Cancer has high incidence and mortality rates
worldwide. Tumor cells can easily develop resistance to traditional
treatments, and survive to result in metastasis and invasion. The
induction of tumor senescence has been proposed as a potential
method for the treatment of tumors. In 1961, Hayflick and Moorhead
(1) cultured human fibroblasts and
found that normal diploid cells proliferated in vitro for
50–70 generations before entering senescence. This limit to cell
proliferation, when cells lose their proliferative ability but
maintain stable metabolic activity, is called the ‘Hayflick Limit’.
After reaching this limit, the cells enter into a senescent state
(2). Senescence is a defensive
mechanism that prevents cells from being damaged. When cells are
senescent, they do not re-enter the cell cycle when exposed to
mitogenic stimuli, but exhibit an enhanced secretory phenotype and
are resistant to cell death. It has been hypothesized that cell
senescence is an important mechanism that may be used to attack
tumorigenic cells. When DNA is damaged, cell senescence becomes the
third pathway, in addition to apoptosis and DNA repair, to defend
against tumorigenesis (3). Some
antitumor drugs have been demonstrated to inhibit tumor cell
proliferation by inducing senescence in vitro and in
vivo (4,5). Therefore, the induction of cancer cell
senescence and the subsequent inhibition of tumorigenesis and
recurrence is a focus of research into novel tumor treatments
Tumor senescence can be divided into two types,
namely replicative senescence and premature senescence. Replicative
senescence is determined by the number of cell divisions (6). Premature senescence is mainly caused by
DNA damage, the loss of tumor suppressor factors, oxidative stress
and malnutrition (7,8).
Tumor cells exhibit unique characteristics when
senescent, the most notable being the loss of ability to
proliferate in an unlimited capacity whilst maintaining metabolic
activity (9). Senescent cells are
usually arrested in the G0 or G1 phase
(10,11). Research has focused on studying the
phenotypical characteristics of cells after they have entered
senescence, as well as the mechanisms of action that activate the
process and the molecular signaling pathways involved (12). In subsequent sections of the present
review, the factors that initiate senescence and induce senescent
phenotypes are discussed.
When tumor cells enter into senescence, they exhibit
morphological changes, including an increase in volume, flattened
shape and increased intercellular space, and they also present with
blocked DNA synthesis, heterochromatin foci, lipofuscin
accumulation, DNA damage-induced foci, loss of lamin B1, satellite
distension, the expression of differentiated embryonic
chondrocyte-expressed 1 and decoy death receptor 2, the
upregulation of certain microRNAs and secretion of numerous
factors, including growth factors, cytokines, chemokines and
proteases, which are collectively known as the
senescence-associated secretory phenotype (SASP) (5,13–17). The
SASP has been shown to contribute to the protective effect of
senescence, and to induce detrimental effects when the pathological
accumulation of senescent cells occurs. One study revealed that
cisplatin can induce HepG2 tumor cells to enter into senescence and
present senescent phenotypes, even when used at a low dose
(18). Such effects of this and
other chemotherapeutic agents have been confirmed in numerous cell
lines, including HCT-116, H460, H1299, HT1080 and A549 (19,20). For
example, Zhang et al (20)
treated HCT-116 colon cancer cells with 20 and 50 nM camptothecin
for 24 h, and the cells entered into senescence 48 h after the
low-dose treatment. Also, when treated with a low level of
DNA-damaging agents, such as cisplatin and doxorubicin, HepG2 cells
clearly progressed into senescence (18,21,22). At
present, several anticancer drugs that are used clinically are
known to mediate therapy-induced senescence, including docetaxel,
bleomycin, cyclophosphamide, doxorubicin, vincristine, etoposide
and cisplatin; all of the aforementioned chemotherapeutic agents
have been shown to induce senescence in various cancer cell lines
(23).
When cells enter senescence, certain markers are
expressed, such as senescence-associated β-galactosidase
(SA-β-gal). SA-β-gal has been identified as a specific marker for
cell senescence, as it is detected by histology in the majority of
senescent cells, but not present in non-senescent cells (24). Senescent cells can also be recognized
using physiological methods, such as measurement of the formation
of senescence-associated heterochromatin foci and SASP-associated
factors (25). Another marker
commonly used to identify senescent cells is the cyclin-dependent
kinase inhibitor 2A (p16INK4a) tumor suppressor protein,
which is expressed at a low level or is undetectable in the
majority of healthy cells and tissues, but is notably upregulated
in most senescent tumor cells (21,26). The
identification of novel markers of senescence may assist in the
prognosis of senescence and cancer. Various other biomarkers of
senescence have been identified and are listed in Table I (27–41).
Regulation of the cell cycle is a complex progress,
which controls basic activities including growth, division and
differentiation (42). Cell division
genes control the initiation and progression of the cell cycle, and
two tumor suppressor genes, namely retinoma inhibitory protein (Rb)
and p53, have important roles in cell cycle arrest and the
maintenance of senescence (43). The
Rb protein (pRb) is critical for the G1/S and
G2/M regulatory points of the cell cycle; when activated
through dephosphorylation it leads to the transcription of S-phase
genes being blocked (44). Tumor
cell senescence is also induced by p53 following treatment with
chemotherapeutic drugs, which results in cell cycle arrest in the
G1/S and G2/M phases (45,46). In
addition, some cyclin-dependent kinase (CDK) 4/6 inhibitors,
including palbociclib and amebaciclib, have also been demonstrated
to induce senescence. Currently, these drugs have been approved for
clinical, to be used alone or in combination, for chemotherapy
(47–49).
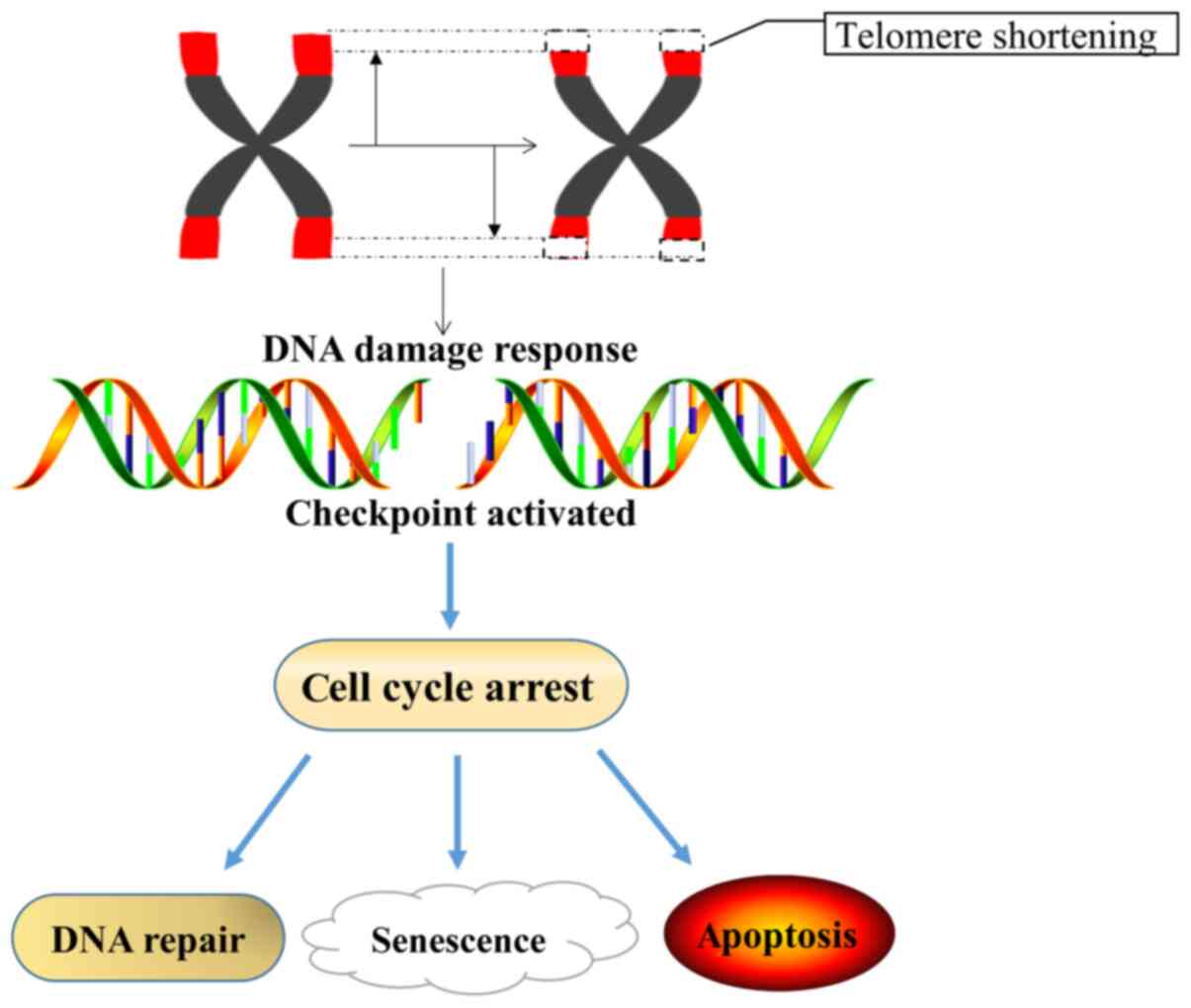
Telomere-induced cell senescence is a component of
replicative senescence. As the telomeres of normal cells shorten,
the cells eventually undergo a stagnation of proliferation or
division and reach senescence. By contrast, tumor cells often
exhibit unlimited proliferative capacity, because the length of
telomeres in tumor cells is stable (Fig.
1) (44,50,51).
Telomerase is a reverse transcriptase, the main
function of which is to add the repeat base sequence TTAGGG to the
end of chromosomes to increase the length of telomeres and the
number of cell divisions (9,52). One study demonstrated that the
telomeres of human primary fibroblasts shorten when the cells lose
their proliferative capacity (53).
However, the cell senescence caused by telomere shortening can be
prevented via the activation of telomerase (51). Telomerase serves an important role in
the process by which cells escape senescence, as its activation can
stabilize the length of telomeres and even prolong the life of
tumor cells. Among the 100 immortalized cell lines tested by Kim
et al (54), telomerase was
highly expressed in 94 tumor-derived cell lines.
Due to the complexity of telomerase, various
strategies for its inhibition have been developed as potential
treatments for cancer. These include the use of antisense
oligonucleotides to target the RNA component of telomerase,
chemical telomerase inhibitors, oligonucleotides and nucleosides,
small-molecule drugs that target human telomerase reverse
transcriptase (TERT), gene therapies, and molecules that target
telomeres and telomerase-related proteins (17,55–59).
Among these, a non-competitive inhibitor of TERT, BIBR1532, has
been demonstrated to shorten telomere length, inhibit cell
proliferation and induce senescence in human cancer cells (60). The aforementioned studies have shown
that telomerase dysfunction can induce cell senescence, damage
organ functions and shorten the human life span. However, the
process of senescence may be reversed by inhibiting telomerase
activation (61).
The levels of active oxygen free radicals within
living organisms increases over time (62). When free radicals are present in
excessive quantities or the antioxidant capacity is inadequate,
oxygen free radicals oxidize unsaturated fatty acids in cells,
causing lipid peroxidation and biofilm damage, thus damaging the
structure and function of organelles (63). The effects of ROS on cells are
positively associated with their concentration. ROS attack
mitochondrial DNA and accelerate the process of tumor cell
senescence. The premature senescence of human fibroblasts occurs in
a high-oxygen environment (40-50% O2), while the cell
cycle is extended in hypoxic conditions (2-3% O2)
(64,65). ROS intervene in tumor senescence
through processes such as lipid peroxidation, DNA damage and
protein destruction (66). Busulfan
has been demonstrated to create DNA-DNA and DNA-protein
cross-links, and induce the senescence of human fibroblasts in a
ROS-dependent manner (67).
When tumor cells are exposed to abnormal external
conditions but are unable to activate their auto-repair mechanisms,
they may undergo senescence or apoptosis (68). Numerous types of antitumor treatment,
including cytotoxic chemotherapy drugs, ionizing radiation and
topoisomerase inhibitors, are DNA-damaging agents that can induce
the senescence of tumor cells and as well as healthy cells
(69–72). It has been shown that
chemotherapeutic compounds, such as doxorubicin, etoposide and
cisplatin, which cause double or single-strand breakages of DNA,
can cause healthy human fibroblasts to become senescent prematurely
(73,74).
The activation of oncogenes in mammalian cells
results in proliferative stress and the induction of senescence,
which limits tumor growth. Genetic mutations can occur at any stage
of development, usually during DNA replication or the interphase of
cell division, and may affect DNA replication, DNA damage repair,
carcinogenesis and senescence (75).
Severe deficiencies in proteins that contribute to the sensing of
DNA damage and its repair have the potential to accelerate
senescence, while milder mutations in these same pathways may
predispose individuals to develop cancer (76,77). In
young human fibroblasts, mutation of the Ras gene has been shown to
induce cell cycle arrest in the G1 phase, a
senescence-like phenotype and SA-β-Gal expression (78,79).
Thus, senescence is a physiological mechanism of tumor suppression
that inhibits the progression from benign tumor lesions to
malignant tumors. In addition to the aforementioned factors,
certain cellular pathways are also able to induce cell
senescence.
Genes such as p53, p21 (CDK inhibitor 1A) and Rb
play important roles in cell senescence, and determine whether
cells are apoptotic or senescent (71). The ATM gene is an important component
of the DNA damage checkpoint pathway, which is critical in cell
cycle regulation, DNA damage response and repair (80). The loss of ATM leads to telomere
shortening and damage. When DNA is damaged, ATM is activated, and
induces the phosphorylation of downstream proteins such as
checkpoint kinase 1 (Chk1), Chk2 and cell division cycle. This
regulates the cell cycle checkpoints, so that DNA damage is
repaired (81).
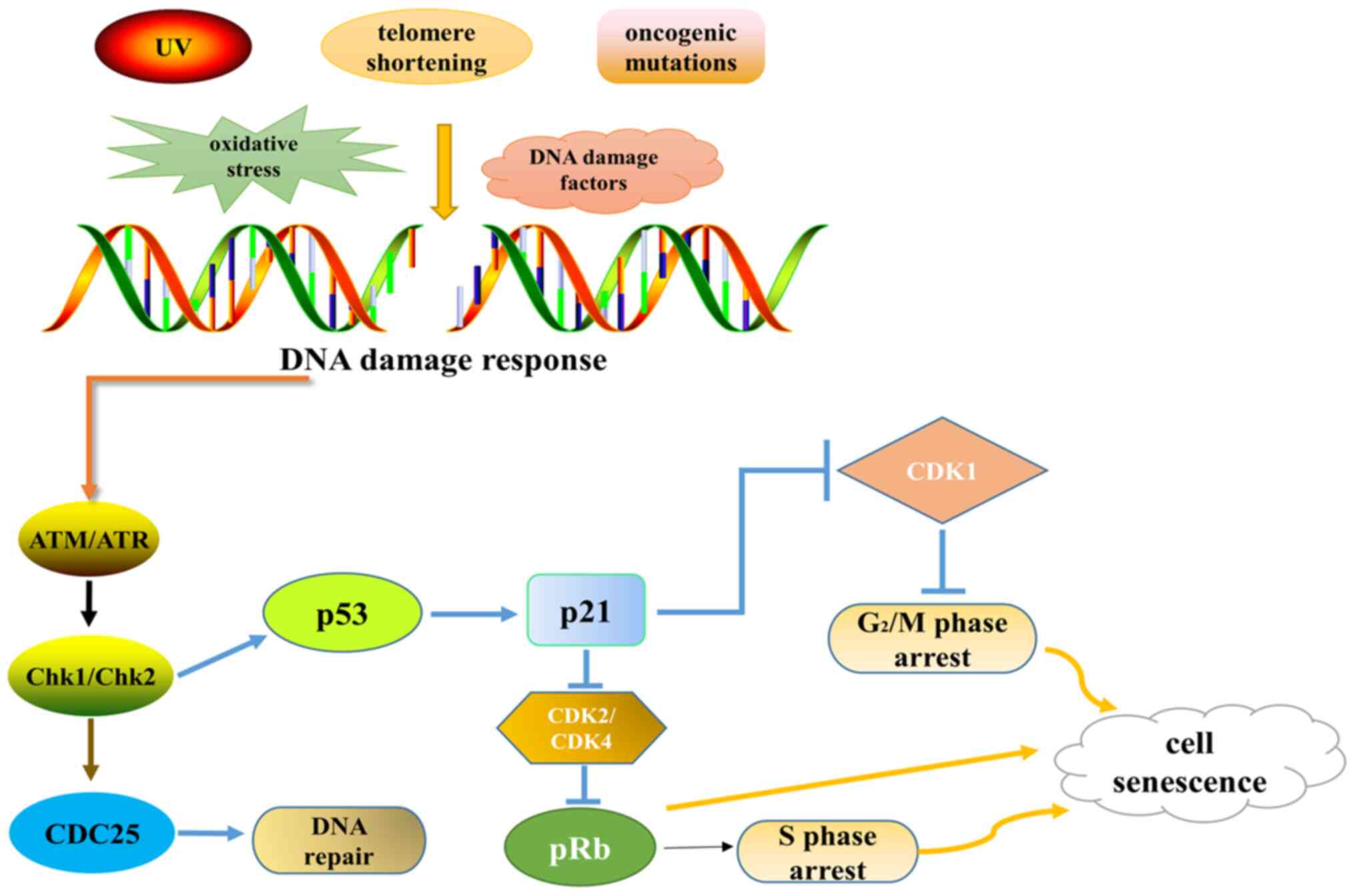
The p53 gene is important in cell carcinogenesis,
senescence, apoptosis and gene repair (82). When cells are exposed to external
stimuli that induce DNA damage, such as chemotherapy drugs,
ionizing radiation, gene mutation or telomerase shorting, the p53
gene is activated (83). p53
activates p21, which inhibits CDK1, causing cells to be arrested in
the G2/M phase and decreasing the phosphorylation level
of pRb by inhibiting the activity of CDK2 and CDK4. This prevents
cells from entering the S phase, and ultimately leads to cell
senescence (Fig. 2). In one study,
mice that were genetically engineered to express altered isoforms
of p53 with increased activity were shown to be resistant to cancer
(84). In human and mouse cells,
inactivation of the p53-p21 pathway leads to cells escaping
senescence (85). The knockdown of
p53 or p21 reduced the drug-induced senescence of HCT116 cell line
several folds, and the same phenomenon was observed when p53
expression was suppressed in HT1080 fibrosarcoma cells (86,87).
Genome-wide analysis has demonstrated that the loss of
p16INK4a expression and/or p53 function is the most
common genetic event in human cancers, and may enable cancers to
evade senescence (88).
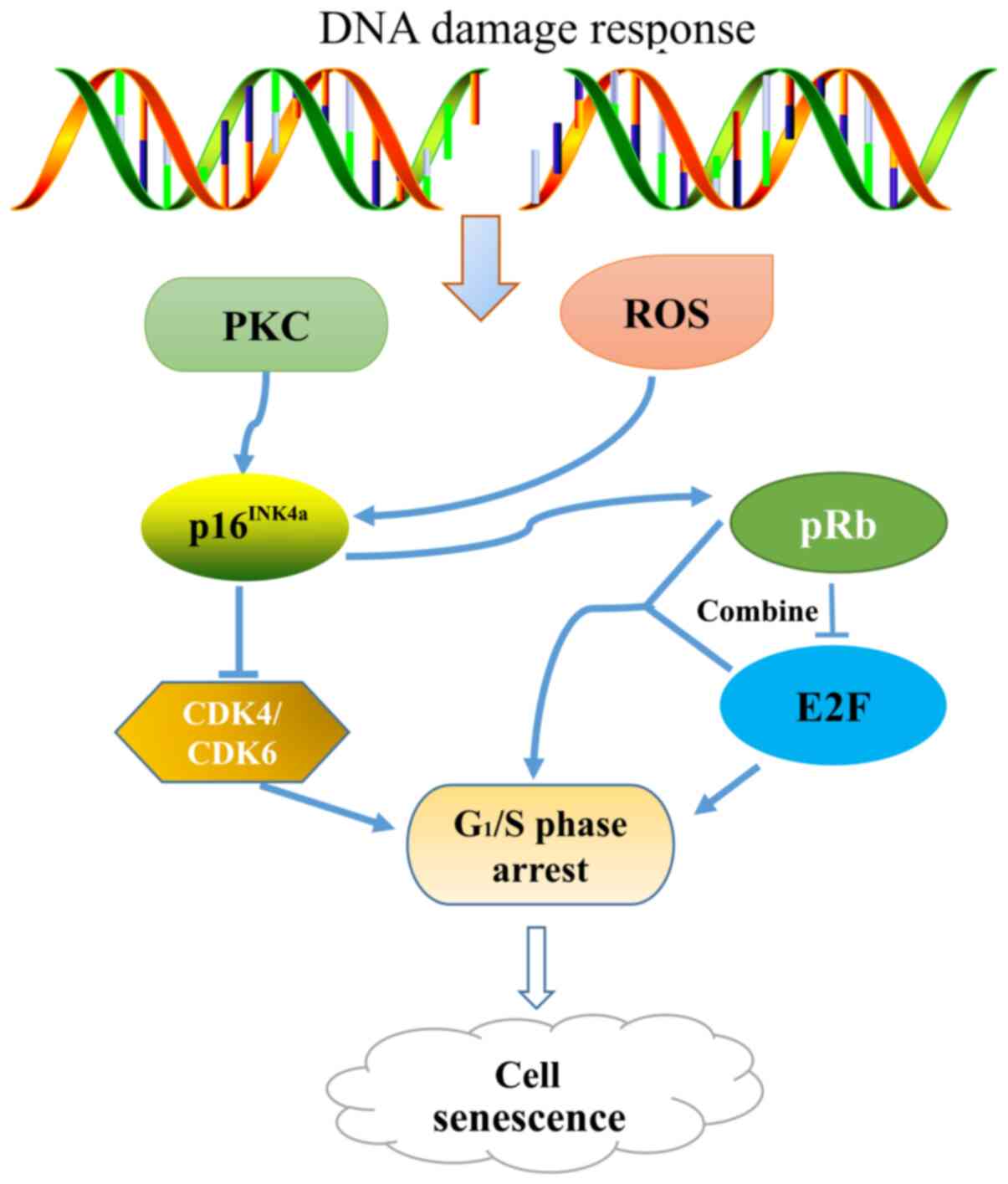
P14 (ARF) is one of the two proteins encoded by the
CDKN2A, the other of which is p16INK4a, and is an
important inducer of cellular senescence (39). The p14 (ARF) protein is expressed at
low levels in normal cells, but is markedly increased as the cells
reach senescence. P14 (ARF) blocks cells in the G1 and
G2/M phases mainly through the p53 pathway, leading to
cell senescence or apoptosis. It has been shown that when the
p16INK4a gene is knocked out, mice are prone to
developing tumors (35). It was
initially considered that the deletion of p16INK4a was
responsible for disturbing of the cell senescence pathway; however,
it has since been demonstrated that ARF-negative mice with normal
expression levels of p16INK4a exhibit the same phenotype
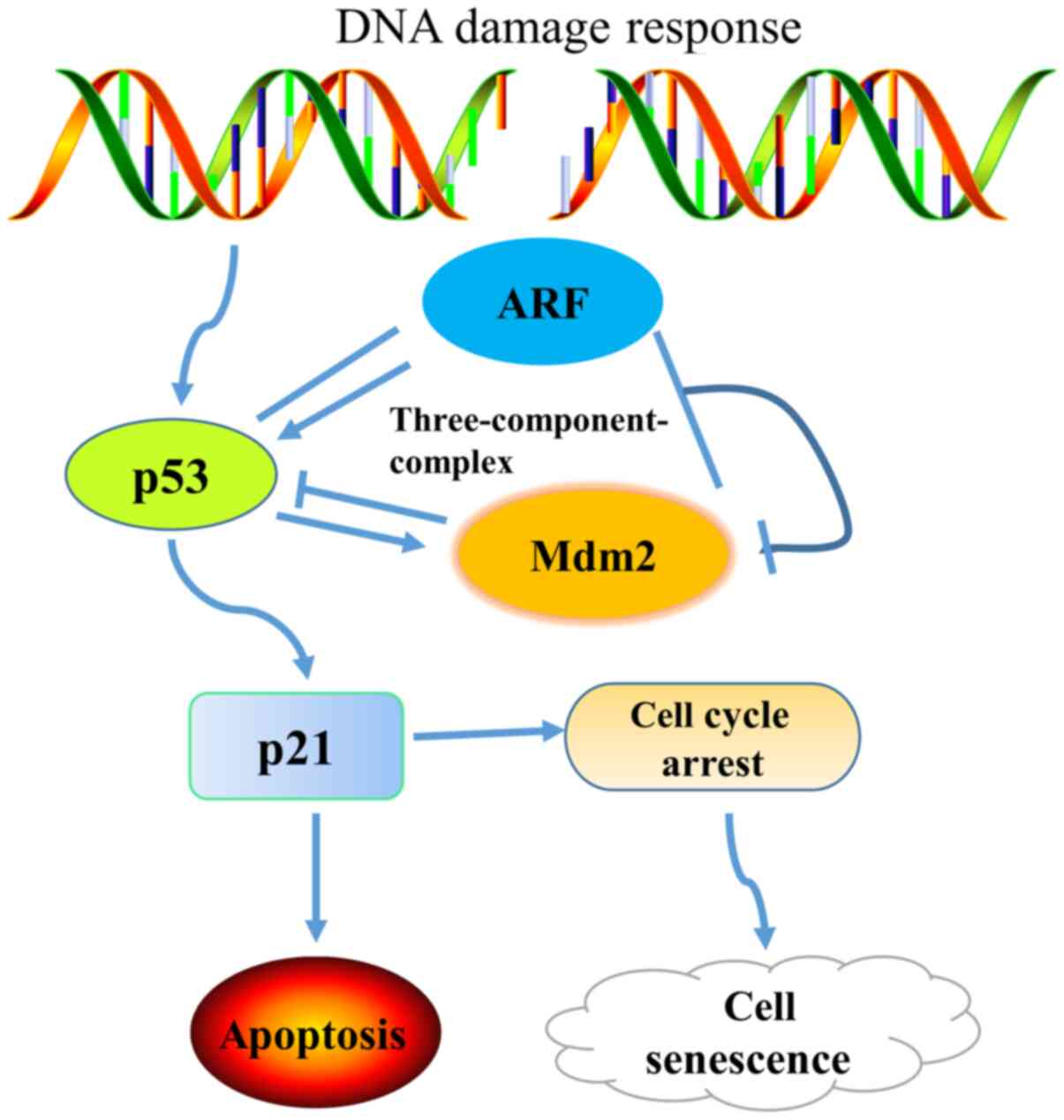
as those with p16INK4a deletion (94). Mdm2, also known as Hdm2, is a
proto-oncogene in human tissues, which induces the degradation of
p53 protein and inhibits its transcriptional activity. In
particular, Mdm2 specifically binds to p53, thereby inhibiting its
transcriptional activity and promoting its extracellular transport,
resulting in the inactivation of the p53 protein (98). Transcription of the Mdm2 gene is
regulated by a p53 transcriptional domain and the RAF-MEK-MAP
kinase signal transduction pathway. The upregulation of Mdm2 by the
RAF-MEK-MAP kinase pathway has been shown to decrease p53-mediated
apoptosis (99–101). The exogenous expression of ARF
stabilizes p53, promotes p53 transcription, p21WAF1/CIP1
expression and activation of the p53-mediated apoptotic pathway,
leading to apoptosis or premature senescence. In the absence of
stimulation, p53 can promote the expression of Mdm2 while,
conversely, Mdm2 reduces the activity of p53. p21 is downstream of
p53, and its expression blocks progression of the cell cycle,
promotes apoptosis and increases the sensitivity of tumor cells to
chemotherapy (102). Mdm2 promotes
the proteasome-mediated degradation of p21 and also reduces the
stability of p21 by binding with it. When Mdm2 expression is
downregulated, the expression of p21 protein increases (Fig. 4). Research has focused on
investigating the involvement of the ARF-Mdm2-p53-p21 pathway in
natural states as well as those involving premature senescence
(103). Notably, one study showed
that the loss of p14 (ARF) expression in patients with prostate
cancer was positively associated with an increased risk of disease
recurrence and metastatic disease (104).
Skp2 is a member of the F-box family, which mediates
cell cycle regulation and cell proliferation by degrading cell
cycle regulatory proteins, including p27, p21, p53, cyclin A,
cyclin E and cyclin D. The Skp2 gene is an important regulator of
the cell cycle, which interacts with the S-phase kinase cyclin
A-CDK2 (cyclin dependent kinase 2) complex. The expression levels
of Skp2 are very low in the G0/G1 phase, but
increase markedly in the S phase. An important substrate of Skp2 in
the cell division cycle is p27 (105). p27 is an important member of the
CDK family; p27kip1 is a protein encoded by the CDKN1B
gene. As a negative regulator of the cell cycle, p27kip1
inhibits the activity of CDK complexes to coordinate the cell
cycle, DNA replication and DNA repair (106,107).
In a study in which 68 cases of non-small cell lung cancer tissues
were compared with normal bronchial epithelial cells using tissue
microarrays and immunohistochemistry, the results revealed that
Skp2 was only expressed in the lung cancer tissues while
p27kip1 was expressed in both normal bronchial
epithelial cells and lung cancer cells (108). In addition, the expression of
p27kip1 was found to be significantly downregulated in
Skp2-positive cells and negatively correlated with Skp2 expression.
In another study, Shapira et al (109) analyzed the expression of Skp2 and
p27kip1 in the tissue sections of 80 patients with
colorectal tumors using immunohistochemistry, and found that Skp2
overexpression was significantly associated with the loss of
p27kip1 expression and cell differentiation.
Skp2 may contribute to a malignant phenotype, and
the overexpression of Skp2 protein results in the accelerated
hydrolysis of p27 and deterioration of tumor cells (110). Since Skp2 can degrade
p27kip1 and promote tumor development, interference with
the Skp2-p27kip1 pathway may induce tumor senescence
(111).
The LBR is an integral membrane protein of the
interphase nuclear envelope. Its N-terminus protrudes into the
nucleoplasm where it binds to lamin B and heterochromatin; these
interactions are disrupted during mitosis (112). A number of studies have indicated
that chromatin and chromatin proteins are involved in cellular
senescence (113,114). For example, the altered expression
of lamins A/C and B, which form the nuclear lamina, as well as
altered heterochromatin structures have been observed in senescent
cells (115,116). In addition, the expression of lamin
B1 (LB1) in WI-38 cells was found to decrease during cellular
senescence, and the silencing of LB1 slowed the proliferation of
these cells and induced premature senescence. These effects were
accompanied by a reduction in p53-dependent ROS, which was repaired
by growth under hypoxic conditions (15).
In addition to the aforementioned senescent
pathways, studies have demonstrated that inhibition of the ERK and
AKT pathways can reduce the number of senescent cells, with ERK and
AKT potentially acting through the ETS variant transcription factor
6 and forkhead box O1 genes (117–120).
As such, drugs that target and disrupt downstream effectors of ERK
and AKT have been proposed as new therapeutic methods. It has also
been demonstrated that oxytocin can alleviate cell senescence
through ERK/nuclear factor erythroid 2-related factor 2 (NrF2)
signaling; this implies that interference with the ERK/NrF2 pathway
may induce cell senescence (121).
Furthermore, the inhibition of mTORC1 has been shown to prolong the
lifespan of yeast, worms, fruit flies and some mice, suggesting
that mTORC1 may also be a novel target for inducing cell senescence
and treating various types of cancer (122).
Cellular senescence and apoptosis are two equally
important endpoints when cells respond to stress. As senescence
acts as a major tumor suppressor mechanism, it has a number of
advantages over apoptosis when dealing with damaged cells. While
apoptosis permanently removes cells, senescence arrests them in a
functional but non-dividing state, which may provide a persistent
signal of oncogenic stress and thereby promote immune surveillance
(123). The absence of senescence
or apoptosis leads to treatment failure. A number of pathways have
been found to activate senescence; however, the phenomenon is
interesting in that although chemotherapeutic drugs induce tumor
cell senescence, they also promote tumor progression by inducing
the secretion of certain matrix metalloproteinases, growth factors
and cytokines (12), which may lead
to tissue remodeling, organ senescence and many age-related
diseases. Processes such as cellular senescence and telomere
shortening, which protect against cancer, may accelerate the aging
process. Various diseases have been found to have an association
with cell senescence, including tumors, idiopathic pulmonary
fibrosis, hypertension, Parkinson's disease and diabetes.
A number of pathways are shared between the
initiation of cell senescence and tumorigenesis in wound healing
and cancer development, such as the activation of proto-oncogenes
(124). Cell senescence and
tumorigenesis have incidence rates that increase with age. The
transient presence of senescent cells is beneficial during normal
tissue repair, but the accumulation of these cells can have an
adverse effect on local tissue homeostasis due to their
pro-inflammatory properties (125–130).
Even though senescence strongly inhibits tumorigenesis when
initially induced (131–135), the prolonged presence of senescent
cells is often associated with malignant cells and supports the
expansion of tumors (69).
Therefore, whether cell senescence promotes or inhibit tumors
varies according to the stage of occurrence, the genetic background
and the tissue. Cellular senescence prevents the development of
tumors in the early stages of life, but also results in the
deterioration of bodily processes. At later life stages, cellular
senescence drives the occurrence of a senescence phenotype and
age-associated diseases, including various degenerative diseases
and a range of hyperplastic diseases. SASP-associated proteins have
conflicting effects on cells within the body, indicating that
cellular senescence has both favorable and unfavorable consequences
during the development of cancer (69,136,137).
These proteins inhibit the development of tumor cells and remove
abnormal and damaged cells from the body. In addition, senescent
cells induce paracrine senescence in neighboring cells through the
SASP, which acts as a barrier against tumor growth. By contrast,
the SASP promotes the occurrence and development of tumors under
certain physiological conditions, by making tumor cells tolerant to
chemotherapeutic drugs (138).
Further studies are required to investigate how the senescence of
tumor cells can be induced or inhibited during treatment. However,
the production of WNT16B and secreted frizzled related protein
produced in an aged or genotoxin-treated tumor microenvironment has
been found to protect cancer cells from chemotherapy in a paracrine
manner (139,140).
Although the inhibition of telomerase activity and
induction of tumor cell senescence is a theoretically feasible
approach for the treatment of tumors, certain issues must be
overcome for its practical application. In particular, the
telomerase activity in certain tumors is very low, and thus the
inhibition of telomerase activity may not be effective for these
tumors. Conversely, some normal cells possess telomerase activity
(141,142). In such cases, the inhibition of
telomerase activity may cause adverse effects and undesirable
results.
Senescence serves as a new strategy for the
prevention tumors and their treatment. However, it would be
beneficial to identify means for inhibiting the senescence of
normal cells and prolonging life span whilst also promoting the
senescence of tumor cells to treat cancer. In addition, the
achievement of an effective balance between the reduction of
abnormal proliferation and slowing down of senescence is important,
to make good use of the double-edged sword of cellular
senescence.
Not applicable.
This review was supported by Yunnan Fundamental
Research Projects (grant no. 202001AU070141) and Yunnan Key
Laboratory of Pharmacology for Natural Products Open grant (grant
no. 2019G005).
All data generated or analyzed during this study are
included in this published article.
GW, YJ and CQ designed and conceived the review and
contributed to critical reading of the manuscript and editing. XC,
YL and JZ participated in drafting the manuscript and performed the
literature review. GW and YJ confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Hayflick L and Moorhead PS: The serial
cultivation of human diploid cell strains. Exp Cell Res.
25:585–621. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Deursen JM: The role of senescent
cells in ageing. Nature. 509:439–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He S and Sharpless NE: Senescence in
health and disease. Cell. 169:1000–1011. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
te Poele RH, Okorokov AL, Jardine L,
Cummings J and Joel SP: DNA damage is able to induce senescence in
tumor cells in vitro and in vivo. Cancer Res. 62:1876–1883.
2002.PubMed/NCBI
|
|
5
|
Wang Z, Liu H and Xu C: Cellular
senescence in the treatment of ovarian cancer. Int J Gynecol
Cancer. 28:895–902. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saretzki G and Von Zglinicki T:
Replicative aging, telomeres, and oxidative stress. Ann N Y Acad
Sci. 959:24–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng C, Yang M, Zhang Y, Lan M, Huang B,
Liu H and Zhou Y: Cyclic mechanical tension reinforces DNA damage
and activates the p53-p21-Rb pathway to induce premature senescence
of nucleus pulposus cells. Int J Mol Med. 41:3316–3326.
2018.PubMed/NCBI
|
|
8
|
Chandeck C and Mooi WJ: Oncogene-induced
cellular senescence. Adv Anat Pathol. 17:42–48. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Falandry C, Bonnefoy M, Freyer G and
Gilson E: Biology of cancer and aging: A complex association with
cellular senescence. J Clin Oncol. 32:2604–2610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baeeri M, Bahadar H, Rahimifard M,
Navaei-Nigjeh M, Khorasani R, Rezvanfar MA, Gholami M and Abdollahi
M: α-Lipoic acid prevents senescence, cell cycle arrest, and
inflammatory cues in fibroblasts by inhibiting oxidative stress.
Pharmacol Res. 141:214–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campisi J: Senescent cells, tumor
suppression, and organismal aging: Good citizens, bad neighbors.
Cell. 120:513–522. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu Y, Armstrong JL, Tchkonia T and
Kirkland JL: Cellular senescence and the senescent secretory
phenotype in age-related chronic diseases. Curr Opin Clin Nutr
Metab Care. 17:324–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Narita M, Nũnez S, Heard E, Narita M, Lin
AW, Hearn SA, Spector DL, Hannon GJ and Lowe SW: Rb-mediated
heterochromatin formation and silencing of E2F target genes during
cellular senescence. Cell. 113:703–716. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Georgakopoulou EA, Tsimaratou K, Evangelou
K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D,
Bartek J, Serrano M and Gorgoulis VG: Specific lipofuscin staining
as a novel biomarker to detect replicative and stress-induced
senescence. A method applicable in cryo-preserved and archival
tissues. Aging (Albany NY). 5:37–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimi T, Butin-Israeli V, Adam SA,
Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel
NS and Goldman RD: The role of nuclear lamin B1 in cell
proliferation and senescence. Genes Dev. 25:2579–2593. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Swanson EC, Manning B, Zhang H and
Lawrence JB: Higher-order unfolding of satellite heterochromatin is
a consistent and early event in cell senescence. J Cell Biol.
203:929–942. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Faget DV, Ren Q and Stewart SA: Unmasking
senescence: Context-dependent effects of SASP in cancer. Nat Rev
Cancer. 19:439–453. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qu K, Lin T, Wei J, Meng F, Wang Z, Huang
Z, Wan Y, Song S, Liu S, Chang H, et al: Cisplatin induces cell
cycle arrest and senescence via upregulating P53 and P21 expression
in HepG2 cells. Nan Fang Yi Ke Da Xue Xue Bao. 33:1253–1259.
2013.PubMed/NCBI
|
|
19
|
Yao GD, Yang J, Li Q, Zhang Y, Qi M, Fan
SM, Hayashi T, Tashiro S, Onodera S and Ikejima T: Activation of
p53 contributes to pseudolaric acid B-induced senescence in human
lung cancer cells in vitro. Acta Pharmacol Sin. 37:919–929. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang JW, Zhang SS, Song JR, Sun K, Zong
C, Zhao QD, Liu WT, Li R, Wu MC and Wei LX: Autophagy inhibition
switches low-dose camptothecin-induced premature senescence to
apoptosis in human colorectal cancer cells. Biochem Pharmacol.
90:265–275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shtutman M, Chang BD, Schools GP and
Broude EV: Cellular model of p21-induced senescence. Methods Mol
Biol. 1534:31–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodenak-Kladniew B, Castro A, Stärkel P,
De Saeger C, García de Bravo M and Crespo R: Linalool induces cell
cycle arrest and apoptosis in HepG2 cells through oxidative stress
generation and modulation of Ras/MAPK and Akt/mTOR pathways. Life
Sci. 199:48–59. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Geng YQ, Guan JT, Xu XH and Fu YC:
Senescence-associated beta-galactosidase activity expression in
aging hippocampal neurons. Biochem Biophys Res Commun. 396:866–869.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aird KM and Zhang R: Detection of
senescence-associated heterochromatin foci (SAHF). Methods Mol
Biol. 965:185–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bernardes de Jesus B and Blasco MA:
Assessing cell and organ senescence biomarkers. Circ Res.
111:97–109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park CW, Bak Y, Kim MJ, Srinivasrao G,
Hwang J, Sung NK, Kim BY, Yu JH, Hong JT and Yoon DY: The novel
small molecule STK899704 promotes senescence of the human A549
NSCLC cells by inducing DNA damage responses and cell cycle arrest.
Front Pharmacol. 9:1632018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi J, Pang L and Jiao S: The response of
nucleus pulposus cell senescence to static and dynamic compressions
in a disc organ culture. Biosci Rep. 38:BSR201800642018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nadeau S, Cheng A, Colmegna I and Rodier
F: Quantifying senescence-associated phenotypes in primary
multipotent mesenchymal stromal cell cultures. Methods Mol Biol.
2045:93–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bernhart E, Damm S, Heffeter P,
Wintersperger A, Asslaber M, Frank S, Hammer A, Strohmaier H,
DeVaney T, Mrfka M, et al: Silencing of protein kinase D2 induces
glioma cell senescence via p53-dependent and -independent pathways.
Neuro Oncol. 16:933–945. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matjusaitis M, Chin G, Sarnoski EA and
Stolzing A: Biomarkers to identify and isolate senescent cells.
Ageing Res Rev. 29:1–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aravinthan A, Mells G, Allison M, Leathart
J, Kotronen A, Yki-Jarvinen H, Daly AK, Day CP, Anstee QM and
Alexander G: Gene polymorphisms of cellular senescence marker p21
and disease progression in non-alcohol-related fatty liver disease.
Cell Cycle. 13:1489–1494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sasaki M, Kuo FY, Huang CC, Swanson PE,
Chen CL, Chuang JH and Yeh MM: Increased expression of
senescence-associated cell cycle regulators in the progression of
biliary atresia: An immunohistochemical study. Histopathology.
72:1164–1171. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marcoux S, Le ON, Langlois-Pelletier C,
Laverdière C, Hatami A, Robaey P and Beauséjour CM: Expression of
the senescence marker p16INK4a in skin biopsies of acute
lymphoblastic leukemia survivors: A pilot study. Radiat Oncol.
8:2522013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pare R, Shin JS and Lee CS: Increased
expression of senescence markers p14 (ARF) and p16(INK4a) in breast
cancer is associated with an increased risk of disease recurrence
and poor survival outcome. Histopathology. 69:479–491. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Valdiglesias V, Giunta S, Fenech M, Neri M
and Bonassi S: γ-H2AX as a marker of DNA double strand breaks and
genomic instability in human population studies. Mutat Res.
753:24–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Noren Hooten N and Evans MK: Techniques to
induce and quantify cellular senescence. J Vis Exp. 555332017.doi:
10.3791/55533. PubMed/NCBI
|
|
38
|
Ko A, Han SY, Choi CH, Cho H, Lee MS, Kim
SY, Song JS, Hong KM, Lee HW, Hewitt SM, et al: Oncogene-induced
senescence mediated by c-Myc requires USP10 dependent
deubiquitination and stabilization of p14ARF. Cell Death Differ.
25:1050–1062. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salama RH, Sayed ZEA, Ashmawy AM, Elsewify
WA, Ezzat GM, Mahmoud MA, Alsanory AA and Alsanory TA:
Interrelations of apoptotic and cellular senescence genes
methylation in inflammatory bowel disease subtypes and colorectal
carcinoma in Egyptians patients. Appl Biochem Biotechnol.
189:330–343. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hernandez-Segura A, de Jong TV, Melov S,
Guryev V, Campisi J and Demaria M: Unmasking transcriptional
heterogeneity in senescent cells. Curr Biol. 27:2652–2660.e4. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Özcan S, Alessio N, Acar MB, Mert E,
Omerli F, Peluso G and Galderisi U: Unbiased analysis of senescence
associated secretory phenotype (SASP) to identify common components
following different genotoxic stresses. Aging (Albany NY).
8:1316–1329. 2016. View Article : Google Scholar
|
|
42
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sage J, Attardi L and Dyke TV: Roles of
p53 and pRB tumor suppressor networks in human cancer: Insight from
studies in the engineered mouse. Genetically Engineered Mice Cancer
Res. 293–308. 2012. View Article : Google Scholar
|
|
44
|
Chellappan SP, Hiebert S, Mudryj M,
Horowitz JM and Nevins JR: The E2F transcription factor is a
cellular target for the RB protein. Cell. 65:1053–1061. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Was H, Czarnecka J, Kowalczyk A, Barszcz
K, Bernas T, Piwocka K and Kaminska B: Some
chemotherapeutics-treated colon cancer cells display a specific
phenotype being a combination of stem-like and senescent cell
features. Cancer Biol Ther. 19:63–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mao Z, Ke Z, Gorbunova V and Seluanov A:
Replicatively senescent cells are arrested in G1 and G2 phases.
Aging (Albany NY). 4:431–435. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tao YF, Wang NN, Xu LX, Li ZH, Li XL, Xu
YY, Fang F, Li M, Qian GH, Li YH, et al: Molecular mechanism of
G1 arrest and cellular senescence induced by LEE011, a
novel CDK4/CDK6 inhibitor, in leukemia cells. Cancer Cell Int.
17:352017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vijayaraghavan S and Keyomarsi K: An
intact G1/S checkpoint determines response to CDK4/6 inhibitor in
breast cancer. Cancer Res. 76:29892016.
|
|
49
|
Vijayaraghavan S and Keyomarsi K: Abstract
P5-08-02: Inhibition of CDK4/6 induces senescence and autophagy in
ER positive breast cancers. Cancer Res. 75:P5–08-02. 2015.
|
|
50
|
Nelson DM, McBryan T, Jeyapalan JC, Sedivy
JM and Adams PD: A comparison of oncogene-induced senescence and
replicative senescence: Implications for tumor suppression and
aging. Age (Dordr). 36:96372014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhu Y, Liu X, Ding X, Wang F and Geng X:
Telomere and its role in the aging pathways: Telomere shortening,
cell senescence and mitochondria dysfunction. Biogerontology.
20:1–16. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Artandi SE and DePinho RA: Role of
telomeres and telomerase in cancer. Carcinogenesis. 31:9–18. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shitara S, Kakeda M, Nagata K, Hiratsuka
M, Sano A, Osawa K, Okazaki A, Katoh M, Kazuki Y, Oshimura M and
Tomizuka K: Telomerase-mediated life-span extension of human
primary fibroblasts by human artificial chromosome (HAC) vector.
Biochem Biophys Res Commun. 369:807–811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim NW, Piatyszek MA, Prowse KR, Harley
CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL and Shay
JW: Specific association of human telomerase activity with immortal
cells and cancer. Science. 266:2011–2015. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Barma DK, Elayadi A, Falck JR and Corey
DR: Inhibition of telomerase by BIBR 1532 and related analogues.
Bioorg Med Chem Lett. 13:1333–1336. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hájek M, Matulová N, Votruba I, Holý A and
Tloust'ová E: Inhibition of human telomerase by diphosphates of
acyclic nucleoside phosphonates. Biochem Pharmacol. 70:894–900.
2005. View Article : Google Scholar
|
|
57
|
Ji XM, Xie CH, Fang MH, Zhou FX, Zhang WJ,
Zhang MS and Zhou YF: Efficient inhibition of human telomerase
activity by antisense oligonucleotides sensitizes cancer cell
storadiotherapy. Acta Pharmacol Sin. 27:1185–1191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kondo Y and Kondo S: Telomerase RNA
inhibition using antisense oligonucleotide against human telomerase
RNA linked to a 2′,5′-Oligoadenylate. Methods Mol Biol. 405:97–112.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shammas MA, Simmons CG, Corey DR and
Shmookler Reis RJ: Telomerase inhibition by peptide nucleic acids
reverses ‘immortality’ of transformed human cells. Oncogene.
18:6191–6200. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pascolo E, Wenz C, Lingner J, Hauel N,
Priepke H, Kauffmann I, Garin-Chesa P, Rettig WJ, Damm K and
Schnapp A: Mechanism of human telomerase inhibition by BIBR1532, a
synthetic, non-nucleosidic drug candidate. J Biol Chem.
277:15566–15572. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Leão R, Apolónio JD, Lee D, Figueiredo A,
Tabori U and Castelo-Branco P: Mechanisms of human telomerase
reverse transcriptase (hTERT) regulation: Clinical impacts in
cancer. J Biomed Sci. 25:222018. View Article : Google Scholar
|
|
62
|
Reczek CR and Chandel NS: ROS promotes
cancer cell survival through calcium signaling. Cancer Cell.
33:949–951. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang W, Huang C, Sun A, Qiao L, Zhang X,
Huang J, Sun X, Yang X and Sun S: Hydrogen alleviates cellular
senescence via regulation of ROS/p53/p21 pathway in bone
marrow-derived mesenchymal stem cells in vivo. Biomed Pharmacother.
106:1126–1134. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zheng H, Huang Q, Huang S, Yang X, Zhu T,
Wang W, Wang H, He S, Ji L, Wang Y, et al: Senescence inducer
Shikonin ROS-dependently suppressed lung cancer progression. Front
Pharmacol. 9:5192018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen Q, Fischer A, Reagan JD, Yan LJ and
Ames BN: Oxidative DNA damage and senescence of human diploid
fibroblast cells. Proc Natl Acad Sci USA. 92:4337–4341. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pan Jing CJ: Advances in study of the
mechanisms of cellular senescence. J Pathogen Biol. 10:672–673.
2015.
|
|
67
|
Probin V, Wang Y and Zhou D:
Busulfan-induced senescence is dependent on ROS production upstream
of the MAPK pathway. Free Radic Biol Med. 42:1858–1865. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mirzayans R, Andrais B, Kumar P and Murray
D: Significance of Wild-type p53 signaling in suppressing apoptosis
in response to chemical genotoxic agents: Impact on chemotherapy
outcome. Int J Mol Sci. 18:9282017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Driscoll DL, Chakravarty A, Bowman D,
Shinde V, Lasky K, Shi J, Vos T, Stringer B, Amidon B, D'Amore N
and Hyer ML: Plk1 inhibition causes post-mitotic DNA damage and
senescence in a range of human tumor cell lines. PLoS One.
9:e1110602014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Klement K and Goodarzi AA: DNA double
strand break responses and chromatin alterations within the aging
cell. Exp Cell Res. 329:42–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yanai M, Makino H, Ping B, Takeda K,
Tanaka N, Sakamoto T, Yamaguchi K, Kodani M, Yamasaki A, Igishi T
and Shimizu E: DNA-PK inhibition by NU7441 enhances
Chemosensitivity to topoisomerase inhibitor in non-small cell lung
carcinoma cells by blocking DNA damage repair. Yonago Acta Med.
60:9–15. 2017.PubMed/NCBI
|
|
73
|
Robles SJ, Buehler PW, Negrusz A and Adami
GR: Permanent cell cycle arrest in asynchronously proliferating
normal human fibroblasts treated with doxorubicin or etoposide but
not camptothecin. Biochem Pharmacol. 58:675–685. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhao W, Lin ZX and Zhang ZQ:
Cisplatin-induced premature senescence with concomitant reduction
of gap junctions in human fibroblasts. Cell Res. 14:60–66. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang JC and Bennett M: Aging and
atherosclerosis: Mechanisms, functional consequences, and potential
therapeutics for cellular senescence. Circ Res. 111:245–259. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Finkel T, Serrano M and Blasco MA: The
common biology of cancer and ageing. Nature. 448:767–774. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lombard DB, Chua KF, Mostoslavsky R,
Franco S, Gostissa M and Alt FW: DNA repair, genome stability, and
aging. Cell. 120:0–512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Benanti JA and Galloway DA: Normal human
fibroblasts are resistant to RAS-induced senescence. Mol Cell Biol.
24:2842–2852. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sheng GY, Yi XR, Ting JS and Ying L:
Current advances of Ras induced senescence and the bypass
mechanism. Progress Bioch Biophysics. 43:652–660. 2016.
|
|
80
|
Balmus G, Zhu M, Mukherjee S, Lyndaker AM,
Hume KR, Lee J, Riccio ML, Reeves AP, Sutter NB, Noden DM, et al:
Disease severity in a mouse model of ataxia telangiectasia is
modulated by the DNA damage checkpoint gene Hus1. Hum Mol Genet.
21:3408–3420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yang CW, Tseng SF, Yu CJ, Chung CY, Chang
CY, Pobiega S and Teng SC: Telomere shortening triggers a feedback
loop to enhance end protection. Nucleic Acids Res. 45:8314–8328.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lima CRDO, Rabelo RE, Vulcani VAS, Cardoso
LD, de Sousa NLM and de Moura VMBD: P53 gene: Major mutations in
neoplasias and anticancer gene therapy. Cienc Rural. 42:845–853.
2012. View Article : Google Scholar
|
|
83
|
Wang C, Jurk D, Maddick M, Nelson G,
Martin-Ruiz C and von Zglinicki T: DNA damage response and cellular
senescence in tissues of aging mice. Aging Cell. 8:311–323. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tyner SD, Venkatachalam S, Choi J, Jones
S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C,
et al: p53 mutant mice that display early ageing-associated
phenotypes. Nature. 415:45–53. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jiang C, Liu G, Luckhardt T, Antony V,
Zhou Y, Carter AB, Thannickal VJ and Liu RM: Serpine 1 induces
alveolar type II cell senescence through activating p53-p21-Rb
pathway in fibrotic lung disease. Aging Cell. 16:1114–1124. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tong Y, Zhao W, Zhou C, Wawrowsky K and
Melmed S: PTTG1 attenuates drug-induced cellular senescence. PLoS
One. 6:e237542011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ling X, Xu C, Fan C, Zhong K, Li F and
Wang X: FL118 induces p53-dependent senescence in colorectal cancer
cells by promoting degradation of MdmX. Cancer Res. 74:7487–7497.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Coppé JP, Rodier F, Patil CK, Freund A,
Desprez PY and Campisi J: Tumor suppressor and aging biomarker
p16(INK4a) induces cellular senescence without the associated
inflammatory secretory phenotype. J Biol Chem. 286:36396–36403.
2011. View Article : Google Scholar
|
|
90
|
Mirzayans R, Andrais B, Hansen G and
Murray D: Role of p16(INK4A) in replicative senescence and DNA
damage-induced premature senescence in p53-deficient human cells.
Biochem Res Int. 2012:9515742012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gao S, Gao Y, He HH, Han D, Han W, Avery
A, Macoska JA, Liu X, Chen S, Ma F, et al: Androgen receptor tumor
suppressor function is mediated by recruitment of retinoblastoma
protein. Cell Rep. 17:966–976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Passegué E and Wagner EF: JunB suppresses
cell proliferation by transcriptional activation of p16(INK4a)
expression. EMBO J. 19:2969–2979. 2000. View Article : Google Scholar
|
|
93
|
Baek MW, Cho HS, Kim SH, Kim WJ and Jung
JY: Ascorbic acid induces necrosis in human laryngeal squamous cell
carcinoma via ROS, PKC, and calcium signaling. J Cell Physiol.
232:417–425. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Min EY, Kim IH, Lee J, Kim EY, Choi YH and
Nam TJ: The effects of fucodian on senescence are controlled by the
p16INK4a-pRb and p14Arf-p53 pathways in hepatocellular carcinoma
and hepatic cell lines. Int J Oncol. 45:47–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Nakade K, Lin CS, Chen XY, Tsai MH,
Wuputra K, Zhu ZW, Pan JZ and Yokoyama KK: Jun dimerization protein
2 controls hypoxia-induced replicative senescence via both the
p16Ink4a-pRb and Arf-p53 pathways. FEBS Open Bio.
7:1793–1804. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Krishnamurthy J, Ramsey MR, Ligon KL,
Torrice C, Koh A, Bonner-Weir S and Sharpless NE: p16INK4a induces
an age-dependent decline in islet regenerative potential. Nature.
443:453–457. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Da Silva-Álvarez S, Picallos-Rabina P,
Antelo-Iglesias L, Triana-Martínez F, Barreiro-Iglesias A, Sánchez
L and Collado M: The development of cell senescence. Exp Gerontol.
128:1107422019. View Article : Google Scholar
|
|
98
|
Zhao Y, Aguilar A, Bernard D and Wang S:
Small-molecule inhibitors of the MDM2-p53 protein-protein
interaction (MDM2 Inhibitors) in clinical trials for cancer
treatment. J Med Chem. 58:1038–1052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zheng S, Huang KE, Pan YL, Zhou Y, Pan SD,
Li X, Jia J, Zheng XL and Tao DY: KIT and BRAF heterogeneous
mutations in gastrointestinal stromal tumors after secondary
imatinib resistance. Gastric Cancer. 18:796–802. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li Z, Fu J, Rew Y, Gribble MW and Medina
JC: Abstract 3663: Discovery of sulfonamide-piperidinones as potent
inhibitors of the MDM2-p53 protein-protein interaction. Cancer Res.
75:3663. 2015.PubMed/NCBI
|
|
101
|
Seipel K, Marques M, Sidler C, Mueller BU
and Pabst T: The cellular p53 inhibitor MDM2 and the growth factor
receptor FLT3 as biomarkers for treatment responses to the
MDM2-inhibitor idasanutlin and the MEK1 inhibitor cobimetinib in
acute myeloid leukemia. Cancers. 10:1702018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yue Z, Rong J, Ping W, Bing Y, Xin Y, Feng
LD and Yaping W: Gene expression of the p16(INK4a)-Rb and
p19(Arf)-p53-p21(Cip/Waf1) signaling pathways in the regulation of
hematopoietic stem cell aging by ginsenoside Rg1. Genet Mol Res.
13:10086–10096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wei W, Hemmer RM and Sedivy JM: Role of
p14(ARF) in replicative and induced senescence of human
fibroblasts. Mol Cell Biol. 21:6748–6757. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Pencik J, Schlederer M, Gruber W, Unger C,
Walker SM, Chalaris A, Marié IJ, Hassler MR, Javaheri T, Aksoy O,
et al: STAT3 regulated ARF expression suppresses prostate cancer
metastasis. Nat Commun. 6:77362015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hershko D, Bornstein G, Ben-Izhak O,
Carrano A, Pagano M, Krausz MM and Hershko A: Inverse relation
between levels of p27(Kip1) and of its ubiquitin ligase subunit
Skp2 in colorectal carcinomas. Cancer. 91:1745–1751. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Sharma SS, Ma L and Pledger WJ: p27Kip1
inhibits the cell cycle through non-canonical G1/S phase-specific
gatekeeper mechanism. Cell Cycle. 14:3954–3964. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ma D, Guo D, Li W and Zhao H: Mdig, a lung
cancer-associated gene, regulates cell cycle progression through
p27(KIP1). Tumour Biol. 36:6909–6917. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Hu X, Liu F, Jiang B and Wang Y: The
expression of Skp2 in human non-small cell lung cancer and its
correlation with expression of p27 protein. Zhongguo Fei Ai Za Zhi.
11:547–550. 2008.(In Chinese). PubMed/NCBI
|
|
109
|
Shapira M, Ben-Izhak O, Linn S, Futerman
B, Minkov I and Hershko DD: The prognostic impact of the ubiquitin
ligase subunits Skp2 and Cks1 in colorectal carcinoma. Cancer.
103:1336–1346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hafez MM, Alhoshani AR, Al-Hosaini KA,
Alsharari SD, Al Rejaie SS, Sayed-Ahmed MM and Al-Shabanah OA:
SKP2/P27Kip1 pathway is associated with advanced ovarian cancer in
Saudi patients. Asian Pac J Cancer Prev. 16:5807–5815. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gstaiger M, Jordan R, Lim M, Catzavelos C,
Mestan J, Slingerland J and Krek W: Skp2 is oncogenic and
overexpressed in human cancers. Proc Natl Acad Sci USA.
98:5043–5048. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Olins AL, Rhodes G, Welch DBM, Zwerger M
and Olins DE: Lamin B receptor: Multi-tasking at the nuclear
envelope. Nucleus. 1:53–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
En A, Takauji Y, Ayusawa D and Fujii M:
The role of lamin B receptor in the regulation of
senescence-associated secretory phenotype (SASP). Exp Cell Res.
390:1119272020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sadaie M, Salama R, Carroll T, Tomimatsu
K, Chandra T, Young AR, Narita M, Pérez-Mancera PA, Bennett DC,
Chong H, et al: Redistribution of the Lamin B1 genomic binding
profile affects rearrangement of heterochromatic domains and SAHF
formation during senescence. Genes Dev. 27:1800–1808. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Solovei I, Wang AS, Thanisch K, Schmidt
CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et
al: LBR and lamin A/C sequentially tether peripheral
heterochromatin and inversely regulate differentiation. Cell.
152:584–598. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Arai R, En A, Takauji Y, Maki K, Miki K,
Fujii M and Ayusawa D: Lamin B receptor (LBR) is involved in the
induction of cellular senescence in human cells. Mech Ageing Dev.
178:25–32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Latorre E, Ostler EL, Faragher RGA and
Harries LW: FOXO1 and ETV6 genes may represent novel regulators of
splicing factor expression in cellular senescence. FASEB J.
33:1086–1097. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
He Q, Xue S, Tan Y, Zhang L, Shao Q, Xing
L, Li Y, Xiang T, Luo X and Ren G: Dual inhibition of Akt and ERK
signaling induces cell senescence in triple-negative breast cancer.
Cancer Lett. 448:94–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lam EW, Francis RE and Petkovic M: FOXO
transcription factors: Key regulators of cell fate. Biochem Soc
Trans. 34:722–726. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Clark O, Daga S and Stoker AW: Tyrosine
phosphatase inhibitors combined with retinoic acid can enhance
differentiation of neuroblastoma cells and trigger ERK- and
AKT-dependent, p53-independent senescence. Cancer Lett. 328:44–54.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wu H, Zhao J, Chen M, Wang H, Yao Q, Fan J
and Zhang M: The Anti-aging effect of erythropoietin via the
ERK/Nrf2-ARE pathway in aging rats. J Mol Neurosci. 61:449–458.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kucheryavenko O, Nelson G, von Zglinicki
T, Korolchuk VI and Carroll B: The mTORC1-autophagy pathway is a
target for senescent cell elimination. Biogerontology. 20:331–335.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shan HY, Bai XJ and Chen XM: Apoptosis is
involved in the senescence of endothelial cells induced by
angiotensin II. Cell Biol Int. 32:264–270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Goruppi S and Dotto GP: Mesenchymal
Stroma: Primary determinant and therapeutic target for epithelial
cancer. Trends Cell Biol. 23:593–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lee S and Schmitt CA: The dynamic nature
of senescence in cancer. Nat Cell Biol. 21:94–101. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Krizhanovsky V, Yon M and Dickins RA:
Senescence of activated stellate cells limits liver fibrosis. Cell.
134:657–667. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Burton DGA and Krizhanovsky V:
Physiological and pathological consequences of cellular senescence.
Cell Mol Life Sci. 71:4373–4386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Demaria M, Desprez PY, Campisi J and
Velarde MC: Cell Autonomous and Non-autonomous effects of senescent
cells in the skin. J Invest Dermatol. 135:1722–1726. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Muller M, Li Z and Maitz PKM: Pseudomonas
pyocyanin inhibits wound repair by inducing premature cellular
senescence: Role for p38 mitogen-activated protein kinase. Burns.
35:500–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Bitar MS, Abdel-Halim SM and Al-Mulla F:
Caveolin-1/PTRF upregulation constitutes a mechanism for mediating
p53-induced cellular senescence: Implications for evidence-based
therapy of delayed wound healing in diabetes. Am J Physiol
Endocrinol Metab. 305:E951–E963. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Michaloglou C, Vredeveld LCW, Soengas MS,
Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi
WJ and Peeper DS: BRAFE600-associated senescence-like cell cycle
arrest of human naevi. Nature. 436:720–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Bartkova J, Rezaei N, Liontos M,
Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E,
Niforou K, Zoumpourlis VC, et al: Oncogene-induced senescence is
part of the tumorigenesis barrier imposed by DNA damage
checkpoints. Nature. 444:633–637. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Chen Z, Trotman LC, Shaffer D, Lin HK,
Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Braig M, Lee S, Loddenkemper C, Rudolph C,
Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T and
Schmitt CA: Oncogene-induced senescence as an initial barrier in
lymphoma development. Nature. 436:660–665. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Collado M, Gil J, Efeyan A, Guerra C,
Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM,
Barbacid M, et al: Tumour biology: Senescence in premalignant
tumours. Nature. 436:6422005. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Campisi J: Aging and cancer: The
double-edged sword of replicative senescence. J Am Geriatr Soc.
45:482–488. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Mavrogonatou E, Pratsinis H and Kletsas D:
The role of senescence in cancer development. Semin Cancer Biol.
62:182–191. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Rao SG and Jackson JG: SASP: Tumor
suppressor or promoter? Yes! Trends Cancer. 2:676–687. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Sun Y, Campisi J, Higano C, Beer TM,
Porter P, Coleman I, True L and Nelson PS: Treatment-induced damage
to the tumor microenvironment promotes prostate cancer therapy
resistance through WNT16B. Nat Med. 18:1359–1368. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sun Y, Zhu D, Chen F, Qian M, Wei H, Chen
W and Xu J: SFRP2 augments WNT16B signaling to promote therapeutic
resistance in the damaged tumor microenvironment. Oncogene.
35:4321–4334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yang B, Shi L, Lei J, Li B and Jin Y:
Advances in optical assays for detecting telomerase activity.
Luminescence. 34:136–152. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Yuan X, Larsson C and Xu D: Mechanisms
underlying the activation of TERT transcription and telomerase
activity in human cancer: Old actors and new players. Oncogene.
38:6172–6183. 2019. View Article : Google Scholar : PubMed/NCBI
|