Background of AhR research
The study of AhR can be discussed from two
standpoints; the first one reflects the reality of current times,
that is, human exposure to synthetic organic compounds and the
consequences that has on human health. During the 1970s, the
studies of several toxicologists, biochemists and molecular
biologists focused on the toxic effects of
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a polychlorinated
dibenzo-p-dioxin that was identified as an unintentional by-product
of the herbicide 2,4,5-trichlorophenoxyacetic acid synthesis
(1). Individuals who worked in the
manufacturing of this herbicide suffered diseases such as porphyria
cutanea tarda and chloracne (2). It
was proven by a later study that TCDD exposure was the cause of
porphyria in such workers, which acted by increasing the activity
of the initial enzyme in heme biosynthesis, δ-aminolevulinic acid
synthetase (3).
The second standpoint is the rather accidental
finding of certain studies from the early 1950s showing that tumor
development was inhibited in rats exposed to the carcinogen
3-methylcholanthrene (3-MC) when it was administrated
simultaneously with other carcinogens (4). It was later proven that this inhibition
of carcinogenesis can be induced not only by 3-MC, but also by a
great variety of polycyclic aromatic hydrocarbons (PAH), as these
compounds impede the action of an enzyme that modifies carcinogens,
nowadays known as cytochrome P450 family 1 subfamily A member 1
(CYP1A1), a member of the cytochrome P450 family (5). Later, in 1969, that modifying activity
was named Ah hydroxylase (AHH) and certain studies revealed that in
some, but not all, syngeneic strains of mice, this enzyme activity
was induced by PAHs (6,7), which suggested the existence of a gene
that controls AHH activity, termed the Ah locus (8,9). The Ah
locus was then found to be involved in the regulation of
carcinogenicity, mutagenicity and toxic responses to PAHs (10). This created an opportunity to examine
other types of toxic compounds, such as TCDD and 3-MC; the results
showed that TCDD was 30,000 times more potent in inducing AHH
activity than 3-MC (11). Therefore,
TCDD became the ideal molecule for testing the activity of the Ah
locus (12).
The study of steroid receptors was also increasing
at the time; with this in mind, the idea of a ‘receptor’ that
controls the Ah locus emerged, which may also explain the greater
affinity for certain compounds, such as TCDD, over others, such as
3-MCA (13). The first radioactively
labeled TCDD [(3H)TCDD)] was synthetized, and finally the existence
of a receptor was confirmed in 1979 and the term AHR was used for
the first time (14). Unexpectedly,
only a fraction of (3H)TCDD bound to the receptor in the cytoplasm
as expected, but another portion bound to the receptor in the
nucleus, as described for the steroid receptors. Shortly after AHR
discovery, it was determined that the weight of the receptor varied
depending on its origin; when it was isolated from the cytoplasm it
was heavier than when found in the nucleus (15,16).
This fact aroused interest regarding other proteins associated with
the receptor, and their role in its function. A few years later, a
protein was discovered that formed a dimer with AHR in the nucleus,
which was named the AHR nuclear receptor translocator (ARNT)
(17). Finally, it was confirmed
that the formation of the TCDD-AHR-ARNT complex was indispensable
for the induction of AHH activity (18). In 1986, a nucleotide sequence,
5′-TNGCGTG-3′, to which the TCDD-AHR-ARNT complex bound to induce
the AHH activity, was identified and named dioxin response element
(19). Subsequently, in Japan,
studies were conducted using other xenobiotic compounds. These
studies found that the xenobiotic-AHR-ARNT complex bound to the
same sequence reported before, which was then renamed xenobiotic
response elements (XRE); today it is also known as Ah response
elements, a term used less often due to its similarity to the
antioxidant response elements (AREs) (20,21).
From that moment forward, the expression of CYP1A1 in response to
natural compounds, drugs and other xenobiotics in general, has been
used as an indirect evaluation of the participation of AHR, and
therefore xenobiotic metabolism. However, it was subsequently
recognized that these XRE sequences were found in a large number of
gene promoters, and not only in CYP1A1. Nowadays, it is known that
the function of AHR extends far beyond xenobiotic metabolism; it
actually functions as a master regulator to control several
biological processes, including cell proliferation, adhesion,
differentiation and death, potentially among others not yet known
(22).
A glance at AHR molecular
features
In 1994, the human AHR promoter was cloned,
and its main characteristics were described. First, this promoter
was not found to contain a TATA box; instead, several binding
motifs were identified, including multiple GC boxes, which act as
binding sites for the transcription factor specificity protein 1
(Sp1). The AHR promoter also possesses binding motifs for
the transcription factor cAMP response elements and E-box, the last
E-box is recognized by c-Myc (23).
Furthermore, it has been described that distal-less 3, a homeobox
transcription factor of importance during development in
vertebrates, also binds to a portion of the AHR promoter and
enhances the transcription factor activity at the XRE sites
(24). In addition, AHR possesses
binding sites for signal transducer and activator of transcription
6 (STAT6), which belongs to the family of the transcription factors
associated with the activity of cytokines such as interleukin
(IL)-4 and IL-13, and growth factors such as transforming growth
factor-β (TGF-β) (25). The
AHR promoter also possesses motifs to bind T-cell
factor/lymphoid enhancer-binding factor (TCF/LEF), factors that are
involved in the Wnt pathway by interacting with β-catenin (26). Finally, the AHR promoter was
also found to have 11 cis nuclear receptor binding sites,
which include progesterone, androgen, glucocorticoid,
proliferation-activated peroxisome, farsenoid X and the vitamin D
receptors. The existence of a complete list of the AHR
promoter characteristics enabled the understanding of the dual
activity of AHR, with the constitutive one being associated with
embryogenesis and fetal development when the receptor activity is
particularly critical, and the second with specific tissue
expression (27).
All these characteristics are conserved among the
human and murine AHR sequences, with the main difference
between them being the mRNA length, which is longer in humans (~6.6
kb) than in mice (5.0–5.4 kb). The open reading frame has 11 exons,
organized to form a mature mRNA, with 28 domains in humans and 26
in mice (28). Focusing on the AHR
domains, this receptor is a member of the basic Helix-Loop-Helix
(bHLH) superfamily of transcriptional regulators. The members of
this family are involved in critical developmental processes,
including sex determination and the development of the nervous
system and muscles. Like other members of this superfamily of
proteins, it contains a binding region to DNA at the amino-terminal
end and an additional Per-Arnt-Sim (PAS) domain at the
carboxy-terminal (29,30). The region of the basic residues is
important for the interaction of AHR with the cis sequence
of the XRE, while the bHLH motif is important for the
heterodimerization between AHR and ARNT (31,32).
AHR-associated proteins
AHR research was initially based only on its
exposure to or interaction with TCDD, but the molecular structure
of the AHR protein was unknown. In the cytosolic fraction, AHR
exhibited a higher sedimentation value, which upon the addition of
TCDD, was found to be decreased and located instead in the nuclear
fraction (33). This finding
revealed the existence of two different forms of the receptor,
depending on cellular localization. It was shown
electrophoretically in subsequent studies that this weight
difference was due to the fact that the cytoplasm receptor was
found in a protein complex that included 2 isoforms of mouse heat
shock protein of 90 kDa (Hsp90) and an X-associated protein 2, also
known as AHR-interacting protein (AIP) or AHR-associated protein 9
(ARA9) (34–36). The proteins in this complex are
important for the function of the AHR. The interaction between
Hsp90 and AHR occurs in the PAS-B motif; this allows ligand binding
to the receptor. In addition, AIP allows for protein-protein
interaction (37). Once in the
nucleus, the AHR protein undergoes degradation by the 26S
proteasome (38,39) (Fig.
1), an important site for the degradation of other
transcription factors, including TGF-β (40) and myoblast determination protein 1
(41).
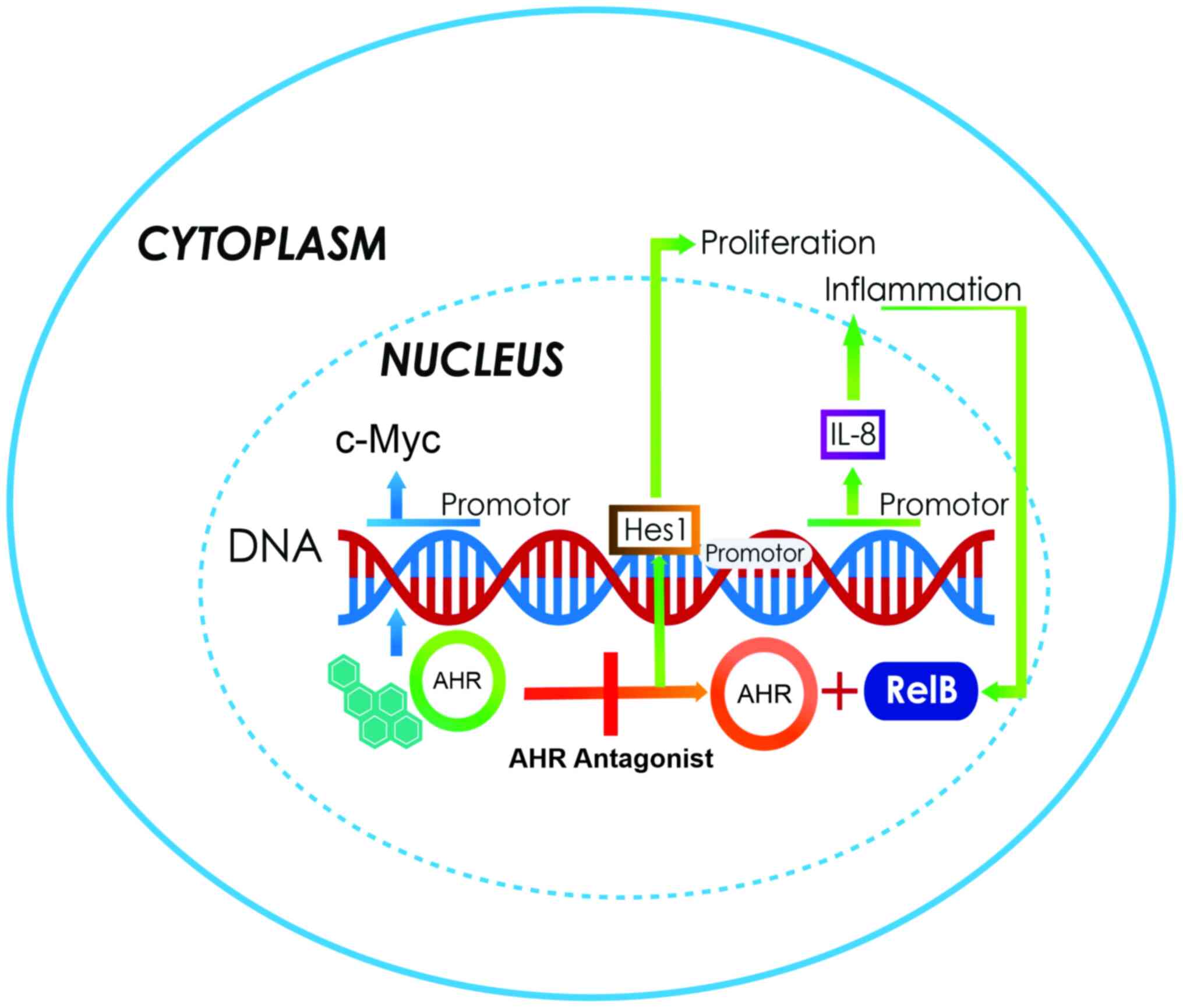
 | Figure 1.Canonical activation of the AhR
pathway. In the cytoplasm, AHR resides in a molecular complex, to
give it stability (A); this complex is formed with two Hsp90
proteins, AIP and p23. Following ligand binding, AHR dissociates
from the complex and translocates to the nucleus (B). Inside the
nucleus, AHR dimerizes with ARNT (green arrows) to form a
heterodimer that binds to the XRE sites on the gene promoters
involved in xenobiotic metabolism (C). Following the activation of
response genes, AHR becomes the target of the ubiquitin 3-ligase
(D) and undergoes degradation by the 26S proteasome in the nucleus
(E). The activation of the non-canonical pathway (orange arrows) is
performed through the binding of AHR to other proteins, such as
pRB, RelA or RelB. In this case, AHR and RelB together bind to
other genes with an XRE cis site in their promoter, and
activate several genes that participate in growth, differentiation,
metabolism, the cell cycle, cell adhesion, apoptosis, immune
response and inflammation (F). AHR, aryl hydrocarbon receptor;
Hsp90, heat shock protein 90; AIP, AHR-interacting protein; ARNT,
AHR nuclear receptor translocator; XRE, xenobiotic response
elements; pRB, retinoblastoma. |
Canonical AhR pathway
To further understand the activation of the AhR
canonical pathway (Fig. 1), a strong
focus must be placed on the detoxification mechanism. This pathway
begins in the cytoplasm with the binding of a ligand to AHR, which
leads not only to a conformational change in AHR that exposes a
nuclear localization signal (NLS), but also to the dissociation of
Hsp90 from the complex, which enables the nuclear translocation
promoted by the action of importins (42). Once in the nucleus, AHR dimerizes
with its partner protein, ARNT, which is also a member of the bHLH
family. The dimerization of AHR and ARNT is performed through the
HLH domains of both proteins (43,44), and
a conformational change in the PAS A region helps stabilize this
union (45). In addition, the
phosphorylation of two regions in the carboxy-terminal of AHR via
the protein kinase C is an important step for DNA binding (46). Once the AHR/ARNT heterodimer is
formed, it binds to promoter regions of target genes that contain
the XRE consensus sequence 5′-TNGCGTG-3′; AHR binds to the
T/NGC5′-half-site, while ARNT binds to the GTG3′-half-site. This
sequence is present in several genes, such as cytochromes; CYP1A1
contains 8 sites, CYP1A2 contains 1 and CYP1B1 contains 3 (47,48).
There is also an exceptional case, the poly/ADP-ribose polymerase,
which contains 16 XRE cis sequences (49). Due to the vast number of studies on
gene expression though AHR activation, several genes with XREs
sequences have now been reported (50). Some of these genes are involved in
xenobiotic metabolism, including phase I genes such as CYP1A1,
CYP1A2, CYP1B1, CYP2A5 and CYP4B1, and phase II genes
such as aldehyde dehydrogenase 3 family member A1,
glutathione-S-transferase (GST), NAD(P)H-quinone
oxidoreductase-1 (NQO1), UDP glucuronosyltransferase 1A1
(UGT1A1) and UGT1A6. Other genes involved in cell
cycle regulation include suppressors such as cyclin dependent
kinase inhibitor 1A (CDKN1; also known as p21), CDKN4
(also known as p27), retinoblastoma tumor suppressor protein
(pRB) and Kruppel-like factor 6 (KLF6), and
activators such as proto-oncogenes c-Jun and c-Myc. Other genes
involved in other signaling pathways include insulin-like growth
factor (IGF) binding protein-1, apoptosis regulator
Bcl-2-associated X, cathepsin D, zinc finger protein slug, nuclear
factor κB subunit 1 and vascular endothelial growth factor A
(VEGF) (51–55).
The first two groups (xenobiotic metabolism genes),
are the most studied in relation to AhR pathway activation. The
action of the cytochromes generates modifications in the xenobiotic
compounds that facilitate their degradation, thus reducing the
ligand concentrations in the cells. Most of the evidence currently
available has demonstrated that AHR can function as an exogenous
ligand sensor, since it belongs to a group of proteins that are
known to be environmental sensors, but several of its ligands are
compounds that appeared only recently in the human ecosystem (the
technosphere). It can therefore be assumed that the canonical AhR
pathway might be a response to the presence of ‘new’ toxic
compounds in the environment (56).
In this context, it is important to recognize the two major
functions of AHR: Xenobiotic metabolism (detoxification) and its
physiological role (development). The high interest in studying
this receptor is not only fueled by its ‘normal’ function, but also
its interaction with several other proteins.
Direct interactions between AHR and other
proteins
pRB
One of the proteins that directly interact with AHR
is pRB. This interaction occurs in the absence of ARNT (57). Two binding sites for pRB could be
identified at the AHR sequence. The AHR-pRB complex functions as a
co-repressor that inhibits the progression of the cell cycle by
displacing the histone acetyl transferase p300 from E2F-dependent
promoters, consequently inhibiting the expression of
S-phase-specific genes (58,59). In addition, AhR activation induces
the expression of the cell cycle suppressors p21 and p27; the
association of these inhibitors with cyclin D1 or E inhibits
phosphorylation of pRB, and as a result, the cycle is blocked at
the G1 phase (60,61).
NF-κβ
NF-κβ is another example of a protein that interacts
directly with AHR in the absence of ARNT. Either NF-κβ subunit
(RelA or RelB) can be involved. Exposure to TCDD induces the
expression of IL-8 through a direct interaction between AHR and
RelB, as this complex binds to a sequence very similar to XRE
(5′-GGGTGCAT-3′) on the IL-8 promoter (62). However, TCDD is also responsible for
the interaction between AHR and RelA (63); this complex induces the expression of
IL-1β, tumor necrosis factor-α, IL-6 (64) and proto-oncogene c-Myc (65).
Nuclear factor-erythroid 2-related
factor 2 (NRF2)
The interaction between AHR and NRF2 has been widely
studied in recent times; however, for several years, these two
pathways were thought to be entirely separate. This was due to NRF2
being a transcription factor that regulates genes containing AREs
in their promoters. Several phase II genes of xenobiotic
metabolism, including NQO1, UGT1A1, UGT1A6 and GST,
have ARE sequences. Therefore, at first glance, exposure to TCDD
appears to activate detoxification via the AhR canonical pathway,
with no involvement of NRF2. The existence of a bidirectional
cross-talk at the genetic level between AHR and NRF2 has now been
well established. The NRF2 promoter has ≥1 XRE sequence and the
AHR promoter has several AREs (66,67).
Since NRF2 is a master regulator of antioxidant responses, the
metabolites generated by the xenobiotic metabolism yield the NRF2
activation to improve the detoxification efficiency.
Estradiol receptors (ERs)
Evidence has revealed the anti-estrogenic action of
TCDD-type ligands. The first indication of this action is the
modification activity of CYP1A1 and CYP1B1 on 17β-estradiol, and
the production of a hormonal ligand with no estrogenic activity
(68). Another indication is the
binding of the AHR/ARNT heterodimer to the cis-inhibiting
regions of the target response genes to the ER (69). Finally, the other molecular event
that can explain the anti-estrogenic activity of AhR activation is
the function of AHR as an E3 ubiquitin ligase towards the ERs that
induces their degradation in the nucleus through the proteasome
pathway (70).
Non-canonical AhR pathway
With great developments in microarray analysis
during the last few decades, new horizons have been opened up in
the field of AhR research. Upon analyzing the cis regions of
promoters and using chromatin immunoprecipitation, it was
discovered that certain genes that were regulated by AHR have
different sequences from those of the classical XREs; these
sequences are known as non-consensus XRE (NC-XRE) (71,72). One
example of these genes is the plasminogen-1 activator inhibitor
(PAI-1) (73). Certain
studies have shown that treatment with TCDD suppresses hepatic
regeneration, as PAI-1 inhibits the urokinase-type plasminogen
activator that is needed to activate the hepatic growth factor
(74). A common characteristic among
these non-consensus promotors is that they contain a repeated
tetranucleotide motif (5′-GGGA-3′); in these cases, the interaction
with ARNT is not necessary (75).
With regards to PAI-1, it is now known that the suppression of
hepatic regeneration is an arrest of cell proliferation caused by
the inhibition of CDK2 activity (76). This blockade depends on the
expression of kinase-dependent cyclin inhibitors such as p21 and
p27, which negatively regulate cell cycle progression by
controlling CDK activity. This regulation can take place due to the
fact that the p21 promoter contains NC-XRE cis regions
(61,77). Recent evidence has suggested that
KLF6 can form a heterodimer with AHR (78), which is able to bind to NC-XRE
cis regions, where several of these family factors can
interact. In fact, KLF4 and KLF6 can also regulate the expression
of CYP1A1 in this manner. Structurally, this interaction takes
place in the carboxy-terminal of AHR (where the bHLH and PAS-A
domains are found), which must bind to the amino terminal of KLF6
(79). This factor also regulates
numerous cellular processes, including proliferation,
differentiation and apoptosis (80).
Alterations in the expression of KLF6 are associated with various
types of cancer, including astrocytomas and gliomas (81). In addition, KLF6 can increase the
expression of p21, affecting cell cycle progression (82), as well as the expression of
E-cadherin genes, TGF-β1 and IGF1 receptor (83).
Potential therapeutic applications of the
crosstalk between AhR pathways and central nervous system (CNS)
tumors
Much of the knowledge regarding tumor growth is
based on stem cell biology and developmental programs, since
several signals are shared among these pathways. The new directed
molecular therapies are designed to inhibit different tumor
signaling pathways (84). For
example, relevant growth factor pathways are known to be involved
in malignant glioma, including platelet-derived growth factor,
epidermal growth factor, VEGF, hepatocyte growth factor (HGF) and
IGF (85). The physiological effects
of AhR activation have been suggested to play an important role in
the modulation of the immune system and carcinogenesis. AHR can
therefore regulate inflammatory response and cell-cycle progression
(86,87). AHR is expressed at high levels and is
chronically active in leukemia and lymphoma (88–90), as
well as in solid tumors such as glioblastoma, ovarian cancer
(91,92), lung cancer (93,94),
liver cancer (95), and head and
neck carcinomas (96). The role of
AhR in cancer is very complex and depends on tumor type. Evidence
has shown that the activated AhR pathway is associated with tumor
growth promotion, but there is also evidence of its
tumor-suppressive activity. Some of the potential therapeutic
applications of AHR activity in the most studied types of CNS
tumors (astrocytomas, medulloblastomas and neuroblastomas) are
explored in the next sections.
Astrocytomas
Also known as gliomas, astrocytomas are a large
group of different types of pediatric and adult tumors that develop
from glial cells; specifically, astrocytomas originate from
astrocytes, which are essential for the structure and support of
neurons. Traditionally, these tumor types were classified by the
World Health Organization (WHO) based only on histopathological
analysis; in fact, based on the presence or absence of marked
mitotic activity, necrosis and microvascular proliferation, tumors
were also classified by a WHO malignancy grading system: Grade II,
low grade; grade III anaplastic; grade IV, glioblastoma. Nowadays,
these tumor types have been reclassified based on their
histological and molecular features (97,98).
AhR research has provided evidence on how this
pathway can be targeted for therapeutic applications. Regarding
astrocytomas in particular, it has been reported that IL-6 induces
the transcriptional activation of VEGF, which, in turn,
participates in the induction of angiogenesis in this type of
tumor. As aforementioned, AHR can form a heterodimer with RelA to
trigger the activation of IL-6, which, finally, fosters de
novo angiogenesis-dependent tumor growth (99). In the context of astrocytic tumors,
for instance, it is well known that glioblastomas are characterized
by high expression of STAT6, a trans-acting factor
that can alter the expression of AHR and other cytokines,
such as IL-6 (Fig. 2). It is also
known that AHR regulates its own expression, which may contribute
not only to an increase in the expression of angiogenic factors,
but also to the production of other cytokines, such as TGF-β, which
also promotes tumor growth (100).
In fact, it is also well known that the high expression of
STAT6 in glioblastoma patients is correlated with lower
survival rates and has been identified as a potential prognostic
marker (101).
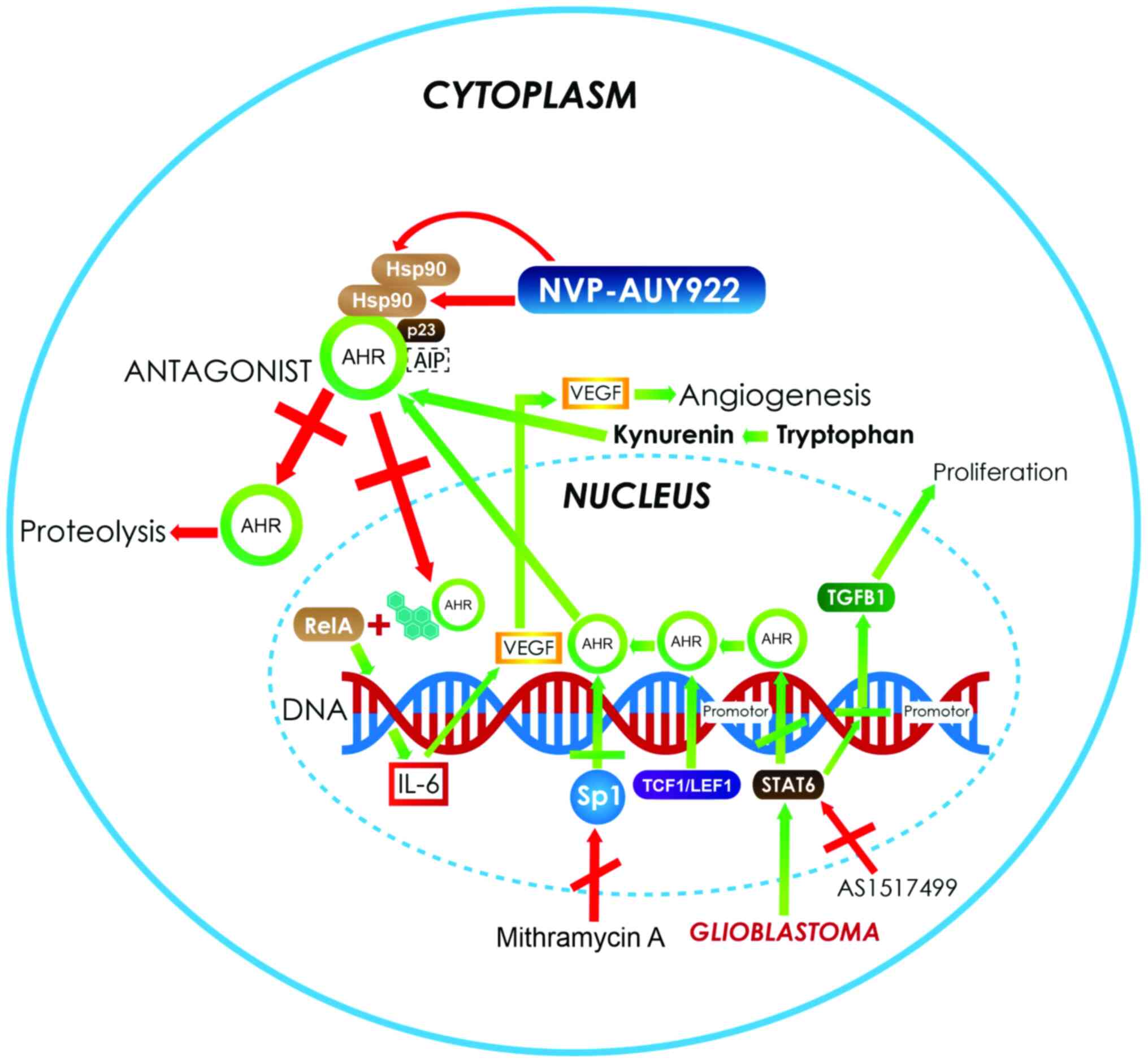 | Figure 2.In astrocytoma and glioblastoma, the
activation of the AhR pathway increases the expression of several
genes, such as VEGF and TGF-β1 (green arrows) that are involved in
angiogenesis and proliferation processes. In addition, the
overexpression of Sp1 activates the transcription of AHR,
increasing its protein levels. Moreover, there are AHR ligands,
such as tryptophan metabolites, produced by the kynurenine pathway
in central nervous system tumors such as astrocytoma (green
arrows), which also bind and activate the AhR pathway. The
strategies used to control the growth of neoplastic cells in
astrocytoma and glioblastoma (red arrows) mainly involve the use of
AHR antagonist. Another target for therapy is the use of
complex-associated protein inhibitors to induce the instability of
the receptor. An example of this is NVP-AUY922, which inhibits
Hsp90 and induces AHR degradation. Another example is the use of
inhibitors such as mithramycin A and AS1517499, which control the
autoinduction of AHR protein expression and stop reactive
responses. AHR, aryl hydrocarbon receptor; VEGF, vascular
endothelial growth factor A; TGF-β1, transforming growth factor-β;
Hsp90, heat shock protein 90; Sp1, specificity protein 1;
TCF1/LEF1, T-cell factor/lymphoid enhancer-binding factor; AIP,
AHR-interacting protein; IL, interleukin. |
In addition to study of the canonical AhR pathway
and its activation by PAH-type ligands, research is underway to
establish an association between the activation produced by the
exposure to xenobiotic compounds and the development of CNS tumors.
The current hypothesis is that the genetic variants of the receptor
can modify and/or increase the risk of glioma development following
exposure to PAH, and thus establish the gene-environment
interaction that leads to the development of glioma (102). The first direct epidemiological
study to provide this type of evidence was that of Gu et al
(102), which consisted of a
case-control study of 384 glioma cases and 384 cancer-free controls
in a Chinese population. It was proposed that two polymorphisms of
the AhR may increase susceptibility to glioma development,
and hence to the formation PAH-DNA adducts, thus consequently
increasing the risk of carcinogenesis in glial cells. This was
evidenced by the staining of DNA-PAH adducts, which revealed an
association between staining intensity and glioma grade (102). A study showed that cigarette
smoking had no effect on glioma development, since smoking is not
the principal factor in PAH-DNA adduct formation; in fact, exposure
to air pollutants or the smoke generated by the burning of organic
material has greater effects on glioma formation than smoking
(103).
As aforementioned, changes in the expression of the
xenobiotic metabolizing genes, such as cytochromes CYP1A1
and CYP1A2, are produced by AhR activation. However,
exposure to an AHR agonist, 3,3′,4,4′,5-pentachlorobiphenyl
(penta-CB), a coplanar polychlorinated biphenyl, unexpectedly does
not cause a significant transcriptional activation of CYP1A1
in C6 glioma cell lines (104). In
addition, when comparing 4HIIB hepatoma and C6 glioma cell lines,
the overexpression of genes that participate in tumor promotion and
progression, protein processing, programmed cell death and/or
metastasis was identified in glioma cells (104). Moreover, in the C6 glioma cell
line, significant overexpression of galectin-1 was observed, which
is known to permit protein-protein interactions and may influence
the progression of the cell cycle and other cellular functions
(104). These data confirmed that
certain AHR ligands, such as penta-CB, causes tissue-selective
chromatin remodeling via histone deacetylase inhibitors; as a
result AhR induces the expression of specialized genes associated
with carcinogenesis in glioma cells (104). On the other hand, it is well known
that AhR and TGF-β signaling are mutually regulated in a
cell-specific manner (105).
Furthermore TGF-β is a crucial factor in the malignant phenotype of
glioblastomas and is also a downstream target of AhR signaling
(105). In addition AHR can
activate the expression of latent TGF-β binding protein 1 (LTBP-1),
a protein characterized as crucial in the TGF-β activation in
gliomas (Fig. 2), such that a
strengthened signal regulated by AHR is doubly established in
glioblastoma cells (105).
Consequently, the LTBP-1/TGF-β pathway in glioma cells promotes
proliferation, clonogenicity and invasiveness, and more
importantly, the use of an AhR antagonists, such as CH-223191 or
AHR gene silencing blocks these effects (105). Therefore, AHR antagonists may be
useful for managing and controlling glioma growth.
In addition, astrocytomas have been associated with
the high expression of Wnt signaling transcription factors such as
TCF-1 and LEF-1 (Fig. 2). It has
also been demonstrated that LEF-1 is capable of distinguishing
grade II and III astrocytomas from glioblastomas, and it may
therefore be considered an important marker of progression
(106). Considering this, along
with the fact that the AHR promoter has TCF/LEF binding
sites, it stands to reason that an increase in the expression of
AHR may also participate in astrocytoma progression
(Fig. 2). In all cases cited herein,
the application of AHR antagonists could have therapeutic effects;
such treatments could have the ability to reduce the synergistic
effects of AhR among other pathways and, perhaps, be able to
improve responses to surgery or chemotherapy. It has been shown,
for example, that the use of Hsp90 inhibitors, such as NVP-AUY922,
increases the cytotoxic effect of ionizing radiation in different
cancer cell lines, including glioblastoma cell lines (107). Notably, two Hsp90 proteins are part
of the AHR complex and play an important role in the stabilization
and structure of the active receptor. The use of this inhibitor
would not allow Hsp90 to bind to AHR in the correct way, thus
leaving the receptor labile in the cytoplasm, where it is a target
for degradation (Fig. 2).
Otherwise, in the context of tans factors
such as Sp1, which is known to be increased in glioblastoma cell
lines, these in fact bind to GC-rich cis regions in the
AHR promoter, therefore increasing receptor transcription
and protein level. Mithramycin A is a chemotherapeutic agent used
in the treatment of solid tumors (Fig.
2); it has been shown to be an inhibitor of Sp1 and to reduce
the secretion of metalloproteinases in astrocytoma cell lines, thus
reducing the production of VEGF and, as a result, decreasing glioma
cell migration (108). This
mechanism may be a consequence of the low expression of AHR,
which, in turn, reduces the levels of IL-6, finally leading to a
decrease in VEGF expression (99).
Another way to control the inductive effect of AhR is through the
use of STAT6 inhibitors such as AS1517499, which, synergistically
with AHR antagonists, reduce the production of the receptor, thus
controlling the effects of angiogenesis and cell proliferation
(Fig. 2) (109). Functional, genomic and molecular
studies have confirmed that the endogenous expression of AHR
protects against glioblastoma cell invasion and growth. Using
CRISPR/Cas9 in the U87 cell line and patient-derived cells to
stably knockout AHR expression or downregulate expression
using RNA interference against AHR, resulted in an increase in cell
invasion in Boyden chamber and 3D tumor spheroid assays, and also
enhanced cell migration in scratch assays. These results confirmed
that AHR exhibits a tumor-suppressive role in glioblastoma cells
and functions as an inhibitor of glioblastoma cell invasion
(110). These findings are of
extreme importance, since they revealed the endogenous function of
AHR when used as a therapeutic target. Notably, ligands are only
used to modify the expression patterns and not to completely block
its activity.
Medulloblastomas
Medulloblastomas are primary cerebellar tumors and
the most common type of malignant brain tumor in children with a
global incidence of 0.49 per 100,000, accounting for ~20% of all
pediatric tumors of the CNS and 64.9% of all embryonal tumors in
children and adolescents (age 0–19 years) in 2008–2016 (111–113).
The medulloblastomas cell origin remains elusive, but is thought
that they originate from abnormally proliferating cerebellar
granule neuron precursors (GNPs) and/or multipotent neural stem
cells (NSCs) (114,115). These types of tumor occur
exclusively in the posterior fossa, and their typical treatment
consists of a combination of chemotherapy and surgical resection
(116,117). Medulloblastoma survivors suffer
sequelae, including cognitive deficits, and problems with
neuroendocrine functions and fertility (118,119).
Different treatment options are therefore required. Recent insights
into the biology of medulloblastoma have revealed molecular
features that improve its categorization into molecular subgroups:
Wnt, sonic hedgehog (Shh), group 3 and group 4 (both classified as
non-Wnt/Shh) (120). However, this
classification has not yet been used for risk stratification in
clinical trials.
GNPs express high levels of AHR in the
external germinal layer of the developing cerebellum, with the
abnormal activation or deletion of AHR leading to the
dysregulation of the GNP cell cycle and maturation. A stable
AHR-knockdown in a DAOY medulloblastoma cell line revealed
an impaired G1 to S transition, decreased DNA synthesis
and reduced proliferation. These effects are also correlated with
the decreased levels of the proliferative gene Hes1 and increased
levels of the cell cycle inhibitor p27. All the alterations were
reversed following the supplementation of human AHR. These results
demonstrated that the abnormal activation or suppression of AHR
could dysregulate the GNP cell cycle and promote the proliferation
of medulloblastoma cells (121).
c-Myc is known to be significantly involved in the generation of
the malignant properties of medulloblastoma cells. This
carcinogenic process also has a synergistic action with HGF
expression, which contributes to the process of becoming malignant.
Despite the fact that AHR contains in its promoter sequence
a cis E-box, it is highly plausible that the overexpression
of c-Myc increases the expression of AHR, exerting effects
that may favor cell growth and proliferation (122). In these cases, the use of AHR
antagonists will be useful for controlling cell proliferation
(Fig. 3).
With regards to Shh medulloblastoma in particular,
some cells are cancer-propagating cells (CPCs) that express SOX2;
this signal is essential for tumor stem cell maintenance. These
cells do not lose their proliferation capacity following
anti-mitotic chemotherapy and are eventually responsible for tumor
relapse. The AHR function has recently been linked to CPCs and
tumor stem cell maintenance; in fact, AHR was shown to regulate the
balance between quiescence and proliferation. This was demonstrated
in AHR-deficient animals, which exhibited decreased quiescence and
increased tumor stem cell proliferation (123). This finding suggested an important
tumor-suppressive role of AHR in mouse Shh medulloblastoma.
Neuroblastomas
Neuroblastoma is a type of cancer that occurs in
young children and starts early during embryonic or fetal
development, the median age at diagnosis is between 16 and 24
months (124,125). Neuroblastoma is the most frequent
type of solid extracranial tumor in children which represented 3–8%
of childhood malignancies worldwide in 2001–2010 (126,127).
This tumor type is derived from early nerve cells called
neuroblasts, most of which develop in the adrenal glands; however,
some of them can expand to other areas, such as the thorax, spinal
column, medulla or abdomen (128,129).
In vitro studies have revealed that the overexpression of
AHR induces cell differentiation, and that the expression of
the receptor is highly correlated with the histological grade of
differentiation of the tumors (130,131).
AHR was recently reported to be expressed in the cerebellar GNPs
during the early postnatal period, where it regulates the growth
and differentiation of granule neuroblasts (132). AHR-deficient mice have been shown
to display a diminished neuronal differentiation in the dentate
gyrus, with the knockout of AHR causing oculomotor and optic nerve
deficits in a mouse model (132).
This suggested that the overexpression of AHR promotes neural
differentiation in neuroblastoma cells. Antecedents in breast
cancer cells reveal that c-Myc, an oncogene whose promoter contains
6 XREs, is repressed by the constitutive expression of AHR
(133). This interaction may occur
due to the action of the E2F1 protein. It is not yet clear which
protein establishes a direct interaction with AHR on the promoter
of the c-Myc gene in neuroblastoma cells to produce this
repression. It is, however, plausible that it forms a co-repressor
complex upon interacting with E2F1, similar to the effect described
in MCF-7 breast cancer cells, where AHR interacted with the pRB
protein (134). A recent study has
shown that AHR plays an important role in neurogenesis and
differentiation, as aforementioned, since its receptor contains
cis binding sites for trans factors expressed in the
early stages of development and differentiation, such as
brain-specific homeobox/POU domain protein 3B (135).
Using an SK-N-SH NB cell line treated with
catabolites of the corticosterone tetrahydrocorticosterone (THB),
5α-THB or 5β-THB, for 3 days, the neuronal differentiation markers,
growth-associated protein 43 neurofilament heavy chain and
neuron-specific enolase were found to be upregulated. The mRNA
expression of SOX10 and MBP, which are early markers of myelinating
cells, was also found to be upregulated in the treated cells. These
results showed that 5α- and 5β-THB promote the expression of
neuronal and myelinating glial differentiation markers in SK-N-SH
NB cells, revealing a potential therapeutic use for 5α- and 5β-THB
in neuroblastoma (135). However,
the presence of a ligand such as TCDD interrupts neurogenesis.
Therefore, in neuroblastoma tumors, AHR acts as a tumor-suppressive
gene and promotes cell differentiation. A study has suggested that
the parents of children suffering from neuroblastoma were probably
exposed to xenobiotic-type AHR ligands during the prenatal period,
and that this suppression of neuronal development was the
consequence of inhibiting the normal function of AHR (131). This could be a new method of
establishing the association between environmental contaminants and
the genesis of tumors such as neuroblastoma (131).
Kynurenine (KYN) pathway
AhR pathway activation by environmental xenobiotic
compounds has already been discussed in the present review;
however, certain endogenous ligands could also activate this
pathway. The tryptophan catabolite kynurenine (KYN) was the first
endogenous ligand described for AHR. KYN is produced by the KYN
pathway among other neuroactive metabolites, including KYN acid,
3-hydroxykynurenine, anthranilic acid, 3-hydroxyanthranilic acid,
picolinic acid (PIC), N-methyl-D-aspartate agonist and quinolinic
acid (QUIN), from which NAD+ is synthetized. In the CNS,
the kynurenine pathway metabolizes ~95% of tryptophan (136). Nowadays, it is well known that, in
CNS tumors, the AhR-kynurenine pathway is active and associated
with malignant progression and poor survival.
Neuroblastoma cells overexpress 2,3-dioxygenase
enzyme and suppress α-amino-β-carboxymuconate-ε-semialdehyde
decarboxylase (137). In addition,
these cells produce more QUIN, a neurotoxin, and less PIC. PIC is a
neuroprotective metabolite with antiproliferative effects (138) that produces the characteristic
neurotoxicity of CNS tumors (139);
this neurotoxicity is comparable to the necrotic effect observed in
multiforme glioblastomas due to the release of glutamate, which is
excessively neurotoxic and causes neuronal death (140). In addition, it is clear that the
KYN produced by these tumors in gliomas acts as an immune
suppressor, and promotes the survival and motility of tumor cells
by activating the AhR pathway. Therefore, there is an association
between tumor progression and low survival rates in patients with
high AHR expression (141).
The KYN-AhR pathway can be used as a target in therapeutic
applications for CNS tumor growth control, as KYN is a proven
ligand for AHR. The use of antagonists, including certain aromatic
compounds such as flavones and polyphenols, could block the pathway
activation and stop tumor growth (142,143).
Conclusions
Based on the analysis of the molecular biology,
biochemistry and physiology of the AhR and its pathway, the
following conclusions can be reached: i) Certain transcription
factor inhibitors could be used to increase the protein levels of
AHR and, as a result, since AHR regulates several cell processes,
it may be possible to achieve the important control of some
cellular processes by inhibiting the activity of AhR pathway in
malignant tumors of the CNS. ii) Compounds that can antagonize the
canonical AhR pathway could also be used as treatment, such as the
flavones. This field has not yet been fully explored, and future
research should be conducted with the objective of progressively
broadening targets and possibilities for the control of tumors,
based on studies of the AhR pathway. iii) It is important to
explore compounds that can inhibit the components of the
cytoplasmic AHR complex, such as Hsp90 (for which one already
exists, NVP-AUY922), AIP and p23. This reduces the stability of the
receptor in the cytoplasm, which is rendered highly labile and can
be degraded, indirectly inhibiting the activator effect of various
cell processes. iv) Another matter that requires attention is the
fact that not all the processes that AHR regulates are directed
towards activating/increasing responses; some are directed towards
inhibiting responses. One such process is the interaction between
AHR and KLF6, which activates transcription and increases the
protein expression of p21, thus blocking the cell cycle
progression. For that reason, it is important to conduct analyses
to confirm these processes and determine whether they involve
activation or repression.
Acknowledgements
This review is a required part of the PhD Graduate
Program in Biological Sciences of the National Autonomous
University of Mexico. The authors would like to acknowledge
scholarship CVU-508581 provided by the Consejo Nacional de Ciencia
y Tecnología (CONACYT) and the support of the University and the
Biological Sciences PhD program of the Universidad Nacional
Autónoma de México.
Funding
The financial support to pay for the publication was
obtained from the Dirección de Investigación of Hospital Infantil
de México Federico Gómez (grant no. HIM-2019-029.SSA1574).
Availability of data and materials
Not applicable.
Authors' contributions
MZO revised and corrected the text and figures and
performed the review of the information. EAV performed the review
of articles, prepared information and designed the figures. FAH
reviewed information, wrote and revised the manuscript. MZO, EAV
and FAH confirm the authenticity of all raw data. All the authors
have made substantive intellectual contributions and meet the
conditions of authorship. All authors have read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schulz KH: Clinical & experimental
studies on the etiology of chloracne. Arch Clinical Exp Dematol.
206:589–596. 1957.(In German).
|
|
2
|
Kimmig J and Schulz KH: Occupational acne
(so-called chloracne) due to chlorinated aromatic cyclic ethers.
Dermatologica. 115:540–546. 1957.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Poland A and Glover E:
2,3,7,8-Tetrachlorodibenzo-p-dioxin: A potent inducer
of-aminolevulinic acid synthetase. Science. 179:476–477. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Richardson HL, Stier AR and
Borsos-Nachtnebel E: Liver tumor inhibition and adrenal histologic
responses in rats to which 3′-methyl-4-dimethylaminoazobenzene and
20-methylcholanthrene were simultaneously administrated. Cancer
Res. 12:356–361. 1952.PubMed/NCBI
|
|
5
|
Conney AH, Miller EC and Miller JA:
Substrate-induced synthesis and other properties of benzpyrene
hydroxylase in rat liver. J Biol Chem. 228:753–766. 1957.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nebert DW and Bausserman L: Genetic
differences in the extent of aryl hydrocarbon hydroxylase induction
in mouse fetal cell cultures. J Biol Chem. 245:6373–6382. 1970.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nebert DW and Gelboin HV: The in vivo and
in vitro induction of aryl hydrocarbon hydroxylase in mammalian
cells of different species, tissues, strains, and developmental and
hormonal states. Achr Biochem Biophys. 134:76–89. 1969. View Article : Google Scholar
|
|
8
|
Nebert DW, Negishi M, Lang MA, Hjelmeland
LM and Eisen HJ: The Ah locus, a multigene family necessary for
survival in a chemically adverse environment: Comparison with the
immune system. Adv Genet. 21:1–51. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nebert DW, Goujon FM and Gielen JE: Aryl
hydrocarbon hydroxylase induction by polycyclic hydrocarbons:
Simple autosomal dominant trait in the mouse. Nat New Biol.
236:107–110. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nebert DW: The 1986 Bernard B. Brodie
award lecture. The genetic regulation of drug-metabolizing enzymes.
Drug Metab Dispos. 16:1–8. 1988.PubMed/NCBI
|
|
11
|
Poland A and Glover E: Comparison of
2,3,7,8-tetrachlorodibenzo-p-dioxin, a potent inducer of aryl
hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol Pharmacol.
10:349–359. 1974.PubMed/NCBI
|
|
12
|
Poland A, Glover E, Robinson JR and Nebert
DW: Genetic expression of aryl hydrocarbon hydroxylase activity.
Induction of monooxygenase activities and cytochrome P1-450
formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice
genetically ‘nonresponsive’ to other aromatic hydrocarbons. J Biol
Chem. 249:5599–5606. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yueh MF, Huang YH, Hiller A, Chen S,
Nguyen N and Tukey RH: Involvement of the xenobiotic response
element (XRE) in Ah receptor-mediated induction of human
UDP-glucuronosyltransferase 1A1. J Biol Chem. 278:15002–15006.
2003. View Article : Google Scholar
|
|
14
|
Poland A, Glover E and Kende AS:
Stereospecific, high affinity binding of
2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence
that the binding species is receptor for induction of aryl
hydrocarbon hydroxylase. J Biol Chem. 251:4936–4946. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gasiewicz TA and Henry E: History of
research on the AhR. Pohjanvirta R: The AH receptor in biology and
toxicology Hoboken, New Jersey: John Wiley & Sons; pp. 3–32.
2012
|
|
16
|
Gasiewicz TA and Bauman PA: Heterogeneity
of the rat hepatic Ah receptor and evidence for transformation in
vitro and in vivo. J Biol Chem. 262:2116–2120. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reyes H, Reisz-Porszasz S and Hankinson O:
Identification of the Ah receptor nuclear translocator proteins
(Arnt) as a component of the DNA binding form of the Ah receptor.
Science. 256:1193–1195. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller AG, Israel D and Whitlock JP Jr:
Biochemical and genetic analysis of variant mouse hepatoma cells
defective in the induction of benzo(a)pyrene-metabolizing enzyme
activity. J Biol Chem. 258:3523–3527. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones PB, Durrin LK, Galeazzi DR and
Whitlock JP Jr: Control of cytochrome P1-450 gene expression:
Analysis of a dioxin-responsive enhancer system. Proc Nat Acad Sci
USA. 83:2802–2806. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujisawa-Sehara A, Yamane M and
Fujii-Kuriyama Y: A DNA-binding factor specific for xenobiotic
responsive elements of P-450c gene exists as a cryptic form in
cytoplasm: Its possible translocation to nucleus. Proc Natl Acad
Sci USA. 85:5859–5863. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pohjanvirta R, Korkalainen M, Moffat ID,
Boutros PC and Okey AB: Role of the AHR and its structure in TCDD
toxicity. Pohjanvirta R: The AH receptor in biology and toxicology
Hoboken, New Jersey: John Wiley & Sons; pp. 179–196. 2012
|
|
22
|
DeGroot D, He G, Fraccalvieri D, Bonati L,
Pandini A and Denison MS: Ahr ligands: Promiscuity in binding and
diversity in response. Pohjanvirta R: The AH receptor in biology
and toxicology Hoboken, New Jersey: John Wiley & Sons; pp.
63–79. 2011, View Article : Google Scholar
|
|
23
|
Eguchi H, Hayashi S, Watanabe J, Gotoh O
and Kawajiri K: Molecular cloning of the human Ah receptor gene
promoter. Biochem Biophys Res Comm. 203:615–622. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shin JH, Haggadone MD and Sunwoo JB:
Transcription factor Dlx3 induces aryl hydrocarbon receptor
promoter activity. Biochem Biophys Rep. 7:353–360. 2016.PubMed/NCBI
|
|
25
|
Tanaka G, Kanaji S, Hirano A, Arima K,
Shinagawa A, Goda C, Yasunaga S, Ikizawa K, Yanagihara Y, Kubo M,
et al: Induction and activation of the aryl hydrocarbon receptor by
IL-4 in B cells. Int Immunol. 17:797–805. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eastman Q and Grosschedl R: Regulation of
LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin
Cell Biol. 11:233–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harper PA, Riddick DS and Okey AB:
Regulating the regulator: Factors that control levels and activity
of the aryl hydrocarbon receptor. Biochem Pharmacol. 72:267–279.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hahn ME: Aryl hydrocarbon receptors:
Diversity and evolution. Chem Biol Interact. 141:131–160. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Burbach KM, Poland A and Bradfield CA:
Cloning of the Ah-receptor cDNA reveals a distinctive
ligand-activated transcription factor. Proc Natl Acad Sci USA.
89:8185–8189. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma Q: Overview of AHR functional domains
and the classical AHR signaling pathway: Induction of drug
metabolizing enzymes. The AH Receptor in Biology and Toxicology.
Pohjanvirta R: John Wiley & Sons; Hoboken, New Jersey: pp.
35–45. 2012
|
|
31
|
Ma Q and Whitlock JP Jr: A novel
cytoplasmic protein that interacts with the Ah receptor, contains
tetratricopeptide repeat motifs, and augments the transcriptional
response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem.
272:8878–8884. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma Q, Dong L and Whitlock JP Jr:
Transcriptional activation by the mouse Ah receptor. Interplay
between multiple stimulatory and inhibitory functions. J Biol Chem.
270:12697–12703. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Murray IA and Perdew GH: Role of the
chaperone proteins in ahr function. The AH Receptor in Biology and
Toxicology. Pohjanvirta R: John Wiley & Sons; Hoboken, New
Jersey: pp. 47–61. 2011, View Article : Google Scholar
|
|
34
|
Chen HS and Perdew GH: Subunit composition
of the heteromeric cytosolic aryl hydrocarbon receptor complex. J
Biol Chem. 269:27554–27558. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meyer BK, Pray-Grant MG, Vanden Heuvel JP
and Perdew GH: Hepatitis B virus X-associated protein 2 is a
subunit of the unliganded aryl hydrocarbon receptor core complex
and exhibits transcriptional enhancer activity. Mol Cel Biol.
18:978–988. 1998. View Article : Google Scholar
|
|
36
|
Carver LA, LaPres JJ, Jain S, Dunham EE
and Bradfield CA: Characterization of the Ah receptor-associated
protein, ARA9. J Biol Chem. 273:33580–33587. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schreiber SL: Chemistry and biology of the
immunophilins and their immunosuppressive ligands. Science.
251:283–287. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma Q and Baldwin KT:
2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl
hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway.
Role of the transcription activation and DNA binding of AhR. J Biol
Chem. 275:8432–8438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roberts BJ and Whitelaw ML: Degradation of
the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin
receptor via the ubiquitin/proteasome pathway. J Biol Chem.
274:36351–36356. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lo RS and Massagué J: Ubiquitin-dependent
degradation of TGF-beta-activated Smad2. Nat Cell Biol. 1:472–478.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Floyd ZE, Trausch-Azar JS, Reinstein E,
Ciechanover A and Schwartz AL: The nuclear ubiquitin-proteasome
system degrades MyoD. J Biol Chem. 276:22468–22475. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ikuta T, Eguchi H, Tachibana T, Yoneda Y
and Kawajiri K: Nuclear localization and export signals of the
human aryl hydrocarbon receptor. J Biol Chem. 273:2895–2904. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fukunaga BN, Probst MR, Reisz-Porszasz S
and Hankinson O: Identification of functional domains of the aryl
hydrocarbon receptor. J Biol Chem. 270:29270–29278. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Reisz-Porszasz S, Probst MR, Fukunaga BN
and Hankinson O: Identification of functional domains of the aryl
hydrocarbon receptor nuclear translocator protein (ARNT). Mol Cell
Biol. 14:6075–6086. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Soshilov A and Denison MS: Role of the
Per/Arnt/Sim domains in ligand-dependent transformation of the aryl
hydrocarbon receptor. J Biol Chem. 283:32995–33005. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mahon MJ and Gasiewicz TA: Ah receptor
phosphorylation: Localization of phosphorylation sites to the
C-terminal half of the protein. Arch Biochem Biophys. 318:166–174.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Swanson H: Dioxin response elements and
regulation of gene transcription. The AH Receptor in Biology and
Toxicology. Pohjanvirta R: John Wiley & Sons; Hoboken, New
Jersey: pp. 81–91. 2012
|
|
48
|
Bacsi S, Reisz-Porszasz S and Hankinson O:
Orientation of the heterodimeric aryl hydrocarbon (dioxin) receptor
complex on its asymmetric DNA recognition sequence. Mol Pharmacol.
47:432–438. 1995.PubMed/NCBI
|
|
49
|
Ma Q: Induction and superinduction of
2,3,7,8-tetrachlorodibenzo-rho-dioxin-inducible poly(ADP-ribose)
polymerase: Role of the aryl hydrocarbon receptor/aryl hydrocarbon
receptor nuclear translocator transcription activation domains and
a labile transcription repressor. Arch Biochem Biophys.
404:309–316. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gasiewicz TA, Henry EC and Collins LL:
Expression and activity of aryl hydrocarbon receptors in
development and cancer. Crit Review Eukaryotic Gene Exp.
18:279–321. 2008. View Article : Google Scholar
|
|
51
|
Barouki R, Coumoul X and
Fernandez-Salguero PM: The aryl hydrocarbon receptor, more than a
xenobiotic-interacting protein. FEBS Lett. 581:3608–3615. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mulero-Navarro S and Fernandez-Salguero
PM: New trends in Aryl hydrocarbon receptor biology. Front Cell Dev
Biol. 4:452016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Larigot L, Juricek L, Dairou J and Coumoul
X: AhR signaling pathways and regulatory functions. Biochim Open.
7:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bock KW: Aryl hydrocarbon receptor (AHR):
From selected human target genes and crosstalk with transcription
factors to multiple AHR functions. Biochem Pharmacol. 168:65–70.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Josyula N, Andersen ME, Kaminski NE, Dere
E, Zacharewski TR and Bhattacharya S: Gene co-regulation and
co-expression in the aryl hydrocarbon receptor-mediated
transcriptional regulatory network in the mouse liver. Arch
Toxicol. 94:113–126. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gu YZ, Hogenesch JB and Bradfield CA: The
PAS superfamily: Sensors of environmental and developmental
signals. Annu Rev Pharmacol Toxicol. 40:519–561. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ge NL and Elferink CJ: A direct
interaction between the aryl hydrocarbon receptor and
retinoblastoma protein. Linking dioxin signaling to the cell cycle.
J Biol Chem. 273:22708–22713. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Huang G and Elferink CJ: Multiple
mechanisms are involved in Ah receptor-mediated cell cycle arrest.
Mol Pharmacol. 67:88–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Marlowe JL, Knudsen ES, Schwemberger S and
Puga A: The aryl hydrocarbon receptor displaces p300 from
E2F-dependent promoters and represses S phase-specific gene
expression. J Biol Chem. 279:29013–29022. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Elferink CJ, Ge NL and Levine A: Maximal
aryl hydrocarbon receptor activity depends on an interaction with
the retinoblastoma protein. Mol Pharmacol. 59:664–673. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jackson DP, Li H, Mitchell KA, Joshi AD
and Elferink C: Ah receptor-mediated suppression of liver
regeneration through NC-XRE-driven p21Cip1 expression. J Mol
Pharmacol. 85:533–541. 2014. View Article : Google Scholar
|
|
62
|
Vogel CF, Sciullo E and Matsumura F:
Involvement of RelB in aryl hydrocarbon receptor-mediated induction
of chemokines. Biochem Biophys Res Commun. 363:722–726. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tian Y, Ke S, Denison M, Rabson A and
Gallo M: Ah receptor and NF-kappaB interactions, a potential
mechanism for dioxin toxicity. J Biol Chem. 274:510–515. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hollingshead BD, Beischlag TV, Dinatale
BC, Ramadoss P and Perdew GH: Inflammatory signaling and aryl
hydrocarbon receptor mediate synergistic induction of interleukin 6
in MCF-7 cells. Cancer Res. 68:3609–3617. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim DW, Gazourian L, Quadri SA,
Romieu-Mourez R, Sherr DH and Sonenshein GE: The RelA NF-kappaB
subunit and the aryl hydrocarbon receptor (AhR) cooperate to
transactivate the c-myc promoter in mammary cells. Oncogene.
19:5498–5506. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yeager RL, Reisman SA, Aleksunes LM and
Klaassen CD: Introducing the ‘TCDD-inducible AhR-Nrf2 gene
battery’. Toxicol Sci. 111:238–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang L, He X, Szklarz GD, Bi Y,
Rojanasakul Y and Ma Q: The aryl hydrocarbon receptor interacts
with nuclear factor erythroid 2-related factor 2 to mediate
induction of NAD(P)H:quinoneoxidoreductase 1 by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch Biochem Biophys.
537:31–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zacharewski TR, Bondy KL, McDonell P and
Wu ZF: Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin
on 17 beta-estradiol-induced pS2 expression. Cancer Res.
54:2707–2713. 1994.PubMed/NCBI
|
|
69
|
Gillesby BE, Stanostefano M, Porter W,
Safe S, Wu ZF and Zacharewski TR: Identification of a motif within
the 5′ regulatory region of pS2 which is responsible for AP-1
binding and TCDD-mediated suppression. Biochemistry. 36:6080–6089.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ohtake F, Fujii-Kuriyama Y and Kato S: AhR
acts as an E3 ubiquitin ligase to modulate steroid receptor
functions. Biochem Pharmacol. 77:474–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Peters JM, Narotsky MG, Elizondo G,
Fernandez-Salguero PM, Gonzalez FJ and Abbott BD: Amelioration of
TCDD-induced teratogenesis in aryl hydrocarbon receptor (AhR)-null
mice. Toxicol Sci. 47:86–92. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dere E, Lo R, Celius T, Matthews J and
Zacharewski TR: Integration of genome-wide computation DRE search,
AhR ChIP-chip and gene expression analyses of TCDD-elicited
responses in the mouse liver. BMC Genomics. 12:365–375. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tijet N, Boutros PC, Moffat ID, Okey AB,
Tuomisto J and Pohjanvirta R: Aryl hydrocarbon receptor regulates
distinct dioxin-dependent and dioxin-independent gene batteries.
Mol Pharmacol. 69:140–153. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mitchell KA, Lockhart CA, Huang G and
Elferink CJ: Sustained aryl hydrocarbon receptor activity
attenuates liver regeneration. Mol Pharmacol. 70:163–170. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huang G and Elferink CJ: A novel
nonconsensus xenobiotic response element capable of mediating aryl
hydrocarbon receptor-dependent gene expression. Mol Pharmacol.
81:338–347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Harper JW, Elledge SJ, Keyomarsi K,
Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley
L, Swindell E, et al: Inhibition of cyclin-dependent kinases by
p21. Mol Biol Cell. 6:387–400. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jackson DP, Joshi AD and Elferink CJ: Ah
receptor pathway intricacies; signaling through diverse protein
partners and DNA-motifs. Toxicol Res (Camb). 4:1143–1158. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wilson SR, Joshi AD and Elferink CJ: The
tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon
receptor DNA binding partner. J Pharmacol Exp Ther. 345:419–429.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang W, Shields JM, Sogawa K,
Fujii-Kuriyama Y and Yang VW: The gut-enriched Krüppel-like factor
suppresses the activity of the CYP1A1 promoter in an Sp1-dependent
fashion. J Biol Chem. 273:17917–17925. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Philipsen S and Suske G: A tale of three
fingers: The family of mammalian Sp/XKLF transcription factors.
Nucleic Acids Res. 27:2991–3000. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jeng YM and Hsu HC: KLF6, a putative tumor
suppressor gene, is mutated in astrocytic gliomas. Int J Cancer.
105:625–629. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Andreoli V, Gehrau RC and Bocco JL:
Biology of Krüppel-like factor 6 transcriptional regulator in cell
life and death. IUBMB Life. 62:896–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Rubinstein M, Idelman G, Plymate SR, Narla
G, Friedman SL and Werner H: Transcriptional activation of the
insulin-like growth factor I receptor gene by the Kruppel-like
factor 6 (KLF6) tumor suppressor protein: Potential interactions
between KLF6 and p53. Endocrinology. 145:3769–3777. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
YangLShiPZhao G, Xu J, Peng W, Zhang J,
Zhang G, Wang X, Dong Z, Chen F and Cui H: Targeting cancer stem
cell pathways for cancer therapy. Sig Transduct Target Ther.
5:82020. View Article : Google Scholar
|
|
85
|
Pearson JRD and Regad T: Targeting
cellular pathways in glioblastoma multiforme. Sig Transduct Target
Ther. 2:170402017. View Article : Google Scholar
|
|
86
|
Murray IA, Patterson AD and Perdew GH:
Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat
Rev Cancer. 14:801–814. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Xue P, Fu J and Zhou Y: The aryl
hydrocarbon receptor and tumor immunity. Front Immunol. 9:2862018.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hayashibara T, Yamada Y, Mori N, Harasawa
H, Sugahara K, Miyanishi T, Kamihira S and Tomonaga M: Possible
involvement of aryl hydrocarbon receptor (AhR) in adult T-cell
leukemia (ATL) leukemogenesis: Constitutive activation of AhR in
ATL. Biochem Biophys Res Commun. 300:128–134. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gentil M, Hugues P, Desterke C, Telliam G,
Sloma I, Souza LEB, Baykal S, Artus J, Griscelli F, Guerci A, et
al: Aryl hydrocarbon receptor (AHR) is a novel druggable pathway
controlling malignant progenitor proliferation in chronic myeloid
leukemia (CML). PLoS One. 13:e02009232018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sanna S, Satta G, Padoan M, Piro S,
Gambelunghe A, Miligi L, Ferri GM, Magnani C, Muzi G, Rigacci L, et
al: Activation of the aryl hydrocarbon receptor and risk of
lymphoma subtypes. Int J Mol Epidemiol Genet. 8:40–44.
2017.PubMed/NCBI
|
|
91
|
Wang K, Li Y, Jiang YZ, Dai CF, Patankar
MS, Song JS and Zheng J: An endogenous aryl hydrocarbon receptor
ligand inhibits proliferation and migration of human ovarian cancer
cells. Cancer Lett. 340:63–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Perez AIL and Bradshaw TD: Exploring new
molecular targets in advanced ovarian cancer: The aryl hydrocarbon
receptor (AhR) and antitumor benzothiazole ligands as potential
therapeutic candidates. Current Trends in Cancer Management.
2018.
|
|
93
|
Tsay JJ, Tchou-Wong KM, Greenberg AK, Pass
H and Rom WN: Aryl hydrocarbon receptor and lung cancer. Anticancer
Res. 33:1247–1256. 2013.PubMed/NCBI
|
|
94
|
Guerrina N, Traboulsi H, Eidelman DH and
Baglole CJ: The aryl hydrocarbon receptor and the maintenance of
lung health. Int J Mol Sci. 19:38822018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liu Z, Wu X, Zhang F, Han L, Bao G, He X
and Xu Z: AhR expression is increased in hepatocellular carcinoma.
J Mol Histol. 44:455–461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
John K, Lahoti TS, Wagner K, Hughes JM and
Perdew GH: The Ah receptor regulates growth factor expression in
head and neck squamous cell carcinoma cell lines. Mol Carcinog.
53:765–776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wesseling P and Capper D: WHO 2016
classification of gliomas. Neuropathol Appl Neurobiol. 44:139–150.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Loeffler S, Fayard B, Weis J and
Weissenberger J: Interleukin-6 induces transcriptional activation
of vascular endothelial growth factor (VEGF) in astrocytes in vivo
and regulates VEGF promoter activity in glioblastoma cells via
direct interaction between STAT3 and Sp1. Int J Cancer.
115:202–213. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Botella LM, Sanz-Rodriguez F, Komi Y,
Fernandez-L A, Varela E, Garrido-Martin EM, Narla G, Friedman SL
and Kojima S: TGF-beta regulates the expression of transcription
factor KLF6 and its splice variants and promotes co-operative
transactivation of common target genes through a Smad3-Sp1-KLF6
interaction. Biochem J. 419:485–495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Merk BC, Owens JL, Lopes MB, Silva CM and
Hussaini IM: STAT6 expression in glioblastoma promotes invasive
growth. BMC Cancer. 11:1842011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gu A, Ji G, Jiang T, Lu A, You Y, Liu N,
Luo C, Yan W and Zhao P: Contributions of aryl hydrocarbon receptor
genetic variants to the risk of glioma and PAH-DNA adducts. Toxicol
Sci. 128:357–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Li H xing, Peng X xiao and Zong Q:
Cigarette smoking and risk of adult glioma: A meta-analysis of 24
observational studies involving more than 2.3 million individuals.
Onco Targets Ther. 9:3511–3523. 2016.PubMed/NCBI
|
|
104
|
Maier MS, Legare ME and Hanneman WH: The
aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl
induces distinct patterns of gene expression between hepatoma and
glioma cells: Chromatin remodeling as a mechanism for selective
effects. Neurotoxicology. 28:594–612. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gramatzki D, Pantazis G, Schittenhelm J,
Tabatabai G, Köhle C, Wick W, Schwarz M, Weller M and Tritschler I:
Aryl hydrocarbon receptor inhibition downregulates the
TGF-beta/Smad pathway in human glioblastoma cells. Oncogene.
28:2593–2605. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pećina-Šlaus N, Kafka A, Tomas D, Marković
L, Okštajner PK, Sukser V and Krušlin B: Wnt signaling
transcription factors TCF-1 and LEF-1 are upregulated in malignant
astrocytic brain tumors. Histol Histopathol. 29:1557–1564.
2014.
|
|
107
|
Djuzenova CS, Blassl C, Roloff K, Kuger S,
Katzer A, Niewidok N, Günther N, Polat B, Sukhorukov VL and Flentje
M: Hsp90 inhibitor NVP-AUY922 enhances radiation sensitivity of
tumor cell lines under hypoxia. Cancer Biol Ther. 13:425–434. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Seznec J, Silkenstedt B and Naumann U:
Therapeutic effects of the Sp1 inhibitor mithramycin A in
glioblastoma. J Neurooncol. 101:365–377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chiba Y, Todoroki M, Nishida Y, Tanabe M
and Misawa M: A novel STAT6 inhibitor AS1517499 ameliorates
antigen-induced bronchial hypercontractility in mice. Am J Respir
Cell Mol Biol. 41:516–524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Jin UH, Karki K, Cheng Y, Michelhaugh SK,
Mittal S and Safe S: The aryl hydrocarbon receptor is a tumor
suppressor-like gene in glioblastoma. J Biol Chem. 294:11342–11353.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
de Robles P, Fiest KM, Frolkis AD,
Pringsheim T, Atta C, St Germaine-Smith C, Day L, Lam D and Jette
N: The worldwide incidence and prevalence of primary brain tumors:
A systematic review and meta-analysis. Neuro Oncol. 17:776–783.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Girardi F, Allemani C and Coleman MP:
Worldwide trends in survival from common childhood brain tumors: A
systematic review. J Glob Oncol. 5:1–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ostrom QT, Cioffi G, Gittleman H, Patil N,
Waite K, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2012–2016. Neuro Oncol. 21 (Suppl
5):v1–v100. 2019. View Article : Google Scholar
|
|
114
|
Gilbertson RJ and Ellison DW: The origins
of medulloblastoma subtypes. Annu Rev Pathol. 3:341–365. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yang ZJ, Ellis T, Markant SL, Read TA,
Kessler JD, Bourboulas M, Schüller U, Machold R, Fishell G, Rowitch
DH, et al: Medulloblastoma can be initiated by deletion of patched
in lineage-restricted progenitors or stem cells. Cancer Cell.
14:135–145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Rossi A, Caracciolo V, Russo G, Reiss K
and Giordano A: Medulloblastoma: From molecular pathology to
therapy. Clin Cancer Res. 14:971–976. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ramaswamy V and Taylor MD:
Medulloblastoma: From myth to molecular. J Clin Oncol.
35:2355–2363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Mulhern RK, Merchant TE, Gajjar A, Reddick
WE and Kun LE: Late neurocognitive sequelae in survivors of brain
tumours in childhood. Lancet Oncol. 5:399–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Doussouki MEI, Gajjar A and Chamdine O:
Molecular genetics of medulloblastoma in children: Diagnostic,
therapeutic and prognostic implications. Future Neurol.
14:FNL82019. View Article : Google Scholar
|
|
120
|
Miranda Kuzan-Fischer C, Juraschka K and
Taylor MD: Medulloblastoma in the molecular era. J Korean Neurosurg
Soc. 61:292–301. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Dever DP and Opanashuk LA: The aryl
hydrocarbon receptor contributes to the proliferation of human
medulloblastoma cells. Mol Pharmacol. 81:669–678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Li Y, Guessous F, Johnson EB, Eberhart CG,
Li XN, Shu Q, Fan S, Lal B, Laterra J, Schiff D and Abounader R:
Functional and molecular interactions between the HGF/c-Met pathway
and c-Myc in large-cell medulloblastoma. Lab Invest. 88:98–111.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Sarić N, Selby M, Ramaswamy V, Kool M,
Stockinger B, Hogstrand C, Williamson D, Marino S, Taylor MD,
Clifford SC and Basson MA: The AHR pathway represses TGFβ-SMAD3
signalling and has a potent tumour suppressive role in SHH
medulloblastoma. Sci Rep. 10:1482020. View Article : Google Scholar
|
|
124
|
Johnsen JI, Dyberg C, Fransson S and
Wickström M: Molecular mechanisms and therapeutic targets in
neuroblastoma. Pharmacol Res. 131:164–176. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Johnsen JI, Dyberg C and Wickström M:
Neuroblastoma-A neural crest derived embryonal malignancy. Front
Mol Neurosci. 12:92019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Steliarova-Foucher E, Colombet M, Ries
LAG, Moreno F, Dolya A, Bray F, Hesseling P, Shin HY and Stiller
CA; IICC-3 contributors, : International incidence of childhood
cancer, 2001-10: A population-based registry study. Lancet Oncol.
8:719–31. 2017. View Article : Google Scholar
|
|
127
|
Panagopoulou P, Georgakis MK, Baka M,
Moschovi M, Papadakis V, Polychronopoulou S, Kourti M,
Hatzipantelis E, Stiakaki E, Dana H, et al: Persisting inequalities
in survival patterns of childhood neuroblastoma in Southern and
Eastern Europe and the effect of socio-economic development
compared with those of the US. Eur J Cancer. 96:44–53. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kholodenko IV, Kalinovsky DV, Doronin II,
Deyev SM and Kholodenko RV: Neuroblastoma origin and therapeutic
targets for immunotherapy. J Immunol Res. 2018:73942682018.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Allen-Rhoades W, Whittle SB and Rainusso
N: Pediatric solid tumors of infancy: An overview. Pediatr Rev.
39:57–67. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Akahoshi E, Yoshimura S and
Ishihara-Sugano M: Over-expression of AhR (aryl hydrocarbon
receptor) induces neural differentiation of Neuro2a cells:
Neurotoxicology study. Environ Health. 5:242006. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wu PY, Liao YF, Juan HF, Huang HC, Wang
BJ, Lu YL, Yu IS, Shih YY, Jeng YM, Hsu WM and Lee H: Aryl
hydrocarbon receptor downregulates MYCN expression and promotes
cell differentiation of neuroblastoma. PLoS One. 9:887952014.
View Article : Google Scholar
|
|
132
|
Latchney SE, Hein AM, O'Banion MK,
DiCicco-Bloom E and Opanashuk LA: Deletion or activation of the
aryl hydrocarbon receptor alters adult hippocampal neurogenesis and
contextual fear memory. J Neurochem. 125:430–445. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yang X, Liu D, Murray TJ, Mitchell G C,
Hesterman EV, Karchner SI, Merson RR, Hahn ME and Sherr DH: The
aryl hydrocarbon receptor constitutively represses c-myc
transcription in human mammary tumor cells. Oncogene. 24:7869–7881.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Puga A, Barnes SJ, Dalton TP, Chang Cy,
Knudsen ES and Maier MA: Aromatic hydrocarbon receptor interaction
with the retinoblastoma protein potentiates repression of
E2F-dependent transcription and cell cycle arrest. J Biol Chem.
275:2943–2950. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Wu PY, Chuang PY, Chang GD, Chan YY, Tsai
TC, Wang BJ, Lin KH, Hsu WM, Liao YF and Lee H: Novel endogenous
ligands of aryl hydrocarbon receptor mediate neural development and
differentiation of neuroblastoma. ACS Chem Neurosci. 10:4031–4042.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Peters JC: Tryptophan nutrition and
metabolism: An overview. Kynurenine and Serotonin Pathways:
Progress in Tryptophan Research. Schwarcz R, Young SN and Brown RR:
Plenum Press Div Plenum Publishing Corp.; New York: pp. 345–358.
1991, View Article : Google Scholar
|
|
137
|
Guillemin GJ, Cullen KM, Lim CK, Smythe
GA, Garner B, Kapoor V, Takikawa O and Brew BJ: Characterization of
the kynurenine pathway in human neurons. J Neurosci.
27:12884–12892. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Coggan SE, Smythe GA, Bilgin A and Grant
RS: Age and circadian influences on picolinic acid concentrations
in human cerebrospinal fluid. J Neurochem. 108:1220–1225. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Adams S, Braidy N, Bessede A, Brew BJ,
Grant R, Teo C and Guillemin GJ: The kynurenine pathway in brain
tumor pathogenesis. Cancer Res. 72:5649–5657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ye ZC and Sontheimer H: Glioma cells
release excitotoxic concentrations of glutamate. Cancer Res.
59:4383–4391. 1999.PubMed/NCBI
|
|
141
|
Opitz CA, Litzenburger UM, Sahm F, Ott M,
Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller
M, et al: An endogenous tumour-promoting ligand of the human aryl
hydrocarbon receptor. Nature. 478:197–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Zhang S, Qin C and Safe SH: Flavonoids as
aryl hydrocarbon receptor agonists/antagonists: Effects of
structure and cell context. Environ Health Perspect. 111:1877–1882.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Kaiser H, Parker E and Hamrick MW:
Kynurenine signaling through the aryl hydrocarbon receptor:
Implications for aging and healthspan. Exp Gerontol.
130:1107972019. View Article : Google Scholar : PubMed/NCBI
|