,49).
Numerous studies have demonstrated the important role of the Hippo
signaling pathway in mammalian organ size and tumorigenesis
(50–52). Changes in gene expression caused by
inhibition of Hippo-associated proteins Yes-associated protein
(YAP)/TAZ can inhibit cell proliferation, promote apoptosis and
ultimately control organ size (52).
Overexpression of YAP/TAZ can result in uncontrolled cell
proliferation, which makes the Hippo signaling pathway one of the
key pathways involved in cancer development (52). Under conditions of low cell density,
YAP and TAZ (both are homologous to Drosophila Yki) control
the transcription program of genes that are the transcription
targets of TEAD and SMAD (53,54).
When the cell density is high, the core kinase cassette is
activated; this activation prevents the nucleocytoplasmic shuttle
of YAP and TAZ, and inhibits their transcriptional activity
(52,55). The vertebrate core kinase cassette
involves Sav, large tumor suppressor 1/2 and macrophage stimulating
1/2, which are homologous to the Drosophila kinases Sav, Wts
and Hippo, respectively (52,54,55).
Some progress has been made regarding the potential
association between FAT cadherin and Hippo signaling in
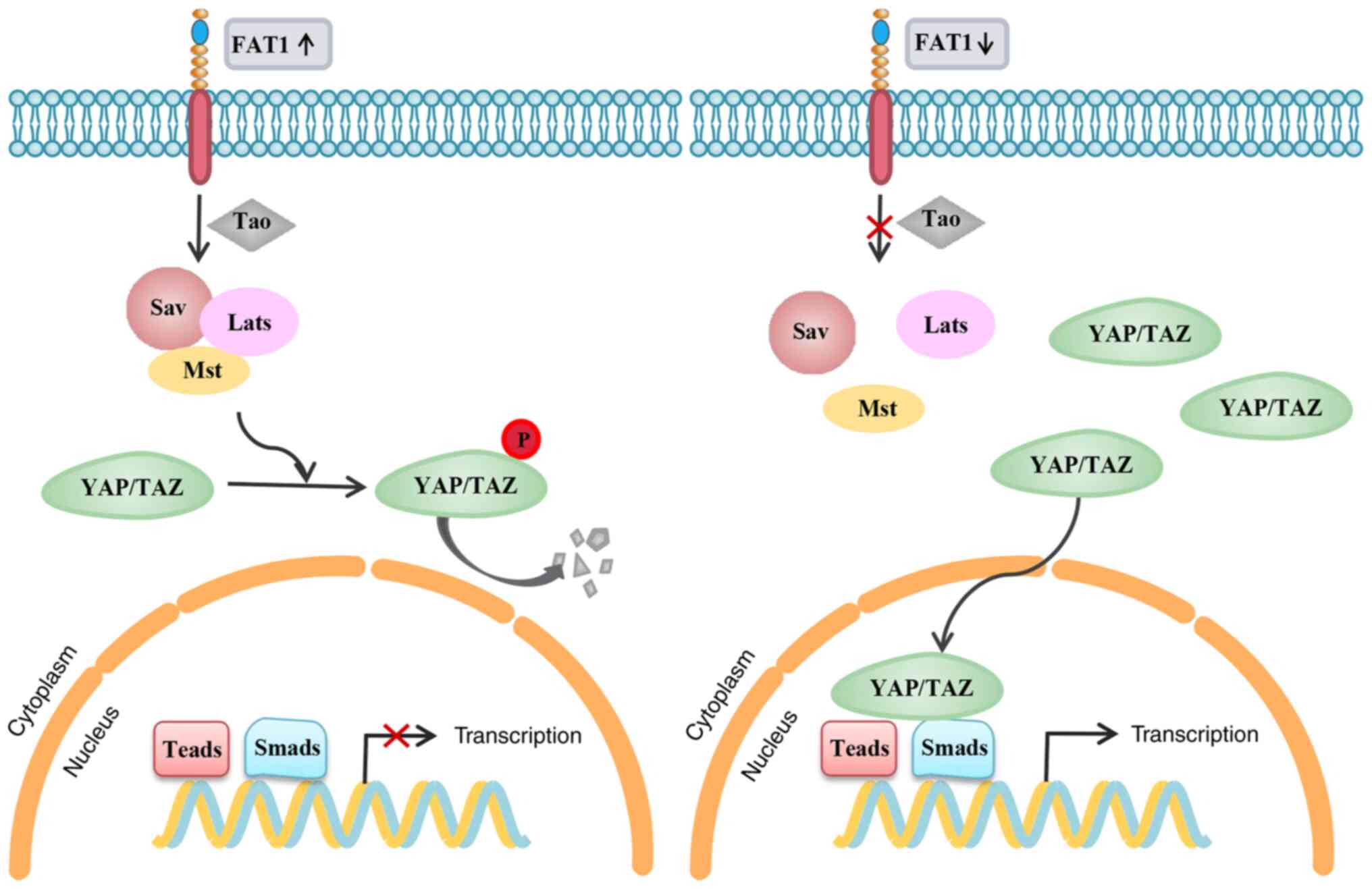
vertebrates. Similar to Drosophila Ft, the FAT1 protein has
been shown to be an upstream factor of the Hippo signaling pathway
(20). FAT1 acts as an upstream
regulator of TAZ, as well as an upstream regulator of YAP in the
process of neuronal differentiation (56). The intracellular domain of FAT1
interacts with a multimeric Hippo signaling protein and
participates in its assembly, which in turn induces the kinase Tao
to activate the Hippo kinase cassette (17). This causes phosphorylation and
inactivation of YAP, a transcriptional coactivator downstream of
the Hippo signaling pathway, and antagonization of the function of
the Hippo effector, TAZ (17).
Knockout of the FAT1 gene increases the nuclear accumulation of YAP
and promotes the nuclear and cytoplasmic shuttling of the
transcriptional activator TAZ, thereby upregulating the
transcription of the Hippo target genes connective tissue growth
factor and ankyrin repeat domain 1, which is also accompanied by an
increase in the nuclear levels of SMAD2/3 (Fig. 2) (57). The role of FAT1 in modulation of
upstream factors of the Hippo signaling pathway provides further
evidence of its important role in development and disease.
FAT1 and the MAPK/ERK signaling
pathway
The MAPK/ERK signaling pathway serves a role in cell
proliferation, differentiation and apoptosis. Uncontrolled MAPK/ERK
activity is commonly observed in tumors (58,59). The
MAPK/ERK signaling pathway is involved in cell migration and
invasion of several types of cancer, such as multiple myeloma, lung
cancer and HCC (58–60). It was revealed that FAT1 mutations in
pituitary spindle cell tumor were associated with increased MAPK
activity, based on the phosphorylation of ERK (p-ERK1/2),
highlighting an association between FAT1 and the MAPK signaling
pathway (61). Another study
suggested that FAT1 may affect esophageal squamous cell carcinoma
(ESCC) cell proliferation through the loss of control of the
MAPK/ERK signaling pathway (21).
Exogenous FAT1 expression can inhibit cell proliferation and colony
formation, and inhibit cell migration and invasion, whereas
FAT1-knockout exerts the opposite effects, both in vivo
(FAT1-knockdown stable ESCC cells and ESCC cells with FAT1-WT
overexpression were subcutaneously injected into the left or right
oxter of female BALB/c-nu mice) and in vitro (ESCC cell
lines) (21). Knockout of FAT1
significantly increased the levels of p-ERK1/2 in multiple
esophageal cancer cells, whereas overexpression of FAT1 decreased
p-ERK1/2 levels (21). When U0126, a
highly selective inhibitor of ERK1/2 phosphorylation, was used to
treat FAT1-knockout cells, it abolished the migratory and invasive
ability of cells (21). The
aforementioned studies indicate that FAT1 affects cell
proliferation in a MAPK/ERK-dependent manner, at least in part.
Wang et al (62) revealed
that FAT1-knockout increased the mRNA expression levels of MAP3K8,
MAP2K2 and MAP2K6 in esophageal squamous cell carcinoma cells, and
decreased the mRNA expression levels of the MAPK inactivator dual
specificity phosphatase 6, resulting in enrichment of the MAPK
signaling pathway. However, additional experimental evidence is
required to understand how FAT1 regulates the MAPK/ERK signaling
pathway.
FAT1 and EMT
EMT is a cellular biological process that promotes
the migration of cancer cells. In addition, EMT is characterized by
stemness, invasion and drug resistance (21,63). The
association between FAT1 and EMT has been confirmed in several
types of cancer, including ESCC (21), HCC (64) and glioblastoma (65). After inhibiting FAT1 expression, the
expression levels of the epithelial marker E-cadherin decreased
significantly, whereas the expression levels of N-cadherin,
vimentin and the stromal marker Snail increased, which in turn
increased cell proliferation, suggesting that FAT1 may be a key
regulator of EMT (21). When
FAT1-knockout cells were treated with U0126, an inhibitor of ERK1/2
phosphorylation, the regulation of E-cadherin, N-cadherin, vimentin
and Snail expression was reversed (21). These results suggest that FAT1
affects cell proliferation and EMT progression, at least in part,
in a MAPK/ERK-dependent manner (21).
Recent studies have suggested that exposure to
partial EMT or mixed EMT can further enhance tumor cell (such as
skin squamous cell carcinoma cells and lung cancer cells) stemness,
migration, invasion and drug resistance (22,63,66). In
mouse models of skin squamous cell carcinoma and lung cancer, it
was revealed that the loss of FAT1 accelerated tumor development
and malignant progression, and promoted the transformation of mixed
EMT phenotype (22). This mixed EMT
state has also been observed in human squamous cell carcinoma with
FAT1 mutations (22). Cutaneous
squamous cell carcinoma with FAT1 deletion exhibited increased
stemness and spontaneous metastasis (22). In mouse and human squamous cell
carcinoma, the loss of FAT1 function promotes tumor initiation,
progression, invasion, stemness and metastasis by inducing the
acquisition of a mixed EMT state (22). The loss of FAT1 function activates
the calcium/calmodulin-dependent protein kinase II/CD44/SRC axis,
and promotes the nuclear translocation of YAP1 and ZEB1 expression,
thus stimulating the interstitial state (22). Additionally, the loss of FAT1
function results in inactivation of enhancer of zeste homolog 2
subunit and promotes SOX2 expression, thus maintaining an
epithelial state (22). In addition,
FAT1-knockout skin squamous cell carcinoma cells were significantly
more resistant to the EGFR inhibitor afatinib and the MEK inhibitor
trametinib (22). Thus, identifying
the role of FAT1 in EMT, as well as the effects of mutations of the
FAT1 gene in the process of EMT, may be of great importance in
improving the prognosis and treatment of patients with cancer
(Fig. 3).
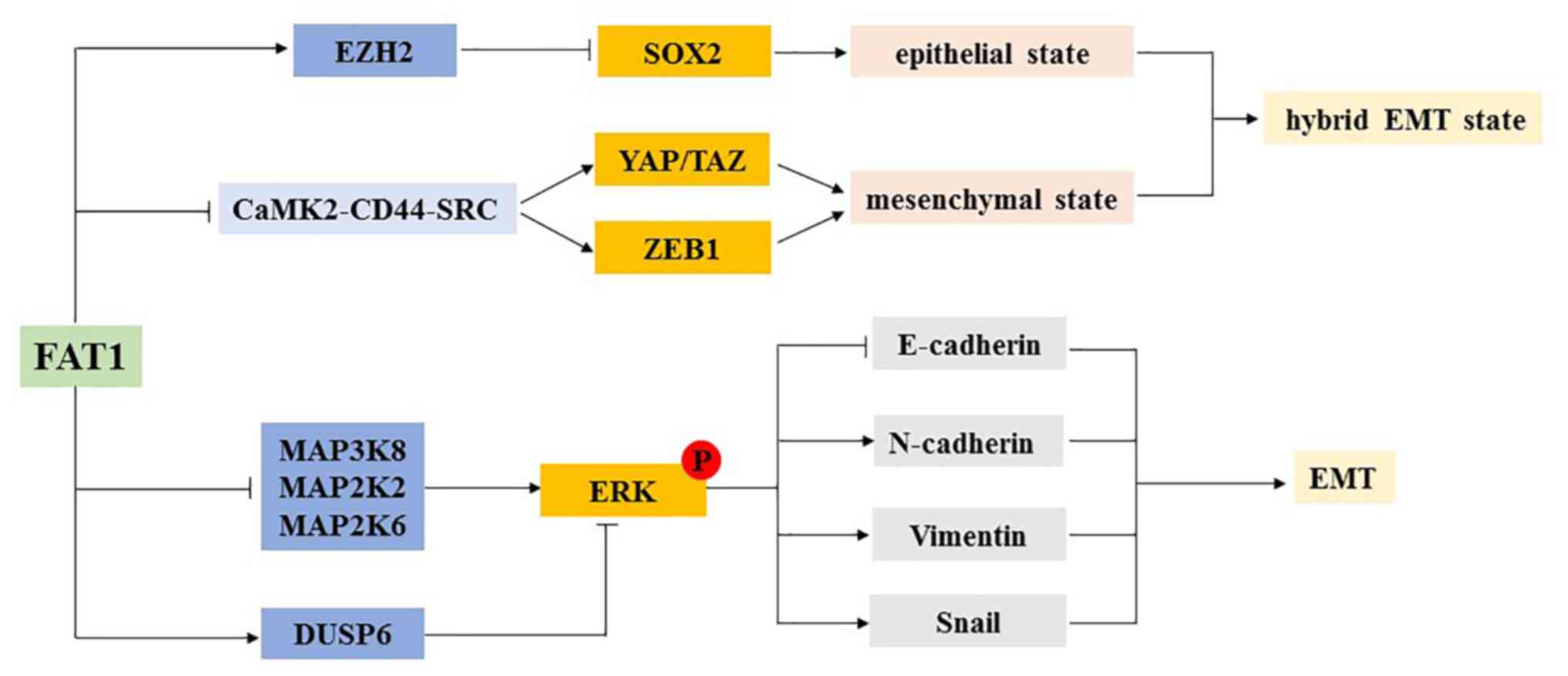 | Figure 3.Association between FAT1 and EMT.
FAT1 controls different transcription procedures via regulating the
expression levels of SOX2 by affecting the expression levels of
EZH2 and the CaMK2-CD44-SRC-YAP-ZEB1 axis, and regulates the
co-expression of epithelial and mesenchymal transcription programs
in cancer cells, leading to the emergence of a hybrid EMT
phenotype. In addition, FAT1 regulates the expression levels of the
epithelial marker E-cadherin and the mesenchymal markers
N-cadherin, vimentin and Snail by affecting the phosphorylation
level of ERK1/2. FAT1, FAT atypical cadherin 1; EMT,
epithelial-mesenchymal transition; p, phosphorylated; ZEB1, zinc
finger E-box binding homeobox 1; CaMK2,
calcium/calmodulin-dependent protein kinase II; DUSP6, dual
specificity phosphatase 6; EZH2, enhancer of zeste homolog 2; YAP,
Yes-associated protein. |
FAT1 in development
Aberrant FAT1 expression or mutations affecting its
function affect several signaling pathways via complex mechanisms,
through which FAT1 physiologically regulates development. FAT1
expression in tissues is higher in certain vertebrates. Ponassi
et al (67) identified FAT1
expression in the brain of developing and adult rats, and revealed
that it was most strongly expressed in areas involved in
neurogenesis. FAT1 serves a role in the early stages of neuronal
cell differentiation, controls the outward growth of neurites
through the Hippo signaling pathway and promotes terminal
differentiation of cells by inhibiting proliferation (20). FAT1 is expressed during the
development of the embryonic brain, kidney and other epithelial
tissues, and similar expression patterns are observed in rats,
chickens, zebrafish and other vertebrates (7). The pups of FAT1-knockout mice resulted
in their death as a result of renal failure within 48 h (68). FAT1 mRNA expression in the gap
junction between podocytes in the glomerular filtration membrane
has been reported (69). Gap
junctions are modified adhesive junctions with wide intercellular
spaces that allow kidney filtration (68). FAT1 creates the necessary
intercellular adhesion between podocyte protrusions, while
maintaining a wider extracellular space than normally found in
adhesion junctions (68). In the
absence of FAT1 mRNA, the physiological orderly array of gap
junctions is not present, and the podocytes become closely opposed
to each other, resulting in significant renal dysfunction, which
may explain the perinatal death of mice lacking FAT1 mRNA (68). Additionally, these mouse pups
exhibited severe midline developmental defects, including total
forebrain deformities, small eyeballs and ocular disorders, with
some mice exhibiting atrophy of the ciliary muscles (68).
There is little knowledge regarding FAT1 in human
tissues, but it appears to serve a more prominent role in embryonic
development. Human tissue in situ hybridization revealed
high levels of FAT1 mRNA transcription in the fetal epithelium
(15). FAT1 is reported to be widely
expressed throughout the nervous system and may control
neuromuscular morphogenesis (36).
Furthermore, FAT1 mRNA expression was identified in embryonic stem
cells and kidney tissues (36).
Although FAT1 expression is downregulated or deleted following
organogenesis and terminal differentiation, there is a low level of
FAT1 expressed in adult tissues, indicating that FAT1 may serve a
role in the maintenance of human organs, as well as in development
(70).
Cadherins are considered to be the primary medium
regulating the dynamic balance of epithelial cells and may serve an
important role in vascular remodeling (71). FAT1 expression in VSMCs is increased
significantly following arterial injury (29). FAT1 expression is increased after
vascular injury, which promotes the migration of VSMCs and inhibits
their proliferation (29). Increased
FAT1 expression provides directional signals and stimulates actin
cytoskeleton remodeling by interacting with Ena/Vasp proteins, thus
promoting the migration of VSMCs, whereas inhibition of
proliferation partly depends on the decrease of nuclear
accumulation of β-catenin (29,30). The
aforementioned studies provide an explanation for the formation of
cell clusters that oppose excessive proliferation in the repair of
vascular injury (36). Although FAT1
expression in human vascular diseases has not been extensively
studied, the loss of negative regulation mediated by FAT1 may lead
to VSMC hyperplasia syndromes, such as restenosis, graft artery
disease or vein graft disease (36).
FAT1 in hereditary diseases
Homozygous frameshift mutations in FAT1 have been
reported to cause the 4q-syndrome characterized by facial
deformities, cutaneous syndactyly and ocular abnormalities, with or
without nephropathy (72). Facial
malformations primarily manifest as an elongated facial appearance,
with a long philtrum and long nose (72). Eye abnormalities in patients with
mutations in the FAT1 gene include ptosis, microphthalmos, retinal
defects and severe amblyopia (72).
The cause may be that the loss of FAT1 function impairs the
adhesion and fusion between epithelial cells, leading to fissures
and neural tube closure defects (7,72). In
addition to the eyes, the key roles of FAT1 cadherin in the
cell-cell connection process is of great importance in the kidney.
Loss of FAT1 function results in decreased adhesion of epithelial
cells and the disappearance of podocyte foot processes, destroying
the lumen formation of renal tubular epithelial cells and causing
kidney disease, such as hormone resistant nephrotic syndrome and
glomerulonephritis (70,72).
Previous studies have revealed that FAT1 expression
is downregulated in FSHD, suggesting that FAT1 may be associated
with FSHD (23,73). The inactivated FAT1 protein results
in apolarized myoblasts, thus altering their morphology and
affecting the overall shapes of certain muscle groups in the face
and shoulders (74,75). Further research is required to
understand the association between FAT1 and FSHD.
Results linking the FAT1 gene with BPAD have also
been published. In a patient-controlled study by Blair et al
(24) in Australia, FAT1 was
identified as a susceptibility gene of BPAD, and single nucleotide
polymorphisms (SNPs) were identified in the FAT1 region that
included the binding sites of EVH1 and β-catenin. An independent
study conducted in German patients (24) revealed that 9 SNPs were identified in
FAT1, of which 8 were located in the haplotype region associated
with BPAD discovered by Blair et al (24). However, although there are several
studies highlighting a genetic component of BPAD (76–78),
relatively little is known regarding its specific cause. However,
FAT1 gene variants may be involved in the etiology of BPAD
(24,79).
FAT1 in cancer
FAT1 in hematological
malignancies
As aforementioned, FAT1 was first cloned from a
human T-ALL cell line, suggesting that FAT1 is expressed in tumors
of the hematopoietic system (15).
However, FAT1 expression is rarely expressed in normal
hematopoietic systems (15). FAT1 is
expressed in 60% of T-ALL, 30% of precursor B-cell ALL and 10% of
acute myeloid leukemia cases, and FAT1 expression is associated
with a more mature immune phenotype of leukemia (19,80).
Compared with patients with early T-ALL, FAT1 expression is higher
in patients with thymic T-ALL and mature T-ALL (80). In addition, high FAT1 mRNA expression
is associated with a poor prognosis and high recurrence rate in
patients with T-ALL (81).
The FAT family members are considered tumor
suppressor candidates, and the FAT1 protein is overexpressed in
human hematopoietic tumors (81).
Overexpression of FAT1 in T-ALL is accompanied by changes in
post-translational processing, resulting in staining being
primarily observed in the membrane at physiological expression
levels to staining being primarily observed in the cytoplasm and
nucleus when overexpressed (15).
This mislocalization is due to changes in the proteolytic pathways
specific to cancer cells (17). FAT1
undergoes S1 proteolytic treatment by proprotein convertase during
the transition process through the trans-Golgi network, resulting
in the expression of heterodimers on the cell surface (17). On the surface of cells expressing
FAT1, FAT1 can be further cleaved by the currently unknown S2
protease that releases the extracellular domain (17). Subsequently, a further cleavage step
catalyzed by γ-secretase releases the cytoplasmic tail (7,17). The
changes in post-translational processing and the resulting
erroneous localization of FAT1 disrupt the normal downstream
function and the apparent copy loss of the signal transduction
pathway of FAT1 (17).
FAT1 in HCC
Liver cancer was the sixth most common cancer and
the third leading cause of cancer-associated death worldwide in
2018 (82). HCC represents 75–85% of
primary liver cancer cases (83).
The incidence of liver cancer continues to increase, and the rate
of metastasis, recurrence and mortality remain high, making liver
cancer a serious threat to human health (83,84). For
example, there were ~850,000 new liver cancer cases in 2018, and an
estimated 800,000 liver cancer-associated deaths per year (85). At present, the mechanisms underlying
the development of HCC have not been fully elucidated, but it is
well-established that HCC is closely associated with liver
fibrosis, and ~90% of HCC cases occur as a result of progression of
liver cirrhosis (86,87). A previous study has revealed that
FAT1 expression increases during liver fibrosis and have confirmed
that activated hepatic stellate cells are the source of increased
FAT1 expression in diseased livers (16). This describes a novel mechanism for
the occurrence of HCC. Valletta et al (16) confirmed that FAT1 expression in
cancer tissues was significantly upregulated compared with in
normal liver tissues by analyzing FAT1 protein expression in 112
human HCC tissues. Strong positive FAT1 expression was
significantly associated with higher tumor stages and proliferation
rates (16). Moreover, FAT1
expression at the mRNA and protein levels in three liver cancer
cell lines (HepG2, PLC and Hep3B) was significantly upregulated
compared with in normal liver cells (16). Knocking down FAT1 expression
decreased the proliferation, migration and anti-apoptotic ability
of HCC cells (16).
FAT1 is a tumor promoter in HCC. FAT1 promotes the
migration of HCC cells by regulating the expression levels of
EMT-associated genes, including Snail, vimentin and E-cadherin
(64). In addition, FAT1 seems to be
a relevant mediator of hypoxia and growth receptor signal
transduction tumorigenic pathways in HCC. Hypoxia often occurs in
tumor tissues and is a key promoter of liver cancer progression
(88). Hepatocyte growth factor
(HGF) also serves a key role in the occurrence of liver cancer
(89). HGF-induced and
hypoxia-mediated activation of hypoxia-inducible factor-1α can
enhance methionine adenosyltransferase (MAT)2A expression and thus
enhance the activity of MATII, whereas the activity of MATI is
inhibited, resulting in decreased levels of methyl donor
S-Adenosyl-l-methionine (SAM) (16).
Lower SAM levels lead to decreased methylation of the FAT1
promoter, thus increasing FAT1 expression (16). Demethylated chemicals can induce FAT1
expression in HCC cells (16). The
use of sorafenib in HCC cells can significantly inhibit FAT1
expression (14).
FAT1 expression is closely associated with the
occurrence and development of HCC. Inhibiting FAT1 expression has
an inhibitory effect on the proliferation and migration of HCC
cells (16). In-depth study of the
mechanism of FAT1 and its association with hypoxia, HGF and related
factors, such as SAM, is of great importance for the diagnosis,
treatment and prognosis of HCC.
FAT1 in head and neck squamous cell
carcinoma (HNSCC)
HNSCC, including oral SCC, is one of the most common
malignant tumors in the world, with ~650,000 new cases and 350,000
deaths each year (89). The primary
risk factors for development of HNSCC are smoking and alcohol abuse
(90). It has been confirmed that
mutations of tumor protein p53 (TP53), NOTCH family genes and
PIK3CA-related elements contribute to the pathogenesis of HNSCC
(91). Additionally, a previous
study has revealed that human papillomavirus (HPV) is closely
associated with the onset and progression of HNSCC (92). However, the role of these factors in
tumors is largely unclear. Despite advances in surgery,
chemotherapy and radiation therapy, the survival rate of patients
with HNSCC has not improved significantly (93). Therefore, understanding the genomic
changes in the carcinogenic process of HNSCC is essential for
correct diagnosis and treatment.
Recently, two large-scale exome sequencing studies
demonstrated that FAT1 mutations occurred frequently in patients
with HNSCC (94,95). FAT1 seems to be the second most
mutated gene in HNSCC, second only to TP53 (92). In ~29% of HNSCC cases, patients
harbor destructive FAT1 mutations, most of which are nonsense and
missense mutations (94,96). These mutations cause a decrease or
loss of FAT1 mRNA and protein expression. Lin et al
(94) revealed that in HNSCC cell
lines (OECM1, HSC3, FaDu, SCC25, SAS and OC3), FAT1 mRNA and
protein expression was decreased or absent compared with in normal
oral mucosal epithelial cells. After FAT1-knockout in cells, the
migration and invasion of cells increased significantly, suggesting
that FAT1 inhibition may enhance the migration and invasiveness of
HNSCC cells (94). Additionally, by
analyzing the clinical data of 96 patients with HNSCC, it was
revealed that the frequency of FAT1 malignant mutations in patients
with lymph node metastasis was higher than in those without
metastasis (92,94). FAT1 gene mutations were significantly
associated with positive lymphatic vascular intravasation and tumor
recurrence (94). Kaplan-Meier
analysis demonstrated that the disease-free survival rate of
patients with laryngeal cancer with FAT1 gene mutations was
significantly lower than that of patients with wild-type FAT1
(94). Martin et al (97) confirmed that the inactivating
mutations of FAT1 caused YAP1 activation through the inactivation
of the Hippo signaling pathway, thereby promoting HNSCC
progression, indicating that FAT1 may act as a tumor suppressor in
HNSCC upstream of YAP1. In addition, FAT1 deficiency may indirectly
promote β-catenin activation through a mechanism downstream of YAP1
(97). There is crosstalk between
the Hippo and Wnt signaling pathways; however, the exact mechanism
of how FAT1 potentially regulates both the Hippo and Wnt signaling
pathways is unclear. The exact signaling pathway is likely to be
cell- or tissue-specific. Therefore, the role of FAT1 expression in
HNSCC should not be underestimated. Frequent changes in FAT1 may
lead to defects in Hippo signaling and uninhibited YAP1 activity.
Molecular therapy targeting FAT1 or its downstream targets may
become an attractive precision therapeutic for the treatment of
HNSCC and other malignant tumors in which FAT1 is mutated.
FAT1 in ESCC
Esophageal cancer is one of the most invasive
tumors (98). Most global esophageal
cancer cases (~70%) occur in China (99), and the majority of cases are ESCC
(98,100). The occurrence of ESCC is associated
with several factors. Unlike Western regions, such as Europe and
the United States, smoking and drinking are not the primary risk
factors for ESCC in China. The risk factors for ESCC in China
include burnt food, sauerkraut, nitrosamines and moldy foods
(101,102). Although progress has been made in
ESCC treatment, the 5-year overall survival rate in patients
remains low, ranging between 15% and 25% (98). Therefore, the identification of new
predictive biomarkers is important for the development of novel
therapeutics for management of ESCC.
A previous study has revealed that FAT1 is one of
the important mutant genes in ESCC (103). FAT1 expression in ESCC tissues is
significantly decreased compared with in normal esophageal tissues
(103). Hu et al (21) compared 76 primary esophageal tumor
tissues with matched adjacent non-tumor tissues, and revealed that
FAT1 expression in non-tumor tissues was significantly higher
compared with in tumor tissues. Additionally, FAT1-knockout
increased the proliferation, shortened the mitotic cycle and
significantly promoted the migration and invasion of ESCC cells
(21). On the other hand,
overexpression of FAT1 inhibited cell proliferation, colony
formation, cell migration and invasion (62). Furthermore, following FAT1-knockout,
the expression levels of the epithelial marker E-cadherin were
significantly decreased, while the expression levels of mesenchymal
markers N-cadherin, vimentin and Snail were significantly
increased, and this was dependent on the MAPK/ERK signaling pathway
(21). The overexpression of FAT1
resulted in the opposite trend, and these changes were abrogated by
treatment with the MEK specific inhibitor U0126 (21). Hou et al (36) analyzed the changes of FAT1 expression
in ESCC and its effect on the proliferation, migration and invasion
of esophageal cancer cells, and the results were consistent with
those of Hu et al (21).
However, Hou et al (36)
revealed that FAT1 was transcriptionally regulated by E2F
transcription factor 1 (E2F1); E2F1 activated the transcription of
FAT1 in ESCC cells by binding to the FAT1 promoter, although the
exact mechanism remains unclear.
In general, FAT1 serves a role in the growth of
ESCC and the occurrence of EMT. Inhibition of FAT1, at least
partially promotes the occurrence and development of ESCC by
increasing the activity of the MAPK/ERK signaling pathway, thereby
exhibiting a tumor suppressor effect in ESCC (21). This is of great importance for
understanding the occurrence and development of ESCC.
FAT1 in breast cancer
Between 2012 and 2016, the breast cancer incidence
rate increased by 0.3% per year worldwide, and the age of incidence
is getting lower, posing a huge threat for women (104). The vast majority of breast cancer
cases are invasive ductal carcinoma (IDC), accounting for ~80% of
cases (105). Abnormal hormone
levels and long-term environmental stimulation are considered high
risk factors for breast cancer (106). Early diagnosis of breast cancer is
difficult as it is usually asymptomatic during the early stages,
and most patients are usually diagnosed at an advanced stage,
leading to a higher mortality rate (105,106).
Wang et al (107) used immunohistochemical analysis to
detect FAT1 and β-catenin protein expression in ductal carcinoma
in situ (DCIS) and IDC, revealing that, compared with in
DCIS, the expression levels of FAT1 and β-catenin in IDC were
significantly decreased. Additionally, loss of FAT1 expression was
associated with higher histological grade and worse lymph node
status in human breast cancer (107,108).
In addition, FAT1 and β-catenin expression in DCIS and IDC were
positively correlated with each other. FAT1(−), β-catenin(−) or
FAT1(−)/β-catenin(−) suggested a worse disease-free survival rate
and prognosis in patients with breast cancer (107). The loss of FAT1 and β-catenin was
associated with breast cancer progression, aggressive behavior and
a poor prognosis, but the underlying mechanisms between the low
expression levels of FAT1 in breast cancer and the Wnt/β-catenin
signaling pathway are incompletely understood (107). FAT1 alone or in combination with
β-catenin may be a valuable biomarker for predicting the prognosis
of patients with breast cancer (107).
Notable progress has been made regarding the role
of FAT1 in breast cancer drug resistance. Cyclin-dependent kinase
4/6 (CDK4/6) inhibitors (CDK4/6i) have been demonstrated to be
effective in treating numerous types of cancer, such as breast
cancer (107), melanoma (109) and pancreatic cancer (110). However, the incidence of drug
resistance in cancer is increasing, and the understanding of the
mechanisms involved remains limited. Li et al (111) analyzed 348 cases of patients with
estrogen receptor-positive breast cancer treated with CDK4/6i and
revealed that FAT1 loss-of-function mutations were associated with
drug resistance. In vivo and in vitro experiments
demonstrated that FAT1 deletion promoted CDK4/6i resistance through
the Hippo signaling pathway (111).
Loss of FAT1 caused YAP and TAZ transcription factors to accumulate
on the CDK6 promoter, leading to a significant increase in CDK6,
whereas overexpression of CDK6 decreased drug sensitivity (111). These findings suggest that
downregulated FAT1 expression may be a means of countering CDK4/6i
(111). This provides a novel
avenue for the study of tumor drug resistance.
FAT1 in other types of cancer
The specific role of FAT1 in tumorigenesis and
development is still being studied. Research on the role of FAT1 in
human cancer has revealed that FAT1 can function as both an
oncogene and a tumor suppressor gene, dependent on the type of
cancer. In addition to its role in HCC, FAT1 exhibits a
carcinogenic role in gastric cancer (GC). FAT1 expression is
upregulated in patients with GC and is associated with cell
migration and invasion, as well as with a poor prognosis (112).
The deletion of FAT1 has been reported in a variety
of tumors. In addition to the aforementioned HNSCC, ESCC and breast
cancer, FAT1 loss has been observed in glioblastoma multiforme
(50), cervical cancer (24), Hodgkin's disease (113), cholangiocarcinoma (114), primary bladder cancer (115), lung cancer (116) and colorectal cancer (117).
After overexpressing the FAT1 gene in a
glioblastoma multiforme cell line, cell cycle progression was
arrested at the G1-S checkpoint, and high FAT1
expression significantly increased the proportion of cells in
G1 phase, while decreasing the proportion of cells in S
phase (19). Chen et al
(118) revealed that the
overexpression of FAT1 in cervical cancer promotes E-cadherin
protein expression and significantly inhibits the proliferation,
invasion and migration of cervical cancer cells, whereas the
knockdown of FAT1 promotes EMT and tumor progression (118). Yu and Li (114) suggested that FAT1 may be a
potential prognostic marker for childhood medulloblastoma. The
proliferation rate of medulloblastoma cells in which FAT1
expression was silenced was significantly increased (114). Low FAT1 expression was associated
with a poor prognosis of childhood medulloblastoma, and the
survival time of children with medulloblastoma with upregulated
FAT1 protein expression was relatively longer compared with those
with downregulated FAT1 protein expression (114) (Table
I).
 | Table I.Functions of FAT atypical cadherin 1
in various diseases. |
Table I.
Functions of FAT atypical cadherin 1
in various diseases.
| Disease | Expression | Protein
interaction/signaling pathways | Function | Refs. |
|---|
| 4q-syndrome | Downregulated | Unknown | Cell adhesion | (65) |
| Facioscapulohumeral
muscular dystrophy | Downregulated | Unknown | Cell polarity | (68,69) |
| Bipolar
disorder | Downregulated | β-catenin | Unknown | (70,71) |
| Acute lymphoblastic
leukemia and acute myeloid leukemia | Upregulated | Unknown | Unknown | (72,73) |
| Hepatocellular
carcinoma | Upregulated | Unknown | Cell proliferation,
migration, invasion and EMT | (14,76–78) |
| Gastric cancer | Upregulated | Unknown | Cell migration and
invasion | (100) |
| Head and neck
squamous cell carcinoma | Downregulated | Hippo and
Wnt/β-catenin signaling pathways | Cell migration and
invasion | (84,85,87) |
| Esophageal squamous
cell carcinoma | Downregulated | MAPK/ERK signaling
pathway | Cell proliferation,
migration, invasion and EMT | (20,31,57) |
| Breast cancer | Downregulated | Hippo and
Wnt/β-catenin signaling pathways | Cell invasion | (96,99) |
| Cervical
cancer | Downregulated | Unknown | Cell proliferation,
migration, invasion and EMT | (106) |
Therapeutic targeting of FAT1
As aforementioned, an increasing number of studies
have confirmed that FAT1 serves an important role in tumors.
Therefore, exploring the therapeutic strategies targeting FAT1 may
be of great importance in the management of several types of
cancer, particularly in patients with FAT1 mutations. Verteporfin
(VP), a drug used to treat macular degeneration, has been shown to
inhibit the proliferation of a variety of GC cell lines (112,119).
In GC cell lines treated with VP, FAT1 expression is significantly
decreased (112). The
proliferative, migratory and invasive abilities of GC cells treated
with VP are also significantly decreased, consistent with those of
cells in which FAT1 expression is knocked down (112). However, the specific mechanisms
remain unclear. When FAT1 exerts a carcinogenic effect, targeting
FAT1 may be an effective method for the treatment of cancer, such
as in patients with GC (112). The
notion of targeting FAT1 may also be relevant to HCC. Xu et
al (120) revealed that low
microRNA (miR)-223-3p expression in HCC cells was negatively
correlated with FAT1 protein expression. Further experiments
confirmed that there was a targeted regulatory association between
miR-223-3p and FAT1; miR-223-3p downregulated FAT1 expression and
inhibited the proliferation, migration, invasion and EMT of HCC
cells by targeting FAT1 (120).
However, the in-depth molecular mechanisms require further
elucidation.
Fan et al (121) examined a novel method of targeted
therapy of FAT1 by developing an anti-FAT1 monoclonal antibody
(clone mAb198.3) that specifically recognized FAT1. The mAb198.3
clone revealed that FAT1 was expressed in 79% of colon
adenocarcinoma cases, but its expression was negative or very low
in most normal tissues (121).
mAb198.3 was conjugated to a skeleton of gold nanoparticles to form
active targeting gold nanoparticles with high payload properties
(121). The coupling of mAb198.3
with gold nanoparticles can bring particles into tumor cells more
effectively, but rarely into normal cells (121). In addition, this active targeted
drug delivery system has high capacity for drug loading and has
great potential for cancer treatment (121).
Conclusions and perspectives
FAT1 cadherin influences the Wnt, Hippo and
MAPK/ERK signaling pathways, as well as EMT, and provides novel
avenues for studying the role of FAT1 in physiological development
and pathogenesis. In cancer, FAT1 appears to act as an oncogenic or
tumor-suppressive molecule depending on the type of tumor (122). However, due to the large size of
the FAT1 cadherin, there are numerous restrictions on molecular
manipulation. Therefore, the understanding of FAT1 remains
incomplete, more research is required to reveal the expression and
role of FAT1 in different types of tumor and studies on the
association between FAT1 and Ena/Vasp protein, as well as the
MAPK/ERK signaling pathway, remain limited. Several key issues
remain to be resolved, including which upstream signals of FAT1
trigger the Wnt, Hippo and MAPK/ERK signaling pathways, which
receptors detect these signals, how the 34 cadherin repeats
regulate intercellular contacts, whether FAT1 is primarily an
adhesion molecule or a signaling protein, and how the association
between these two functions is coordinated. In addition, the
transcription factors and target genes that mediate FAT1 function,
as well as the molecular mechanisms underlying the dysregulated
FAT1 expression, remain to be identified.
In the present review, the effects of FAT1 on cell
proliferation, polarization, migration and invasion via modulation
of several signaling pathways and protein interactions were
discussed. Improving the understanding of the role of FAT1 in
certain diseases may highlight novel therapeutic targets for
diagnosis and/or treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from The
National Natural Science Foundation of China (grant no.
81472449).
Availability of data and materials
Not applicable.
Authors' contributions
XL conceived the presented idea and supervised the
project. ZP and YG collected all the references and data. ZP wrote
the manuscript. ZP and XL were responsible for confirming the
authenticity of the data. All authors discussed, contributed
towards, read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Haugan K, Marcussen N, Kjølbye A, Nielsen
M, Hennan J and Petersen J: Treatment with the gap junction
modifier rotigaptide (ZP123) reduces infarct size in rats with
chronic myocardial infarction. J Cardiovasc Pharmacol. 47:236–242.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsuchida S, Arai Y, Kishida T, Takahashi
KA, Honjo K, Terauchi R, Inoue H, Oda R, Mazda O and Kubo T:
Silencing the expression of connexin 43 decreases inflammation and
joint destruction in experimental arthritis. J Orthop Res.
31:525–530. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El-Bahrawy M, Talbot I, Poulsom R, Jeffery
R and Alison M: The expression of E-cadherin and catenins in
colorectal tumours from familial adenomatous polyposis patients. J
Pathol. 198:69–76. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nollet F, Kools P and van Roy F:
Phylogenetic analysis of the cadherin superfamily allows
identification of six major subfamilies besides several solitary
members. J Mol Biol. 299:551–572. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tanoue T and Takeichi M: New insights into
Fat cadherins. J Cell Sci. 118:2347–2353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bryant PJ, Huettner B, Held LI Jr, Ryerse
J and Szidonya J: Mutations at the fat locus interfere with cell
proliferation control and epithelial morphogenesis in
Drosophila. Dev Biol. 129:541–554. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sadeqzadeh E, de Bock C and Thorne R:
Sleeping giants: Emerging roles for the fat cadherins in health and
disease. Med Res Rev. 34:190–221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bennett F and Harvey K: Fat cadherin
modulates organ size in Drosophila via the
Salvador/Warts/Hippo signaling pathway. Curr Biol. 16:2101–2110.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Silva E, Tsatskis Y, Gardano L, Tapon N
and McNeill H: The tumor-suppressor gene fat controls tissue growth
upstream of expanded in the hippo signaling pathway. Curr Biol.
16:2081–2089. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oh H and Irvine K: In vivo regulation of
Yorkie phosphorylation and localization. Development.
135:1081–1088. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang C, Axelrod J and Simon M: Regulation
of Frizzled by fat-like cadherins during planar polarity signaling
in the Drosophila compound eye. Cell. 108:675–688. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hill E, Broadbent I, Chothia C and Pettitt
J: Cadherin superfamily proteins in caenorhabditis elegans and
Drosophila melanogaster. J Mol Biol. 305:1011–1024. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Down M, Power M, Smith SI, Ralston K,
Spanevello M, Burns GF and Boyd AW: Cloning and expression of the
large zebrafish protocadherin gene, Fat. Gene Expr Patterns.
5:483–490. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Liu J, Liang X, Chen J, Hong J,
Li L, He Q and Cai X: History and progression of Fat cadherins in
health and disease. Onco Targets Ther. 9:7337–7343. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dunne J, Hanby AM, Poulsom R, Jones TA,
Sheer D, Chin WG, Da SM, Zhao Q, Beverley PC and Owen MJ: Molecular
cloning and tissue expression of FAT, the human homologue of the
Drosophila fat gene that is located on chromosome 4q34-q35
and encodes a putative adhesion molecule. Genomics. 30:207–223.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Valletta D, Czech B, Spruss T, Ikenberg K,
Wild P, Hartmann A, Weiss TS, Oefner PJ, Müller M, Bosserhoff AK
and Hellerbrand C: Regulation and function of the atypical cadherin
FAT1 in hepatocellular carcinoma. Carcinogenesis. 35:1407–1415.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sadeqzadeh E, de Bock CE, Zhang XD,
Shipman KL, Scott NM, Song C, Yeadon T, Oliveira CS, Jin B, Hersey
P, et al: Dual processing of FAT1 cadherin protein by human
melanoma cells generates distinct protein products. J Biol Chem.
286:28181–28191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schreiner D, Müller K and Hofer H: The
intracellular domain of the human protocadherin hFat1 interacts
with Homer signalling scaffolding proteins. FEBS Lett.
580:5295–5300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morris LGT, Kaufman AM, Gong Y, Ramaswami
D, Walsh LA, Turcan S, Eng S, Kannan K, Zou Y, Peng L, et al:
Recurrent somatic mutation of FAT1 in multiple human cancers leads
to aberrant Wnt activation. Nat Genet. 45:253–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmed AF, de Bock CE, Lincz LF, Pundavela
J, Zouikr I, Sontag E, Hondermarck H and Thorne RF: FAT1 cadherin
acts upstream of Hippo signalling through TAZ to regulate neuronal
differentiation. Cell Mol Life Sci. 72:4653–4669. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu X, Zhai Y, Kong P, Cui H, Yan T, Yang
J, Qian Y, Ma Y, Wang F, Li H, et al: FAT1 prevents epithelial
mesenchymal transition (EMT) via MAPK/ERK signaling pathway in
esophageal squamous cell cancer. Cancer Lett. 397:83–93. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pastushenko I, Mauri F, Song Y, de Cock F,
Meeusen B, Swedlund B, Impens F, Van Haver D, Opitz M, Thery M, et
al: Fat1 deletion promotes hybrid EMT state, tumour stemness and
metastasis. Nature. 589:448–455. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mariot V, Roche S, Hourdé C, Portilho D,
Sacconi S, Puppo F, Duguez S, Rameau P, Caruso N, Delezoide AL, et
al: Correlation between low FAT1 expression and early affected
muscle in facioscapulohumeral muscular dystrophy. Ann Neurol.
78:387–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Blair IP, Chetcuti AF, Badenhop RF,
Scimone A, Moses MJ, Adams LJ, Craddock N, Green E, Kirov G, Owen
MJ, et al: Positional cloning, association analysis and expression
studies provide convergent evidence that the cadherin gene FAT
contains a bipolar disorder susceptibility allele. Mol Psychiatry.
11:372–383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Krause M, Bear J, Loureiro J and Gertler
F: The Ena/VASP enigma. J Cell Sci. 115:4721–4726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Krause M, Leslie JD, Stewart M, Lafuente
EM, Valderrama F, Jagannathan R, Strasser GA, Rubinson DA, Liu H,
Way M, et al: Lamellipodin, an Ena/VASP ligand, is implicated in
the regulation of lamellipodial dynamics. Dev Cell. 7:571–583.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lafuente EM, van Puijenbroek AFL, Krause
M, Carman CV, Freeman GJ, Berezovskaya A, Constantine E, Springer
TA, Gertler FB and Boussiotis VA: RIAM, an Ena/VASP and profilin
ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion.
Dev Cell. 7:585–595. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Renfranz P and Beckerle M: Doing
(F/L)PPPPs: EVH1 domains and their proline-rich partners in cell
polarity and migration. Curr Opin Cell Biol. 14:88–103. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moeller MJ, Soofi A, Braun GS, Li X, Watzl
C, Kriz W and Holzman LB: Protocadherin FAT1 binds Ena/VASP
proteins and is necessary for actin dynamics and cell polarization.
EMBO J. 23:3769–3779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanoue T and Takeichi M: Mammalian Fat1
cadherin regulates actin dynamics and cell-cell contact. J Cell
Biol. 165:517–528. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nobes CD and Hall A: Rho GTPases control
polarity, protrusion, and adhesion during cell movement. J Cell
Biol. 144:1235–1244. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang P, Yan R, Zhang X, Wang L, Ke X and
Qu Y: Activating Wnt/β-catenin signaling pathway for disease
therapy: Challenges and opportunities. Pharmacol Ther. 196:79–90.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nusse R and Clevers H: Wnt/β-catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Perugorria MJ, Olaizola P, Labiano I,
Esparza-Baquer A, Marzioni M, Marin JJG, Bujanda L and Banales JM:
Wnt-β-catenin signalling in liver development, health and disease.
Nat Rev Gastroenterol Hepatol. 16:121–136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hou R, Liu L, Anees S, Hiroyasu S and
Sibinga NES: The Fat1 cadherin integrates vascular smooth muscle
cell growth and migration signals. J Cell Biol. 173:417–429. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kaplan D, Meigs T and Casey P: Distinct
regions of the cadherin cytoplasmic domain are essential for
functional interaction with Galpha 12 and beta-catenin. J Biol
Chem. 276:44037–44043. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rockman SP, Currie SA, Ciavarella M,
Vincan E, Dow C, Thomas RJ and Phillips WA: Id2 is a target of the
beta-catenin/T cell factor pathway in colon carcinoma. J Biol Chem.
276:45113–45119. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kolligs FT, Nieman MT, Winer I, Hu G, Van
Mater D, Feng Y, Smith IM, Wu R, Zhai Y, Cho KR and Fearon ER:
ITF-2, a downstream target of the Wnt/TCF pathway, is activated in
human cancers with beta-catenin defects and promotes neoplastic
transformation. Cancer Cell. 1:145–155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Staley B and Irvine K: Hippo signaling in
Drosophila: Recent advances and insights. Dev Dyn. 241:3–15.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pan D: The hippo signaling pathway in
development and cancer. Dev Cell. 19:491–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oh H and Irvine K: Yorkie: The final
destination of Hippo signaling. Trends Cell Biol. 20:410–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wei X, Shimizu T and Lai Z: Mob as tumor
suppressor is activated by Hippo kinase for growth inhibition in
Drosophila. EMBO J. 26:1772–1781. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
McCartney BM, Kulikauskas RM, LaJeunesse
DR and Fehon RG: The neurofibromatosis-2 homologue, Merlin, and the
tumor suppressor expanded function together in Drosophila to
regulate cell proliferation and differentiation. Development.
127:1315–1324. 2000.PubMed/NCBI
|
|
50
|
Camargo FD, Gokhale S, Johnnidis JB, Fu D,
Bell GW, Jaenisch R and Brummelkamp TR: YAP1 increases organ size
and expands undifferentiated progenitor cells. Curr Biol.
17:2054–2060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dong J, Feldmann G, Huang J, Wu S, Zhang
N, Comerford SA, Gayyed MF, Anders RA, Maitra A and Pan D:
Elucidation of a universal size-control mechanism in
Drosophila and mammals. Cell. 130:1120–1133. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao B, Li L, Lei Q and Guan K: The
Hippo-YAP pathway in organ size control and tumorigenesis: An
updated version. Genes Dev. 24:862–874. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Beyer TA, Weiss A, Khomchuk Y, Huang K,
Ogunjimi AA, Varelas X and Wrana JL: Switch enhancers interpret
TGF-β and Hippo signaling to control cell fate in human embryonic
stem cells. Cell Rep. 5:1611–1624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wrighton K, Dai F and Feng X: A new kid on
the TGFbeta block: TAZ controls Smad nucleocytoplasmic shuttling.
Dev Cell. 15:8–10. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nishioka N, Inoue KI, Adachi K, Kiyonari
H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki
N, et al: The Hippo signaling pathway components Lats and Yap
pattern Tead4 activity to distinguish mouse trophectoderm from
inner cell mass. Dev Cell. 16:398–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ahmed A, de Bock C, Sontag E, Hondermarck
H, Lincz L and Thorne R: FAT1 cadherin controls neuritogenesis
during NTera2 cell differentiation. Biochem Biophys Res Commun.
514:625–631. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Skouloudaki K, Puetz M, Simons M, Courbard
JR, Boehlke C, Hartleben B, Engel C, Moeller MJ, Englert C, Bollig
F, et al: Scribble participates in Hippo signaling and is required
for normal zebrafish pronephros development. Proc Natl Acad Sci
USA. 106:8579–8584. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ye CY, Zheng CP, Ying WW and Weng SS:
Up-regulation of microRNA-497 inhibits the proliferation, migration
and invasion but increases the apoptosis of multiple myeloma cells
through the MAPK/ERK signaling pathway by targeting Raf-1. Cell
Cycle. 17:2666–2683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yan P, Zhu H, Yin L, Wang L, Xie P, Ye J,
Jiang X and He X: Integrin αvβ6 promotes lung cancer proliferation
and metastasis through upregulation of IL-8-mediated MAPK/ERK
signaling. Transl Oncol. 11:619–627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang WM, Xu Y, Wang YH, Sun HX, Sun YF, He
YF, Zhu QF, Hu B, Zhang X, Xia JL, et al: HOXB7 promotes tumor
progression via bFGF-induced activation of MAPK/ERK pathway and
indicated poor prognosis in hepatocellular carcinoma. Oncotarget.
8:47121–47135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Miller MB, Bi WL, Ramkissoon LA, Kang YJ,
Abedalthagafi M, Knoff DS, Agarwalla PK, Wen PY, Reardon DA,
Alexander BM, et al: MAPK activation and HRAS mutation identified
in pituitary spindle cell oncocytoma. Oncotarget. 7:37054–37063.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang Y, Wang G, Ma Y, Teng J, Wang Y, Cui
Y, Dong Y, Shao S, Zhan Q and Liu X: FAT1, a direct transcriptional
target of E2F1, suppresses cell proliferation, migration and
invasion in esophageal squamous cell carcinoma. Chin J Cancer Res.
31:609–619. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Saxena K, Jolly M and Balamurugan K:
Hypoxia, partial EMT and collective migration: Emerging culprits in
metastasis. Transl Oncol. 13:1008452020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Meng P, Zhang YF, Zhang W, Chen X, Xu T,
Hu S, Liang X, Feng M, Yang X and Ho M: Identification of the
atypical cadherin FAT1 as a novel glypican-3 interacting protein in
liver cancer cells. Sci Rep. 11:402021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Srivastava C, Irshad K, Dikshit B,
Chattopadhyay P, Sarkar C, Gupta DK, Sinha S and Chosdol K: FAT1
modulates EMT and stemness genes expression in hypoxic
glioblastoma. Int J Cancer. 142:805–812. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pastushenko I, Brisebarre A, Sifrim A,
Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D,
Moers V, Lemaire S, et al: Identification of the tumour transition
states occurring during EMT. Nature. 556:463–468. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ponassi M, Jacques TS, Ciani L and ffrench
Constant C: Expression of the rat homologue of the
Drosophila fat tumour suppressor gene. Mech Dev. 80:207–212.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ciani L, Patel A, Allen N and
ffrench-Constant C: Mice lacking the giant protocadherin mFAT1
exhibit renal slit junction abnormalities and a partially penetrant
cyclopia and anophthalmia phenotype. Mol Cell Biol. 23:3575–3582.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Inoue T, Yaoita E, Kurihara H, Shimizu F,
Sakai T, Kobayashi T, Ohshiro K, Kawachi H, Okada H, Suzuki H, et
al: FAT is a component of glomerular slit diaphragms. Kidney Int.
59:1003–1012. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yaoita E, Kurihara H, Yoshida Y, Inoue T,
Matsuki A, Sakai T and Yamamoto T: Role of Fat1 in cell-cell
contact formation of podocytes in puromycin aminonucleoside
nephrosis and neonatal kidney. Kidney Int. 68:542–551. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Uglow EB, Slater S, Sala-Newby GB,
Aguilera-Garcia CM, Angelini GD, Newby AC and George SJ:
Dismantling of cadherin-mediated cell-cell contacts modulates
smooth muscle cell proliferation. Circ Res. 92:1314–1321. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lahrouchi N, George A, Ratbi I, Schneider
R, Elalaoui SC, Moosa S, Bharti S, Sharma R, Abu-Asab M, Onojafe F,
et al: Homozygous frameshift mutations in FAT1 cause a syndrome
characterized by colobomatous-microphthalmia, ptosis, nephropathy
and syndactyly. Nat Commun. 10:11802019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Puppo F, Dionnet E, Gaillard MC, Gaildrat
P, Castro C, Vovan C, Bertaux K, Bernard R, Attarian S, Goto K, et
al: Identification of variants in the 4q35 gene FAT1 in patients
with a facioscapulohumeral dystrophy-like phenotype. Hum Mutat.
36:443–453. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Park HJ, Lee W, Kim SH, Lee JH, Shin HY,
Kim SM, Park KD, Lee JH and Choi YC: FAT1 gene alteration in
facioscapulohumeral muscular dystrophy type 1. Yonsei Med J.
59:337–340. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Caruso N, Herberth B, Bartoli M, Puppo F,
Dumonceaux J, Zimmermann A, Denadai S, Lebossé M, Roche S, Geng L,
et al: Deregulation of the protocadherin gene FAT1 alters muscle
shapes: implications for the pathogenesis of facioscapulohumeral
dystrophy. PLoS Genet. 9:e10035502013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Stahl EA, Breen G, Forstner AJ, McQuillin
A, Ripke S, Trubetskoy V, Mattheisen M, Wang Y, Coleman JRI, Gaspar
HA, et al: Genome-wide association study identifies 30 loci
associated with bipolar disorder. Nat Genet. 51:793–803. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bipolar Disorder and Schizophrenia Working
Group of the Psychiatric Genomics Consortium. Electronic address:
douglas.ruderfer@vanderbilt.edu; Bipolar Disorder and Schizophrenia
Working Group of the Psychiatric Genomics Consortium: Genomic
dissection of bipolar disorder and schizophrenia, including 28
subphenotypes, . Cell. 173:1705–1715.e1716. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Psychiatric GWAS Consortium Bipolar
Disorder Working Group, : Large-scale genome-wide association
analysis of bipolar disorder identifies a new susceptibility locus
near ODZ4. Nat Genet. 43:977–983. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Abou Jamra R, Becker T, Georgi A, Feulner
T, Schumacher J, Stromaier J, Schirmbeck F, Schulze TG, Propping P,
Rietschel M, et al: Genetic variation of the FAT gene at 4q35 is
associated with bipolar affective disorder. Mol Psychiatry.
13:277–284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Neumann M, Seehawer M, Schlee C, Vosberg
S, Heesch S, von der Heide EK, Graf A, Krebs S, Blum H, Gökbuget N,
et al: FAT1 expression and mutations in adult acute lymphoblastic
leukemia. Blood Cancer J. 4:e2242014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
de Bock CE, Ardjmand A, Molloy TJ, Bone
SM, Johnstone D, Campbell DM, Shipman KL, Yeadon TM, Holst J,
Spanevello MD, et al: The Fat1 cadherin is overexpressed and an
independent prognostic factor for survival in paired
diagnosis-relapse samples of precursor B-cell acute lymphoblastic
leukemia. Leukemia. 26:918–926. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
GBD 2015 Mortality and Causes of Death
Collaborators, . Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980–2015: A systematic analysis for the global burden of
disease study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Shariff MI, Cox IJ, Gomaa AI, Khan SA,
Gedroyc W and Taylor-Robinson SD: Hepatocellular carcinoma: Current
trends in worldwide epidemiology, risk factors, diagnosis and
therapeutics. Expert Rev Gastroenterol Hepatol. 3:353–367. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Dhanasekaran R, Limaye A and Cabrera R:
Hepatocellular carcinoma: Current trends in worldwide epidemiology,
risk factors, diagnosis, and therapeutics. Hepat Med. 4:19–37.
2012.PubMed/NCBI
|
|
85
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bataller R and Brenner D: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Farazi P and DePinho R: Hepatocellular
carcinoma pathogenesis: From genes to environment. Nat Rev Cancer.
6:674–687. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rosmorduc O and Housset C: Hypoxia: A link
between fibrogenesis, angiogenesis, and carcinogenesis in liver
disease. Semin Liver Dis. 30:258–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Argiris A, Karamouzis M, Raben D and
Ferris R: Head and neck cancer. Lancet. 371:1695–1709. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pai SI and Westra WH: Molecular pathology
of head and neck cancer: Implications for diagnosis, prognosis, and
treatment. Annu Rev Pathol. 4:49–70. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kim KT, Kim BS and Kim JH: Association
between FAT1 mutation and overall survival in patients with human
papillomavirus-negative head and neck squamous cell carcinoma. Head
Neck. 38 (Suppl 1):S2021–S2029. 2016. View Article : Google Scholar
|
|
93
|
Gupta S, Kong W, Peng Y, Miao Q and
Mackillop W: Temporal trends in the incidence and survival of
cancers of the upper aerodigestive tract in Ontario and the United
States. Int J Cancer. 125:2159–2165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lin SC, Lin LH, Yu SY, Kao SY, Chang KW,
Cheng HW and Liu CJ: FAT1 somatic mutations in head and neck
carcinoma are associated with tumor progression and survival.
Carcinogenesis. 39:1320–1330. 2018.PubMed/NCBI
|
|
95
|
Katoh Y and Katoh M: Comparative
integromics on FAT1, FAT2, FAT3 and FAT4. Int J Mol Med.
18:523–528. 2006.PubMed/NCBI
|
|
96
|
Liu CJ, Liu TY, Kuo LT, Cheng HW, Chu TH,
Chang KW and Lin SC: Differential gene expression signature between
primary and metastatic head and neck squamous cell carcinoma. J
Pathol. 214:489–497. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Martin D, Degese MS, Vitale-Cross L,
Iglesias-Bartolome R, Valera JLC, Wang Z, Feng X, Yeerna H, Vadmal
V, Moroishi T, et al: Assembly and activation of the Hippo
signalome by FAT1 tumor suppressor. Nat Commun. 9:23722018.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pennathur A, Gibson M, Jobe B and Luketich
J: Oesophageal carcinoma. Lancet. 381:400–412. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Tran GD, Sun XD, Abnet CC, Fan JH, Dawsey
SM, Dong ZW, Mark SD, Qiao YL and Taylor PR: Prospective study of
risk factors for esophageal and gastric cancers in the Linxian
general population trial cohort in China. Int J Cancer.
113:456–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ohashi S, Miyamoto S, Kikuchi O, Goto T,
Amanuma Y and Muto M: Recent advances from basic and clinical
studies of esophageal squamous cell carcinoma. Gastroenterology.
149:1700–1715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li WQ, Park Y, Wu JW, Ren JS, Goldstein
AM, Taylor PR, Hollenbeck AR, Freedman ND and Abnet CC: Index-based
dietary patterns and risk of esophageal and gastric cancer in a
large cohort study. Clin Gastroenterol Hepatol. 11:1130–1136.e1132.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang L, Zhou Y, Cheng C, Cui H, Cheng L,
Kong P, Wang J, Li Y, Chen W, Song B, et al: Genomic analyses
reveal mutational signatures and frequently altered genes in
esophageal squamous cell carcinoma. Am J Hum Genet. 96:597–611.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Sauer AG, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Britt KL, Cuzick J and Phillips KA: Key
steps for effective breast cancer prevention. Nat Rev Cancer.
20:417–436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang L, Lyu S, Wang S, Shen H, Niu F, Liu
X, Liu J and Niu Y: Loss of FAT1 during the progression from DCIS
to IDC and predict poor clinical outcome in breast cancer. Exp Mol
Pathol. 100:177–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lee S, Stewart S, Nagtegaal I, Luo J, Wu
Y, Colditz G, Medina D and Allred DC: Differentially expressed
genes regulating the progression of ductal carcinoma in situ to
invasive breast cancer. Cancer Res. 72:4574–4586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Guo L, Qi J, Wang H, Jiang X and Liu Y:
Getting under the skin: The role of CDK4/6 in melanomas. Eur J Med
Chem. 204:1125312020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Salvador-Barbero B, Álvarez-Fernández M,
Zapatero-Solana E, El Bakkali A, Menéndez MDC, López-Casas PP, Di
Domenico T, Xie T, VanArsdale T, Shields DJ, et al: CDK4/6
inhibitors impair recovery from cytotoxic chemotherapy in
pancreatic adenocarcinoma. Cancer Cell. 37:340–353.e346. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li Z, Razavi P, Li Q, Toy W, Liu B, Ping
C, Hsieh W, Sanchez-Vega F, Brown DN, Da Cruz Paula AF, et al: Loss
of the FAT1 tumor suppressor promotes resistance to CDK4/6
inhibitors via the hippo pathway. Cancer Cell. 34:893–905.e898.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kang MH, Jeong GS, Smoot DT, Ashktorab H,
Hwang CM, Kim BS, Kim HS and Park YY: Verteporfin inhibits gastric
cancer cell growth by suppressing adhesion molecule FAT1.
Oncotarget. 8:98887–98897. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Döhner H, Bloomfield C, Frizzera G,
Frestedt J and Arthur D: Recurring chromosome abnormalities in
Hodgkin's disease. Genes Chromosomes Cancer. 5:392–398. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Yu J and Li H: The expression of FAT1 is
associated with overall survival in children with medulloblastoma.
Tumori. 103:44–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Polascik TJ, Cairns P, Chang WY,
Schoenberg MP and Sidransky D: Distinct regions of allelic loss on
chromosome 4 in human primary bladder carcinoma. Cancer Res.
55:5396–5399. 1995.PubMed/NCBI
|
|
116
|
Shivapurkar N, Virmani AK, Wistuba II,
Milchgrub S, Mackay B, Minna JD and Gazdar AF: Deletions of
chromosome 4 at multiple sites are frequent in malignant
mesothelioma and small cell lung carcinoma. Clin Cancer Res.
5:17–23. 1999.PubMed/NCBI
|
|
117
|
Shivapurkar N, Maitra A, Milchgrub S and
Gazdar AF: Deletions of chromosome 4 occur early during the
pathogenesis of colorectal carcinoma. Hum Pathol. 32:169–177. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Chen M, Sun X, Wang Y, Ling K, Chen C, Cai
X, Liang X and Liang Z: FAT1 inhibits the proliferation and
metastasis of cervical cancer cells by binding β-catenin. Int J
Clin Exp Pathol. 12:3807–3818. 2019.PubMed/NCBI
|
|
119
|
Zhang H, Ramakrishnan SK, Triner D,
Centofanti B, Maitra D, Győrffy B, Sebolt-Leopold JS, Dame MK,
Varani J, Brenner DE, et al: Tumor-selective proteotoxicity of
verteporfin inhibits colon cancer progression independently of
YAP1. Sci Signal. 8:ra982015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Xu J, Wang B, Liu ZT, Lai MC, Zhang ML and
Zheng SS: miR-223-3p regulating the occurrence and development of
liver cancer cells by targeting FAT1 gene. Math Biosci Eng.
17:1534–1547. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Fan L, Campagnoli S, Wu H, Grandi A, Parri
M, De Camilli E, Grandi G, Viale G, Pileri P, Grifantini R, et al:
Negatively charged AuNP modified with monoclonal antibody against
novel tumor antigen FAT1 for tumor targeting. J Exp Clin Cancer
Res. 34:1032015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Katoh M: Function and cancer genomics of
FAT family genes (review). Int J Oncol. 41:1913–1918. 2012.
View Article : Google Scholar : PubMed/NCBI
|