Introduction
p53 is a tumor suppressor protein (1). Mutations within p53 have been
demonstrated to lead to its inactivation, which is associated with
~50% of all types of human cancer (2). The p53 protein contains an amino
N-terminal transactivation domain, a proline-rich domain, a central
DNA-binding domain (DBD), a tetramerization domain and a
carboxy-terminal regulatory domain (CRD) (3). The majority of the cancer-associated
mutations are missense mutations located within the DBD (4). R175H, Y220C, G245S, R248Q/W, R249S,
R273C/H and R282W are high-frequency mutations that alter protein
conformation and attenuate sequence-specific binding to proteins
(5). This results in the inhibition
of wild-type (WT) p53 function, and the mutated p53 interacts with
inappropriate proteins and stimulates oncogenic genes (6). p53 is also an important transcription
factor involved in the regulation of DNA repair, cell cycle arrest
and apoptosis (7). Under normal
conditions, p53 is present in a steady state, whereas in cells
undergoing DNA damage or abnormal oncogenic events, p53 is
activated through post-translational modifications, such as
phosphorylation, ubiquitination and acetylation (8). Therefore, the levels of p53 are
increased, resulting in the transactivation of the downstream
target genes, which are involved in cell cycle arrest, DNA repair,
autophagy and cellular metabolism (4). Furthermore, p53 activates genes such as
AMP-activated protein kinase β, tuberin and PTEN to suppress the
mTOR (nutrient sensor) signaling pathway, which participates in
aerobic glycolysis and oxidative phosphorylation (9). In addition, cytosolic p53 exerts
transcription-independent functions, including triggering apoptosis
by interacting with the apoptotic effector proteins BAX and BAK,
and repressing autophagy by inhibiting the positive autophagy
regulator AMP-dependent kinase (10–12).
In eukaryotic cells, genomic DNA is packaged into
chromatin and encapsulates histone octamers to form nucleosomes
(13). The N- and C-termini of
histones can be covalently modified by methylation, acetylation and
ubiquitination (14). Histone
ubiquitination is an important epigenetic modification widely
involved in the regulation of chromatin structure, gene
transcription, the cell cycle and other physiological processes
(15). Ubiquitination is one of the
covalent post-translational modifications, during which the 8-kD
ubiquitin molecule (mono-ubiquitination) or a poly-ubiquitin chain
(poly-ubiquitination) conjugate to a protein substrate (16). The ubiquitin E3 ligase functions in
the last step of the ubiquitination cascade (17). The ring finger (RNF) E3 enzyme,
RNF20/RNF40 dimer, catalyzes the histone H2B mono-ubiquitination
(H2Bub1) at lysine 120 in the C-terminus (18). H2Bub1 is essential for maintaining
functionality of the p53 tumor suppressor protein (19). The loss of RNF20 and RNF40 attenuates
the p53-dependent cell response to cellular stress or toxicity; for
example, RNF20-knockdown by RNA interference in HeLa cells leads to
decreased apoptosis and impaired cell cycle arrest compared with
those in control-transfected HeLa cells (20). In addition, WAC has been reported to
act as a functional partner of the RNF20/RNF40 dimer, which
mediates the interaction of the RNF20/RNF40 dimer with RNA
polymerase II (21,22). Furthermore, p53 has been reported to
interact with the RNF20/RNF40/WAC complex directly and to recruit
the complex for H2Bub1 to target the p53 gene loci (21,23).
An increasing number of studies have demonstrated
that the RNF20/RNF40/WAC complex is associated with genomic
stability and tumorigenesis (8,24–26).
Therefore, it is important to investigate the underlying molecular
mechanism by which the RNF20/RNF40/WAC complex and p53
synergistically regulate target gene transcription. A previous
study by Wu et al (27)
reported that the CRD of p53 is a key binding region with the
RNF20/RNF40 dimer; however, only partial deletion mutants of the
p53 DBD were constructed in their study. There is currently no
evidence of specific sites interacting with the RNF20/RNF40 dimer
close to the C-terminal region of the p53 DBD. In the present
study, we hypothesized that, in addition to the CRD region
interacting with RNF20, the amino acid (AA)201-AA300 region of the
p53 DBD may also interact with RNF20/RNF40, and that one of the
cancer-related hotspot sites of p53 may be a potential site for the
interaction with the RNF20/RNF40 dimer. The aim of the present
study was to detect the specific binding site of p53 with the
RNF20/RNF40 dimer and to investigate the gene regulation function
of these sites.
Materials and methods
Cell culture
The 293T, U2OS and HCT116 cell lines were obtained
from the Shanghai Institute of Biochemistry and Cell Biology, and
cultured in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck KGaA) with 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), and 1% streptomycin and
penicillin. The cells were cultured at 37°C in a humidified
incubator with 5% CO2. All cell lines were confirmed to
be free from mycoplasma contamination.
Co-immunoprecipitation (Co-IP) and
western blot assays
After washing with 1X PBS, the cells were lysed with
the NETN-100 buffer [150 mM NaCl, 0.2 mM EDTA, 1% NP-40 and 50 mM
Tris-HCl (pH 7.5)] for 10 min on ice, supplemented with 1X protease
inhibitor cocktail (Merck KGaA) and 1 mM phenylmethylsulfonyl
fluoride, and centrifuged at 12,000 × g at 4°C for 10 min. For
Co-IP, the cell lysates were incubated with 50 µl protein A/G
agarose (Sangon Biotech Co., Ltd.) and the following antibodies
(dilution, 1:1,000 for western blotting and 1:500 for Co-IP):
Anti-HA (cat. no. D199961-0100; Sangon Biotech Co., Ltd.), anti-p53
(cat. no. ab26; Abcam), anti-FLAG (cat. no. 14793; Cell Signaling
Technology, Inc.), anti-WAC (cat. no. ABE471; Merck KGaA), anti-rat
IgG (cat. no. ab172730; Abcam), anti-RNF20 (cat. no. A300-714A;
Bethyl Laboratories, Inc.) and anti-RNF40 (cat. no. Q2680124;
Bethyl Laboratories, Inc.) overnight at 4°C. Following incubation,
precipitates were washed three times with PBS and added to the
NETN-100 buffer. Whole cell lysates and washed precipitates were
incubated at 100°C for 8 min in a dry bath incubator (Tiangen
Biotech Co., Ltd) and separated by 10% SDS-PAGE. The membranes were
incubated with the aforementioned antibodies, followed by goat
anti-mouse IgG (H+L) HRP-conjugated (cat. no. AP308P;
Sigma-Aldrich; Merck KGaA) and goat anti-rabbit IgG H&L
HRP-conjugated (cat. no. ab205718; Abcam) secondary antibodies
(dilution, 1:1,000) at room temperature for 1.5 h. The membranes
were incubated with SuperSignal West Pico Chemiluminescent
Substrate (Thermo Fisher Scientific, Inc.) and visualized using an
X-ray film or an automatic digital imaging system. A total of three
independent experiments were performed. The semi-quantitative
analysis of the western blotting results was performed using ImageJ
1.42q software Java 1.6.0_12(64-bit) (National Institutes of
Health) and GraphPad Prism version 6.02 for Windows (GraphPad
Software, Inc.).
Mutant construction and plasmid
transfection
Human full-length RNF20, RNF40, RNF20 deletion 1
(D1)-D8 and RNF40 D1-D14 were cloned into the HA-tagged vector
modified from pCDNA 3.1 (Thermo Fisher Scientific, Inc.). Human
full-length, internal deletion (Δaa. 101-200 and Δaa. 201-300) and
missense mutation sequences of p53 (R248W, R248Q, R249S, R273C,
R273H, R282W and R248QR273C) were cloned into the pCDNA3.1-HA
vector. Human full-length and mutated WAC deletion (WD)1-7 were
cloned into the pS-FLAG-SBP vector (Addgene, Inc.). Deletion
mutations within RNF20, RNF40 and WAC, and missense mutations in
p53, were generated using the QuikChange site-directed mutagenesis
kit (Agilent Technologies, Inc.). Constructed plasmids (5 µg for
cells incubated in a 10-cm dish) with the indicated mutations were
transfected into U2OS cells (for RNF20/RNF40/WAC deletion mutations
generation) and 293T cells (for p53 deletion and missense mutations
generation) at 70–80% confluence using ViaFect (Promega
Corporation) and Opti-MEM (Thermo Fisher Scientific, Inc.) without
FBS for 4–6 h according to the manufacturers' instructions.
Subsequently, the cells were washed with PBS and cultured in DMEM
with 10% FBS and 1% streptomycin and penicillin for 24 h prior to
co-IP and western blot assays. The single amino acid mutations were
constructed according to previous studies by Tzin et al
(28) and Kitzman et al
(29), whereas the deletion
mutations were constructed according to previous studies by Zhang
and Yu (21).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from WT, RNF20−/−
and RNF40−/− HCT116 cell lines using TRIzol®
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. RNA concentration was measured using a
microplate reader. RT of the total RNA was performed using the
PrimeScript™ 1st Strand cDNA Synthesis kit (Takara Bio, Inc.). qPCR
was performed with gene-specific primers and the QuantiNova
SYBR® Green PCR kit (Qiagen, Inc.) on a CFX96 Real-Time
PCR Detection system (Bio-Rad Laboratories, Inc.) The primers used
were as follows: GAPDH forward, 5′-ACCCACTCCTCCACCTTTGA-3′ and
reverse, 5′-CTGTTGCTGTAGCCAAATTCGT-3′; p53 forward,
5′-AGATGGGGTCTCACAGTGTTGC-3′ and reverse,
5′-ATGTTGACCCTTCCAGCTCCAC-3′; p21 forward,
5′-CATGCCAGCTACTTCCTCCT-3′ and reverse, 5′-CAGGTCTGAGTGTCCAGGAA-3′;
p53-upregulated modulator of apoptosis (PUMA) forward,
5′-GACGACCTCAACGCACAGTA-3′ and reverse, 5′-CTAATTGGGCTCCATCTCG-3′;
Achaete-Scute homolog 1 (mash1) forward,
5′-CGACTTCACCAACTGGTTCTG-3′ and reverse, 5′-ATGCAGGTTGTGCGATCA-3′;
and octamer-binding protein 4 (Oct4) forward,
5′-CGCAAGCCCTCATTTCAC-3′ and reverse, 5′-CATCACCTCCACCACCTG-3′. The
following thermocycling conditions were used: Initial denaturation
at 95°C for 2 min, followed by 35–40 cycles at 95°C for 5 sec and
60°C for 10 sec. The 2−∆∆Cq method was used for
quantification (30). A total of
three independent experiments were performed.
Cell line construction using
CRISPR-Cas9
The primers for the deletion of RNF20 or RNF40 were
designed from the CRISPR website (http://crispr.mit.edu/) and confirmed for accuracy
using the Cas OFFinder website (http://www.rgenome.net/cas-offinder/). The following
primers were used: CRISPR-RNF20-CDS2 forward,
5′-CACCGTATTGATTGTCAACCGATAC-3′ and reverse,
5′-AAACGTATCGGTTGACAATCAATAC-3′; and CRISPR-RNF40-CDS2 forward,
5′-CACCGTCCTCATCGTCAATCGCTAC-3′ and reverse,
5′-AAACGTAGCGATTGACGATGAGGAC-3′. The pSpCas9(BB)-2A-Pure V2.0
(PX49) vector (Addgene, Inc.) was used, while Lipofectamine 2000
(Thermo Fisher Scientific, Inc.) was used for plasmid transfection.
A total of 2.5 µg/well plasmid was transfected into HCT116 cells at
70–80% confluence in a 6-well plate for 4–6 h at 37°C.
Subsequently, the cells were cultured using a concentration
gradient and treated with 1 µg/ml puromycin for 3 days to screen
the successfully transfected cells. Following 2-week culture, 12
monoclones with expected RNF20 deletion and 12 monoclones with
expected RNF40 deletion were selected and expanded for further
culture. Finally, the protein knockout effect was detected using
western blot analysis.
DNA damage-inducing drug
treatment
WT, RNF20−/− and RNF40−/−
HCT116 cell lines were treated with DNA damage-inducing drugs
doxorubicin (Dox; 0.5 µM) and etoposide (VP-16; 100 µM) for 0, 2.5
and 5 h prior to western blot analysis. WT, RNF20−/−,
RNF40−/−, p53−/− HCT116 cell lines and
p53−/− HCT116 transfected with pCDNA3.1-HA-p53 R282W or
pCDNA3.1-HA-p53 WT cells were treated with Dox (0.25 µM) for 12 h
prior to RT-qPCR detection.
Statistical analysis
Data are presented as the mean ± SD. One-way ANOVA
followed by Bonferroni's correction was used to determine the
statistical differences among the experimental groups. GraphPad
Prism version 6.02 for Windows (GraphPad Software, Inc.) and ImageJ
1.42q software with Java 1.6.0_12 (64-bit) (National Institutes of
Health) were used for data and statistical analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
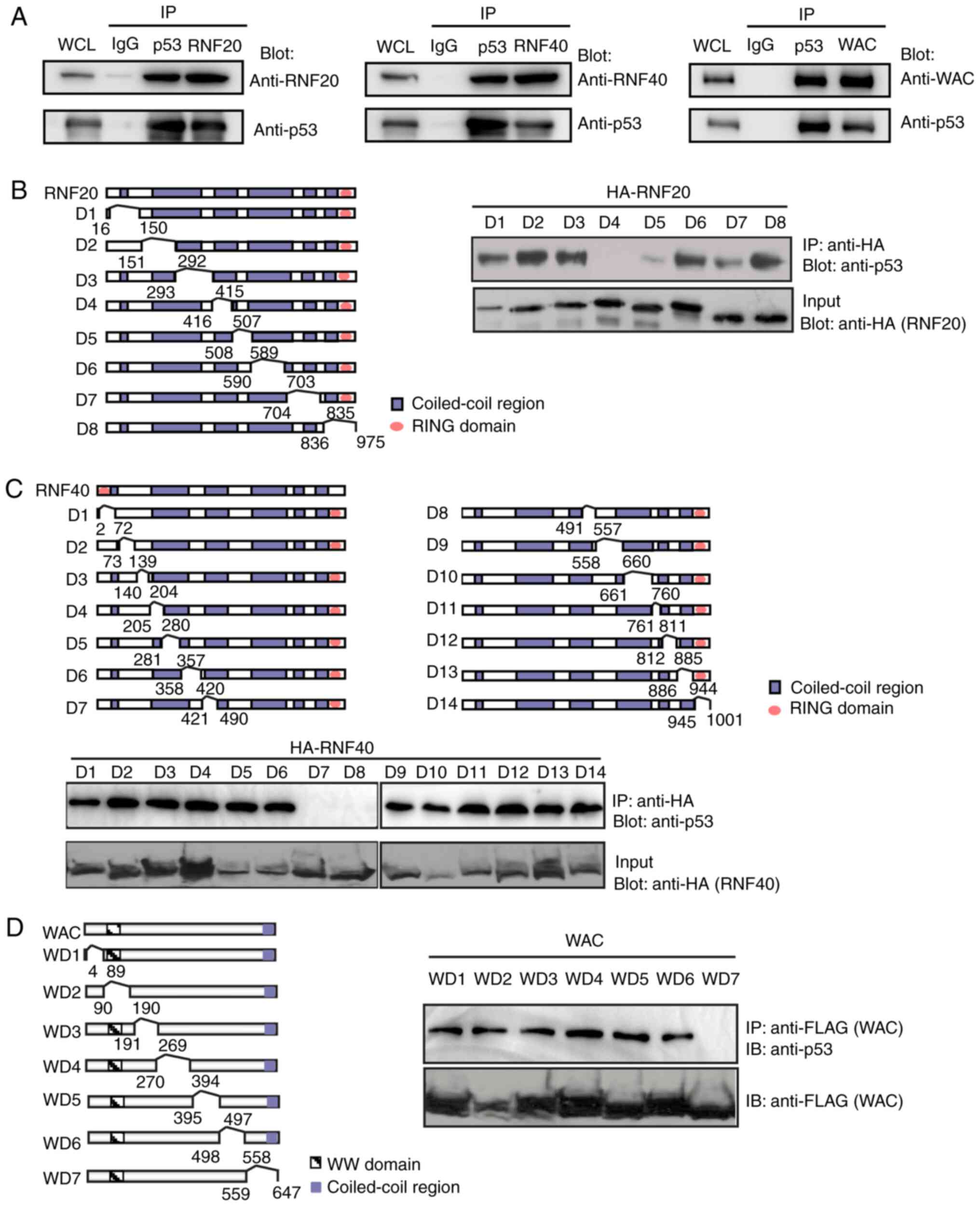
RNF20, RNF40 and WAC interacts with
p53 through the coiled-coil domain
p53 has been demonstrated to be functionally
associated with genomic DNA and involved in chromatin modifications
(31). Furthermore, the chromatin
RNF20/RNF40/WAC remodeler complex has been reported to mediate the
protein expression levels of p53 (32,33).
Therefore, endogenous co-IP and western blot assays were performed
in U2OS cell lysates to detect the interaction between p53 and the
RNF20/RNF40/WAC complex. The results revealed that p53 interacted
with RNF20, RNF40 and WAC (Fig. 1A).
Next, a series of internal deletion mutations within RNF20, RNF40
and WAC were created, as indicated in the structure images to map
the interaction regions in RNF20, RNF40 and WAC with p53 (Fig. 1B-D). As demonstrated in Fig. 1B-D, the D4 mutant of RNF20, the D7
and D8 mutants of RNF40 and the D7 mutant of WAC disrupted the
interaction with p53, indicating that the coiled-coil motif of
RNF20 and RNF40 in the corresponding areas recognized the p53
protein. However, p53, RNF20 and RNF40 all interacted with WAC
through the WD7 mutant; therefore, it was hypothesized that either
p53 interacted with WAC directly, or the RNF20/RNF40 dimer
functioned as a bridge to connect p53 and WAC.
Mapping the specific p53 binding site
with RNF20 and RNF40
Subsequently, the missense mutation in the DBD,
which has been associated with ~50% of all types of cancer, was
investigated. To determine whether the mutation sites in the p53
DBD mediated the interaction with RNF20 and RNF40, a WT recombinant
plasmid, pCDNA3.1-HA-p53, was used as a template, and two partial
deletion mutants in DBD domain including Δaa. 101-200 and Δaa.
201-300 were constructed (Fig. 2A and
B) and transfected into the U2OS cell line. The cell lysates
were examined using co-IP and western blot assays. The results
demonstrated that the Δaa. 201-300 mutation prevented p53 from
interacting with RNF20 and RNF40 (Fig.
2B), suggesting that the p53 C-terminal of DBD may be crucial
for the interaction with RNF20 and RNF40. Next, a series of
plasmids with high frequency cancer-related p53 missense mutations
within the p53 C-terminal of DBD were constructed (Fig. 2C). A total of eight plasmids,
including HA-p53WT, HA-p53R248W, HA-p53R248Q, HA-p53R249S,
HA-p53R273C, HA-p53R273H, HA-p53R282W and p53R248QR273C, were
transfected into the 293T cell line, and the interactions between
p53 and RNF20 were examined using co-IP and western blot assays.
The results demonstrated that the R282W mutation in the p53 DBD
prevented p53 from interacting with RNF20 (Fig. 2B). Thus, the key site for the p53
interaction with RNF20 was R282.
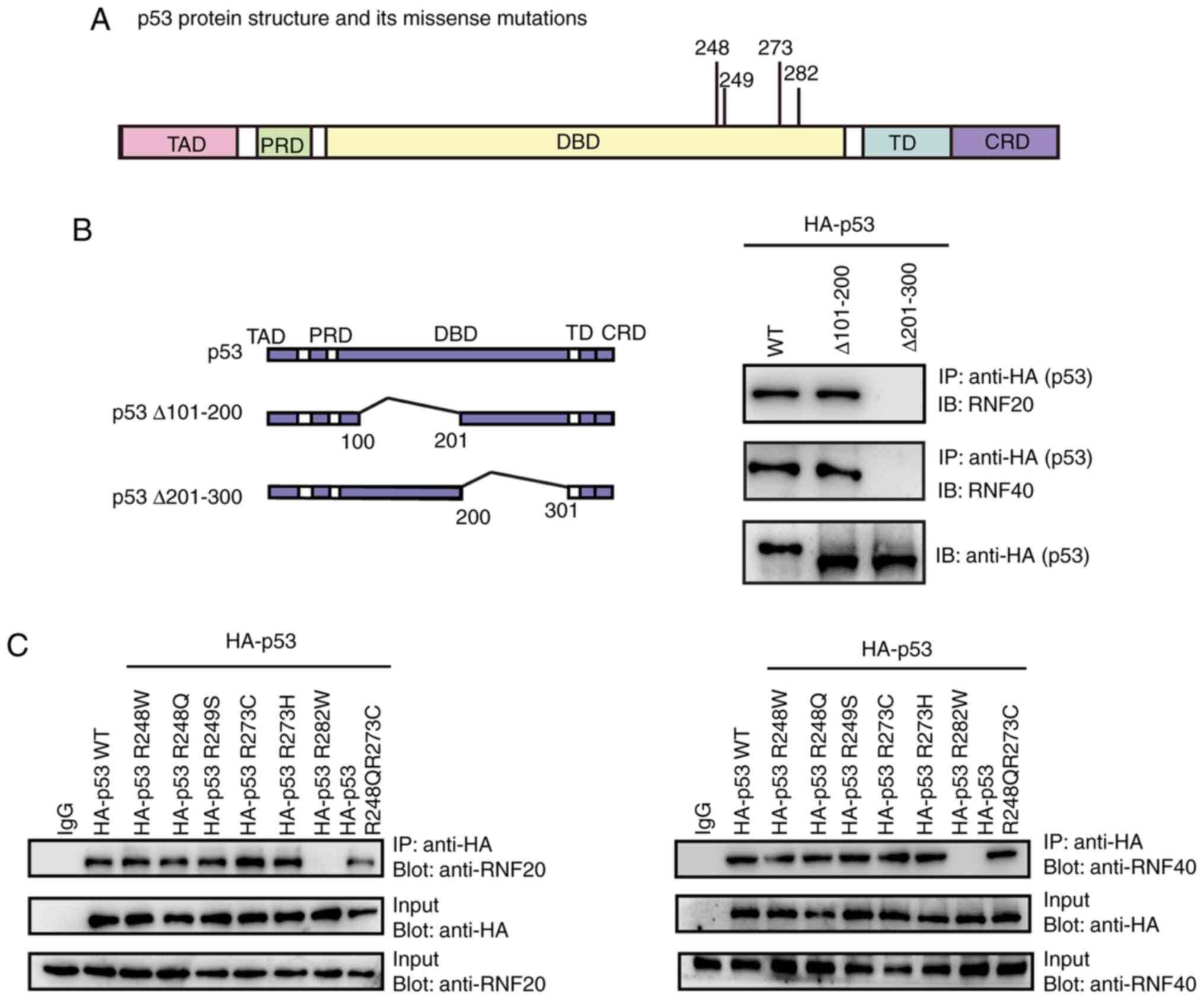 | Figure 2.Mapping the key binding site in p53
with RNF20 and RNF40. (A) The location of the p53 missense
mutations associated with cancer in the DNA-binding domain. (B and
C) The 293T cell line was transfected with pCDNA3.1-HA-p53 plasmids
with WT, partial internal deletion and the indicated missense
mutations. Co-IP and western blot analyses were used to determine
the exact site in p53, which interacted with RNF20 and RNF40. RNF,
ring finger; co-IP, co-immunoprecipitation; IB, immunoblot; WT,
wild-type. TAD, transcriptional activation domain; PRD,
proline-rich domain; DBD, DNA-binding domain; TD, tetramerization
domain; CRD, carboxy-terminal regulatory domain. |
RNF20/RNF40 complex maintains the
activities of p53 and its target genes
To further investigate the role of RNF20 and RNF40
in p53 stabilization and transcription of p53 target genes, p53
protein expression levels were analyzed using western blot
analysis, and the relative mRNA expression levels of p53, p21,
PUMA, mash1 and Oct4 genes were determined using RT-qPCR in RNF20-
or RNF40-knockout cells. CRISPR/Cas9 was used to knock out RNF20
and RNF40 in the HCT116 WT cell line, and the RNF20−/−
and RNF40−/− HCT116 cell lines were successfully
constructed (Fig. 3A). The HCT116
WT, RNF20−/− and RNF40−/− HCT116 cell lines
were treated with DNA damage-inducing drugs Dox (0.5 µM) and VP-16
(100 µM) for 0, 2.5 and 5 h. Western blot analysis results
demonstrated that following RNF20- or RNF40-knockout in the HCT116
cell lines, the protein expression levels of p53 were significantly
reduced compared with those in the WT cells. Following DNA damage
induced by Dox and VP-16, the p53 protein expression levels were
increased in all the cell lines compared with those in the cells
without Dox or VP-16 treatment; however, the p53 protein expression
levels were lower in the RNF20−/− and
RNF40−/− HCT116 cells lines compared with those in the
WT cell line (Fig. 3B and C). Thus,
RNF20 and RNF40 stabilized the p53 protein expression level. To
determine the effects of the RNF20/RNF40 complex on the mRNA
expression levels of p53 and its downstream genes, the HCT116 WT,
RNF20−/− and RNF40−/− cell lines were treated
with Dox (0.25 µM; 12 h), and the relative mRNA expression levels
of the transcription targets of p53 were detected using RT-qPCR.
The results demonstrated that following RNF20- and RNF40-knockout,
the mRNA expression levels of p53, p21, PUMA, mash1 and Oct4 in
these cell lines without Dox treatment were significantly lower
compared with those in the WT cells without Dox treatment.
Following induction of DNA damage, the increases in the levels of
p53 and its target genes in RNF20−/− and
RNF40−/− HCT116 cells lines treated with Dox were
inhibited compared with those in WT HCT116 cells treated with Dox
(Fig. 3D). Taken together, these
results suggested that the RNF20/RNF40 complex stabilized the p53
protein expression levels and maintained the mRNA expression of its
p53 target genes.
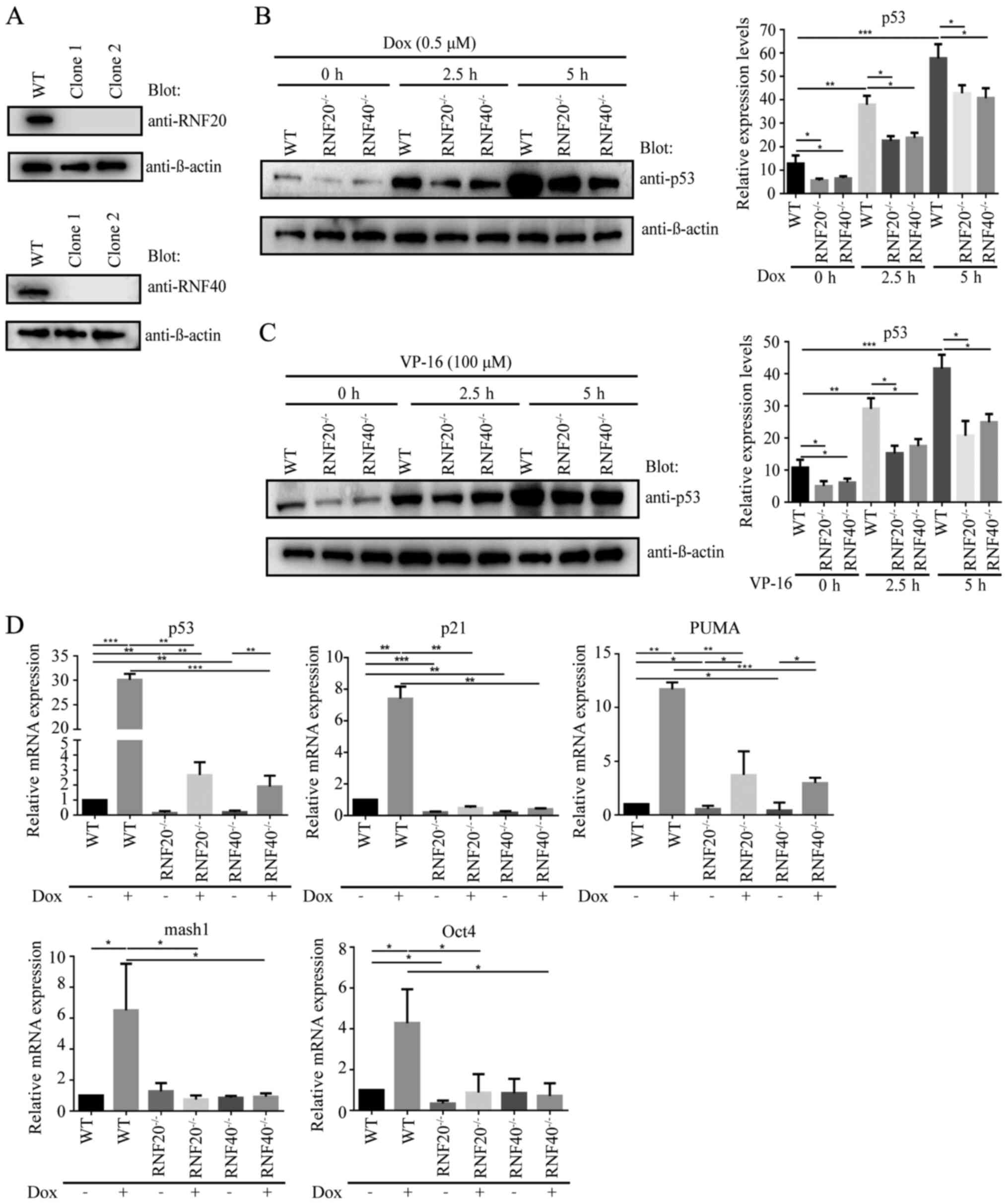 | Figure 3.RNF20/RNF40 dimer maintains the
activity of p53 and its target genes. (A) RNF20−/− and
RNF40−/− HCT116 cell lines were successfully constructed
using CRISPR/Cas9. Clones 1 and 2 were two samples from the
RNF20−/− HCT116 cell line, and clones 3 and 4 were from
the RNF40−/− HCT116 cell line. (B and C) The protein
expression levels of p53 were analyzed using western blot analysis
in the HCT116 WT, RNF20−/− and RNF40−/− cell
lines following treatment with 0.5 µM Dox and 100 µM VP-16 for 0,
2.5 and 5 h. (D) The relative mRNA expression levels of p53, p21
PUMA, mash1 and Oct4 in the WT, RNF20−/− and
RNF40−/− HCT116 cell lines with or without doxorubicin
treatment from three independent experiments were quantified using
reverse transcription-quantitative PCR. The data are presented as
the mean ± SD. n=3. *P<0.05, **P<0.01 and ***P<0.001. RNF,
ring finger; WT, wild-type; Dox, doxorubicin; VP-16, etoposide;
PUMA, p53-upregulated modulator of apoptosis; mash1, Achaete-Scute
homolog 1; Oct4, octamer-binding protein 4. |
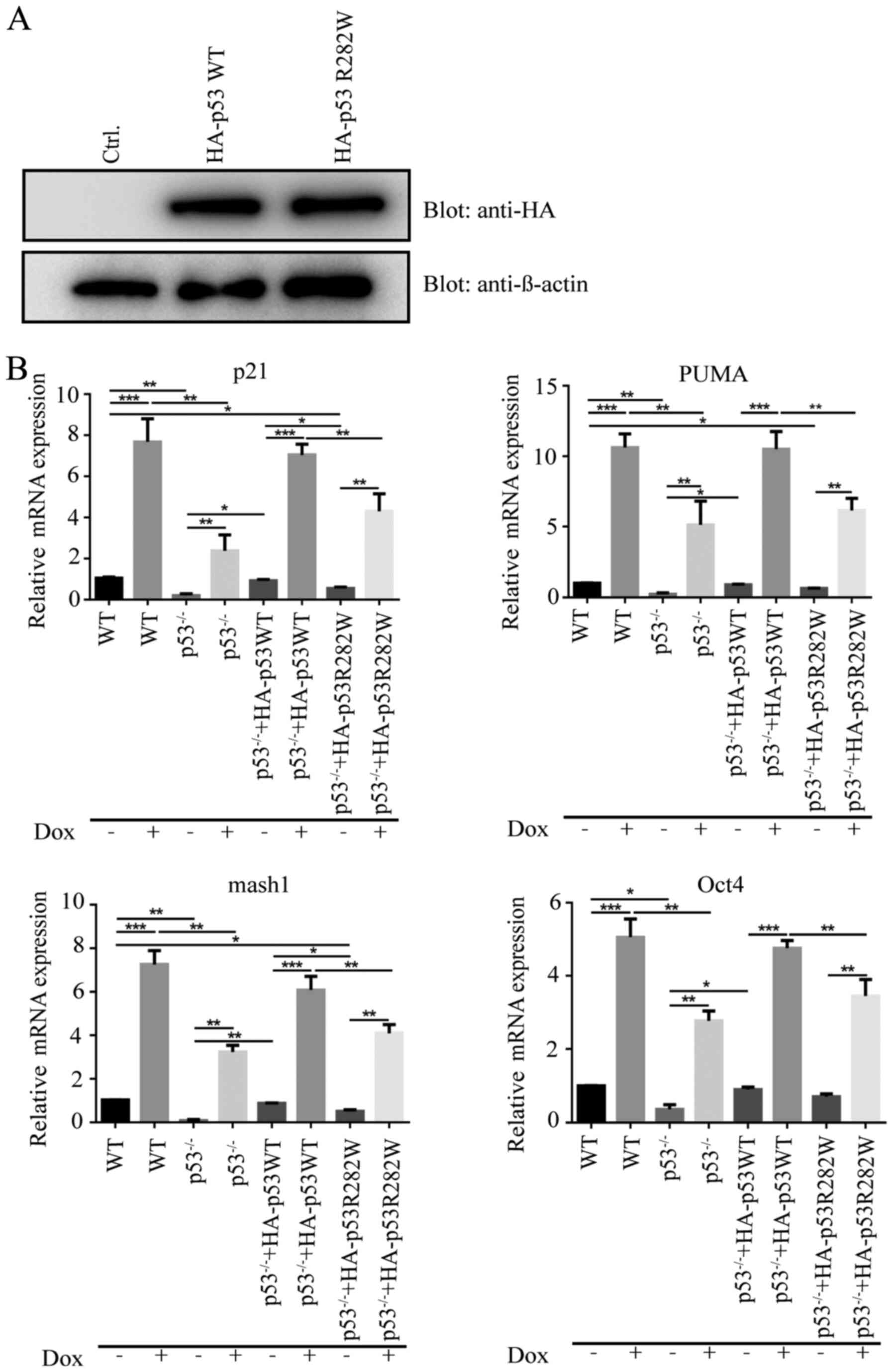
Mutation in p53 associated with RNF20
interaction suppresses the mRNA expression levels of p53 target
genes
The loss of the RNF20/RNF40 dimer downregulated the
mRNA expression levels of the p53 target genes both in normal
conditions and following DNA damage; however, it was unclear
whether the p53 key binding site for RNF20, R282, mediated the
inhibition of the p53 target gene expression following DNA damage.
To investigate this, the pCDNA3.1-HA-p53 R282W plasmid was
transfected into the p53−/− HCT116 cell line (Fig. 4A). Following Dox (0.5 µM) treatment
for 12 h, the mRNA expression levels of p21 in the WT cells were
significantly increased compared with those in the untreated WT
cells. The mRNA expression levels of p21 in the p53−/−
HCT116 cells were markedly reduced compared with those in the WT
cells, and the p21 levels of p53−/− HCT116 cells treated
with Dox were reduced compared with those in WT cells treated with
Dox; however, the transfection of the pCDNA3.1-HA-p53 WT plasmid in
p53−/− HCT116 cells rescued the mRNA expression level of
p21 compared with p53−/− HCT116 cells. In addition, the
mRNA expression levels of p21 were lower in the pCDNA3.1-HA-p53
R282W-transfected p53−/− HCT116 cells compared with
those in cells transfected with pCDNA3.1-HA-p53 WT, which indicated
that the p53 R282 site mediated the mRNA expression of p21
(Fig. 4B). The results of the PUMA,
Oct4 and Mash1 mRNA expression level analysis were consistent with
those of p21 (Fig. 4B). These
results demonstrated that the R282W mutation resulted in the
inability of p53, to bind to RNF20, which further inhibited the
downstream target genes of p53.
Discussion
p53 activates a variety of transcriptional targets
in response to cellular stress or DNA damage (4). It has been reported that the
RNF20/RNF40/WAC complex regulates the transcription of the p53
target genes by regulating H2Bub1 under genotoxic stress (14). H2Bub1 is conserved in the evolution
process from yeast to mammals (34).
Mammalian RNF20 and RNF40 have been demonstrated to be homologous
to the E3 ubiquitin ligase Bre1 (BRE1) gene in yeast, forming a
protein dimer, which synergistically interacts with E2 ubiquitinase
RAD6 to catalyze histone H2B ubiquitination (35). WAC is a functional partner of
RNF20/RNF40, which interacts with the coiled-coil region of
RNF20/RNF40 to mediate H2Bub1 (21).
The present study aimed to investigate the interaction domain
between the RNF20/RNF40/WAC complex and p53, as well as the role of
this complex gene transcription regulation.
The coiled-coil domains in RNF20, RNF40 and WAC have
been reported to be highly conserved regions, as well as the WW
domain of WAC, suggesting that these areas may serve important
biological functions (33). Notably,
the interaction between RNF20/40 and WAC is also mediated through
the coiled-coil motif (21). The
results of the present study demonstrated that p53 not only
interacted with RNF20 and RNF40, consistent with a previous report
by Wu et al (27), but also
with WAC directly or through the RNF20/RNF40 dimer. p53 bound to
the coiled-coil regions of RNF20, RNF40 and WAC. In addition, a
series of deletion mutants of p53 were constructed in the present
study, and the results demonstrated that other than the CTD region
interacting with RNF20, the C-terminal part of the p53 DBD also
interacted with RNF20/RNF40.
p53 initiates the transcription of genes associated
with cell cycle arrest, cell senescence, apoptosis, metabolism, DNA
repair and other processes under cell stress (36,37). The
loss of p53 function is primarily attributed to mutations,
including gene fragment deletions, insertions, missense mutations
and loss of heterozygosity (38).
Missense mutations caused by single nucleotide polymorphisms
account for >80% of the total p53 mutations (39). Among these p53 missense mutations,
97% are point mutations and occur in the DBD (40). Mutations at the following positions
occur at high frequency in cancer and are also termed hotspot
mutations: R175H, R248Q, R248W, R249S, R273H, R273C and R282W
(41). The present study focused on
the specific sites interacting with the RNF20/RNF40 dimer close to
the C-terminal region of the p53 DBD. These mutations were screened
by generating a series of missense mutants, and the results of the
co-IP assay revealed that R282W was a key site for interaction with
the RNF20/RNF40/WAC complex. Since human RNF20 and RNF40 share
sequence homology with BRE1 in Saccharomyces cerevisiae, the key
binding sites of p53 interacting with RNF20 and RNF40 are the same
(42).
p53 activity is regulated by numerous
post-translational modifications associated with protein
chaperones, regulatory factors and chromatin remodelers (43). During the genotoxic stress response,
p53 induces the transcription of a series of target genes, such as
p21, growth arrest and DNA damage 45, Mdm2 proto-oncogene and PUMA
(44). In the present study, RNF20
and RNF40 were demonstrated to be essential for maintaining p53
protein and mRNA expression levels, as well as those of its
downstream genes, following DNA damage induction. Therefore, the
RNF20/RNF40/WAC complex may be an important regulator of gene
transcription associated with DNA damage response. Mutated p53
loses the function of cell cycle arrest, induction of apoptosis,
mediation of cell senescence, repair of mismatched DNA bases and
preservation of genomic stability (31). In addition, mutated p53 acquires a
series of functions similar to those of oncogenes, such as
transcription of target genes to accelerate cancer progression,
enhancement of cancer cell chemical resistance and prevention of
apoptosis; such mutations are termed gain-of-function (GOF)
mutations (45). The results of the
present study demonstrated that the R282 mutation, which was
identified to be at the key binding site of p53 to RNF20 and RNF40,
eliminated the induction of the p53 target genes. Thus, RNF20
cannot adequately maintain the stability of p53 and its downstream
genes. However, the detailed mechanism remains unclear.
Previous studies have demonstrated that the R282W
mutation is an important cause of p53 GOF, since it alters the
protein-protein interaction ability and DNA-binding function of p53
(46–48). This p53 GOF mutant interacts with p63
and p73, which is involved in chemoresistance and anticancer drug
metabolism through cytochrome P450 3A4 induction (49). The R282W mutation also contributes to
the epithelial-mesenchymal transition by suppressing Kruppel-like
factor 17 and promotes cancer cell invasion by microRNA-155
induction (50,51). The R282 mutation has also been
identified in patients with Li-Fraumeni syndrome and is enriched in
bone tumors compared with bone tissues from healthy subjects
(52).
Post-translational modification of proteins is a
common mechanism in various cell signaling pathways in eukaryotic
cells (43). For instance, poly(ADP)
ribose polymerase 1-mediated poly-ribosylation is an essential
component of base excision repair pathways, and ataxia
telangiectasia and Rad3-related protein/ataxia telangiectasia
mutated-mediated phosphorylation of protein kinase checkpoint
kinase 1 (CHK1)/CHK2 are required in DNA double-strand break repair
pathways (43,53). Ubiquitination serves a crucial role
in the cell cycle and proliferation, regulating the cellular levels
of cytokines and coordinating oncogene transcription and the DNA
damage response (22). According to
a recent study, defects in ubiquitination are associated with human
malignancies, such as ovarian, breast and colorectal cancer
(26,54,55).
Hooda et al (55) have
reported that RNF20 deficiency contributes to high-grade serous
ovarian carcinoma initiation, and a study by Tarcic et al
(26) revealed that RNF20 represses
NF-κB and its downstream inflammatory cytokines, which inhibits
cancer cells proliferation and migration in basal-like breast
tumors. Tarcic et al (54)
have also reported that RNF20+/− mice are predisposed to
inflammation-associated colorectal cancer, and tissues from human
colorectal tumors exhibit downregulation of RNF20/RNF40 and H2Bub1
in both the epithelium and the stroma. Therefore, mutations or low
expression levels of the RNF20/RNF40/WAC complex, along with the
dysregulation of deubiquitinases and cyclin-dependent kinases have
been reported to affect tumorigenic pathways (55). Taken together, the R282W mutation and
the RNF20/RNF40/WAC complex are associated with the clinical
prognosis of patients with colorectal or breast cancer (47,56).
Previous studies have hypothesized that decitabine may maintain
RNF20 expression to restore H2Bub1 expression levels for treating
primary breast carcinoma (22,57).
Furthermore, proteasome inhibitors may be considered for the
treatment of early stage tumors (58,59). In
addition, the p53 R282 mutation has been predicted to be a
potential biomarker for cancer prognosis, as it is associated with
radioresistance, and patients with bladder cancer harboring the p53
R282 mutation have been shown to exhibit a shorter survival time
compared with that in patients with nonsense mutations (47,60,61).
However, whether the p53 R282W mutant and the RNF20/RNF40/WAC
complex collaboratively contribute to tumor development and
progression requires further investigation.
Acknowledgements
The authors would like to thank Miss Jie Ren and
Miss Yuyu Jiang (Shanghai Normal University, Shanghai, China) for
revising the manuscript.
Funding
This study was supported by The National Natural
Science Foundation of China (grant nos. 81572775 and 81773004 to
FZ) and the Program for Professor of Special Appointment (Eastern
Scholar) at Shanghai Institutions of Higher Learning (grant no.
TP2014055 to FZ).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding authors on reasonable
request.
Authors' contributions
DM, KG and DZ performed the experiments and created
the figures. DM wrote the manuscript. CZ contributed to the
analysis and interpretation of the data and the revision of the
manuscript. CS and FZ conceived the project, designed the
experiments, and analyzed and interpreted the data. DM, KG and FZ
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–370. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53. Annu Rev Biochem. 77:557–582.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53 and cancer-associated mutants.
Adv Cancer Res. 97:1–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ozaki T and Nakagawara A: Role of p53 in
Cell Death and Human Cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Joerger AC and Fersht AR:
Structure-function-rescue: The diverse nature of common p53 cancer
mutants. Oncogene. 26:2226–2242. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Allen MA, Andrysik Z, Dengler VL, Mellert
HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X,
Kraus WL, et al: Global analysis of p53-regulated transcription
identifies its direct targets and unexpected regulatory mechanisms.
eLife. 3:e022002014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dai C and Gu W: p53 post-translational
modification: Deregulated in tumorigenesis. Trends Mol Med.
16:528–536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bernard M, Yang B, Migneault F, Turgeon J,
Dieudé M, Olivier MA, Cardin GB, El-Diwany M, Underwood K, Rodier
F, et al: Autophagy drives fibroblast senescence through MTORC2
regulation. Autophagy. 16:2004–2016. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ho CJ, Lin RW, Zhu WH, Wen TK, Hu CJ, Lee
YL, Hung TI and Wang C: Transcription-independent and -dependent
p53-mediated apoptosis in response to genotoxic and non-genotoxic
stress. Cell Death Discov. 5:1312019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Follis AV, Llambi F, Ou L, Baran K, Green
DR and Kriwacki RW: The DNA-binding domain mediates both nuclear
and cytosolic functions of p53. Nat Struct Mol Biol. 21:535–543.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mrakovcic M and Frohlich LF: p53-mediated
molecular control of autophagy in tumor cells. Biomolecules.
8:142018. View Article : Google Scholar
|
|
13
|
Clapier CR and Cairns BR: The biology of
chromatin remodeling complexes. Annu Rev Biochem. 78:273–304. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cole AJ, Clifton-Bligh R and Marsh DJ:
Histone H2B monoubiquitination: Roles to play in human malignancy.
Endocr Relat Cancer. 22:T19–T33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chandrasekharan MB, Huang F and Sun ZW:
Histone H2B ubiquitination and beyond: Regulation of nucleosome
stability, chromatin dynamics and the trans-histone H3 methylation.
Epigenetics. 5:460–468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hammond-Martel I, Yu H and Affar B: Roles
of ubiquitin signaling in transcription regulation. Cell Signal.
24:410–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Buetow L and Huang DT: Structural insights
into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev
Mol Cell Biol. 17:626–642. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shiloh Y, Shema E, Moyal L and Oren M:
RNF20-RNF40: A ubiquitin-driven link between gene expression and
the DNA damage response. FEBS Lett. 585:2795–2802. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang K, Wang J, Tong TR, Wu X, Nelson R,
Yuan YC, Reno T, Liu Z, Yun X, Kim JY, et al: Loss of H2B
monoubiquitination is associated with poor-differentiation and
enhanced malignancy of lung adenocarcinoma. Int J Cancer.
141:766–777. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shema E, Tirosh I, Aylon Y, Huang J, Ye C,
Moskovits N, Raver-Shapira N, Minsky N, Pirngruber J, Tarcic G, et
al: The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a
putative tumor suppressor through selective regulation of gene
expression. Genes Dev. 22:2664–2676. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang F and Yu X: WAC, a functional
partner of RNF20/40, regulates histone H2B ubiquitination and gene
transcription. Mol Cell. 41:384–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnsen SA: The enigmatic role of H2Bub1
in cancer. FEBS Lett. 586:1592–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sethi G, Shanmugam MK, Arfuso F and Kumar
AP: Role of RNF20 in cancer development and progression - a
comprehensive review. Biosci Rep. 38:BSR201712872018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chernikova SB, Razorenova OV, Higgins JP,
Sishc BJ, Nicolau M, Dorth JA, Chernikova DA, Kwok S, Brooks JD,
Bailey SM, et al: Deficiency in mammalian histone H2B ubiquitin
ligase Bre1 (Rnf20/Rnf40) leads to replication stress and
chromosomal instability. Cancer Res. 72:2111–2119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JH, Jeon YG, Lee KH, Lee HW, Park J,
Jang H, Kang M, Lee HS, Cho HJ, Nam DH, et al: RNF20 Suppresses
Tumorigenesis by Inhibiting the SREBP1c-PTTG1 Axis in Kidney
Cancer. Mol Cell Biol. 37:e00265–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tarcic O, Granit RZ, Pateras IS, Masury H,
Maly B, Zwang Y, Yarden Y, Gorgoulis VG, Pikarsky E, Ben-Porath I,
et al: RNF20 and histone H2B ubiquitylation exert opposing effects
in Basal-Like versus luminal breast cancer. Cell Death Differ.
24:694–704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu C, Cui Y, Liu X, Zhang F, Lu LY and Yu
X: The RNF20/40 complex regulates p53-dependent gene transcription
and mRNA splicing. J Mol Cell Biol. 12:113–124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tzin V, Rogachev I, Meir S, Moyal Ben Zvi
M, Masci T, Vainstein A, Aharoni A and Galili G: Tomato fruits
expressing a bacterial feedback-insensitive
3-deoxy-D-arabino-heptulosonate 7-phosphate synthase of the
shikimate pathway possess enhanced levels of multiple specialized
metabolites and upgraded aroma. J Exp Bot. 64:4441–4452. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kitzman JO, Starita LM, Lo RS, Fields S
and Shendure J: Massively parallel single-amino-acid mutagenesis.
Nat Methods. 12:203–206, 4 p following 206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stiewe T and Haran TE: How mutations shape
p53 interactions with the genome to promote tumorigenesis and drug
resistance. Drug Resist Updat. 38:27–43. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shema E, Tirosh I, Aylon Y, Huang J, Ye C,
Moskovits N, Raver-Shapira N, Minsky N, Pirngruber J, Tarcic G, et
al: Corrigendum: The histone H2B-specific ubiquitin ligase
RNF20/hBRE1 acts as a putative tumor suppressor through selective
regulation of gene expression. Genes Dev. 31:19262017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hwang WW, Venkatasubrahmanyam S,
Ianculescu AG, Tong A, Boone C and Madhani HD: A conserved RING
finger protein required for histone H2B monoubiquitination and cell
size control. Mol Cell. 11:261–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakamura K, Kato A, Kobayashi J,
Yanagihara H, Sakamoto S, Oliveira DV, Shimada M, Tauchi H, Suzuki
H, Tashiro S, et al: Regulation of homologous recombination by
RNF20-dependent H2B ubiquitination. Mol Cell. 41:515–528. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kao CF, Hillyer C, Tsukuda T, Henry K,
Berger S and Osley MA: Rad6 plays a role in transcriptional
activation through ubiquitylation of histone H2B. Genes Dev.
18:184–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang Q, Su Z, Gu W and Rustgi AK: Mutant
p53 on the path to metastasis. Trends Cancer. 6:62–73. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vousden KH and Prives C: Blinded by the
Light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Zhang J, Tong JHM, Chan AWH, Yu J,
Kang W and To KF: Targeting the oncogenic p53 mutants in colorectal
cancer and other solid tumors. Int J Mol Sci. 20:59992019.
View Article : Google Scholar
|
|
39
|
Hanel W, Marchenko N, Xu S, Yu SX, Weng W
and Moll U: Two hot spot mutant p53 mouse models display
differential gain of function in tumorigenesis. Cell Death Differ.
20:898–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cho Y, Gorina S, Jeffrey PD and Pavletich
NP: Crystal structure of a p53 tumor suppressor-DNA complex:
Understanding tumorigenic mutations. Science. 265:346–355. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kumar P and Wolberger C: Structure of the
yeast Bre1 RING domain. Proteins. 83:1185–1190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shanmugam MK, Arfuso F, Arumugam S,
Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS,
Sethi G, et al: Role of novel histone modifications in cancer.
Oncotarget. 9:11414–11426. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Menendez D, Nguyen TA, Freudenberg JM,
Mathew VJ, Anderson CW, Jothi R and Resnick MA: Diverse stresses
dramatically alter genome-wide p53 binding and transactivation
landscape in human cancer cells. Nucleic Acids Res. 41:7286–7301.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stein Y, Rotter V and Aloni-Grinstein R:
Gain-of-function mutant p53: All the roads lead to tumorigenesis.
Int J Mol Sci. 20:61972019. View Article : Google Scholar
|
|
46
|
Zhang Y, Coillie SV, Fang JY and Xu J:
Gain of function of mutant p53: R282W on the peak? Oncogenesis.
5:e1962016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu J, Wang J, Hu Y, Qian J, Xu B, Chen H,
Zou W and Fang JY: Unequal prognostic potentials of p53
gain-of-function mutations in human cancers associate with
drug-metabolizing activity. Cell Death Dis. 5:e11082014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Calhoun S and Daggett V: Structural
effects of the L145Q, V157F, and R282W cancer-associated mutations
in the p53 DNA-binding core domain. Biochemistry. 50:5345–5353.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Deyoung MP and Ellisen LW: p63 and p73 in
human cancer: Defining the network. Oncogene. 26:5169–5183. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ali A, Shah AS and Ahmad A:
Gain-of-function of mutant p53: Mutant p53 enhances cancer
progression by inhibiting KLF17 expression in invasive breast
carcinoma cells. Cancer Lett. 354:87–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Neilsen PM, Noll JE, Mattiske S, Bracken
CP, Gregory PA, Schulz RB, Lim SP, Kumar R, Suetani RJ, Goodall GJ,
et al: Mutant p53 drives invasion in breast tumors through
up-regulation of miR-155. Oncogene. 32:2992–3000. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu J, Qian J, Hu Y, Wang J, Zhou X, Chen H
and Fang JY: Heterogeneity of Li-Fraumeni syndrome links to unequal
gain-of-function effects of p53 mutations. Sci Rep. 4:42232014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Y and Hunter T: Roles of Chk1 in
cell biology and cancer therapy. Int J Cancer. 134:1013–1023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tarcic O, Pateras IS, Cooks T, Shema E,
Kanterman J, Ashkenazi H, Boocholez H, Hubert A, Rotkopf R,
Baniyash M, et al: RNF20 Links histone H2B ubiquitylation with
inflammation and inflammation-associated cancer. Cell Rep.
14:1462–1476. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hooda J, Novak M, Salomon MP, Matsuba C,
Ramos RI, MacDuffie E, Song M, Hirsch MS, Lester J, Parkash V, et
al: Early loss of histone H2B monoubiquitylation alters chromatin
accessibility and activates key immune pathways that facilitate
progression of ovarian cancer. Cancer Res. 79:760–772. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun
S and Pang D: Elevated expression of USP22 in correlation with poor
prognosis in patients with invasive breast cancer. J Cancer Res
Clin Oncol. 137:1245–1253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Minsky N, Shema E, Field Y, Schuster M,
Segal E and Oren M: Monoubiquitinated H2B is associated with the
transcribed region of highly expressed genes in human cells. Nat
Cell Biol. 10:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Prenzel T, Begus-Nahrmann Y, Kramer F,
Hennion M, Hsu C, Gorsler T, Hintermair C, Eick D, Kremmer E,
Simons M, et al: Estrogen-dependent gene transcription in human
breast cancer cells relies upon proteasome-dependent
monoubiquitination of histone H2B. Cancer Res. 71:5739–5753. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mimnaugh EG, Chen HY, Davie JR, Celis JE
and Neckers L: Rapid deubiquitination of nucleosomal histones in
human tumor cells caused by proteasome inhibitors and stress
response inducers: Effects on replication, transcription,
translation, and the cellular stress response. Biochemistry.
36:14418–14429. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Skinner HD, Sandulache VC, Ow TJ, Meyn RE,
Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK and Myers JN:
TP53 disruptive mutations lead to head and neck cancer treatment
failure through inhibition of radiation-induced senescence. Clin
Cancer Res. 18:290–300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Okaichi K, Ide-Kanematsu M, Izumi N,
Morita N, Okumura Y and Ihara M: Variations in sensitivity to
ionizing radiation in relation to p53 mutation point. Anticancer
Res. 28((5A)): 2687–2690. 2008.PubMed/NCBI
|