Introduction
Historically, human cancer cell lines have been
widely used to study cancer biology or as preclinical models to
evaluate anti-cancer agents. However, these cell lines may not
necessarily preserve the quality of their source tumor tissues'
characteristics, because their genome sequence, gene expression
profile, and morphology can change while passaging culture over
long periods. Additionally, most of these cell lines are cultured
in a monolayer or used as murine xenograft, neither of which are
physically representative of tumor tissues (1,2). Thus,
the clinical efficacy of anti-cancer drugs is not identical to that
obtained during evaluations in cancer cell lines. Approximately 85%
of approved preclinical drugs tested in cancer clinical trials have
not demonstrated sufficient efficacy or safety to warrant
regulatory approval (3–5).
Patient-derived tumor xenograft (PDX) models have
been used as preclinical cancer models since they closely mimic
human cancer tissue (6–11). Increasing evidence suggests that PDX
predicts patient response to drugs by being directly comparable to
the corresponding cancer tissue. However, the evaluation of
anti-cancer agents using these models is challenging due to their
low throughput and high cost. Therefore, in vitro systems
such as ex vivo assays using PDX, patient-derived tumor
organoids (PDOs), or spheroid models that accurately recapitulate
tissue architecture and function have been developed recently.
These in vitro systems have been established for different
types of tumor tissues (e.g., bladder, breast, brain, colon,
endometrium, kidney, liver, lung, pancreatic, prostate, kidney, and
stomach), and associated high-throughput assay systems for drug
screening have also been developed (11–22). In
addition, heterogeneous ex vivo organoid cultures of primary
tumors obtained from patients or PDX have gained considerable
traction in recent years due to the ease of culturing and its
ability to maintain stromal cellular complexity (23–25).
These models are expected to enhance our understanding of cancer
biology and facilitate the evaluation of drug efficacy in
vitro.
To date, we have constructed a series of novel PDOs
from different tumor tissue types under the Fukushima Translational
Research Project, designated as F-PDO. F-PDOs form large cell
clusters with a morphology similar to the original tumor and can be
cultured for more than six months (26). In addition, our comparative
histological and comprehensive gene expression analyses have shown
that the characteristics of F-PDOs were similar to their source
tumors, even after long-term growth in culture conditions.
Moreover, we have generated a novel series of PDX models from
numerous cancer tissues, including hematopoietic tumors, which we
designated as Fukushima (F)-PDX. F-PDX models also have similar
characteristics to their source tissue. Furthermore, we have
constructed suitable high-throughput assay systems for each F-PDO
in 96- and 384-well plates. We used these assay systems to evaluate
several molecular targeted drugs and antibody drugs. Lastly, we
analyzed the changes in the higher-order structure of F-PDOs caused
by anti-cancer drugs using the 3D cell analysis system (27). The evaluation of anti-cancer drugs
using F-PDO-based in vitro assay systems was comparable to
the evaluation of anti-cancer drugs in clinical use.
Immunotherapy is one of the most significant
paradigm shifts in the history of cancer therapy. Immunotherapeutic
approaches include adoptive cell therapies, monoclonal antibodies,
immune checkpoint inhibitors, bispecific T-cell engagers (BiTEs),
cytokines, and vaccines used against various cancers to date.
However, immunotherapeutic approaches have resulted in a wide
variation in the degree and duration of patient responses and
adverse effects. Numerous cancers remain entirely refractory to
immunotherapy (28–31); thus, further improvements are needed.
Besides, there are currently several reports on the construction of
assay systems for immunotherapeutic agents using PDO (32). However, to our knowledge, there are
no reports of simple and high-throughput assay systems for drug
screening. Although many efficient and simple in vitro assay
systems are available for determining clinically efficacious
immunotherapy potency, such as the chromium 51 release assay,
cytokine release assay, flow cytometry-based detection of crucial
biomarkers, lactate dehydrogenase release assay, these are limited
in throughput and simplicity of use (33). To address this issue, we aimed to
construct simpler and more accurate in vitro assays to
evaluate anti-cancer drugs and immunotherapy potency using the
F-PDO and F-PDX models, and predict the clinical efficacy of
anti-cancer drugs.
Materials and methods
Compounds and antibodies
Twenty-one anti-cancer agents tested in this study
(Table SI) were dissolved in
dimethyl sulfoxide (final concentration; 20 mM) and stored at −80°C
until use. The purity and integrity of the agents were measured
using ultra-performance liquid chromatography-mass spectrometry
(Waters Corporation) as follows: a 1-µl injection volume, a Waters
CORTECS C18 column (particle size: 1.6 µm; column size: 2.1×50 mm;
Waters Corporation), linear aqueous acetonitrile (MeCN) gradient
containing 0.1% formic acid (5–90% MeCN, 1.6 min; flow rate, 1
ml/min, 40°C), and the components of the significant ultraviolet
adsorption peaks were identified using mass spectrometry (Table SI).
Cetuximab and bevacizumab were obtained from Bristol
Myers Squibb and Merck & Co., respectively. Blinatumomab was
provided by Amgen.
Cells
F-PDOs were established in our previous study
(26), and three lung F-PDOs
(RLUN001, RLUN021, and RLUN023), each established from a different
patient, were used in this study. Patient-derived hematopoietic
tumor cells were provided by Fukushima Medical University Hospital,
the Japanese Pediatric Cancer Group ALL-R14 study, ProteoGenex, or
AllCells. Peripheral blood mononuclear cells (PBMCs; HPBMC
Peripheral Blood Mono, cry amp) were provided by LONZA. Natural
killer (NK)-Lymphokine-activated killer (LAK) cells were produced
from PBMCs using the AdoptCell-NK kit (Kohjin Bio), following the
manufacturer's instructions. Cytotoxic T lymphocytes (CTLs) were
cultured from PBMCs in the ALyS505N-175 medium (Cell Science and
Technology Institute, Inc.) containing IL-2 using anti-CD-3
antibody-solidified flasks following the manufacturer's
instructions. ADCC Bioassay Effector Cell V variant (High Affinity)
was obtained from BPS Bioscience.
Cell culture
F-PDOs were cultured and maintained in 15 ml of
Cancer Cell Expansion Medium plus (Fujifilm Wako Pure Chemical,
Ltd.) using ultra-low attachment 75 cm2 flasks (Corning,
Inc.) at 37°C in a humidified incubator with 5% CO2 according to
previous reports (26,27). The number and viability of cells were
measured using trypan blue dye exclusion and the Vi-Cell XR Cell
Viability Analyzer (Beckman Coulter).
PDX
These experiments were performed with the approval
of the Institutional Animal Care and Use Committee of Fukushima
Medical University. To produce F-PDX, immunodeficient NOG
(NOD.Cg-PrkdcscidIl2rgtm1Sug/ShiJic)
mice (34) were used. Male NOG mice
(6- to 8-weeks old) were provided by the Central Institute for
Experimental Animals (Kawasaki, Japan) and housed in plastic cages
(136×208×115 mm) within a safety rack (CLEA Japan, Inc., Tokyo,
Japan) in a pathogen-free state, at 22±2°C with 55±5% humidity and
a 12-h light/12-h dark cycle. The plastic cages, bedding, and
filter caps sterilized in an autoclave or via gas sterilization
were used. The mice could to feed ad libitum on a commercial
30 kGy gamma-irradiated sterilized diet (CE-2; CLEA Japan, Inc.)
and ultra-filtered membrane water. Hematopoietic tumor cells
(0.25–2.5×106 cells) were suspended in 0.3 ml of Minimum
Essential Medium α without nucleosides (cat. no. 12561056; Thermo
Fisher Scientific, Inc.). They were injected via the tail vein of
busulfan-treated or untreated mice. Proliferated tumor cells were
extracted from the mouse spleen and bone marrow and frozen in
CELLBANKER 1 (Zenoaq Resource) under liquid nitrogen until they
were used for assays.
Cell viability assay
The cell viability assay for F-PDO was performed by
modifying a method reported previously (26,27).
Three lung F-PDOs were minced using a CellPet FT (JTEC Corporation)
with a filter holder containing a 70- or 100-µm mesh filter. The
cell clusters in each of the selected F-PDO suspension had sizes
ranging from 140 to 160 µm, and 10 cell clusters per well were
seeded into a 384-well, round-bottomed, ultra-low-attachment
microplate (Corning, Inc.) in 40 µl medium, using CELL HANDLER™
(Yamaha Motor). Twenty-four hours after seeding, F-PDOs were
treated with 40 nl of compound solutions at final concentrations
ranging from 1.0 nM to 20 µM, using a series of 10 concentrations
(serially diluted 3-fold) using an Echo 555 Liquid Handler
(Labcyte, Inc.). After 144 h, 10 µl of the CellTiter-Glo 3D Cell
Viability Assay kit solution (Promega Corporation) was added to
F-PDOs in each well, and the plates were incubated for 15 min at
25°C after agitation of the plate using a mixer. Luminescence by
luciferase was measured using the EnSpire Plate Reader
(PerkinElmer, Inc.). Cell viability was calculated by dividing the
ATP content in the test wells by that in the vehicle-control wells
after subtracting the background levels. The 6-day growth rate was
determined by dividing the ATP content in the wells without
anti-cancer agents by those in the vehicle-control wells at 24 h
after seeding.
Cell growth inhibition assays using hematopoietic
tumor-derived PDX were performed in a similar manner as described
above, except for the sample preparation. The frozen cells were
thawed in a 37°C water bath and suspended in 10 ml of RPMI-1640
medium (Fujifilm Wako Pure Chemicals, Ltd.) containing 10% fetal
bovine serum (FBS) and penicillin/streptomycin, or Iscove's
modified Dulbecco's medium (IMDM; Fujifilm Wako Pure Chemicals,
Ltd.). The cells were centrifuged for 5 min at 400 × g. After
removing the medium, the cells were resuspended in 15 ml of
RPMI-1640 medium or StemSpan Leukemic Cell Culture Kit (STEMCELL
Technologies) corresponding to each PDX. The cells were then seeded
at 0.5 or 1×104 cells per well in 384-well plates.
Twenty-four hours after seeding, the cells were treated with
anti-cancer agents for 48 h, and a plate reader measured the ATP
content.
The area under the activity curve (AUC) and
half-maximal inhibitory concentration (IC50) were calculated from
the dose-response curves adjusted to the luminescence signal
intensities using a 4-parameter sigmoid model or a sigmoidal
fixed-slope model without the Hill equation and analyzed using
Morphit software, version 6.0 (The Edge Software Consultancy,
Ltd.). The data are presented as the average ± standard deviation
of triplicate experiments. The Z′-factor, a dimensionless parameter
that ranges between 1 (infinite separation) and <0, was defined
as Z′ = 1-(3σc+ +
3σc−)/|µc+-µc−|, where
σc+, σc−, µc+ and µc−
are the standard deviations (σ) and averages (µ) of the high
(c+) and low (c−) controls (35). Cluster analysis was conducted using
the unweighted pair group method with the arithmetic mean
hierarchical clustering method by TIBCO Spotfire software, version
7.11 (TIBCO Software Inc.).
Antibody-dependent cellular
cytotoxicity (ADCC) assay using reporter cells
ADCC Bioassay Effector Cell expressing a firefly
luciferase gene was thawed in RPMI-1640 medium (Fujifilm Wako Pure
Chemical, Ltd.) containing 10% FBS and penicillin-streptomycin and
cultured in a 5% CO2 incubator at 37°C. The medium was changed to
the growth medium containing 1 mg/ml Geneticin and 200 µg/ml
Hygromycin B (Fujifilm Wako Pure Chemical, Ltd.) after the first
passage, following the manufacturer's instructions.
Minced RLUN021 using CellPet FT with a 100-µm mesh
filter was diluted 2.5- or 5-fold using Cancer Cell Expansion
Medium plus, and 20 µl of the suspension was seeded in 384-well
black plates (Greiner Bio-One GmbH). Subsequently, 10 µl of the
cetuximab solution was added to RLUN021 at a final concentration of
1, 10 or 100 µg/ml. During the incubation of RLUN021 with
cetuximab, the medium of the ADCC Bioassay Effector Cell, a
reporter effector cell line, was changed to Cancer Cell Expansion
Medium plus. The reporter effector cells were then seeded at
5×104 cells per well 60 min post-antibody treatment.
After 5 h, the luminescence was measured using the ONE-Glo
Luciferase Assay System (Promega Corporation) and the EnSpire Plate
Reader.
Real-time potency assessment using the
xCELLigence RCTA system
The xCELLigence RTCA System (ACEA Bioscience) was
used to evaluate F-PDO's cytolysis with CTLs and NK cells. E-plate
96 (ACEA Bioscience), specifically designed to perform cell-based
assays with the xCELLigence RTCA System, was coated with
fibronectin solution (0.5 µg/well; Fujifilm Wako Pure Chemical,
Ltd.) at 4°C overnight. After removing the fibronectin solution, 50
µl of the culture medium was added to each well to measure the
background impedance. Prior to seeding, RLUN021 was filtered
through a 40-µm cell strainer (Corning, Inc.). The cell suspension
(50 µl) were seeded at 2.5×103 cells per well in E-Plate
96. The plate was incubated at approximately 24°C for 30 min and
transferred to the xCELLigence RTCA instrument in a CO2 incubator
at 37°C. Each well contained a final volume of 100 µl. Forty-eight
hours after seeding, 50 µl of culture media was removed from each
well, and 50 µl of effector cell suspension was added at an
effector: target cell ratio of 5:1 or 10:1. Changes in impedance
signals were measured every 15 min as the cell index. Cytolysis
values were converted from the cell index to percent cytolysis
values using the xCELLigence immunotherapy software, version 2.3
(ACEA Bioscience). ‘Percent cytolysis’ refers to the proportion of
target cells that were killed by effector cells compared to F-PDO
alone as a control. The cell indexes of the wells containing only
effector cells were subtracted from the index of the sample wells
at each time point. Then, each value was normalized to the cell
index immediately before antibody addition. The normalized index
was converted to percent cytolysis according to the following
equation: % cytolysis = (1-normalized cell index [sample
wells])/normalized cell index (target alone wells) ×100.
Three-dimensional cell analysis
Images were captured using a FLUOVIEW FV3000
Confocal Laser Scanning Microscope (Olympus) with a UCPLNFLN20X
objective lens. All imaging data were analyzed using the NoviSight
3D Cell Analysis System (Olympus). NucView 530 Caspase-3 Substrate
(Biotium, Inc,) was used to quantify apoptotic cells. In detail,
RLUN021 was suspended in Cancer Cell Expansion Medium plus
containing 2 µM NucView 530 Caspase-3 Substrate. The suspension
(100 µl) was seeded in a well of a 96-well, round-bottomed,
ultra-low-attachment microplate (Corning, Inc.) and incubated for
30 min at 37°C in a CO2 incubator. After incubation, 50 µl of the
medium was removed from each well, and 50 µl of the medium
containing NK cells (0.25, 0.5, 1.0 or 2.0×105 cells per
well) were added into the well. The plate was incubated for 30 min
and 3 h at 37°C in a CO2 incubator. Cells were then collected by
centrifugation (200 × g, 2 min) and were fixed overnight at 4°C in
a phosphate buffer solution containing 4% paraformaldehyde
(Fujifilm Wako Pure Chemical, Ltd.) and 1% Triton X-100 (Fujifilm
Wako Pure Chemical, Ltd.). Subsequently, the RLUN021 cells were
washed twice with D-PBS (−), and the nucleus was stained with
Hoechst 33342 (Dojindo Laboratories, Kumamoto, Japan) in D-PBS (−)
for 30 min at approximately 20–25°C. Finally, F-PDO was washed
twice with D-PBS (−) and photographed under a microscope. In the
NoviSight analysis, the nuclei in F-PDO were recognized with
Hoechst 33342 signals. The dead cells were defined based on the
NucView530 signals in the nucleus.
Cytotoxicity assay using calcein
CTL cells were cultured in ALyS505N-175 medium
containing 175 IU/ml of IL-2 for 2 days. Frozen acute lymphocytic
leukemia (ALL)-derived F-PDX cells were thawed in phenol red-free
RPMI-1640 (Fujifilm Wako Pure Chemicals, Ltd.) on the day of the
assays. The ALL cells were stained with 10 µM membrane-permeant
calcein-acetoxymethyl (calcein-AM; PromoCell GmbH) for 30 min at
37°C and then washed twice with fresh medium. Twenty-five
microliters of stained ALL cell suspension were seeded onto 96-well
plates (Sumitomo Bakelite Co., Ltd.) at 3×104 cells per
well. Next, CTL cells were centrifuged to change the medium from
ALyS505N-175 to RPMI-1640, and 25 µl of CTL cell suspension was
added to each well at a CTL: ALL ratio of 5:1. Finally, 10
concentrations of blinatumomab from 1 pM to 100 nM were added to
the CTL and ALL cell mixtures. Each well contained a final volume
of 100 µl. The plates were incubated at 37°C for 2 h and then
centrifuged for 5 min at 400 × g. After 10 min of centrifugation,
the wells of ALL cells without CTL cells and blinatumomab were
treated with a lysis solution (Promega Corporation) to release all
calcein from the stained ALL cells (cell lytic fluorescence of
calcein-stained cells). Fifty microliters of the supernatant in all
wells was transferred to a 96-well half-area black plate (Corning,
Inc.). The fluorescence of calcein was measured at λex=495 nm and
λem=530 nm using the EnSpire Plate Reader. Dead cell ratio was
defined as the fluorescence of dead ALL cells using blinatumomab
(the test wells with antibody - the test wells without
antibody)/fluorescence of total ALL cells (cell lytic fluorescence
of calcein-stained cells-background fluorescence of calcein-stained
cells) ×100, expressed as a percentage.
Statistical analysis
Statistical analyses were performed with Microsoft
Excel 2016 (Microsoft Corporation, Redmond, WA, USA), MEPHAS
(http://www.gen-info.osaka-u.ac.jp/testdocs/tomocom/mokuji1-e.html),
xCELLigence immunotherapy software, version 2.3 or js-STAR XR
version 1.0.3j (http://www.kisnet.or.jp/nappa/software/star/index.htm)
software. Statistical differences between different test conditions
were determined using one-way ANOVA followed by Dunnetts test. Holm
test was used for multiple comparison with js-STAR XR software.
Statistical significance was defined as P<0.05 was considered
statistically significant. The data are shown as means and the
standard deviation from three or four replicate samples.
Results
Development of a cell growth
inhibition assay using F-PDO
To evaluate anti-cancer agents using a
patient-derived tumor model, we aimed to develop a high-accuracy
and high-throughput screening (HTS) assay using F-PDOs and 384-well
microplates. We have previously reported the construction of an HTS
assay for each F-PDO (26,27). In this experiment, we further
attempted to develop an accurate HTS assay by aligning the cell
clusters' size to exclude cell debris from the assay system. We
examined the use of F-PDO (RLUN001 and RLUN023) established from
lung tumors, which is difficult to assay using reported methods. To
seed the cell clusters in a 384-well plate, a cell picking and
imaging system, CELL HANDLER™, which allows for accurate cell
picking without damaging the cells, was used. HTS performance was
evaluated by computing the coefficients of variation (CV) and the
Z′-factor. The Z′-factor has been widely accepted to validate assay
quality and performance, and the assay is suitable for HTS if this
value is >0.5 (35). The control
data points in the 384-well plate assay showed little variability,
with CV values of 5.4 and 6.4% and calculated Z′-factors of 0.84
and 0.81, for RLUN001 and RLUN023, respectively (Table I). These results imply that this
assay has high performance for HTS. In the method without CELL
HANDLER™, CV values were 8.1 and 26.0%, and the Z′-factor values
were 0.76 and 0.23 for RLUN001 and RLUN023, respectively (data not
shown). Thus, the use of CELL HANDLER™ made it possible to perform
assays with high accuracy, even when using 384-well plates.
 | Table I.High-throughput screening performance
using F-PDO and F-PDX models. |
Table I.
High-throughput screening performance
using F-PDO and F-PDX models.
| A, F-PDO |
|---|
|
|---|
| Model | Growth rate | CV, % | Z′-factor |
|---|
| RLUN001 | 2.6 | 5.4 | 0.84 |
| RLUN023 | 2.6 | 6.4 | 0.81 |
|
| B, F-PDX
hematopoietic tumor |
|
| Model | Growth
rate | CV, % |
Z′-factor |
|
| DLEU002 | 1.2 | 4.0 | 0.88 |
| DLEU003 | 1.6 | 12.2 | 0.63 |
| DLEU005 | 1.0 | 1.3 | 0.96 |
| DLEU006 | 1.0 | 3.7 | 0.88 |
| DLEU009 | 1.2 | 3.9 | 0.88 |
| DLEU012 | 1.0 | 2.0 | 0.94 |
| DLEU016 | 1.5 | 3.7 | 0.89 |
| DLEU026 | 1.7 | 3.2 | 0.90 |
| DLEU031 | 1.4 | 2.2 | 0.94 |
| DLEU011 | 1.3 | 4.5 | 0.87 |
| DLEU018 | 0.9 | 3.4 | 0.90 |
| DLEU020 | 1.1 | 4.7 | 0.86 |
| DLEU022 | 1.6 | 2.1 | 0.94 |
| DLEU027 | 1.4 | 3.1 | 0.91 |
| DLEU030 | 0.9 | 4.7 | 0.86 |
| DLEU013 | 1.4 | 7.8 | 0.77 |
| DLEU028 | 0.8 | 2.6 | 0.92 |
| DLEU029 | 1.1 | 4.0 | 0.88 |
To investigate the sensitivity of F-PDOs to
anti-cancer agents using our HTS system, growth inhibition was
assessed using RLUN001 and RLUN023 treated with eight anti-cancer
agents, specifically, epidermal growth factor receptor (EGFR)
inhibitors (erlotinib, gefitinib, afatinib, lapatinib, osimertinib,
and rociletinib), and paclitaxel, which are standard clinical
treatments for non-small cell lung cancer, and mitomycin C as a
positive control. RLUN001 and RLUN023, which possess a sensitivity
mutation (p.E746-A750del) to EGFR inhibitors, may indicate a high
sensitivity. F-PDOs were treated with the drugs 24 h after seeding
and were subsequently incubated for six days. The IC50 and AUC
values of the anti-cancer agents in each F-PDO are shown in
Table II. RLUN001 and RLUN023
showed high sensitivity (IC50 <250 nM) to all EGFR inhibitors.
We have previously reported that the IC50 values of erlotinib and
gefitinib for RLUN021 without EGFR mutations were very high at
>20 and 6 µM, respectively (27).
These results indicate that an F-PDO with susceptibility mutations
is highly sensitive to EGFR inhibitors, consistent with general
clinical results (36).
| Table II.IC50 and AUC values of anticancer
agents against each Fukushima-patient-derived tumor organoid
model. |
Table II.
IC50 and AUC values of anticancer
agents against each Fukushima-patient-derived tumor organoid
model.
|
| RLUN001 | RLUN023 |
|---|
|
|
|
|
|---|
| Anticancer
agent | IC50,
nM | AUC | IC50,
nM | AUC |
|---|
| Mitomycin C | 716 | 66 | 201 | 175 |
| Paclitaxel | 3 | 383 | 105 | 854 |
| Afatinib | 4 | 6 | <1 | 52 |
| Lapatinib | 239 | 106 | 168 | 247 |
| Erlotinib | 292 | 121 | 186 | 353 |
| Gefitinib | 33 | 32 | 15 | 433 |
| Osimertinib | 6 | 0 | 5 | 367 |
| Rociletinib | 163 | 25 | 68 | 400 |
Development of a cell growth
inhibition assay using F-PDX
Conducting simple in vitro assays using PDX
to evaluate anti-cancer drug candidates before conducting costly
and time-consuming animal studies is desirable. In this study, we
attempted to construct an HTS assay system using F-PDX derived from
hematopoietic tumors. We constructed an in vitro assay
system with 384-well plates using hematopoietic tumor-derived
F-PDX: nine types of ALL-derived cells, six types of acute myeloid
leukemia (AML)-derived cells, one type of multiple myeloma
(MM)-derived cells, and two types of mixed phenotype acute leukemia
(MPAL)-derived cells were used in the study. The growth rate of
F-PDXs in the plates was approximately 0.8- to 1.7-fold in two days
of culture (Table I). As shown in
Table I, CV and Z′-factor values
were 1.3 to 12.2% and 0.63 to 0.96, respectively. These results
imply that this assay has satisfactory performance for HTS. To
investigate the sensitivity of F-PDXs to anti-cancer agents using
our HTS system, growth inhibition was assessed using treatment with
13 anti-cancer agents. The IC50 values of the anti-cancer agents in
each F-PDX are shown in Tables
III–V. The sensitivity of
hematopoietic tumors to anti-cancer agents varied regardless of the
type of cancer. In particular, the sensitivity of ALL cells to
anti-cancer agents was significant.
| Table III.IC50 values of anticancer agents
against each ALL-derived F-PDX model. |
Table III.
IC50 values of anticancer agents
against each ALL-derived F-PDX model.
| ALL-derived F-PDX
Medium | DLEU002 RPMI | DLEU003
StemSpan | DLEU005 RPMI | DLEU006 RPMI | DLEU009 RPMI | DLEU012 RPMI | DLEU016 RPMI | DLEU026 RPMI | DLEU031
StemSpan |
|---|
| Anticancer
agent |
|
|
|
|
|
|
|
|
|
|
Prednisolone | 0.165 | >20 | >20 | 0.183 | 0.045 | 0.234 | 0.038 | >20 | 1.656 |
|
Cytarabine | 0.833 | 2.632 | 15.210 | >20 | 4.868 | 3.312 | 0.015 | >20 | 0.263 |
|
Doxorubicin | 0.019 | 0.385 | 0.034 | 0.020 | 0.041 | 0.105 | 0.017 | 1.092 | 0.064 |
|
Mitoxantrone | 0.001 | 0.191 | 0.002 | 0.001 | 0.002 | 0.037 | 0.001 | 0.189 | 0.005 |
|
Bleomycin | 9.760 | >20 | 9.991 | 0.146 | 0.151 | >20 | 3.785 | >20 | 5.103 |
|
Clofarabine | 0.045 | >20 | 0.069 | >20 | 0.039 | >20 | 0.024 | >20 | 0.040 |
|
Dasatinib | 4.647 | >20 | 11.144 | 0.069 | 3.047 | 19.186 | 0.361 | >20 | >20 |
|
Daunorubicin | 0.004 | 0.164 | 0.012 | 0.004 | 0.009 | 0.194 | 0.006 | 0.431 | 0.025 |
|
Idarubicin | 0.002 | 0.119 | 0.006 | 0.002 | 0.004 | 0.056 | 0.004 | 0.090 | 0.013 |
|
Tretinoin | >20 | >20 | >20 | >20 | >20 | 17.978 | 14.233 | >20 | >20 |
|
Vincristine | 0.012 | 0.822 | 0.032 | 17.235 | 0.028 | 0.125 | 0.001 | >20 | 0.114 |
|
Imatinib | 19.322 | >20 | >20 | 16.640 | 14.286 | >20 | 13.756 | >20 | >20 |
|
Nelarabine | >20 | >20 | >20 | >20 | >20 | >20 | >20 | >20 | >20 |
| Table V.IC50 values of anticancer agents
against MM- and MPAL-derived F-PDX models. |
Table V.
IC50 values of anticancer agents
against MM- and MPAL-derived F-PDX models.
| F-PDX Medium | MM/DLEU013
StemSpan | MPAL/DLEU028
RPMI | MPAL/DLEU029
StemSpan |
|---|
| Anticancer
agent |
|
|
|
|
Prednisolone | >20 | 0.022 | >20 |
|
Cytarabine | 7.548 | 3.986 | 0.551 |
|
Doxorubicin | 0.041 | 0.022 | 0.037 |
|
Mitoxantrone | 0.071 | 0.002 | 0.002 |
|
Bleomycin | 19.556 | 0.775 | 1.649 |
|
Clofarabine | 4.287 | 0.528 | 0.029 |
|
Dasatinib | 0.990 | 14.456 | >20 |
|
Daunorubicin | 0.031 | 0.006 | 0.017 |
|
Idarubicin | 0.031 | 0.004 | 0.005 |
|
Tretinoin | >20 | >20 | >20 |
|
Vincristine | 0.028 | 0.026 | 0.072 |
|
Imatinib | >20 | 9.831 | >20 |
|
Nelarabine | >20 | >20 | >20 |
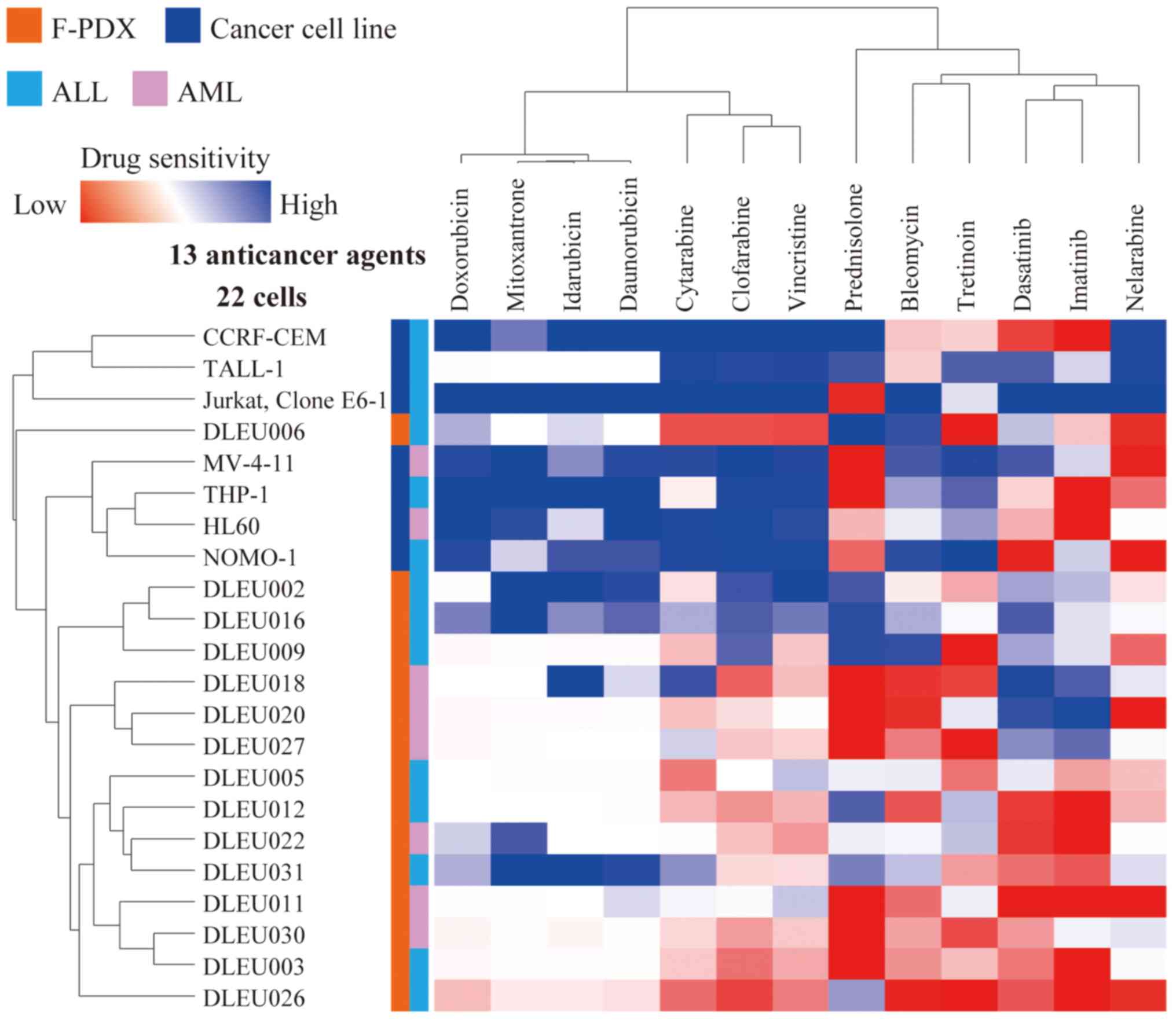
Cluster analysis was performed for five ALL- and two
AML-derived cancer cell lines and nine ALL- and six AML-derived
F-PDX cells to determine sensitivity to anti-cancer drugs (Fig. 1). The sensitivity profiles of ALL and
AML cells to anti-cancer drugs could not be distinguished. In
contrast, the sensitivity profiling of cancer cell lines and F-PDX
differed significantly. F-PDX was more resistant to anti-cancer
drugs than cancer cell lines.
In vitro evaluation of ADCC with
antibody drugs
We developed a simple system to measure ADCC
activity using a patient-derived cancer model. ADCC is a defense
mechanism involving cellular immunity. Immune effector cells have
an active effect on the immune system cells that lyse cancer cells,
whose membrane-surface antigens are bound by antibodies. The
antibody's Fc region binds to effector cells (NK cells, monocytes,
etc.) that have the FcγR receptor (FcγRIIIa). Therefore, effector
cells are activated, and granzyme and other substances are released
to lyse the target cells. To measure ADCC activity against F-PDO,
we used the ADCC Bioassay Effector Cell as a reporter effector cell
line and cetuximab, a monoclonal antibody targeting EGFR. RLUN021
cells, which have high expression levels of EGFR, were selected as
the target cancer cells (27). The
ADCC Bioassay Effector Cells were Jurkat T cells expressing firefly
luciferase under the control of the nuclear factor of activated T
cells response elements with constitutive expression of human
FcγRIIIa, a high affinity (V158) variant.
ADCC activity was measured as the luciferase
activity from reporter effector cells. Because RLUN021 formed
significant cell clusters and an accurate number of the cells could
not be counted, it was challenging to determine the target cell:
effector cell ratio. Therefore, we set two dilution conditions
(2.5- and 5-fold) of target cells for the numbers of
5×104 effector cells. No luminescence was detected in
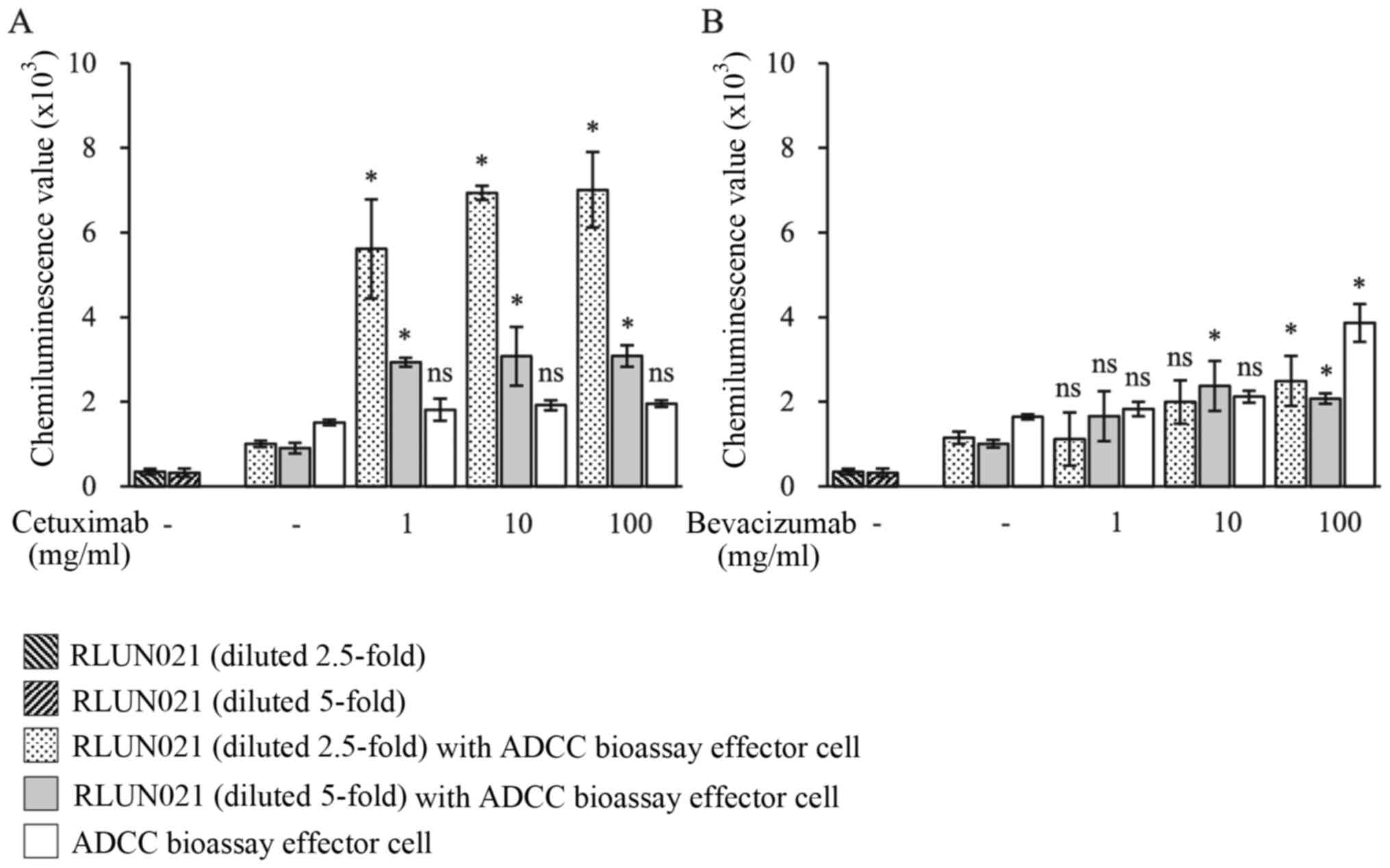
RLUN021 cells alone or only incubated with effector cells. As
expected, when RLUN021 diluted 2.5-fold were incubated with
cetuximab for 1 h and then were treated with effector cells for 5
h, a significantly high level of luminescence was observed that
referred to ADCC activity (Fig. 2A).
Even though ADCC activity was decreased with an increase in
dilution rate of the target cells, its levels were higher than
those in the effector cells alone (white bars). No ADCC activity
was observed even with 100 µg/ml bevacizumab (anti-VEGF antibody)
as a negative control compared with the effector cells alone group
(Fig. 2B).
Immune response activity using CTL and
NK cells
To investigate the cytolysis of F-PDO with CTL and
NK cells using the xCELLigence system, which monitor the number,
morphology, and attachment of cells for a long duration, changes in
impedance signals (cell index) were assessed using RLUN021 treated
with CTL and NK cells in 96-well plates. RLUN021 had a high
expression of the major histocompatibility complex (MHC) class I
chain-related gene A (MICA). MICA, expressed on cancer cells, binds
to the receptor for MICA/B and NKG2D (CD314) on NK cells, and the
NK cells subsequently cause cytotoxicity in cancer cells (37). Therefore, RLUN021 is suitable for the
measurement of cytolysis with NK cells. PBMCs are cultured with
IL-2 to induce LAK cells that cause cytotoxicity. No tumor antigen
stimulation is required to induce LAK cells. LAK cells are not
defined by MHC and widely induce immune cell-mediated cytotoxicity
against tumor cells. LAK cells are a heterogeneous population of
killer cells primarily divided into two groups: CTL and NK cells,
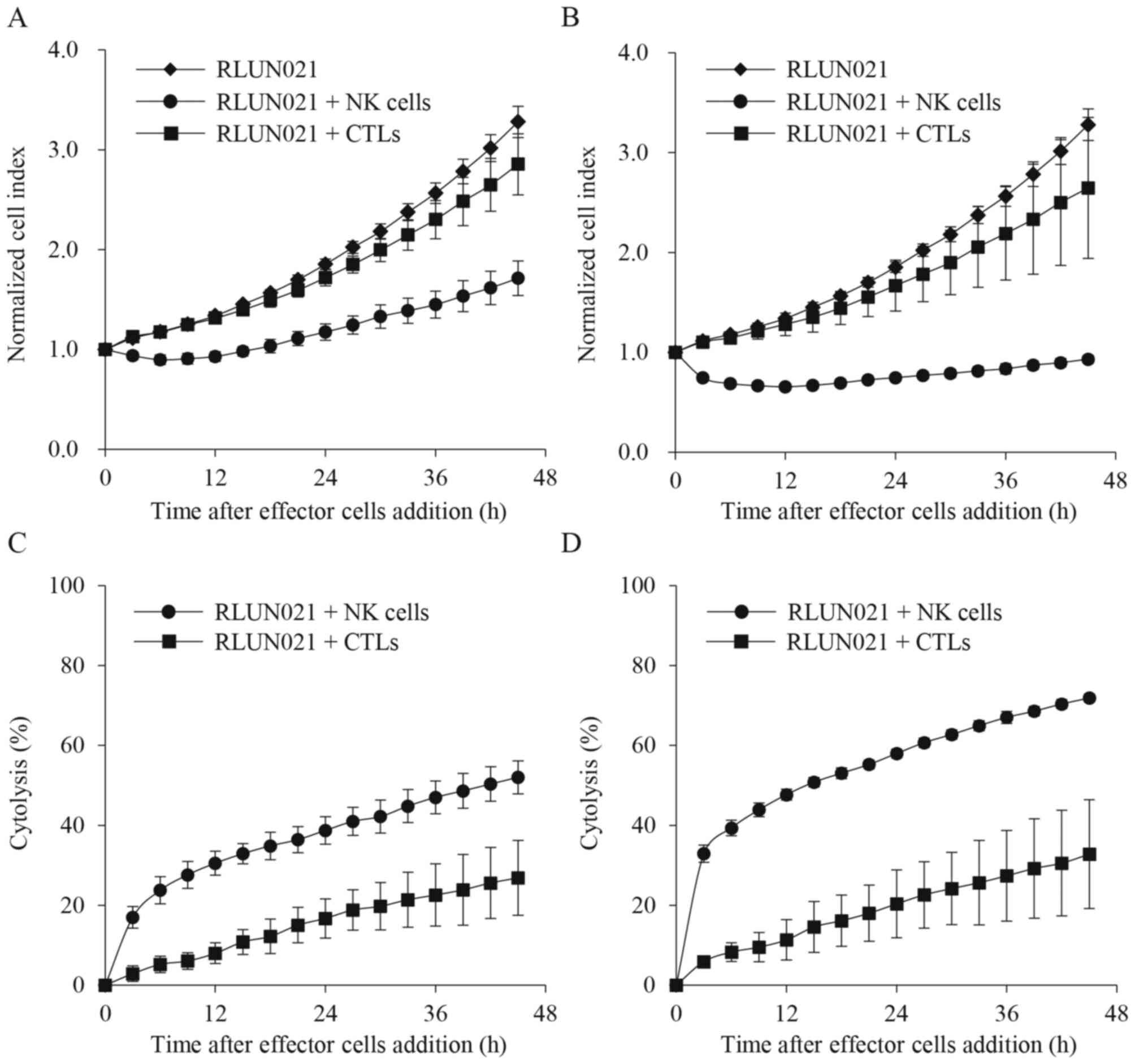
which are not restricted to MHC. Thus, RLUN021 was treated with CTL
and NK cells. Compared to target cell alone, following treatment
with 1:5 (Fig. 3A and C) and 1:10
(Fig. 3B and D) ratios of F-PDO
cells to effector cells, NK cells immediately suppressed an
increase in cell index (Fig. 3A and
B). NK cell-mediated cytolysis was approximately 50 and 30% at
a ratio of 10:1 (Fig. 3D) and 5:1
(Fig. 3C), respectively, after 12
h.
In contrast, CTL-mediated cytolysis was slower, and
approximately 10% of the cells were lysed at 12 h (Fig. 3C and D). There was also no
significant difference in cytolysis induction when the number of
effector cells was doubled (Fig. 3).
These results indicate that the F-PDO assay system can evaluate
immune response activity using real-time impedance-based
technology.
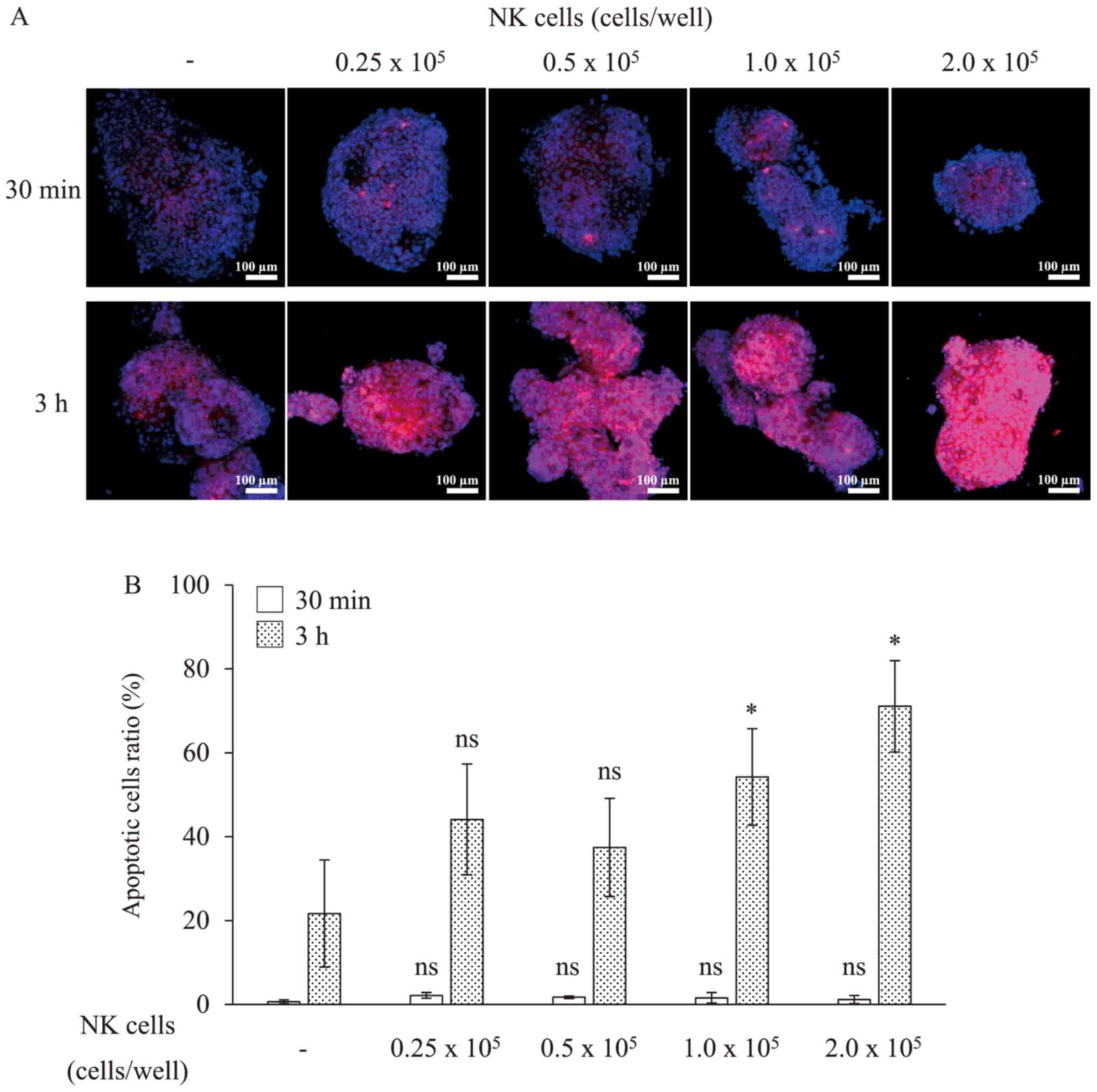
To quantify the induction of cell death, we
subsequently observed the cytolysis of RLUN021 by NK cells as an
indicator of caspase-3 activation caused by the induction of
apoptosis on cytolysis. Thirty minutes after NK cells treatment,
apoptotic cells were not detected in cell clusters (Fig. 4). After 3 h, apoptosis was observed
in approximately 70% of the RLUN021 cells treated with
2×105 NK cells per well. Meanwhile, apoptosis was
observed in approximately 50% of RLUN021 treated with
1.0×105 NK cells per well. However, RLUN021 treated with
0.25 or 0.5×105 NK cells per well did not showed
significant difference compared to the no NK cells treatment group.
Thus, apoptosis was induced depending on the number of NK cells
treated. These results were consistent with the results using the
xCELLigence system.
Immune response of BiTE using
F-PDX
A bispecific antibody, blinatumomab, and a
patient-derived tumor model, ALL-derived F-PDXs, were used to
construct a BiTE assay system. Blinatumomab is a bispecific
antibody drug approved in 2014 for the treatment of B cell-derived
ALL. Blinatumomab simultaneously binds to CD19 on the surface of B
cells and CD3 on the surface of CTL cells. It causes cytotoxicity
to ALL cells by engaging CTL cells with ALL cells. Six kinds of
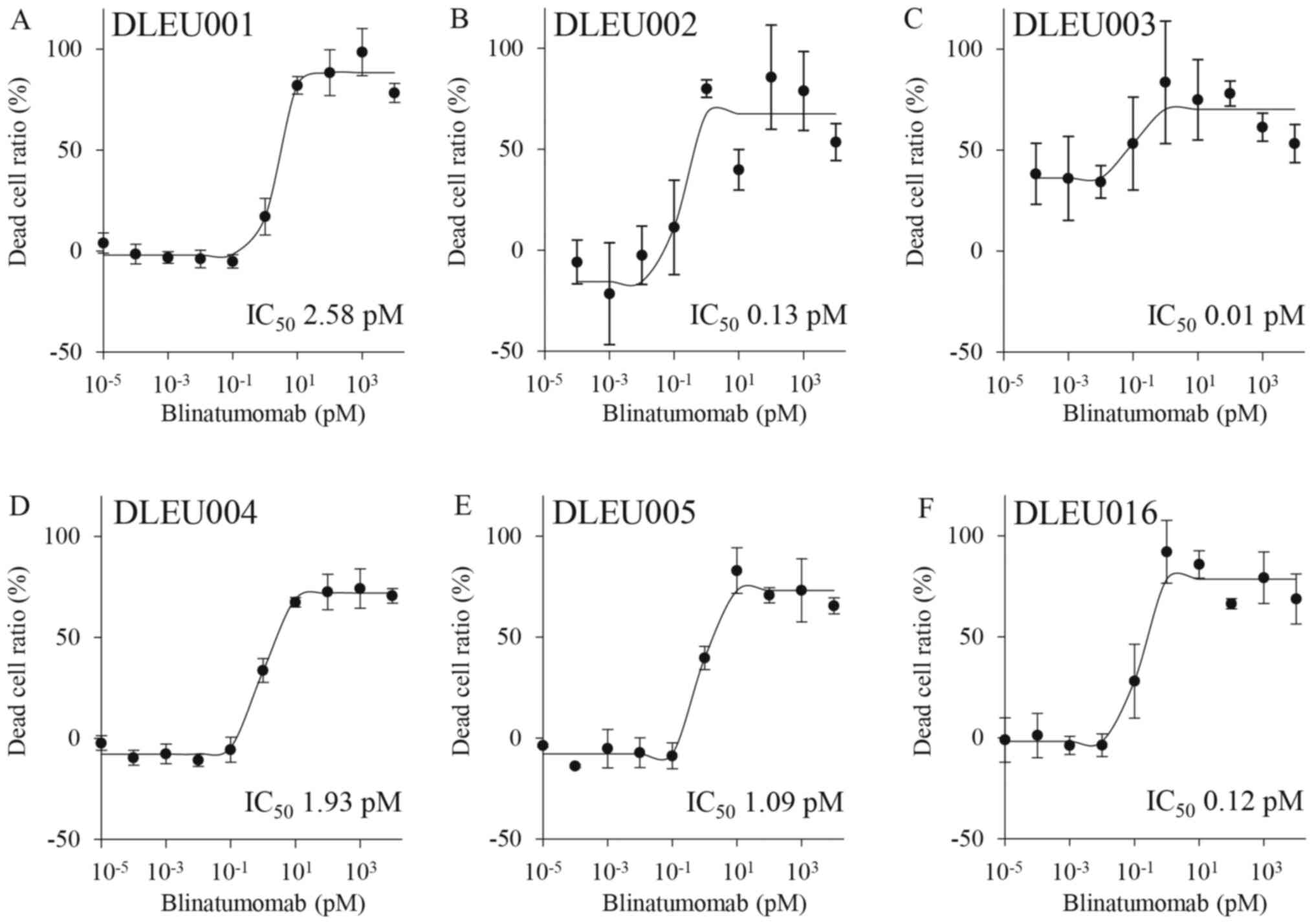
ALL-derived F-PDXs (DLEU001, 002, 003, 004, 005, 016) and CTL cell
mixtures were treated with blinatumomab (Fig. 5). The data points for dead cells
showed little variation and low CV values. The assay system was
stable and satisfactory as an evaluation system for BiTE. Because
all the ALL cells used in this experiment were CD19 positive,
blinatumomab induced cell death in all ALL cells and showed low
IC50 values (approximately 0.01–3 pM). It was interesting to
observe that there was a difference in the effect of blinatumomab
despite the lack of significant differences in the expression of
CD19 in ALL cells. There may be differences in susceptibility to
CTLs owing to varying patient characteristics.
Discussion
Here, we have developed a high accuracy and
throughput assay system for anti-cancer drug evaluation using
patient-derived models, F-PDO and F-PDX. Recently, in vitro
assays using PDO and PDX have been reported (11–23).
These culture and assays use extracellular matrixes such as
Matrigel® to create tumor tissue scaffolds or enzymes
such as trypsin and collagenase to disrupt the organs. Organoid
cancer models (15,38), widely available from the American
Type Culture Collection in support of the Human Cancer Models
Initiative, also use extracellular matrix and enzymes. There are
few reports of HTS systems using PDO with 384-well plates, and
these HTS systems also utilize Matrigel or other matrices (19,39). Our
method's advantage is that it does not involve extracellular matrix
utilization and enzymatic treatment during culture and assay,
significantly reducing labor requirements and cost. Therefore, it
is also relatively easy to be adapted to HTS assay systems and
various measurement systems. Furthermore, the absence of enzymatic
treatment has the advantage of maintaining cell-to-cell
interactions and the conditions of heterogeneous source tumors.
However, because the extracellular matrix can act as a scaffold for
cells and affect tissue morphogenesis, differentiation, and
homeostasis, it is desirable to use extracellular matrix for
research purposes.
PDO characteristics are unsuitable for HTS or cell
analysis using 96-well or 384-well formats for anti-cancer drugs
because these structures display heterogeneous sizes and also form
large clusters in culture. Thus, CELL HANDLER™, which
can isolate cell spheroids of a precise size without damage, and
NoviSight, which can quantify the specific cells present in cell
spheroids with complex structures, were used to improve the
accuracy of anti-cancer drug assays and 3D cell analysis using
384-well plates as well as to automate the process. This resulted
in uniform cell sizes, and when applied, we succeeded in developing
a precise HTS platform using F-PDOs. The assays using F-PDO and
hematopoietic tumor-derived F-PDX were performed with high
accuracy. In contrast, the solid tumor-derived F-PDX assay did not
(data not shown) because it was challenging to mince the solid
tumor tissue uniformly. Collecting a large minced tumor tissue
using CELL HANDLER™ was also challenging. By developing a uniform
mincing method for tumor tissues and using CELL HANDLER™ to collect
large tissues, we plan to establish a more accurate assay system
using solid cancer-derived F-PDX in the future.
In the EGFR inhibitor sensitivity test of lung
tumor-derived F-PDOs, those with mutant EGFRs that are clinically
sensitive to EGFR inhibitors were more sensitive than the wild
type. F-PDX derived from hematopoietic tumors showed different
sensitivity to anti-cancer agents compared with the existing cancer
cell lines. Furthermore, F-PDO and F-PDX showed different
sensitivities owing to varying patient characteristics. These
results suggest that F-PDOs reflect the clinical status of the
source tumors in terms of responsiveness to drugs. Therefore, this
assay system enables the evaluation of anti-cancer agents under
conditions that reflect clinical conditions more accurately than
conventional methods do and may be useful in identifying markers
that predict the pharmacological effects of anti-cancer drugs.
Several attempts have been made to evaluate
immunotherapy potency, particularly the development of
patient-derived tumor models, including PDX. However, PDX is not
suitable for evaluating tumor immunology and drugs targeting the
immune system because of severely immunocompromised host animals.
Thus, humanized mice harboring a human immune system have been
used, although problems with the graft-versus-host disease severely
limit its implementation (40,41). In
addition, in vivo culture is hindered by labor and financial
burdens due to the lengthy process of establishing tumor
engraftment and generating cohorts for experimentation. Thus, we
developed an in vitro assay system using F-PDO or F-PDX to
evaluate immunotherapy potency. By co-culturing F-PDO and immune
cells, cytotoxicity to target tumor cells could be measured in
real-time using xCELLigence. In addition, NoviSight enabled us to
detect the number of tumor cells that were dying in specific
locations in cell clusters. Furthermore, in vitro evaluation
of the bispecific antibody could also be performed using
hematopoietic tumor-derived F-PDX and immune cells. Compared with
the animal model, this system is straightforward and cost-effective
to develop and can evaluate immunotherapy potency.
Finally, this study demonstrated that F-PDO and
F-PDX models, which retain tumor tissue characteristics, are
superior in evaluating potential novel cancer immunotherapies and
anti-cancer drugs. Therefore, they present opportunities for drug
assessment and advances in personalized medicine approaches.
Although the assay system developed by us is suitable for the
initial screening of drugs, it does not reproduce the tumor
microenvironment and thus cannot evaluate the efficacy of drugs
in vivo. Therefore, we will construct an in vitro
system that can mimic human tumor tissue by co-culturing with
vascular endothelial cells and other stromal cells or
organ-on-a-chip technology in the future. Our ultimate goal is to
develop a system that can mimic cancer tissues in vivo and
evaluate the efficacy of anti-cancer drugs without animal
models.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Goto Hiroaki
(Kanagawa Children's Medical Center, Yokohama, Japan) and Dr
Chitose Ogawa (National Cancer Center, Tokyo, Japan) in the
Japanese Pediatric Cancer Group for providing ALL cells from the
patients.
Funding
The present study was supported by grants from the
Translational Research Programs from Fukushima Prefecture.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MT, SW, KK, HS and KS conceptualized and designed
the experiments. NT, AH, GH, HT, HH, YD, HI, KT, KG, NO and SM
performed the experiments. NT, AH, HT, GH, HH and MT analyzed the
data and wrote the paper. MT revised the paper. MT and NT confirmed
the authenticity of the data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All experiments involving materials derived from
humans were performed following the guidelines of the Declaration
of Helsinki and were approved in advance by the Ethics Committee of
the Fukushima Medical University (Fukushima, Japan; approval nos.
1953 and 2192; approval dates March 18, 2020, and May 26, 2016,
respectively). The participants provided written informed consent.
Furthermore, animal experiments were performed with the approval of
the Institutional Animal Care and Use Committee of Fukushima
Medical University (approval nos. 27011, 29035 and 2019027;
approval dates April 1, 2015, April 1, 2017, and April 1, 2019,
respectively).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PDO
|
patient-derived tumor organoid
|
|
PDX
|
patient-derived tumor xenograft
|
|
BiTEs
|
bispecific T-cell engagers
|
|
ADCC
|
antibody-dependent cellular
cytotoxicity
|
|
CTLs
|
cytotoxic T lymphocytes
|
|
NK cells
|
natural killer cells
|
|
ALL
|
acute lymphocytic leukemia
|
|
AML
|
acute myeloid leukemia
|
|
LAK
|
lymphokine-activated killer
|
|
HTS
|
high-throughput screening
|
References
|
1
|
Sharma SV, Haber DA and Settleman J: Cell
line-based platforms to evaluate the therapeutic efficacy of
candidate anticancer agents. Nat Rev Cancer. 10:241–253. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shamir ER and Ewald AJ: Three-dimensional
organotypic culture: Experimental models of mammalian biology and
disease. Nat Rev Mol Cell Biol. 15:647–664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arrowsmith J and Miller P: Trial watch:
Phase II and phase III attrition rates 2011–2012. Nat Rev Drug
Discov. 12:5692013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arrowsmith J: Trial watch: Phase II
failures: 2008–2010. Nat Rev Drug Discov. 10:328–329. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DiMasi JA, Reichert JM, Feldman L and
Malins A: Clinical approval success rates for investigational
cancer drugs. Clin Pharmacol Ther. 94:329–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tentler JJ, Tan AC, Weekes CD, Jimeno A,
Leong S, Pitts TM, Arcaroli JJ, Messersmith WA and Eckhardt SG:
Patient-derived tumour xenografts as models for oncology drug
development. Nat Rev Clin Oncol. 9:338–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siolas D and Hannon GJ: Patient-derived
tumor xenografts: Transforming clinical samples into mouse models.
Cancer Res. 73:5315–5319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosfjord E, Lucas J, Li G and Gerber HP:
Advances in patient-derived tumor xenografts: From target
identification to predicting clinical response rates in oncology.
Biochem Pharmacol. 91:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hidalgo M, Amant F, Biankin AV, Budinská
E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo
GM, et al: Patient-derived xenograft models: An emerging platform
for translational cancer research. Cancer Discov. 4:998–1013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao H, Korn JM, Ferretti S, Monahan JE,
Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al:
High-throughput screening using patient-derived tumor xenografts to
predict clinical trial drug response. Nat Med. 21:1318–1325. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maru Y and Hippo Y: Current status of
patient-derived ovarian cancer models. Cells. 8:5052019. View Article : Google Scholar
|
|
12
|
Weeber F, Ooft SN, Dijkstra KK and Voest
EE: Tumor organoids as a pre-clinical cancer model for drug
discovery. Cell Chem Biol. 24:1092–1100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crespo M, Vilar E, Tsai SY, Chang K, Amin
S, Srinivasan T, Zhang T, Pipalia NH, Chen HJ, Witherspoon M, et
al: Colonic organoids derived from human induced pluripotent stem
cells for modeling colorectal cancer and drug testing. Nat Med.
23:878–884. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato T, Stange DE, Ferrante M, Vries RG,
Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J,
Siersema PD, et al: Long-term expansion of epithelial organoids
from human colon, adenoma, adenocarcinoma, and Barrett's
epithelium. Gastroenterology. 141:1762–1772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van de Wetering M, Francies HE, Francis
JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J,
Taylor-Weiner A, Kester L, et al: Prospective derivation of a
living organoid biobank of colorectal cancer patients. Cell.
161:933–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boj SF, Hwang CI, Baker LA, Chio II, Engle
DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al:
Organoid models of human and mouse ductal pancreatic cancer. Cell.
160:324–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao D, Vela I, Sboner A, Iaquinta PJ,
Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora
VK, et al: Organoid cultures derived from patients with advanced
prostate cancer. Cell. 159:176–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Girda E, Huang EC, Leiserowitz GS and
Smith LH: The use of endometrial cancer patient-derived organoid
culture for drug sensitivity testing is feasible. Int J Gynecol
Cancer. 27:1701–1707. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Broutier L, Mastrogiovanni G, Verstegen
MM, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes-Benito R,
Sidorova O, Gaspersz MP, et al: Human primary liver cancer-derived
organoid cultures for disease modeling and drug screening. Nat Med.
23:1424–1435. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pauli C, Hopkins BD, Prandi D, Shaw R,
Fedrizzi T, Sboner A, Sailer V, Augello M, Puca L, Rosati R, et al:
Personalized in vitro and in vivo cancer models to guide precision
medicine. Cancer Discov. 7:462–477. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kondo J, Endo H, Okuyama H, Ishikawa O,
Iishi H, Tsujii M, Ohue M and Inoue M: Retaining cell-cell contact
enables preparation and culture of spheroids composed of pure
primary cancer cells from colorectal cancer. Proc Natl Acad Sci
USA. 108:6235–6240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshida T, Okuyama H, Endo H and Inoue M:
Spheroid cultures of primary urothelial cancer cells: Cancer
tissue-originated spheroid (CTOS) method. Methods Mol Biol.
1655:145–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meijer TG, Naipal KA, Jager A and van Gent
DC: Ex vivo tumor culture systems for functional drug testing and
therapy response prediction. Future Sci OA. 3:FSO1902017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Inoue A, Deem AK, Kopetz S, Heffernan TP,
Draetta GF and Carugo A: Current and future horizons of
patient-derived xenograft models in colorectal cancer translational
research. Cancers (Basel). 11:13212019. View Article : Google Scholar
|
|
25
|
Hum NR, Sebastian A, Gilmore SF, He W,
Martin KA, Hinckley A, Dubbin KR, Moya ML, Wheeler EK, Coleman MA,
et al: Comparative molecular analysis of cancer behavior cultured
in vitro, in vivo, and ex vivo. Cancers (Basel). 12:6902020.
View Article : Google Scholar
|
|
26
|
Tamura H, Higa A, Hoshi H, Hiyama G,
Takahashi N, Ryufuku M, Morisawa G, Yanagisawa Y, Ito E, Imai JI,
et al: Evaluation of anticancer agents using patient-derived tumor
organoids characteristically similar to source tissues. Oncol Rep.
40:635–646. 2018.PubMed/NCBI
|
|
27
|
Takahashi N, Hoshi H, Higa A, Hiyama G,
Tamura H, Ogawa M, Takagi K, Goda K, Okabe N, Muto S, et al: An in
vitro system for evaluating molecular targeted drugs using lung
patient-derived tumor organoids. Cells. 8:4812019. View Article : Google Scholar
|
|
28
|
Pan C, Liu H, Robins E, Song W, Liu D, Li
Z and Zheng L: Next-generation immuno-oncology agents: Current
momentum shifts in cancer immunotherapy. J Hematol Oncol.
13:292020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hegde PS and Chen DS: Top 10 challenges in
cancer immunotherapy. Immunity. 52:17–35. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Farkona S, Diamandis EP and Blasutig IM:
Cancer immunotherapy: The beginning of the end of cancer? BMC Med.
14:732016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andrews MC and Wargo JA: Immunotherapy
resistance: The answers lie ahead - not in front - of us. J
Immunother Cancer. 5:102017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu H, Lyu X, Yi M, Zhao W, Song Y and Wu
K: Organoid technology and applications in cancer research. J
Hematol Oncol. 11:1162018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cerignoli F, Abassi YA, Lamarche BJ,
Guenther G, Santa Ana D, Guimet D, Zhang W, Zhang J and Xi B: In
vitro immunotherapy potency assays using real-time cell analysis.
PLoS One. 13:e01934982018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ito M, Hiramatsu H, Kobayashi K, Suzue K,
Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et
al: NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse
model for engraftment of human cells. Blood. 100:3175–3182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang JH, Chung TD and Oldenburg KR: A
simple statistical parameter for use in evaluation and validation
of high throughput screening assays. J Biomol Screen. 4:67–73.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shah R and Lester JF: Tyrosine Kinase
Inhibitors for the treatment of EGFR mutation-positive
non-small-cell lung cancer: A clash of the generations. Clin Lung
Cancer. 21:e216–e228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Salih HR, Antropius H, Gieseke F, Lutz SZ,
Kanz L, Rammensee HG and Steinle A: Functional expression and
release of ligands for the activating immunoreceptor NKG2D in
leukemia. Blood. 102:1389–1396. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Palechor-Ceron N, Krawczyk E, Dakic A,
Simic V, Yuan H, Blancato J, Wang W, Hubbard F, Zheng YL, Dan H, et
al: Conditional reprogramming for patient-derived cancer models and
next-generation living biobanks. Cells. 8:13272019. View Article : Google Scholar
|
|
39
|
Du Y, Li X, Niu Q, Mo X, Qui M, Ma T, Kuo
CJ and Fu H: Development of a miniaturized 3D organoid culture
platform for ultra-high-throughput screening. J Mol Cell Biol.
12:630–643. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cassidy JW, Caldas C and Bruna A:
Maintaining tumor heterogeneity in patient-derived tumor
xenografts. Cancer Res. 75:2963–2968. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shultz LD, Brehm MA, Garcia-Martinez JV
and Greiner DL: Humanized mice for immune system investigation:
Progress, promise and challenges. Nat Rev Immunol. 12:786–798.
2012. View Article : Google Scholar : PubMed/NCBI
|