Lung cancer is a malignant disease with high
morbidity and mortality rates, and non-small cell lung cancer
(NSCLC) accounts for 80–85% of all lung cancer cases. Clinically,
only a small percentage of patients with NSCLC are diagnosed at an
early stage (I or II), at which tumors can be surgically removed.
The majority of patients with NSCLC present with locally advanced
or metastatic disease at the time of diagnosis, leaving
chemotherapy, targeted therapy, and immunotherapy as the primary
treatment strategies (1). However,
primary resistance and acquired resistance after long-term drug
usage are inevitable problems (2).
Previous data suggest that the 5-year survival rate of patients
with advanced NSCLC is <5% (3).
Moreover, the occurrence of drug resistance is a major obstacle to
successful treatment, which requires urgent medical attention
(3,4).
Existing therapeutic approaches primarily counter
drug resistance by targeting tumor cells and sparing those of the
tumor microenvironment (TME). Since the concept of ‘seed and soil’
was proposed, the role of the TME in tumor drug resistance has
received increasing attention. For example, a hypoxic
microenvironment was found to induce cisplatin resistance in NSCLC
(5,6). Collagen, a component of the
extracellular matrix (ECM), induces NSCLC resistance to epidermal
growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) by
binding to the collagen receptor integrin α11β1 (7). In addition to these physical factors,
stromal cells that surround the tumor, such as cancer stem cells
(CSCs) (8) and stromal fibroblasts
(9), can induce therapeutic
resistance in NSCLC. Cancer-associated fibroblasts (CAFs) are an
important component of the TME (10), which serve a primary role in drug
resistance. Compared with tumor cells, CAFs are considered to be
genetically stable with few mutations (11), and can influence tumor progression
through the secretion of ECM proteins, proteases, cytokines,
chemokines and growth factors (12,13). In
addition, CAFs are involved in drug resistance in various
malignancies, such as head and neck (14), breast (15), ovarian (13), gastrointestinal (16), pancreatic (17) and colorectal cancer (18), though the underlying mechanisms
differ between tumor types. Thus, the mechanisms of CAF-mediated
NSCLC resistance have gained considerable attention (19). The present review describes the
functions and mechanisms of CAFs in NSCLC drug resistance, as well
as potential strategies to reverse this effect.
Numerous types of stromal cell, including
fibroblasts, are present in the TME. Fibroblasts are activated in
response to cancer cells, after which they are referred to as CAFs
or myofibroblasts. CAFs are spindle-shaped cells of variable size
and proliferative capacity (20,21).
Moreover, CAFs exhibit high heterogeneity in terms of origin,
surface markers and resident organs, which determines their
functions in tumor progression. During the early stages of tumor
development, CAFs play an antitumor role by promoting tissue
repair. However, as the tumor progresses, CAFs promote tumor
growth, metastasis and drug resistance.
Studies have demonstrated that cells, including
resident tissue fibroblasts, bone marrow (BM)-derived mesenchymal
stromal cells (MSCs), epithelial cells, endothelial cells, CSCs,
hematopoietic stem cells (HSCs), vascular smooth muscle cells
(VSMCs) and pericytes may act as the predecessors of CAFs (22–30).
When healthy tissue is damaged and malignancy develops, immune
cells are recruited to the site of injury, and via the release of
specific mediators, activate the differentiation of resident
fibroblasts into CAFs (22). In this
manner, human breast fibroblasts gradually differentiate into CAFs,
promoting tumor progression by establishing TGF-β and
stromal-derived factor autocrine signals (23). TGF-β1 is the primary factor that
activates the conversion of resident fibroblasts into CAFs.
Moreover, hypoxia also promotes this process via the accumulation
of reactive oxygen species (ROS) and the activation of the
hypoxia-inducible factor (HIF)-1α-mediated signaling pathway
(24). In addition, VSMCs and
pericytes can differentiate into CAFs in breast cancer (25). BM-MSCs can also differentiate into
CAFs. For example, CAFs with the phenotype and functional
characteristics identical to those of BM-MSCs were isolated from
primary human neuroblastoma tumors (26). In colon tumors, CAFs are generated
via the activation of native mesenchymal cell populations and the
recruitment of BM-MSCs (27).
Furthermore, mouse-induced pluripotent stem cells were treated with
conditioned media to generate CSC-like cells, which are a
heterogeneous population surrounded by myofibroblast-like cells. At
the same time, the expression of fibroblast activation protein
(FAP), alpha-smooth muscle actin (α-SMA), and other key CAF markers
was significantly increased, and for the first time, CSCs were
confirmed to be the key origin of CAFs in the TME (28). Studies in two different pancreatic
cancer mouse models also revealed that endothelial-mesenchymal
transition transforms fibroblasts into CAFs after exposure to
TGF-H1 (29). Moreover,
fibroblast-specific protein-1+fibroblastscan be derived from
epithelial-mesenchymal transformation (EMT) in the local
environment (30). McDonald et
al (31) found that CAFs derived
from HSCs promoted the generation of tumor blood vessels. The
origins of different CAFs populations are listed in Table I.
The expression of CAF markers can be determined by
immunofluorescence and immunohistochemical staining, and
quantitatively detected by western blotting. As a heterogeneous
cell population, different markers can be used to identify CAFs,
the most common of which are podoplanin (PDPN), platelet-derived
growth factor receptor (PDGF-R), vimentin, α-SMA and FAP. However,
in isolation, none of these markers can specifically identify CAFs
(32). PDPN+ CAFs are able to
promote tumor formation (33). The
expression of PDPN was detected in the CAFs of 177 patients with
lung adenocarcinoma, and PDPN+ CAFs were found only in invasive
rather than non-invasive adenocarcinoma (34). The expression of PDPN promotes
platelet aggregation and contributes to cancer cell invasiveness
(34). Therefore, PDPN+ CAFs are
closely associated with the aggressiveness of various cancer types,
including lung adenocarcinoma (33,34),
breast cancer (35) and squamous
cell carcinoma (36). PDGF-Rs can be
categorized as PDGFR A and PDGFR B. PDGFR ligands include the PDGFs
(PDGF-aa, PDGF-bb, PDGF-ab, PDGF-cc and PDGF-dd), and their
expression is closely related to tumor occurrence and CAFs function
(37). Vimentin is involved in the
formation of cytoskeletal networks, especially in
mesenchymal-derived cells. Due to their strong mesenchymal
phenotype, vimentin is highly expressed in all types of
fibroblasts, and has therefore been widely used for the
identification of CAFs (38–40). Park et al (41) demonstrated that vimentin promotes
lung cancer invasion and metastasis by promoting the recruitment of
CAFs. CAFs are divided into two distinct clusters, namely C1-type
and C2-type CAFs. Notably, the expression of α-SMA is lower in
C1-type compared with the C2-type CAFs, though C1-type CAFs inhibit
the self-renewal of oral cancer cells by releasing bone
morphogenetic protein 4 (42). α-SMA
is expressed by various cell types, including fibroblasts, making
it impossible to use alone as a marker for CAF recognition. The
upregulation of FAP is associated with poor prognosis in >90% of
epithelial cancer types (43–45). Due
to its high expression level in the tumor stroma, FAP has been used
as a CAF identification marker in numerous studies (46). FAP+ CAFs control tumor progression by
secreting chemokine (C-X-C motif) ligand 12 (CXCL12) and binding to
its receptor chemokine (C-X-C motif) receptor 4 (CXCR4). FAP+CAFs
increase T cell recruitment and promote the antitumor effect by
mediating CXCL12/CXCR4 axis deletion in pancreatic ductal
adenocarcinoma (47,48). In addition, FAP-activated prodrugs
have been demonstrated as a feasible strategy for treating cancer,
such as prostate cancer and breast cancer (49). Thapsigargin (TG) is a highly toxic
natural plant product; a cytotoxic TG analog was coupled to
FAP-selective peptide substrates to create inactive prodrugs, and
when the prodrugs were activated, they led to apoptosis of prostate
and breast cancer cells, but had no obvious toxicity to host cells
(49). No single marker can mark all
the CAFs, and not all CAFs express all potential marker proteins.
Therefore, there is still a need to investigate CAFs-specific
markers. In addition to the common CAFs markers, there are other
less commonly used markers, such as microfibrillar-associated
protein 5 (MFAP5), collagen type XI alpha 1 chain, tenascin-C,
PDPN, integrin α11β1, neural/glial antigen, collagen 11-α1 and
asporin (40). However, collagen
11-α1, MFAP5 and asporin are expressed only by CAFs, which can
improve the specificity of their identification (50). Currently, a combination of markers,
as well as cellular phenotype, is the most reliable method for the
identification of CAFs.
Though CAFs lack specific markers, literature
reports that CAF markers may possess organs heterogeneity, and that
CAFs expressing the same marker may possess different functions in
different organs. For example, in ovarian cancer, PDGF-R+ CAFs
promote tumor progression by remodeling the ECM (51). However, CAFs expressing PDGF increase
the levels of the Puma in myofibroblasts, which subsequently
activates Bak, a pro-apoptotic protein that induce
cholangiocarcinoma cell apoptosis (52). Similarly, CAFs may express different
markers in different organs. PDGF-R is expressed by a variety of
different CAFs; however, those originating from BM-MSCs do not
express PDGF-Rα in breast tumors and lung metastases (53). At present, CD200 is only known to be
highly expressed in CAFs derived from NSCLC and can promote the
sensitivity of NSCLC to EGFR-TKIs (54). Su et al (8) demonstrated that CD10+/GPR77+CAFs
continually promote p65 phosphorylation and acetylation by binding
GPR77 receptor C5a, thereby promoting the self-renewal of tumor
stem cells and enhancing drug resistance in patients with lung and
breast cancer.
Heterogeneity between origins, markers and resident
organs determines the different functions of CAFs. Genetically
engineered mouse models and clinical studies have demonstrated that
at least two types of CAFs with different functions exist, namely
pro-cancer CAFs (pCAF) and anticancer CAFs (rCAF) (55). CAFs also exhibit different functions
in tumor progression. In NSCLC, the functional heterogeneity of
CAFs primarily results from differences in expression markers, and
there are few studies on the functional differences caused by the
heterogeneity of origins. CAFs that exert pro-tumor effects include
α-SMA+PDPN+, FAP+, CD34+ and CD10+/GPR77+CAFs. CAFs with antitumor
properties include CD200+ and CD99+CAFs. The pro/anti-tumor effect
of CAFs in NSCLC progression are summarized in Table II.
CAFs promote tumor growth and metastasis, as well as
tumor cell drug resistance. Tissue analysis revealed a high
mortality rate among 220 patients with NSCLC and high α-SMA
expression, indicating that α-SMA is associated with poor survival
time (56). Immunohistochemical
analysis of 304 patients with pTNM stage I–III NSCLC revealed that
CAF-associated CD34 expression was an independent prognostic factor
for stage I–III NSCLC, and that SMA+CAFs were associated with
higher tumor stages and promoted tumor progression (57). The 28 patients with NSCLC were
divided into two CAF subgroups, the high desmoplastic CAFs
(HD-CAFs) and low desmoplastic CAFs (LD-CAFs), according to the
obtained scores and classification based on desmoplasia. Compared
with LD-CAFs, HD-CAFs exhibited a higher collagen matrix remodeling
rate and promoted tumor invasion and growth (58). The immunohistochemical analysis of
tumor samples from 536 patients with NSCLC indicated that CAFs
expressing FAP-1 were associated with poor patient prognosis, which
has also been demonstrated in patients with pancreatic cancer. In
addition, FAP+CAFs have been associated with reduced survival time
(59,60). Yoshida et al (61) demonstrated that compared with the
control group, lung adenocarcinoma cells co-cultured with PDPN+
CAFs possessed greater drug resistance properties. In patients with
postoperative recurrence, compared with the PDPN-CAF group, the
PDPN+ group showed a lower treatment response to EGFR-TKIs. These
results suggest that PDPN+ CAFs are involved in primary drug
resistance to these compounds. Using the collagen invasion assay
model of co-culture between cancer cells and CAFs, CAFs were found
to invade local tissues through ECM remodeling, followed by
subsequent cancer cell invasion. Furthermore, the down regulation
of CAF-associated PDPN reduced the invasiveness of CAFs and cancer
cells (62). In addition,
CD10+/GPR77+CAFs maintain the stemness of CSCs and promote drug
resistance in patients with lung cancer (8). Therefore, it is undeniable that
heterogeneity of origin is an important influencing factor of CAF
function, which is a valuable future research direction.
In addition to their tumor-promoting functions, CAFs
also possess antitumor properties. For example, CD200+ CAFs
enhanced the sensitivity of lung cancer to gefitinib. Moreover,
individuals with CD200+ CAFs exhibit longer progression-free
survival after gefitinib treatment following post-surgical relapse.
The binding of CD200 to its receptor CD200R1, which is expressed by
immune cells, triggers an immunosuppressive response, leading to an
antitumor effect (54). In addition,
CD99 is a newly discovered CAF marker, the overexpression of which
may inhibit tumor progression (63).
However, the tumor-suppressive mechanism of CAFs remains unclear,
and requires further investigation.
CAFs influence tumor formation by promoting drug
resistance, though the associated underlying mechanisms remain
unclear. Clarifying these mechanisms may help to prevent the
occurrence of drug resistance in NSCLC. Next, the mechanisms by
which CAFs mediate the resistance of NSCLC to chemotherapy,
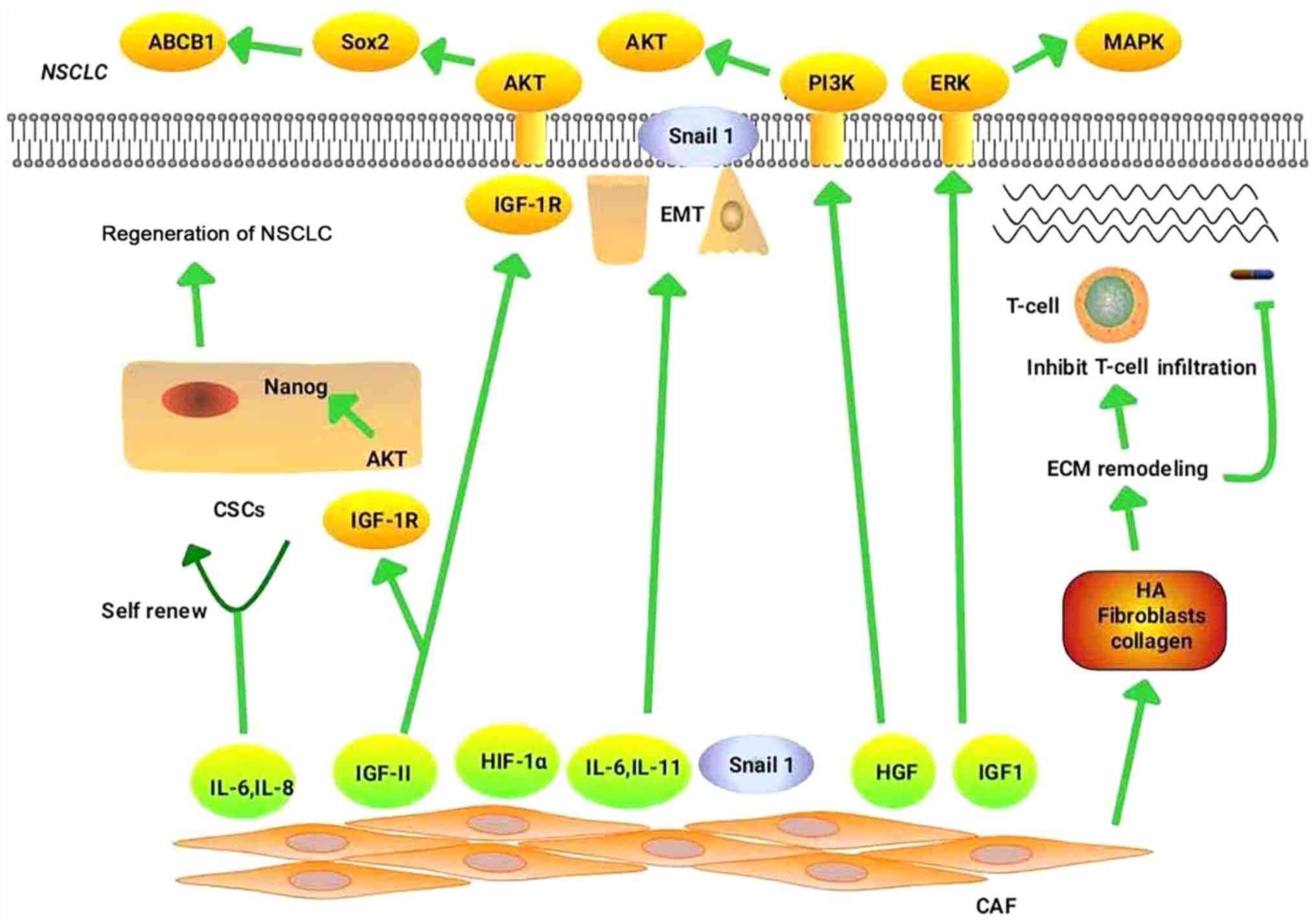
targeted therapy and immunotherapy, are explored. Fig. 1 illustrates the mechanisms by which
CAFs regulate drug resistance in NSCLC.
CAFs promote the resistance of NSCLC to chemotherapy
primarily by mediating EMT (19),
remodeling the ECM (7,11), maintaining the stemness of CSCs
(8) and promoting metabolic
reprogramming (64–66).
EMT is an important developmental process that is
closely associated with drug resistance (67,68).
During EMT, epithelial cell markers, such as N-and E-cadherin, are
downregulated, while mesenchymal cell markers, such as vimentin and
fibronectin, are upregulated. Previous studies have reported that
EMT is associated with drug resistance in pancreatic (69), bladder (70), breast (71) and colorectal cancer (72). Compared with the control group, the
expression of E-cadherin in the indirect CAF co-culture group was
reduced, the expression of vimentin was enhanced, and migration and
invasion ability were correspondingly augmented (20). Therefore, CAFs are among the factors
mediating EMT. Studies have shown that CAFs regulate EMT and
promote drug resistance by secreting IL-6 and hepatocyte growth
factor (HGF) (73). For example,
CAFs significantly increased TGF-β1-induced EMT in cancer cells by
secreting IL-6, thereby contributing to cisplatin resistance in
NSCLC (74). This process involves
the expression of TGF-β1, and silencing TGF-β1 reverses EMT and
thus increases the sensitivity of NSCLC to cisplatin (74). HGF, also known as scatter factor, is
a member of the fibrinogen family that which functions to activate
EMT (75). Ying et al
(19) investigated the functions of
HGF in the paclitaxel resistance of NSCLC by constructing a
three-dimensional microfluidic chip. The high levels of HGF
secreted by CAFs enhanced the phosphatidylinositol 3 kinase/protein
kinase B (PI3K/AKT) activation, as well as the expression of GRP78,
and promoted the resistance of NSCLC to paclitaxel. CAFs were also
found to induce EMT in NSCLC cells, inducing resistance to
chemotherapy. Therefore, targeting CAFs may enhance the therapeutic
effect of drugs towards NSCLC.
Studies have reported the presence of white blood
cells with stem cell-like properties in patients with acute myeloid
leukemia, which are designated as CSCs (76,77).
CSCs are a subgroup of tumor cells that exhibit strong resistance
to chemotherapy (77,78), of which there are two primary
underlying mechanisms. The first outlines that in a hypoxic
microenvironment, CSCs remain quiescent in a non-permanent dormant
state, and that chemotherapeutic drugs primarily target rapidly
dividing cancer cells, allowing quiescent stem cells to survive and
regenerate to form tumors at a later point in time (79). Another mechanism is the use of
ATP-binding box (ABC) transporters to expel chemotherapeutic drugs,
resulting in drug resistance (80).
CAFs primarily promote NSCLC drug resistance by maintaining the
stemness of CSCs, and stimulating their self-renewal. When CAFs are
co-cultured with CSCs, CAF-associated insulin-like growth factor-II
(IGF-II) activates the insulin-like growth factor 1 receptor
(IGF1R) on CSCs, thereby activating the IGF-II/IGF1R/Nanog
signaling pathway to maintain CSCs stemness, both in vivo
and in vitro. In turn, CSCs promote CAF-associated IGF-II
secretion via cytokines such as basic fibroblast growth factor. The
IGF-II/IGF1R axis promotes the expression of Nanog in cancer cells,
and blocking the IGF-II/IGF1R/Nanog pathway reduces the stemness of
CSCs (12). CAFs exhibit high CD44
expression in tumor hypoxic and avascular areas, and CAF CD44
expression is significantly increased following treatment with an
angiogenesis inhibitor. Through co-cultures and tumor sphere
formation assays, CAFs were found to maintain the stemness of CSCs
and enhance the resistance of tumor cells to anticancer drugs,
properties that were not exhibited by CD44-deficient CAFs (81). In addition, CD10+/GPR77+CAFs can
maintain the stemness of CSCs by secreting IL-6 and IL-8, thereby
promoting drug resistance in patients with lung cancer (8). According to these studies, CAFs can
promote the chemotherapeutic drug resistance of NSCLC by regulating
CSCs.
Under normal physiological conditions, the ECM
supports the proliferation and migration of surrounding cells.
Cancer tissue is generally stiffer than normal tissue. The
stiffness of the ECM is primarily attributed to the accumulation of
hyaluronic acid (HA) at its core, which can withstand the
compressive stress of the tumor, while the accumulation of collagen
and fibronectin in the periphery promotes resistance to tensile
stress. ECM stiffness functions as a barrier to tumor cell drug
absorption (82). CAFs promote tumor
resistance by increasing matrix stiffness through the enhancement
of ECM components such as HA and collagen. Collagen is resistant to
tensile stress, as it becomes harder on stretching (11). HA is also resistant to stress.
Integrin α11β1 is a specific collagen receptor associated with
increased collagen stiffness. CAFs can affect the stiffness of
interstitial collagen by expressing high levels of integrin α11,
which promotes the progression of NSCLC tumors (7). NSCLC cells cultured on a semi-solid
growth substrate (to simulate the stiffness of the matrix in the
TME) can promote the resistance of NSCLC to chemotherapeutic drugs
(83). In lung adenocarcinoma, PDPN+
CAFs physically remodel the ECM (62). Therefore, CAFs can promote NSCLC
resistance to chemotherapy via ECM remodeling.
Due to mitochondrial defects, cancer cell metabolism
is altered, the ability to oxidize glucose to CO2 is
inhibited, and the propensity to convert glucose into lactic acid
increases. These phenomena are collectively known as the Warburg
effect (64,84), which is mediated by pyruvate kinase
M2 (PKM2). PKM2 is upregulated in NSCLC cell lines and can promote
NSCLC resistance to cisplatin (65).
Under hypoxic conditions, cisplatin-resistant cells secrete
exosomes containing high concentrations of PKM2, which are absorbed
by cisplatin-sensitive cells. Exosomal PKM2 also regulates
glycolysis in treatment-sensitive cells, promoting cell survival
and inhibiting apoptosis. Secondly, in the tumor microenvironment,
exosomes secreted by the cisplatin-resistant cells deliver PKM2 to
CAFs, and the metabolically reprogrammed CAFs release pyruvate and
lactate, promoting chemotherapeutic resistance (66). In addition, CAF autophagy releases
lactic acid, ketone bodies and glutamine to create a nutrient-rich
microenvironment that supports tumor growth (64).
EMT is a reversible process regulated by several
EMT-related transcription factors (EMT-TFs), such as Snail, Slug,
Twist and zinc finger E-box-binding homeobox 1 (ZEB1). EMT enhances
the migration and invasiveness, as well as the resistance of cancer
cells to targeted therapy (85). For
example, the A549 lung cancer cell line developed drug resistance
after long-term treatment with gefitinib. These gefitinib-resistant
cells showed reduced expression of E-cadherin and vimentin,
indicating the occurrence of EMT (86). Moreover, the expression of the EMT
regulator ZEB1 was increased in the HCC4006ER erlotinib-resistant
cell line. HCC4006ER cells acquired an EMT phenotype and were able
to activate the TGF-β1/SMAD pathway (87). Snail, a key transcription factor for
EMT, is closely associated with chemotherapy resistance. CAFs
deliver Snail to lung cancer cells through exosomes, which induce
EMT in these cells and promote drug resistance (88). However, whether CAFs can also promote
NSCLC drug resistance by regulating other EMT-TFs remains to be
elucidated. EMT is associated with NSCLC-targeted drug resistance,
which is primarily achieved through extracellular signal-regulated
kinases/mitogen-activated protein kinase (ERK/MAPK), Hedgehog (Hh)
and other related signaling pathways. CAFs can activate
corresponding receptors in NSCLC through the upregulation of growth
factors such as HGF and IGF-1, and also regulate EMT and gefitinib
resistance in a paracrine manner (89). IGF1R induces EMT in NSCLC cells and
increases their resistance to EGFR-TKIs by enhancing the ERK/MAPK
signaling pathway, small interfering (si)RNAs targeting IGF1R
reversed the EMT phenotype and resistance to EGFR-TKIs (90). Choe et al (91) reported that co-culturing CAFs with
NSCLC stimulated CAFs to induce EMT by activating the Hh signaling
pathway, making PC9 cells resistant to erlotinib. A combination of
the cell surface molecules Patched and Smoothened with the ligands
sonic hedgehog, Indian hedgehog and desert hedgehog activates the
transcription factor GLI1, thereby activating the Hh pathway and
mediating tumor cell resistance to EGFR-TKIs by inducing EMT
(92). In summary, CAFs can promote
NSCLC resistance to targeted therapy by regulating EMT-TFs, and by
activating multiple pathways, which also indicates that CAFs play
an important role in the resistance of NSCLC to targeted
therapy.
Hypoxia is a hallmark feature of the TME, and is
considered to be one of the key factors for drug resistance in
tumors (97). Cancer cells are often
in a state of hypoxia that promotes tumor growth (98). In rapidly growing tumors, the
distance between cells and blood vessels increases, which in turn
impedes drug absorption into the tumor, especially in a hypoxic
environment (99). For example,
EGFR-mutated NSCLC cell lines exposed to high concentrations of
gefitinib under low oxygen conditions acquired drug-resistant
cells, known as gefitinib-resistant persistent cells (GRPs).
Moreover, stem cell-associated genes are highly expressed in GRPs.
This process is primarily mediated by an upregulation in IGF1
expression by HIF1, which in turn activates IGF1R on GRPs, thereby
promoting NSCLC resistance to gefitinib and increasing CSCs numbers
(93). The expression level of
HIF-1α is upregulated in CAFs (94),
indicating that these cells may promote NSCLC drug resistance by
regulating the hypoxic microenvironment. In addition, HIF-1 can
promote NSCLC drug resistance by inducing the expression of ABC
transporters (99). EBC-1R is an
NSCLC cell line resistant to the EMT inhibitors PHA-665752 and
crizotinib, which possesses the characteristics of CSCs, and forms
spheres (95) in which the
expression of ATP-binding cassette sub-family B member 1 (ABCB1) is
upregulated. Drug resistance is reversed following treatment with
the ABCB1 inhibitor elacridar (95).
IGF-II is an insulin-like hormone that plays an important role in
regulating cellular proliferation, differentiation, senescence and
drug resistance. CAFs regulate NSCLC cell drug resistance by
secreting IGF-II and binding to the membrane receptor IGF-1R, in
addition to activating the IGF-II/IGF-1R/AKT/Sox2/ABCB1 pathway in
cancer cells, which in turn upregulates the expression of
P-glycoprotein (96).
Over the past few years, immune checkpoint
inhibitors have played an important role in clinical trials, and
have been approved as the standard therapy for advanced NSCLC
(100). For instance, nivolumab and
pembrolizumab targeting programmed cell death protein 1 (PD-1),
atezolizumab targeting programmed cell death ligand 1 (PD-L1), and
tremelimumab targeting cytotoxic T-lymphocyte antigen 4 have been
approved by the United States Food and Drug Administration for
NSCLC treatment (101). However,
only 15–25% of patients with NSCLC respond to immune checkpoint
inhibitors, the majority of which experience primary drug
resistance (102). At present, only
a limited number of studies have reported CAF-mediated NSCLC
resistance to immune checkpoint inhibitors, although this has also
been reported for other tumor species. In NSCLC, CAFs primarily
prevent the infiltration and migration of immune cells by
remodeling the ECM and preventing the binding of immune checkpoint
inhibitors to their receptors, thus prompting immune escape. The
density and direction of the ECM influence the behavior and
migration of T cells in human lung cancer. T cells generally
accumulate in areas with loose stromal fibers (103), and a dense ECM serves as a contact
barrier between T cells and the tumor cells (104). It also prevents T cells from
binding PD-1 inhibitors, and thus promotes the resistance of tumor
cells to immune checkpoint inhibitors (103). The ECM includes collagen, laminin
and fibronectin. Lysyl oxidase crosslinks collagen molecules into
fibers to form a dense ECM, which inhibits the migration of T cells
and reduces the effect of PD-1 inhibitors (104). In a xenograft model of NSCLC, CAFs
overexpressing lysyl oxidase like-1 were found to remodel the
collagen matrix in vivo (105), suggesting that CAFs promote NSCLC
resistance to immunotherapeutic drugs through the ECM. CAFs are the
primary producers of TGF-β (106)
and can influence T cell infiltration via TGF-β. TGF-β signaling
was demonstrated to inhibit T cell infiltration in breast mouse
tumor models (104). CAFs
specifically inhibit CD8+ T cell infiltration, thereby promoting
tumor resistance to different immunosuppressive agents (107). In addition, CAFs can function as
antigen-presenting cells and induce T-cell death in an
antigen-dependent manner via PD-L2 and FASL (108). Compared with patients with
PD-L1-CAFs, those with PD-L1+ CAFs exhibited significantly
prolonged relapse-free survival, and the expression of PD-L1 in
CAFS was affected by IFN-γ (109).
At present, literature reporting the correlation between CAFs and
immune checkpoint inhibitors is limited. The correlation between
CAF-associated surface markers and immune markers was studied in
536 patients with NSCLC, and the results indicated that CAFs had
little effect on immune cell infiltration in NSCLC (110). Therefore, whether CAFs also promote
the drug resistance of tumor cells by inhibiting T cell
infiltration requires investigated further.
There are currently two available strategies for
reversing drug resistance by targeting CAFs, one of which is to
inhibit the production of CAFs, while the other is to block the
pathways downstream of them.
Fibroblast to CAFs transformation relies on the
expression of TGF-β. Treatment with TGF-β rapidly activates the
TGF-β signaling pathway, resulting in the transformation of
fibroblasts into myofibroblast phenotype. Myofibroblast
transdifferentiation requires the production of ROS, and the
expression of NAD(P)H Oxidase-4 (NOX4) is associated with the CAF
marker α-SMA. A NOX4 inhibitor (GKT137831) or targeted
NOX4-knockout (short hairpin RNA and siRNA) reduced the
accumulation of CAFs. Therefore, CAF generation can be inhibited by
decreasing NOX4 expression, which may reduce the occurrence of
NSCLC drug resistance (111).
Pirfenidone is a pyridine compound that inhibits fibroblast
proliferation and CAF differentiation and activation (112). FAP is expressed by the majority of
CAFs, and T cells can be genetically modified to express
FAP-specific chimeric antigen receptors. These FAP-specific T cells
recognize and destroy FAP+CAFs with subsequent antitumor effects
(113). Some CAFs possess
myofibroblast characteristics and express α-SMA, which can
significantly promote NSCLC resistance to chemotherapy via the
expression of high levels of inflammatory cytokines and chemokines
(114). Plasminogen activator
inhibitor-1 (PAI-1) can promote the MF characteristics of CAFs, and
the expression of PAI-1 in CAFs is correlated with the expression
of α-SMA (114). PAI-1 inhibitors
also decrease the expression levels of α-SMA and inhibit the MF
characteristics of CAFs, improving chemotherapeutic efficacy in
NSCLC (114). Thus inhibiting the
MF properties of CAFs may be a novel therapeutic strategy for the
treatment of chemotherapy-resistant NSCLC (114).
Currently, the primary methods of reversing NSCLC
drug resistance are via the inhibition CAF downstream pathways.
According to literature, resistance can be reversed by targeted
inhibition of the cytokines secreted by CAFs, such as IL-6
(115), IGFII (96), HGF (2), Annexin A3 (ANXA3) (116) and γ-glutamyl transferase 5 (GGT5)
(117). CAFs express IL-6 to
upregulate Bcl-2 and Mcl-1, reduce the sensitivity of NSCLC to
cisplatin, and protect NSCLC cells from apoptosis (115). A combination of IL-6-targeted
inhibitors and cisplatin can either reduce or inhibit the cisplatin
resistance in NSCLC (115). CAFs
regulate NSCLC cell drug resistance through the secretion of IGF2
and by binding IGF-1R, activating the AKT/Sox2/P-GP pathway in
cancer cells. Traditional chemotherapeutic regimes, combined with
IGF2-targeted inhibitors, may serve as an innovative therapeutic
strategy for NSCLC (96). CAFs
activate ERK by secreting HGF, which contributes to the resistance
of NSCLC cells to EGFR-TKIs, and combination therapy with
HGF-targeted drugs restores the sensitivity of cancer cells to
EGFR-TKIs (2). In addition, the
expression of ANXA3 is higher in CAFs than in normal fibroblasts
(NFs). Furthermore, the overexpression of ANXA3 increased the
cisplatin resistance of lung cancer cells. The underlying mechanism
was that CAFs enhanced chemotherapeutic resistance by activating
the ANXA3/JNK signaling pathway to inhibit cisplatin-induced
apoptosis. The resistance of cancer cells to cisplatin can also be
decelerated using JNK-targeting inhibitors (116). GGT5 is a member of the γ-glutamyl
transpeptidase family that is abundantly expressed by CAFs,
promoting NSCLC resistance to paclitaxel and cisplatin. NSCLC
regains its sensitivity to chemotherapy drugs when GGT5 is blocked
(117). Furthermore, CAFs secrete
IL-11, which activates the IL-11R/STAT3 anti-apoptotic signaling
pathway by binding to IL-11R, thereby promoting the
chemotherapeutic resistance of NSCLC. STAT3 inhibitors can obstruct
this process and reverse drug resistance (118). With further understanding of the
roles of CAFs in NSCLC drug resistance, targeted inhibition of CAFs
and their secreted cytokines can serve as suitable candidates for
the treatment of drug resistance.
CAFs secrete several types of cytokines, such as
Snail and IL-6, to remodel the EMT (73,88). The
secretion of IL-6 by CAFs induces EMT and promotes cisplatin
resistance in NSCLC cells (73).
Co-culturing of NSCLC with CAFs results in the secretion of IL-6
and oncostatin-M (OSM) from CAFs, which in turn activates STAT3.
The activated OSM receptors (OSMR)/JAK1/STAT3 pathway contributes
to NSCLC cell resistance to chemotherapy drugs. However, this
process can be effectively blocked by the JAK1 inhibitor filgotinib
(119). In addition, CAFs deliver
Snai1 to cancer cells through exosomes to induce cancer cell EMT.
However, CAFs can also inhibit EMT when treated with the exosome
release inhibitor GW4869, restoring NSCLC drug sensitivity
(88). CAFs also promote EMT by
secreting TGF-β, indicating that the ability of the TEM to support
tumor cells can be reduced by inhibiting TGF-β (120).
According to the aforementioned findings, T cell
migration, and the efficacy of anti-PD-1 blockers, can be improved
by reducing ECM content and matrix stiffness, which can improve the
sensitivity of NSCLC cells to chemotherapy and immunotherapy.
Matrix metalloproteinases (MMPs), ERK1/2, JNK, and HIF-1 have been
proven to promote ECM degradation (82). However, CAFs primarily degrade the
ECM by secreting MMPs, and CAFs co-cultured with NSCLC cells
promote the expression of MMP1 and MMP9 (121), effectively reversing drug
resistance.
CAFs can facilitate CSC-induced drug resistance in
NSCLC in various ways. Therefore, targeting CSCs can improve the
therapeutic effect on tumors. For example, CD10+/GPR77+CAFs can
promote the self-renewal of CSCs and enhance drug resistance in
patients with lung cancer. According to these findings, GPR77
monoclonal antibody therapy may destroy the ecological niche of
CSCs, thus retarding the formation of tumors and reversing
chemotherapeutic resistance (8). In
addition, as aforementioned, CAFs maintain the stemness of CSCs and
promote NSCLC drug resistance through the IGF-II/IGF1R/Akt/Nanog
signaling pathway. However, the inhibition of this pathway reverses
drug resistance to NSCLC (12).
CAFs can promote NSCLC drug resistance by inducing
EMT, increasing CSC stiffness, remodeling the ECM, and creating a
hypoxic microenvironment. These functions are crucial for the role
of CAFs in NSCLC drug resistance. The heterogeneity of CAFs is an
important factor in the failure of cancer treatment. The lack of
reliable markers to identify CAF cell populations has further
hindered our understanding of the relationship between CAFs and
therapeutic resistance. Therefore, identifying CAF-specific surface
markers is key for the future direction of this research field. Due
to the heterogeneity of CAFs, targeted inhibitors have yet to be
discovered. However drug resistance can be reversed by reducing the
accumulation of CAFs, as well as targeted inhibition of their
downstream pathways. In addition, the drug sensitivity of NSCLC can
be restored by inhibiting EMT, degrading the ECM and destroying the
ecological niche of CSCs.
Not applicable.
No funding was received.
Not applicable.
CC was involved in conceptualization, collection and
review of the literature, interpretation, drafting the manuscript,
writing and critical revision. JH and SY were involved in
critically revising the manuscript for important intellectual
content. WL, XW, HS, TQ and FXC were involved in drafting the
manuscript or revising it critically for important intellectual
content. HG and ZL were involved in conceptualization, figure
preparation, writing and critical revision, resource provision and
supervision. All authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rotow J and Bivona TG: Understanding and
targeting resistance mechanisms in NSCLC. Nat Rev Cancer.
17:637–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arbour KC and Riely GJ: Systemic therapy
for locally advanced and metastatic non-small cell lung cancer: A
review. JAMA. 322:764–774. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holohan C, Schaeybroeck SV, Longley DB and
Johnston PG: Cancer drug resistance: An evolving paradigm. Nat Rev
Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salem A, Asselin MC, Reymen B, Jackson A,
Lambin P, West CM, OConnor JP and Faivre-Finn C: Targeting hypoxia
to improve non-small cell lung cancer outcome. J Natl Cancer Inst.
110:2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fischer C, Leithner K, Wohlkoenig C,
Quehenberger F, Bertsch A, Olschewski A, Olschewski H and Hrzenjak
A: Panobinostat reduces hypoxia-induced cisplatin resistance of
non-small cell lung carcinoma cells via HIF-1α destabilization. Mol
Cancer. 14:42015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Navab R, Strumpf D, To C, Pasko E, Kim KS,
Park CJ, Hai J, Liu J, Jonkman J, Barczyk M, et al: Integrin α11β1
regulates cancer stromal stiffness and promotes tumorigenicity and
metastasis in non-small cell lung cancer. Oncogene. 35:1899–1908.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su S, Chen J, Yao H, Liu J, Yu S, Lao L,
Wang M, Luo M, Xing Y, Chen F, et al:
CD10+GPR77+ cancer-associated fibroblasts
promote cancer formation and chemoresistance by sustaining cancer
stemness. Cell. 172:841–856.e16. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang W, Li Q, Yamada T, Matsumoto K,
Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S and
Yano S: Crosstalk to stromal fibroblasts induces resistance of lung
cancer to epidermal growth factor receptor tyrosine kinase
inhibitors. Clin Cancer Res. 15:6630–6638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Radisky DC: Fibroblasts act as
co-conspirators for chemotherapy resistance. Cancer Biol Ther.
7:1348–1349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii G, Ochiai A and Neri S: Phenotypic
and functional heterogeneity of cancer-associated fibroblast within
the tumor microenvironment. Adv Drug Deliv Rev. 99:186–196. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA,
Ling TY, Yu SL, Yuan SS, Chen YJ, Lin CY, et al: Cancer-associated
fibroblasts regulate the plasticity of lung cancer stemness via
paracrine signalling. Nat Commun. 5:34722014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leung CS, Yeung TL, Yip KP, Wong KK, Ho
SY, Mangala LS, Sood AK, Lopez-Berestein G, Sheng J, Wong ST, et
al: Cancer-associated fibroblasts regulate endothelial adhesion
protein LPP to promote ovarian cancer chemoresistance. J Clin
Invest. 128:589–606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
New J, Arnold L, Ananth M, Alvi S,
Thornton M, Werner L, Tawfik O, Dai H, Shnayder Y, Kakarala K, et
al: Secretory autophagy in cancer-associated fibroblasts promotes
head and neck cancer progression and offers a novel therapeutic
target. Cancer Res. 77:6679–6691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Allaoui R, Bergenfelz C, Mohlin S,
Hagerling C, Salari K, Werb Z, Anderson RL, Ethier SP, Jirström K,
Påhlman S, et al: Cancer-associated fibroblast-secreted CXCL16
attracts monocytes to promote stroma activation in triple-negative
breast cancers. Nat Commun. 7:130502016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun Y, Wang R, Qiao M, Xu Y, Guan W and
Wang L: Cancer associated fibroblasts tailored tumor
microenvironment of therapy resistance in gastrointestinal cancers.
J Cell Physiol. 233:6359–6369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
von Ahrens D, Bhagat TD, Nagrath D, Maitra
A and Verma A: The role of stromal cancer-associated fibroblasts in
pancreatic cancer. J Hematol Oncol. 10:762017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen
S, Wang Y, Wang T and Hou Y: Carcinoma-associated fibroblasts
promote the stemness and chemoresistance of colorectal cancer by
transferring exosomal lncRNA H19. Theranostics. 8:3932–3948. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ying L, Zhu Z, Xu Z, He T, Li E, Guo Z,
Liu F, Jiang C and Wang Q: Cancer associated fibroblast-derived
hepatocyte growth factor inhibits the paclitaxel-induced apoptosis
of lung cancer A549 cells by up-regulating the PI3K/Akt and GRP78
signaling on a microfluidic platform. PLoS One. 10:e01295932015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shan T, Chen S, Chen X, Lin WR, Li W, Ma
J, Wu T, Ji H, Li Y, Cui X and Kang Y: Prometastatic mechanisms of
CAF-mediated EMT regulation in pancreatic cancer cells. Int J
Oncol. 50:121–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lai D, Ma L and Wang F: Fibroblast
activation protein regulates tumor-associated fibroblasts and
epithelial ovarian cancer cells. Int J Oncol. 41:541–550. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Foster DS, Jones RE, Ransom RC, Longaker
MT and Norton JA: The evolving relationship of wound healing and
tumor stroma. JCI Insight. 3:e999112018. View Article : Google Scholar
|
|
23
|
Kojima Y, Acar A, Eaton EN, Mellody KT,
Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg
RA and Orimo A: Autocrine TGF-beta and stromal cell-derived
factor-1 (SDF-1) signaling drives the evolution of tumor-promoting
mammary stromal myofibroblasts. Proc Natl Acad Sci USA.
107:20009–20014. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Louault K, Li R and DeClerck YA:
Cancer-associated fibroblasts: Understanding their heterogeneity.
Cancers (Basel). 12:31082020. View Article : Google Scholar
|
|
25
|
An Y, Liu F, Chen Y and Yang Q: Crosstalk
between cancer-associated fibroblasts and immune cells in cancer. J
Cell Mol Med. 24:13–24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Borriello L, Nakata R, Sheard MA,
Fernandez GE, Sposto R, Malvar J, Blavier L, Shimada H, Asgharzadeh
S, Seeger RC and DeClerck YA: Cancer-associated fibroblasts share
characteristics and protumorigenic activity with mesenchymal
stromal cells. Cancer Res. 77:5142–5157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koliaraki V, Pallangyo CK, Greten FR and
Kollias G: Mesenchymal cells in colon cancer. Gastroenterology.
152:964–979. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nair N, Calle AS, Zahra MH, Prieto-Vila M,
Oo AKK, Hurley L, Vaidyanath A, Seno A, Masuda J, Iwasaki Y, et al:
A cancer stem cell model as the point of origin of
cancer-associated fibroblasts in tumor microenvironment. Sci Rep.
7:68382017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeisberg EM, Potenta S, Xie L, Zeisberg M
and Kalluri R: Discovery of endothelial to mesenchymal transition
as a source for carcinoma-associated fibroblasts. Cancer Res.
67:10123–10128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iwano M, Plieth D, Danoff TM, Xue C, Okada
H and Neilson EG: Evidence that fibroblasts derive from epithelium
during tissue fibrosis. J Clin Invest. 110:341–350. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McDonald LT, Russell DL, Kelly RR, Xiong
Y, Motamarry A, Patel RK, Jones JA, Watson PM, Turner DP, Watson
DK, et al: Hematopoietic stem cell-derived cancer-associated
fibroblasts are novel contributors to the pro-tumorigenic
microenvironment. Neoplasia. 17:434–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gascard P and Tlsty TD:
Carcinoma-associated fibroblasts: Orchestrating the composition of
malignancy. Genes Dev. 30:1002–1019. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hoshino A, Ishii G, Ito T, Aoyagi K,
Ohtaki Y, Nagai K, Sasaki H and Ochiai A: Podoplanin-positive
fibroblasts enhance lung adenocarcinoma tumor formation: Podoplanin
in fibroblast functions for tumor progression. Cancer Res.
71:4769–4779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawase A, Ishii G, Nagai K, Ito T, Nagano
T, Murata Y, Hishida T, Nishimura M, Yoshida J, Suzuki K and Ochiai
A: Podoplanin expression by cancer associated fibroblasts predicts
poor prognosis of lung adenocarcinoma. Int J Cancer. 123:1053–1059.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schoppmann SF, Berghoff A, Dinhof C,
Jakesz R, Gnant M, Dubsky P, Jesch B, Heinzl H and Birner P:
Podoplanin-expressing cancer-associated fibroblasts are associated
with poor prognosis in invasive breast cancer. Breast Cancer Res
Treat. 134:237–244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ono S, Ishii G, Nagai K, Takuwa T, Yoshida
J, Nishimura M, Hishida T, Aokage K, Fujii S, Ikeda N, Ochiai A, et
al: Podoplanin-positive cancer-associated fibroblasts could have
prognostic value independent of cancer cell phenotype in stage I
lung squamous cell carcinoma: Usefulness of combining analysis of
both cancer cell phenotype and cancer-associated fibroblast
phenotype. Chest. 143:963–970. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Heldin CH: Targeting the PDGF signaling
pathway in tumor treatment. Cell Commun Signal. 11:972013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hsia LT, Ashley N, Ouaret D, Wang LM,
Wilding J and Bodmer WF: Myofibroblasts are distinguished from
activated skin fibroblasts by the expression of AOC3 and other
associated markers. Proc Natl Acad Sci USA. 113:E2162–E2171. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Herrera M, Islam AB, Herrera A, Martín P,
García V, Silva J, Garcia JM, Salas C, Casal I, de Herreros AG, et
al: Functional heterogeneity of cancer-associated fibroblasts from
human colon tumors shows specific prognostic gene expression
signature. Clin Cancer Res. 19:5914–5926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nurmik M, Ullmann P, Rodriguez F, Haan S
and Letellier E: In search of definitions: Cancer-associated
fibroblasts and their markers. Int J Cancer. 146:895–905. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Park SY, Kim HM and Koo JS: Differential
expression of cancer-associated fibroblast-related proteins
according to molecular subtype and stromal histology in breast
cancer. Breast Cancer Res Treat. 149:727–741. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Patel AK, Vipparthi K, Thatikonda V, Arun
I, Bhattacharjee S, Sharan R, Arun P and Singh S: A subtype of
cancer-associated fibroblasts with lower expression of alpha-smooth
muscle actin suppresses stemness through BMP4 in oral carcinoma.
Oncogenesis. 7:782018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Öhlund D, Elyada E and Tuveson D:
Fibroblast heterogeneity in the cancer wound. J Cell Biol.
211:1503–1523. 2014.
|
|
44
|
Brennen WN, Isaacs JT and Denmeade SR:
Rationale behind targeting fibroblast activation protein-expressing
carcinoma-associated fibroblasts as a novel chemotherapeutic
strategy. Mol Cancer Ther. 11:257–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huber MA, Kraut N, Park JE, Schubert RD,
Rettig WJ, Peter RU and Garin-Chesa P: Fibroblast activation
protein: Differential expression and serine protease activity in
reactive stromal fibroblasts of melanocytic skin tumors. J Investig
Dermatol. 120:182–188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Berdiel-Acer M, Sanz-Pamplona R, Calon A,
Cuadras D, Berenguer A, Sanjuan X, Paules MJ, Salazar R, Moreno V,
Batlle E, et al: Differences between CAFs and their paired NCF from
adjacent colonic mucosa reveal functional heterogeneity of CAFs,
providing prognostic information. Mol Oncol. 8:1290–1305. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fearon DT: The carcinoma-associated
fibroblast expressing fibroblast activation protein and escape from
immune surveillance. Cancer Immunol Res. 2:187–193. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Feig C, Jones JO, Kraman M, Wells RJ,
Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL,
et al: Targeting CXCL12 from FAP-expressing carcinoma-associated
fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic
cancer. Proc Natl Acad Sci USA. 110:20212–20217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brennen WN, Rosen DM, Wang H, Isaacs JT
and Denmeade SR: Targeting carcinoma-associated fibroblasts within
the tumor stroma with a fibroblast activation protein-activated
prodrug. J Natl Cancer Inst. 104:1320–1334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yoshida GJ: Regulation of heterogeneous
cancer-associated fibroblasts: The molecular pathology of activated
signaling pathways. J Exp Clin Cancer Res. 39:1122020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Raghavan S, Snyder CS, Wang A, McLean K,
Zamarin D, Buckanovich RJ and Mehta G: Carcinoma-associated
mesenchymal stem cells promote chemoresistance in ovarian cancer
stem cells via PDGF signaling. Cancers (Basel). 12:20632020.
View Article : Google Scholar
|
|
52
|
Rizvi S, Mertens JC, Bronk SF, Hirsova P,
Dai H, Roberts LR, Kaufmann SH and Gores GJ: Platelet-derived
growth factor primes cancer-associated fibroblasts for apoptosis. J
Biol Chem. 289:22835–22849. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Raz Y, Cohen N, Shani O, Bell RE,
Novitskiy SV, Abramovitz L, Levy C, Milyavsky M, Leider-Trejo L,
Moses HL, et al: Bone marrow-derived fibroblasts are a functionally
distinct stromal cell population in breast cancer. J Exp Med.
215:3075–3093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishibashi M, Neri S, Hashimoto H,
Miyashita T, Yoshida T, Nakamura Y, Udagawa H, Kirita K, Matsumoto
S, Umemura S, et al: CD200-positive cancer associated fibroblasts
augment the sensitivity of epidermal growth factor receptor
mutation-positive lung adenocarcinomas to EGFR tyrosine kinase
inhibitors. Sci Rep. 7:466622017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mizutani Y, Kobayashi H, Iida T, Asai N,
Masamune A, Hara A, Esaki N, Ushida K, Mii S, Shiraki Y, et al:
Meflin-positive cancer-associated fibroblasts inhibit pancreatic
carcinogenesis. Cancer Res. 79:5367–5381. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Alcaraz J, Carrasco JL, Millares L, Luis
IC, Fernández-Porras FJ, Martínez-Romero A, Diaz-Valdivia N, De Cos
JS, Rami-Porta R, Seijo L, et al: Stromal markers of activated
tumor associated fibroblasts predict poor survival and are
associated with necrosis in non-small cell lung cancer. Lung
Cancer. 135:151–160. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schulze AB, Schmidt LH, Heitkötter B, Huss
S, Mohr M, Marra A, Hillejan L, Görlich D, Barth PJ, Rehkämper J
and Evers G: Prognostic impact of CD34 and SMA in cancer-associated
fibroblasts in stage I–III NSCLC. Thorac Cancer. 11:120–129. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hao J, Zeltz C, Pintilie M, Li Q,
Sakashita S, Wang T, Cabanero M, Martins-Filho SN, Wang DY, Pasko
E, et al: Characterization of distinct populations of
carcinoma-associated fibroblasts from non-small cell lung carcinoma
reveals a role for ST8SIA2 in cancer cell invasion. Neoplasia.
21:482–493. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kilvaer TK, Khanehkenari MR, Hellevik T,
Al-Saad S, Paulsen EE, Bremnes RM, Busund LT, Donnem T and Martinez
IZ: Cancer associated fibroblasts in stage I–IIIA NSCLC: Prognostic
impact and their correlations with tumor molecular markers. PLoS
One. 10:e01349652015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cohen SJ, Alpaugh RK, Palazzo I, Meropol
NJ, Rogatko A, Xu Z, Hoffman JP, Weiner LM and Cheng JD: Fibroblast
activation protein and its relationship to clinical outcome in
pancreatic adenocarcinoma. Pancreas. 37:154–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yoshida T, Ishii G, Goto K, Neri S,
Hashimoto H, Yoh K, Niho S, Umemura S, Matsumoto S, Ohmatsu H, et
al: Podoplanin-positive cancer-associated fibroblasts in the tumor
microenvironment induce primary resistance to EGFR-TKIs in lung
adenocarcinoma with EGFR mutation. Clin Cancer Res. 21:642–651.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Neri S, Ishii G, Hashimoto H, Kuwata T,
Nagai K, Date H and Ochiai A: Podoplanin-expressing
cancer-associated fibroblasts lead and enhance the local invasion
of cancer cells in lung adenocarcinoma. Int J Cancer. 137:784–796.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Edlund K, Lindskog C, Saito A, Berglund A,
Pontén F Göransson-Kultima H, Isaksson A, Jirström K, Planck M,
Johansson L, et al: CD99 is a novel prognostic stromal marker in
non-small cell lung cancer. Int J Cancer. 131:2264–2273. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mitchell MI and Engelbrecht AM: Metabolic
hijacking: A survival strategy cancer cells exploit? Crit Rev Oncol
Hematol. 109:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Suzuki A, Puri S, Leland P, Puri A,
Moudgil T, Fox BA, Puri RK and Joshi BH: Subcellular
compartmentalization of PKM2 identifies anti-PKM2 therapy response
in vitro and in vivo mouse model of human non-small-cell lung
cancer. PLoS One. 14:e02171312019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang D, Zhao C, Xu F, Zhang A, Jin M,
Zhang K, Liu L, Hua Q, Zhao J, Liu J, et al: Cisplatin-resistant
NSCLC cells induced by hypoxia transmit resistance to sensitive
cells through exosomal PKM2. Theranostics. 11:2860–2875. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mallini P, Lennard T, Kirby J and Meeson
A: Epithelial-to-mesenchymal transition: What is the impact on
breast cancer stem cells and drug resistance. Cancer Treat Rev.
40:341–348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ye Q, Su L, Chen D, Zheng W and Liu Y:
Astragaloside IV Induced miR-134 expression reduces EMT and
increases chemotherapeutic sensitivity by suppressing CREB1
signaling in colorectal cancer cell line SW-480. Cell Physiol
Biochem. 43:1617–1626. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ding X, Ji J, Jiang J, Cai Q, Wang C, Shi
M, Yu Y, Zhu Z and Zhang J: HGF-mediated crosstalk between
cancer-associated fibroblasts and MET-unamplified gastric cancer
cells activates coordinated tumorigenesis and metastasis. Cell
Death Dis. 9:8672018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shintani Y, Fujiwara A, Kimura T, Kawamura
T, Funaki S, Minami M and Okumura M: IL-6 secreted from
cancer-associated fibroblasts mediates chemoresistance in NSCLC by
increasing epithelial-mesenchymal transition signaling. J Thorac
Oncol. 11:1482–1492. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lapidot T, Sirard C, Vormoor J, Murdoch B,
Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA and
Dick JE: A cell initiating human acute myeloid leukaemia after
transplantation into SCID mice. Nature. 367:645–648. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Leon G, MacDonagh L, Finn SP, Cuffe S and
Barr MP: Cancer stem cells in drug resistant lung cancer: Targeting
cell surface markers and signaling pathways. Pharmacol Ther.
158:71–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Shafee N, Smith CR, Wei S, Kim Y, Mills
GB, Hortobagyi GN, Stanbridge EJ and Lee EY: Cancer stem cells
contribute to cisplatin resistance in Brca1/p53-mediated mouse
mammary tumors. Cancer Res. 68:3243–3250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Schöning JP, Monteiro M and Gu W: Drug
resistance and cancer stem cells: The shared but distinct roles of
hypoxia-inducible factors HIF1α and HIF2α. Clin Exp Pharmacol
Physiol. 44:153–161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen Z, Shi T, Zhang L, Zhu P, Deng M,
Huang C, Hu T, Jiang L and Li J: Mammalian drug efflux transporters
of the ATP binding cassette (ABC) family in multidrug resistance: A
review of the past decade. Cancer Lett. 370:153–164. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kinugasa Y, Matsui T and Takakura N: CD44
expressed on cancer-associated fibroblasts is a functional molecule
supporting the stemness and drug resistance of malignant cancer
cells in the tumor microenvironment. Stem Cells. 32:145–156. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Najafi M, Farhood B and Mortezaee K:
Extracellular matrix (ECM) stiffness and degradation as cancer
drivers. J Cell Biochem. 120:2782–2790. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Keeratichamroen S, Lirdprapamongkol K and
Svasti J: Mechanism of ECM-induced dormancy and chemoresistance in
A549 human lung carcinoma cells. Oncol Rep. 39:1765–1774.
2018.PubMed/NCBI
|
|
84
|
De Rosa V, Iommelli F, Monti M, Fonti R,
Votta G, Stoppelli MP and Del Vecchio S: Reversal of warburg effect
and reactivation of oxidative phosphorylation by differential
inhibition of EGFR signaling pathways in non-small cell lung
cancer. Clin Cancer Res. 21:5110–5120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Iderzorig T, Kellen J, Osude C, Singh S,
Woodman JA, Garcia C and Puri N: Comparison of EMT mediated
tyrosine kinase inhibitor resistance in NSCLC. Biochem Biophys Res
Commun. 496:770–777. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yoshida T, Song L, Bai Y, Kinose F, Li J,
Ohaegbulam KC, Muñoz-Antonia T, Qu X, Eschrich S, Uramoto H, et al:
ZEB1 mediates acquired resistance to the epidermal growth factor
receptor-tyrosine kinase inhibitors in non-small cell lung cancer.
PLoS One. 11:e01473442016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
You J, Li M, Cao LM, Gu QH, Deng PB, Tan Y
and Hu CP: Snail1-dependent cancer-associated fibroblasts induce
epithelial-mesenchymal transition in lung cancer cells via
exosomes. QJM. 112:581–590. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yi Y, Zeng S, Wang Z, Wu M, Ma Y, Ye X,
Zhang B and Liu H: Cancer-associated fibroblasts promote
epithelial-mesenchymal transition and EGFR-TKI resistance of
non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim
Biophys Acta Mol Basis Dis. 1864:793–803. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhou J, Wang J, Zeng Y, Zhang X, Hu Q,
Zheng J, Chen B, Xie B and Zhang WM: Implication of
epithelial-mesenchymal transition in IGF1R-induced resistance to
EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget.
6:44332–44345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Choe C, Shin YS, Kim C, Choi SJ, Lee J,
Kim SY, Cho YB and Kim J: Crosstalk with cancer-associated
fibroblasts induces resistance of non-small cell lung cancer cells
to epidermal growth factor receptor tyrosine kinase inhibition.
Oncol Targets Ther. 8:3665–3678. 2015. View Article : Google Scholar
|
|
92
|
Della Corte CM, Bellevicine C, Vicidomini
G, Vitagliano D, Malapelle U, Accardo M, Fabozzi A, Fiorelli A,
Fasano M, Papaccio F, et al: SMO gene amplification and activation
of the hedgehog pathway as novel mechanisms of resistance to
anti-epidermal growth factor receptor drugs in human lung cancer.
Clin Cancer Res. 21:4686–4697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Murakami A, Takahashi F, Nurwidya F,
Kobayashi I, Minakata K, Hashimoto M, Nara T, Kato M, Tajima K,
Shimada N, et al: Hypoxia increases gefitinib-resistant lung cancer
stem cells through the activation of insulin-like growth factor 1
receptor. PLoS One. 9:e864592014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Petrova V, Annicchiarico-petruzzelli M,
Melino G and Amelio I: The hypoxic tumour microenvironment.
Oncogenesis. 7:102018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sugano T, Seike M, Noro R, Soeno C, Chiba
M, Zou F, Nakamichi S, Nishijima N, Matsumoto M, Miyanaga A, et al:
Inhibition of ABCB1 overcomes cancer stem cell-like properties and
acquired resistance to MET inhibitors in non-small cell lung
cancer. Mol Cancer Ther. 14:2433–2440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang Q, Yang J, Bai J and Ren J: Reverse
of non-small cell lung cancer drug resistance induced by
cancer-associated fibroblasts via a paracrine pathway. Cancer Sci.
109:944–955. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Li F, Mei H, Gao Y, Xie X, Nie H, Li T,
Zhang H and Jia L: Co-delivery of oxygen and erlotinib by
aptamer-modified liposomal complexes to reverse hypoxia-induced
drug resistance in lung cancer. Biomaterials. 145:56–71. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bridgford JL, Xie SC, Cobbold SA, Pasaje
CFA, Herrmann S, Yang T, Gillett DL, Dick LR, Ralph SA, Dogovski C,
et al: Artemisinin kills malaria parasites by damaging proteins and
inhibiting the proteasome. Nat Commun. 9:38012018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chou CW, Wang CC, Wu CP, Lin YJ, Lee YC,
Cheng YW and Hsieh CH: Tumor cycling hypoxia induces
chemoresistance in glioblastoma multiforme by upregulating the
expression and function of ABCB1. Neuro Oncol. 14:1227–1238. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Raju S, Joseph R and Sehgal S: Review of
checkpoint immunotherapy for the management of non-small cell lung
cancer. Immunotargets Ther. 7:63–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kloten V, Lampignano R, Krahn T and
Schlange T: Circulating tumor Cell PD-L1 expression as biomarker
for therapeutic efficacy of immune checkpoint inhibition in NSCLC.
Cells. 8:8092019. View Article : Google Scholar
|
|
102
|
Pu X, Wu L, Su D, Mao W and Fang B:
Immunotherapy for non-small cell lung cancers: Biomarkers for
predicting responses and strategies to overcome resistance. BMC
Cancer. 18:10822018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Salmon H, Franciszkiewicz K, Damotte D,
Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F and
Donnadieu E: Matrix architecture defines the preferential
localization and migration of T cells into the stroma of human lung
tumors. J Clin Invest. 122:899–910. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nicolas-Boluda A, Vaquero J, Barrin S,
Kantari-Mimoun C, Ponzo M, Renault G, Deptuła P, Pogoda K, Bucki R,
Cascone I, et al: Tumor stiffening reversion through collagen
crosslinking inhibition improves T cell migration and anti-PD-1
treatment. Cold Spring Harbor. 2020.
|
|
105
|
Zeltz C, Pasko E, Cox TR, Navab R and Tsao
MS: LOXL1 is regulated by integrin α11 and promotes non-small cell
lung cancer tumorigenicity. Cancers (Basel). 11:7052019. View Article : Google Scholar
|
|
106
|
Saunier EF and Akhurst RJ: TGF beta
inhibition for cancer therapy. Curr Cancer Drug Targets. 6:565–578.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ford K, Hanley CJ, Mellone M,
Szyndralewiez C, Heitz F, Wiesel P, Wood O, Machado M, Lopez MA,
Ganesan AP, et al: NOX4 inhibition potentiates immunotherapy by
overcoming cancer-associated fibroblast-mediated CD8 T-cell
exclusion from tumors. Cancer Res. 80:1846–1860. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lakins MA, Ghorani E, Munir H, Martins CP
and Shields JD: Cancer-associated fibroblasts induce
antigen-specific deletion of CD8 + T Cells to protect
tumour cells. Nat Commun. 9:9482018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Teramoto K, Igarashi T, Kataoka Y, Ishida
M, Hanaoka J, Sumimoto H and Daigo Y: Clinical significance of
PD-L1-positive cancer-associated fibroblasts in pN0M0 non-small
cell lung cancer. Lung Cancer. 137:56–63. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kilvaer TK, Rakaee M, Hellevik T, Østman
A, Strell C, Bremnes RM, Busund LT, Dønnem T and Martinez-Zubiaurre
I: Tissue analyses reveal a potential immune-adjuvant function of
FAP-1 positive fibroblasts in non-small cell lung cancer. PLoS One.
13:e01921572018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hanley CJ, Mellone M, Ford K, Thirdborough
SM, Mellows T, Frampton SJ, Smith DM, Harden E, Szyndralewiez C,
Bullock M, et al: Targeting the myofibroblastic cancer-associated
fibroblast phenotype through inhibition of NOX4. J Natl Cancer
Inst. 110:109–120. 2018. View Article : Google Scholar
|
|
112
|
Fujiwara A, Funaki S, Fukui E, Kimura K,
Kanou T, Ose N, Minami M and Shintani Y: Effects of pirfenidone
targeting the tumor microenvironment and tumor-stroma interaction
as a novel treatment for non-small cell lung cancer. Sci Rep.
10:109002020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kakarla S, Chow K, Mata M, Shaffer DR,
Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K and
Gottschalk S: Antitumor effects of chimeric receptor engineered
human T cells directed to tumor stroma. Mol Ther. 21:1611–1620.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Masuda T, Nakashima T, Namba M, Yamaguchi
K, Sakamoto S, Horimasu Y, Miyamoto S, Iwamoto H, Fujitaka K,
Miyata Y, et al: Inhibition of PAI-1 limits chemotherapy resistance
in lung cancer through suppressing myofibroblast characteristics of
cancer-associated fibroblasts. J Cell Mol Med. 23:29842019.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Duan S, Tsai Y, Keng P and Chen Y, Lee SO
and Chen Y: IL-6 signaling contributes to cisplatin resistance in
non-small cell lung cancer via the up-regulation of anti-apoptotic
and DNA repair associated molecules. Oncotarget. 6:27651–27660.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang L, Li X, Ren Y, Geng H, Zhang Q, Cao
L, Meng Z, Wu X, Xu M and Xu K: Cancer-associated fibroblasts
contribute to cisplatin resistance by modulating ANXA3 in lung
cancer cells. Cancer Sci. 110:1609–1620. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wei JR, Dong J and Li L: Cancer-associated
fibroblasts-derived gamma-glutamyltransferase 5 promotes tumor
growth and drug resistance in lung adenocarcinoma. Aging (Albany
NY). 12:13220–13233. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Tao L, Huang G, Wang R, Pan Y, He Z, Chu
X, Song H and Chen L: Cancer-associated fibroblasts treated with
cisplatin facilitates chemoresistance of lung adenocarcinoma
through IL-11/IL-11R/STAT3 signaling pathway. Sci Rep. 6:384082016.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Shien K, Papadimitrakopoulou VA, Ruder D,
Behrens C, Shen L, Kalhor N, Song J, Lee JJ, Wang J, Tang X, et al:
JAK1/STAT3 activation through a proinflammatory cytokine pathway
leads to resistance to molecularly targeted therapy in non-small
cell lung cancer. Mol Cancer Ther. 16:2234–2245. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Foster JG, Wong SC and Sharp TV: The
hypoxic tumor microenvironment: Driving the tumorigenesis of
non-small-cell lung cancer. Future Oncol. 10:2659–2674. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Rebelo SP, Pinto C, Martins TR, Harrer N,
Estrada MF, Loza-Alvarez P, Cabeçadas J, Alves PM, Gualda EJ,
Sommergruber W and Brito C: 3D-3-culture: A tool to unveil
macrophage plasticity in the tumour microenvironment. Biomaterials.
163:185–197. 2018. View Article : Google Scholar : PubMed/NCBI
|