Introduction
Glioblastoma (GBM) is the most common and fatal
primary brain tumor, characterized by poor survival, with a median
survival time of ~1–2 years (1,2). The
current standard treatment for GBM includes maximal surgical
resection followed by an effective combination of radiotherapy and
chemotherapy (1). Despite this
multimodal approach, due to the intricate pathophysiology of GBM,
the prognosis and survival of patients remain poor (2). The development of resistance by tumor
cells and the inability of drugs to effectively cross the
blood-brain and blood-tumor barriers are often suggested as major
impediments to therapeutic development (3). Thus, novel and effective therapies are
urgently required to treat patients with GBM.
Herpes simplex virus type 1 (HSV-1), a common
natural human pathogen, can cause serious disease, from
asymptomatic viral shedding to fatal encephalitis and disseminated
disease (4). The virus is a
double-stranded DNA (dsDNA) virus containing a 152 kbp dsDNA
molecule encoding ~80 proteins (4).
The HSV genome encompasses two components, the long and short
regions, both containing a unique region flanked by inverted repeat
regions (terminal long/internal long and internal short/terminal
short) (5). HSV genes can be divided
into three groups according to the sequence of viral gene
expression, namely immediate early, early and late genes (5). The products of immediate early genes
regulate gene transcription, including the product of the ribosomal
protein S23 gene, infected-cell polypeptide 47 (ICP47), which
decreases major histocompatibility complex (MHC) class I expression
in infected cells by inhibiting transporter associated with antigen
presentation (6). The large subunit
of ribonucleotide reductase (ICP6) encoded by the early gene, UL39,
serves an essential role in HSV replication in noncycling cells
(7). The product of the late gene
γ34.5, ICP34.5, is considered a major mediator of HSV
neuropathogenicity, which enables the replication and propagation
of viruses in the brain (8). HSV
strains lacking ICP34.5 maintain their ability to replicate in
tumor cells; however, they fail to effectively replicate in normal
neurons (8).
HSV-1 has several features that make the development
of novel oncolytic (o)HSVs possible using genetic engineering
techniques (9). For example, its
large genome is stable and can be manipulated as multiple
nonessential genes, including those responsible for pathogenicity,
can be deleted, mutated or replaced with therapeutic transgenes,
and its genome does not integrate (9). HSV replication is affected by several
nucleotide-metabolizing enzymes, such as thymidine kinase, ICP34.5,
ICP6 and ICP0 (6). Only rapidly
proliferating tumor cells, which do not express the precursors for
viral DNA synthesis found in normal cells, can promote HSV
replication without the presence of nucleotide-metabolizing
enzymes, explaining why oncolytic viruses (OVs) can target tumors
(6). Engineered oHSVs can directly
destroy tumor cells by selectively replicating in them (oncolysis),
altering the tumor microenvironment and stimulating antitumor
immune responses, which are the main mechanisms by which OVs kill
cancer cells (10). Conversely,
anti-HSV agents, including ganciclovir and acyclovir, can ensure
the safety of oHSV in clinical use (6). The first genetically engineered HSV
capable of undergoing selective replication and killing GBM cells
was reported by Martuza et al in 1991, providing novel
insights into the application of ‘virotherapy’ for treating GBM
(11). It has been reported that
oHSVs, which selectively infect and kill tumor cells, are effective
in treating different types of cancer, including GBM, in
preclinical and clinical trials (12).
The present review summarizes different strategies
used in preclinical and clinical trials to improve the therapeutic
efficacy of oHSVs in GBM. In addition, findings from completed
clinical trials evaluating the potential use of oHSVs for GBM are
discussed. Limitations of the current clinical use of oHSVs for GBM
are highlighted to propose a promising future direction for
promoting the clinical application of virotherapy.
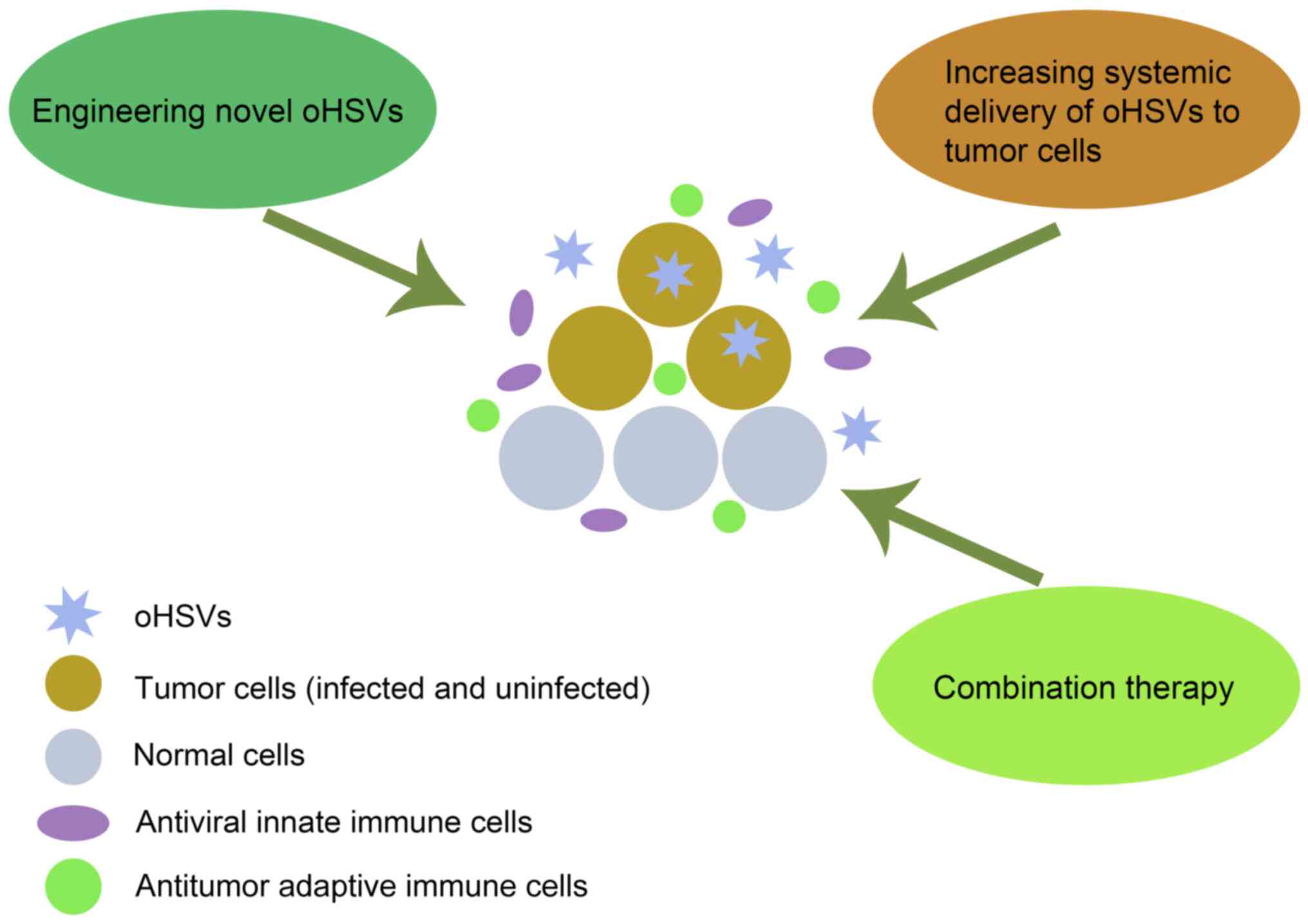
Multiple strategies to improve the
therapeutic efficacy of oHSV
Although oHSVs have exhibited promising results at
the clinical stage, several studies have demonstrated that viruses
alone are unlikely to completely cure GBM (6). In recent years, multiple strategies
have tried to improve the efficacy of oHSVs in the treatment of
GBM, including engineering novel oHSVs, developing combination
therapies and increasing systemic delivery of oHSVs to tumor cells
(Fig. 1).
Engineering novel oHSVs
As a biological antitumor agent for cancers, oHSV
has gone through several generations of genetic manipulation.
Traditionally, the main genetic engineering techniques used to
generate HSV mutants include homologous recombination, cell
transfection and bacterial artificial chromosome technology
(6). Initially, genetically
engineered viruses are constructed by deleting or mutating one or
more of the HSV-1 genes (10).
Conversely, there are double copies of multiple genes, such as
ICP34.5, ICP4 and ICP0, which complicate the genetic manipulation
of HSV-1 and limit the development of oHSVs but also provide more
possibilities, such as oHSVs with a single copy of γ34.5, ensuring
the safety of normal nerve cells while maintaining the antitumor
effect (8). Due to the development
of bioengineering technology and the improved understanding of
HSV-1 genes, additional novel oHSVs with improved efficacy and
safety have been engineered (Table
I).
 | Table I.List of typical oncolytic herpes
simplex viruses associated with the treatment of glioblastoma. |
Table I.
List of typical oncolytic herpes
simplex viruses associated with the treatment of glioblastoma.
| Classification | Virus | Mutation
characteristic | Stage | (Refs.) |
|---|
| Unarmed | Dlsptk | TK- (UL23) | P | (11) |
|
| HSV1716 |
γ34.5− | C (I) | (13,84–86) |
|
| C101 |
γ34.5− | P | (71) |
|
| C130 | γ34.5−,
HCMV-TRS1+ | P | (19) |
|
| C134 | γ34.5−,
HCMV–IRS1+ | C (I) | (18–20) |
|
| MG18L | US3−,
UL39−, LacZ+ | P | (47) |
|
| G207 | γ34.5−,
UL39−, LacZ+ | C (I) | (13,87–90) |
|
| rHSVQ1 | γ34.5−,
UL39−, EGFP+ | P | (71) |
|
| NG34 | γ34.5−,
UL39−, GADD34+ | P | (17) |
|
| G47Δ | γ34.5−,
US12−, UL39−, LacZ+ | C (I/IIa) | (13,91,92) |
|
| rQNestin34.5 | γ34.5−
(one copy of the γ34.5+), UL39−,
EGFP+ | C (I) | (15,17) |
| Retargeted | R-LM113 |
hHER2-retargeted | P | (24,25) |
|
| R-115 | hHER2-retargeted,
mIL-12 | P | (71) |
|
| R-337 | hHER2-retargeted,
mIL-12 | P | (22) |
|
| KNE |
EGFR-retargeted | P | (27) |
|
| KGE-4: T124 | EGFR-retargeted,
ICP4 regulated by miR-124 | P | (28) |
| Armed | HSV–VAE | γ34.5−,
UL39−, VAE+ | P | (35) |
|
| HSV1-CD | γ34.5−,
UL39−, LacZ+, CD+ | P | (38) |
|
| M032 | γ34.5−,
expressing hIL-12 | C (I) | (30,71) |
|
| M002 | γ34.5−,
expressing mIL-12 | P | (30) |
|
| OV-ChaseM | Expressing secreted
ChaseM | P | (44) |
|
| MGH2.1 | γ34.5−,
UL39−, CPA+, CYP2B1+ | P | (30,95) |
|
| AdFlt3L | γ34.5−,
LacZ+, expressing Flt3L | P | (30) |
|
| oHSV-ULBP3 | γ34.5−,
miR-124+, expressing ULBP3 | P | (33) |
|
| NG34scFvPD-1 | γ34.5−,
UL39−, GADD34+, scFVPD-1+ | P | (32) |
|
| OV-CDH1 | γ34.5−,
UL39−, CDH1+ (encoding E-cadherin) | P | (34) |
|
| G47Δ-TRAIL | γ34.5−,
US12−, UL39−, LacZ+, expressing
TRAIL | P | (31) |
|
| G47Δ-IL12 | γ34.5−,
US12−, UL39−, LacZ+, expressing
mIL-12 | P | (30,45) |
|
| G47Δ-mAngio | γ34.5−,
US12−, UL39−, LacZ+, expressing
mAngio | P | (95) |
Promoting the safety and viability of
oHSVs
A series of trials demonstrated the safety and
potential efficacy of mutants, such as HSV1716 (an ICP34.5 null
mutant), G207 (deletion of both copies of the γ134.5 gene locus and
a lacZ insertion into the UL39 locus) and G47Δ (generated from G207
with an additional deletion of the US12 gene) (13,14). The
multifunctional viral protein, ICP34.5 plays a key role in the host
interferon (IFN) response, which suppresses viral protein synthesis
(8). Even in tumor cells,
replication of oHSV without ICP34.5 is severely limited (14). To improve the replication of oHSV
lacking γ34.5, another mutant (rQNestin34.5) with greater
replication and reproduction capacity and cytotoxicity was
generated, in which one copy of the γ34.5 gene was reinserted into
the UL39-deleted, γ34.5-deleted viral genome under the control of a
tumor-specific promoter (15). The
carboxyl-terminus of GADD34 exhibits sequence homology with the
virulence factor ICP34.5 of HSV, which inhibits the neuropeptide Y
receptor Y4-catalyzed dephosphorylation of eukaryotic initiation
factor 2α (16). To further optimize
the virus and decrease its potential neurotoxicity to normal nerve
cells, Nakashima et al (17)
generated a novel weakened HSV-1 strain (NG34) by replacing ICP34.5
with GADD34 based on rQNestin34.5. NG34 expressing human GADD34 was
demonstrated to be significantly less neurovirulent in the brains
of non-tumor-bearing HSV-1-susceptible mice and exerted
considerable therapeutic potency in human GBM panels compared with
rQNestin34.5 (17).
Increasing evidence suggest that pathophysiological
hypoxia promotes the glioma stem-like cells (GSCs) phenotype and is
associated with tumor progression and poor prognosis (18). However, recent study has revealed
that the efficacy of oHSVs lacking γ34.5 was diminished in hypoxic
conditions (18). C130 and C134 are
two types of chimeric HSV-1 with deletion of the γ34.5 gene, which,
however, express the human cytomegalovirus tRNA-Ser (anticodon TGA)
2–1 and insulin receptor substate 1 genes, whose protein product
counteracts with host protein kinase R (19). A study demonstrated that the
cytotoxicity and viral recovery of C134 strain were significantly
improved under both hypoxic and normoxic conditions compared with
γ34.5-deleted strains, and its ability to infect and kill CD133+
GSCs was similar to that of wild-type (WT) HSV-1 (18). A preclinical evaluation of C134 in
mice and non-human primates confirmed its safety, thus providing a
strong basis for further clinical trials (20).
Receptor mediated retargeted
oHSVs
The majority of engineered OVs exhibit high safety
at the expense of virulence (13).
However, the ability of oHSVs to specifically infect tumor cells is
not ideal. The solution to this problem is the development of an
oHSV that specifically targets human GBM cells (21). It is currently known that HSV entry
is a multistep process that requires the essential glycoproteins,
gD, gH/gL and gB (22). gD is
activated by interacting with its natural receptors, nectin 1 or
HSV entry mediator (HVEM) (22).
This is an essential interaction for the entry of HSV. In addition,
gD activation is propagated to gH/gL and eventually to gB, a type
of fusogenic glycoprotein that mediates the fusion of virus
envelope with cell membrane (22).
Thus, the re-targeting strategy for HSV can be implemented via
modifying not only gD, but also gH (21).
Human epidermal growth factor receptor 2 (HER2), a
member of the EGFR family, is often overexpressed in GBM and other
types of cancer (23). R-LM113 is a
recombinant HSV strain generated by insertion of a single-chain
variable fragment (scFv) specific for HER2 in the region encoding
the viral envelope glycoprotein gD (24). This HSV is fully retargeted to HER2,
which is frequently expressed in GBMs (25). A study demonstrated that mice
injected with HER2-engineered GBM cells infected with R-LM113 can
survive twice as long as mice injected with uninfected cells
(25). EGFR is another attractive
target given that it is frequently mutated or overexpressed in GBM
(26). A study revealed that an
EGFR-retargeted oHSV (containing the gB:NT allele, KNE) can
efficiently and accurately enter cells expressing EGFR; this oHSV
was generated by introducing human EGFR-specific scFv into mutant
gD, which was demonstrated to be safe and effective in an
orthotopic mouse model of human GBM (27). Mazzacurati et al (28) generated a new EGFR-retargeted virus
strain (KGE-4:T124) by inserting four copies of the microRNA
(miR)-124 identification sequence into the 3′-untranslated region
of the ICP4 gene. Kge-4: T124 exhibited similar therapeutic effects
and had significantly reduced neurovirulence in an orthotopic human
GBM xenograft model (28). In
addition, several cell surface proteins, such as folic acid
receptor and CD44, are upregulated in tumor cells, broadening the
potential for the development of retargeted oHSVs (28). Tropism manipulation through
retargeting provides an avenue to increase cell-targeted viral
infectivity and specificity; however, further clinical trials are
required to verify the safety and efficacy of these novel
engineered oHSVs.
Armed oHSVs carrying therapeutic
genes
Although several types of oHSVs have achieved good
results in preclinical or clinical trials, the results have
demonstrated that it is difficult to completely eliminate tumors
using the virus alone (6). Some
genes are not required for HSV viral replication, and their
presence allows oHSVs to accommodate relatively large amounts of
foreign DNA molecules (5). Thus, to
enhance the oncolytic effect of HSV, several genes with therapeutic
potential on tumors can be introduced on the basis of gene deletion
or mutation to generate the so-called ‘armed oHSVs’. These genes
include immunomodulatory, tumor suppressor, antiangiogenic and
prodrug-activating genes.
Beyond direct oncolysis, the efficacy of GBM
virotherapy depends on the activation of antitumor immune
responses. The immunosuppressive environment of GBM is a
contributing factor to GBM development and progression (29). To overcome the immunosuppressive
barriers in GBM, several immune-stimulating genes, such as
interleukin (IL)-4, IL-12, Fms-related tyrosine kinase 3 ligand,
immune checkpoint inhibitors, particularly cytotoxic T
lymphocyte-associated antigen-4 and programmed death ligand 1, and
immune stimulators, which can effectively inhibit or even kill
tumor cells via antitumor immune responses, can be inserted into
various oHSVs (30). Some
preclinical trials demonstrated the safety and therapeutic efficacy
of these armed viruses, such as oHSV-UL16 binding protein 3,
G47Δ-tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) and NG34scFv-programmed cell death 1) (31–33). In
addition, treatment with OV-CDH1, an oHSV engineered to express
CDH1 encoding E-cadherin (34), can
substantially prolong the survival of GBM-bearing mice via the
OV-CDH1-expressed E-cadherin, which in turn can interact with
Killer cell lectin-like receptor G1 on natural killer (NK) cells,
thus allowing evasion of NK cell-mediated cytotoxicity and
improving viral spread (34).
The initiation of tumor neoangiogenesis and
vasculogenesis play key roles in the GBM microenvironment (3). It has been reported that angiogenesis
and vascular permeability stimulate tumor-associated macrophages
and microglia, resulting in limited replication and spread of oHSVs
in GBM cells (12). Zhu et al
(35) demonstrated that an oncolytic
HSV, namely γ34.5−, UL39−, carrying an
endostatin-angiostatin fusion gene (VAE) can significantly
attenuate the activity of GSCs in vitro. In addition, the
expression of exogenous VAE can inhibit human brain microvascular
endothelial cell proliferation (35). Furthermore, the antitumor effects of
G47Δ-IL12 include activation of the innate and adaptive immune
systems and inhibition of angiogenesis (30).
Prodrug-activating gene therapy can cause cell death
by specifically converting a non-toxic prodrug into one or more
cytotoxic metabolites (36). Yeast
cytosine deaminase (CD) is a peculiar system that converts the
nontoxic antifungal agent 5-fluorocytosine (5-FC) to the cytotoxic
chemotherapeutic agent 5-fluorouracil (5-FU) (37). HSV1-CD combined with systemic 5-FC
administration can increase the 5-FU concentration preferentially
within tumors, thus potentially decreasing systemic side effects
and increasing therapeutic efficacy (38).
Developing combination therapies
The hallmarks of GBM are closely associated with
multiple changes in the tumor microenvironment, including the
maintenance of proliferative signaling and the potential for
replicative immortality, invasion and metastasis (39). Thus, improved treatment for GBM, with
their multiple oncogenic pathways and refractory nature, requires a
multimodal approach. Recent studies have reported that the
combination of an oHSV and multiple anticancer modalities is often
more effective than any single treatment alone for GBM, as with
other malignancies (10,17).
Combining oHSVs with standard of care
therapy for GBM
Both radiation and temozolomide (TMZ) chemotherapy
are genotoxic, while oHSV infection also causes cellular DNA damage
responses (40). Synergy between TMZ
and oHSVs in GBM treatment occurs primarily through oHSV-mediated
manipulation of the DNA damage response, a universal mechanism of
cancer cell resistance to radiation and chemotherapy (40). A previous study suggested that TMZ
combined with oHSV G207 and G47Δ can kill human glioma cells by
mediating the coordinated manipulation of DNA damage responses
(40,41). Chondroitin sulfate proteoglycans
(CSPGs), commonly overexpressed in GBM, serves a critical role in
cell-cell and cell-extracellular matrix interactions. Increasing
evidence suggest that their overexpression is closely associated
with GBM cell proliferation and invasion (42). ChaseM is a mutant humanized version
of the Chase ABC enzyme that can remove chondroitin sulfate
glycosaminoglycans from CSPGs (43).
Combination therapy with OV-ChaseM, an oncolytic HSV-1 expressing
secreted Chase (44), and TMZ
resulted in a notable synergistic increase in glioma cell death
accompanied by enhanced apoptotic cell death (44). Recently, another study revealed that
TMZ exerts a negative effect on oHSV immunotherapy for GBM in
addition to its synergistic effect (45). Due to its negative effect on the
number and activity of T cells and macrophages, which is closely
associated with the therapeutic efficacy of G47Δ-IL12, TMZ cannot
improve the overall survival of orthotopic tumor-bearing mice
treated with G47Δ-IL12; instead, it eliminates certain beneficial
effects of G47Δ-IL12 when both are administered concurrently
(45). Therefore, understanding the
effects of TMZ on different types of oHSVs in different GBM models
is crucial for the clinical application of TMZ in combination with
oHSVs. In addition, the combination of an oHSV with ionizing
radiation exhibited synergistic therapeutic effects by inhibiting
the repair of DNA double strand damage following treatment of GBM
cells with ionizing radiation and by enhancing viral replication by
exposure to ionizing radiation (46).
Combining oHSV with other anticancer
agents
In addition to standard of care therapy, the
combination of oHSV with other anticancer agents can also be
promising for treating GBM. In vivo, the combination of
MG18L, an oHSV containing a US3 deletion and LacZ insertion in the
UL39 gene, and the PI3K/Akt inhibitors LY294002, triciribine,
GDC-0941 and BEZ235, can significantly prolong the overall survival
time of mice compared with mice treated with either single agent,
achieving a long-term survival rate of 50% in GBM-bearing mice
(47). Cheema et al (48) investigated a novel combinatorial
approach of G47Δ with low-doses of etoposide and revealed that this
combination can increase the survival of mice-bearing intracranial
human GSC-derived tumors, while no significant adverse side effects
were observed. In addition, the combination notably increased the
sub-G1 apoptotic population and significantly decreased
the G2-M phase population (48). It has been reported that the
enhancing effect of antiangiogenic agents on the antitumor efficacy
of OVs is associated with increased antiangiogenesis, viral spread,
decreased macrophage populations and enhanced oncolytic effects
(49,50). Combination treatment with bevacizumab
and an oHSV exerted significant effects in a glioma-bearing model
compared with those induced by either method alone (51). Another study suggested that
bortezomib, a peptide-based reversible proteasome inhibitor, can
cooperate with oHSV to treat tumors by increasing the expression of
heat shock protein 90, which in turn can mediate the nuclear
translocation of the oHSV polymerase enhancing viral replication
(52). This combination therapy can
also promote the activation of receptor interacting
serine/threonine kinase 1 and c-JNK, resulting in tumor cell death
and activation of the antitumor immune response by upregulating
IFN-γ and TRAIL in NK cells (52,53). In
addition, the combination of an oHSV with a poly(ADP-ribose)
polymerase inhibitor (PARP i), a new anticancer strategy, can
sensitize GSCs to PARP i therapy, thus promoting DNA damage, cell
death and apoptosis (54). Treatment
of mice bearing either PARP i-sensitive or PARP i-resistant
GSC-derived brain tumors with this combination therapy can markedly
extend the median survival time compared with mice treated with
either single agent (54).
OS2966 was the first clinical-ready humanized
monoclonal antibody blocking integrin β1 (55). In a preclinical model of therapy
resistant GBM, OS2966 exerted significant antitumor efficacy
(55). A recent study has
demonstrated that treatment with OS2966 attenuates the secretion of
proinflammatory cytokines and IFN signaling in oHSV-treated tumor
cells, and inhibits macrophage migration, resulting in enhanced
oHSV replication (56). Furthermore,
OS2966 can significantly inhibit the activation of the oHSV-induced
focal adhesion kinase/Akt signaling pathway, thus increasing the
cleavage of PARP and resulting in enhanced oHSV cytotoxicity
(56). Notch signaling is highly
active in GSCs, inhibiting cell differentiation and maintaining
stem-like characteristics, thus contributing to GBM tumorigenesis
and resistance to conventional therapies (57). Several γ secretase inhibitors (GSIs)
blocking the Notch signaling have been used in clinical trials
against different types of tumor, including glioma, and were
demonstrated to exhibit antitumor properties by inhibiting or
slowing tumor growth (58,59). Increasing evidence suggest that
several viral infections activate the Notch signaling. For example,
both oncolytic and WT HSV-1-infected glioma cells can activate the
Notch signaling in the surrounding cells via miR-H16-mediated
factors to inhibit hypoxia-inducible factor-1 downregulation
(60,61). Thus, treatment of GBM with oHSV can
further activate the Notch signaling, possibly promoting tumor
growth (61). Notably, a recent
study demonstrated that the combination of an oHSV with a
Notch-blocking GSI can affect previously unresponsive GBM cells
sensitive to GSIs, and demonstrated a therapeutic advantage over
treatment with oHSV in two different models of intracranial glioma
without affecting the safety profile of the virus in vivo
(61). Trametinib, an orally
administered mitogen-activated protein kinase (MEK) inhibitor, can
be beneficial for the treatment of GBM by targeting the
RAS-RAF-MEK-ERK signaling pathway; however, due to its poor central
nervous system penetrance, the use of the MEK inhibitor for GBM is
limited (62). A recent study
suggested that oHSV therapy can significantly increase the
blood-brain barrier penetration of trametinib and reverse the
trametinib-mediated feedback reactivation of the MEK signaling
(62). In addition, trametinib
treatment decreases oHSV therapy-mediated inflammatory tumor
necrosis factor-α secretion and enhances the T cell-dependent
antitumor immunity (63). These
findings suggest that more in-depth studies from different
perspectives are required to support the application of oHSV in GBM
treatment to provide additional benefits to patients with brain
tumors.
Combining oHSVs with suppression of
innate immunity
The antiviral immune response is the body's immunity
against the virus, which can effectively resist infection and
virus-mediated damage in the body (5). The prominent feature of tumor cells in
immunology is the emergence of new antigen markers that are not
expressed by the corresponding normal cells, which trigger the
immune system to eliminate these tumor cells (3). Antiviral and antitumor immune responses
are important guarantees for the adaptation of the organism to
changes and ensuring normal survival (10). oHSVs, with tumor selectivity and
immunogenicity, are considered the ideal choice when combined with
immunotherapy to improve the specificity and effectiveness of tumor
treatment (64). When a virus
infects the body, the activation of NK cells, macrophages and
microglia can hinder viral replication and spread; this process has
been recognized as a crucial defense mechanism against viruses
(65). HSV can escape the host's
immune response by various mechanisms, including inactivating
immunoglobulin, inhibiting the production of cytokines/chemokines,
blocking the maturation of antigen presenting cells, decreasing the
expression of MHC class I in infected cells and inhibiting
cytotoxic T lymphocyte mediated cell death (66). To prevent oHSV from replicating into
normal cells and to adapt its function to the tumor
microenvironment, a series of genes involved in host immune escape
need to be deleted or mutated (6).
Although an immunosuppressive state of the tumor microenvironment
allows oHSVs to enter and replicate, the replication of oHSVs is
not ideal (12). It is possible that
inhibition of innate immune response can decrease the antiviral
immunity and further ensure the replication of oHSVs in tumor
cells. In addition, oHSVs in tumor cells can enhance the
immunogenicity of tumor by promoting the infiltration of
inflammatory cells (10). For
example, G207 can induce systemic antitumor immunity by activating
cytotoxic T lymphocytes (66). It
has been reported that cyclophosphamide-induced inhibition of the
systemic immune response and oxindole/imidazole-induced disruption
of the STAT1/3-dependent oHSV barrier can substantially increase
viral survival and propagation, eventually leading to neoplastic
regression (67,68). In addition, transforming growth
factor (TGF)-β can regulate innate immune responses by inhibiting
the intracranial recruitment and activation of NK, macrophages and
microglia (66). Han et al
(69) demonstrated that combination
therapy with TGF-β and oHSV significantly increases the survival of
mice in both syngeneic and xenograft GBM models. Cellular
communication network factor 1 (CCN1) plays a key role in
coordinating the destruction and clearance of pathogens by
enhancing the innate macrophage-mediated antiviral immune response
(70). Anti-CCN1 antibody combined
with oHSV exhibits a tendency to improve tumor control, which is
associated with increased viral replication caused via suppression
of the antiviral immune response (71). Several studies have suggested that
innate immune cells, including NK cells, macrophages and microglia,
not only contribute to the early elimination of oHSV, but also play
an antitumor role in oncolytic therapy through different mechanisms
of antitumor immune responses (10,66).
However, the complex and apparently contradictory functions of
combination therapies require careful thought and explanation. In
addition, for all oHSV-based combination therapies, a series of
problems caused by adverse reactions, long-term side effects and
immune system dysregulation need to be resolved.
Increasing systemic delivery of oHSVs
to tumor cells
Achieving systemic delivery of therapeutic viruses
is the primary condition for the success of oHSV therapy. There are
some problems and defects that can limit the efficacy of oHSV
delivery into tumor cells by either intratumoral or intravenous
injection (71). The main reason for
the poor efficacy of the direct injection into the resection cavity
or into the tumor is that the cerebrospinal fluid and the secondary
postoperative bleeding into the cavity may rinse out the injected
virus (72). Systemic intravenous
injection is considered the best method for clinical delivery of
OVs (71). There are still many
obstacles that need to be addressed when trying to deliver OVs
specifically to tumors through intravenous injection, including
poor penetration into tumors and rapid immune-mediated
neutralization (73). The
blood-brain barrier is the first barrier to intravenous injection
of oHSV (74). To overcome this
limitation, it has been reported that destroying the blood-brain
barrier through a hypertonic mannitol solution and focused
ultrasound can increase the number of viruses reaching the tumor
site (73,74).
Cell carriers
To overcome the limitation of immune-mediated
neutralization in the systemic delivery of OVs, the use of cells as
delivery vectors for OVs seems to be a reliable method (75). Cell carriers can not only protect the
virus from being neutralized by various immune cells in the blood,
but also display inherent tumor tropism (75). Briefly, the OVs are inserted into
cell carriers, and when they target the tumor cells, OVs are
released to infect and destroy the tumor cells. Transformed cells,
immune cells and stem cells can be used as delivery vehicles
(75). Transformed cells were the
first type of cell vectors used for oHSV delivery to tumors since
they were more readily infected with OVs compared with normal cells
(76). The ability of transformed
cells to locate specific anatomical and tumor sites is limited
(75). In addition, the overall
feasibility of immune cells is limited since they are expensive,
and their clinical application is challenging. Factors released by
mesenchymal stem cells (MSCs) are known to exert antitumor effects,
which inhibits the proliferation of glioma cancer cells (77). In addition, MSCs have great potential
for treating a variety of diseases associated with immune responses
(77). It is highly attractive to
use MSCs as carriers to extend the current therapeutic strategies.
Several preclinical studies have demonstrated that stem cell-based
cell carriers can deliver a variety of OVs to tumors (75,78). Due
to tumor specificity and given that different carrier cells may
have different reactions with tumor cells, different cell carriers
may be required to achieve tumor specific delivery (76). Although this approach is promising,
there are still limitations and several obstacles for the
therapeutic application of cell carriers, such as the cost,
manufacturing and regulatory requirements (77,78).
Improving intratumoral delivery
The release of viral progeny from the killed solid
cancer cells is an important mechanism for OVs in destroying tumors
(10). However, due to the uneven
morphology and special physiological characteristics of tumors, the
spread of chemotherapeutic drugs or OVs between tumor cells is
limited during the delivery phase (79). In addition to the transport of OVs
into the solid tumor, the method to improve the delivery of OVs in
solid tumor is another important factor for improving the
therapeutic effect of OVs (76).
Neovascularization is considered as an important characteristic of
tumors; however, these microvascular networks are abnormal and
irregular (80). Previous studies
have demonstrated that reconstructing the tumor vasculature and
changing the tumor's pressure gradient can improve perfusion, thus
ameliorating drug penetration into the tumor (81,82). In
addition, ultrasound and magnetic drug are also considered
attractive and safe technologies (83).
oHSV clinical trials for GBM
Several clinical trials have assessed the
effectiveness of oHSVs in treating a range of tumors, including GBM
(84–86). HSV1716 is one of the ICP34.5 null
mutants (13). The first study
demonstrated the safety and feasibility of using a reproducible
oHSV in human therapy (84). The
results demonstrated that intratumoral injection of HSV1716 at
doses of 103−105 plaque-forming units (pfu)
for the treatment of nine cases of recurrent malignant glioma did
not cause notable adverse reactions and reactivation of latent HSV
(84). Furthermore, two separate
phase I clinical trials completed by the same UK group revealed
replication of HSV1716 without toxicity and supported the safety of
delivering HSV1716 into the resection cavity (85,86). The
most recent phase I trial of HSV1716 (NCT02031965) for recurrent
high-grade glioma was terminated in 2016, and no report has been
published (71).
G207 is another HSV-1 mutant with deletion of both
copies of the γ134.5 gene and lacZ insertion into the UL39 locus.
Based on the promising results of preclinical trials, four phase I
trials have been completed in the United States using G207 alone or
in combination with radiation (87–90). The
first trial commenced with 106 pfu dosage injected to a
single enhancing location. This was completed with the 21st patient
who had progressive or recurrent malignant gliomas despite being
inoculated with standard therapy (3×109 pfu at five
sites) (87). No toxic or serious
adverse events, particularly HSV encephalitis, can be clearly
attributed to G207, while radiological and neuropathologic evidence
support the antitumor activity (87). In the following phase IB clinical
trial, six patients with recurrent GBM each received two doses of
G207, at a total dose of 1.15×109 pfu. This study
demonstrated viral replication and antitumor activity of G207, and
a good overall safety profile of multiple-dose administration of
G207, including direct injection into the brain tissue surrounding
the tumor resection cavity (88).
The same group subsequently performed a phase I trial with G207,
where nine patients with progressive and recurrent malignant glioma
each inoculated with one dose of G207 at multiple sites of the
tumor margin, followed by local treatment with 5 Gy of radiation
(NCT00157703) (89). This study
supported the safety and potential clinical efficacy of combining
G207 with radiation therapy for treating malignant glioma.
Recently, this group further designed a phase I trial with
continuous infusion of G207 via intratumoral catheters to treat
progressive or recurrent malignant brain tumors (90). The results of this trial demonstrated
that intratumoral catheter can be used as a potentially effective
means of delivering OVs, and that the frameless stereotactic
technique was safe and feasible (90).
G47Δ, additional deletion of the ICP47 gene from
G207, is currently the only third-generation oHSV to be evaluated
in humans via a series of clinical trials completed in Japan
(91). After the phase I–IIa
clinical trial of G47Δ was completed in 2014, suggesting the safety
of G47Δ injection into the human brain (UMIN000002661), a
subsequent phase II clinical trial was performed in 2015 at the
University of Tokyo to evaluate the efficacy of G47Δ in patients
with recurrent or residual GBM. However, currently no results have
been published (UMIN000015995) (92). These clinical trials adopt several
strategies to improve efficacy, including engineering of novel
oHSVs, developing combination therapies and improving the delivery
efficiency of oHSVs into tumor cells. Thus, the combination of
multiple strategies may remain the focus of follow-up research.
In addition to oHSVs, scientists from the Duke
University completed another phase I clinical trial and announced
promising results (93). According
to this trial, 21% of a total of 61 patients with recurrent World
Health Organization grade IV GBM, who were treated with
intratumoral infusion of recombinant non-pathogenic
polio-rhinovirus chimera, survived for >3 years (NCT01491893)
(93). Several novel oHSVs, such as
rQNestin34.5, C134 and M032, have exhibited encouraging antitumor
efficacy and safety in the treatment of GBM in preclinical studies;
thus, clinical trials or preparations for trials are underway. The
clinical trials for treating malignant glioma with oHSVs are
summarized in Table II.
| Table II.oHSV clinical trials for malignant
glioma. |
Table II.
oHSV clinical trials for malignant
glioma.
| oHSV | Country | Stage | Target disease | Delivery | Trial number |
|---|
| HSV1716 | UK | I | Recurrent
high-grade glioma |
Intratumoral/Peritumoral | NCT02031965 |
| G207 | US | I | Progressive and
recurrent malignant glioma | Intratumoral
injection | NCT00157703 |
| G47Δ | Japan | II | Recurrent or
residual glioblastoma | Intratumoral
injection | UMIN000015995 |
| rQNestin34.5 | US | I | Recurrent
high-grade glioma | Peritumoral | NCT03152318 |
| C134 | US | I | Recurrent
high-grade glioma | Intratumoral
injection | NCT03657576 |
| M032 | US | I | Progressive and
current high-grade glioma | Intratumoral
injection | NCT02062827 |
Future perspectives
Due to the complex microenvironment and physiology
of GBM, the development of highly effective and novel therapies for
patients with GBM is difficult and slow. The continuous efforts and
investigation of oHSV-based therapy in preclinical and clinical
trials have provided valuable experiences and optimistic results,
offering hope for the successful clinical application of oHSVs for
treating GBM. Initially, the main emphasis was on oHSV safety by
means such as deletion of the ICP34.5 gene. Some novel oHSVs were
evaluated for the treatment of GBM in clinical trials. In these
trials, the use of these oHSVs in patients' brains was generally
well tolerated, but no significant improvement in overall patient
survival was observed. All OVs have the characteristics of the
parental WT virus, and their overall antitumor efficacy is not
ideal; thus, the development of novel viruses that are both safe
and exhibit excellent antitumor effects, particularly in GBM,
remains a top priority. Novel oHSVs with multigene mutations and
armed with specific foreign genes require further investigation. In
addition, given that the virus is easily neutralized by circulating
antibodies when it enters the bloodstream, the most effective
method for delivering oHSVs may not be intravenous infusion.
After penetrating tumor cells, the virus replicates
and eventually triggers cell lysis, which releases new viral
progeny that attack and kill neighboring cells (94). Tumor resection following viral
inoculation is associated with a low virus replication rate that
may indicate ineffective replication and limited transmission of
oHSV in tumors, a limitation that can directly compromise its
efficacy in GBM therapy (95).
Modification of intratumoral physiology may solve the problem of
poor tumor cell penetration. In addition, determining how to
decrease antiviral immunity and allow the virus to induce stronger
antitumor immunity deserves further investigation. Recently,
additional oHSV-based treatment strategies, such as arming the
virus with genes that encode proteins with therapeutic effects and
combining oHSV with other agents, have been investigated (33,56).
Thus, more clinical and preclinical trials are required to
determine the best combination of multiple strategies to achieve
unexpected results. Several challenges remain that require joint
efforts in the clinical translation of oHSV therapy to improve the
prognosis of patients with GBM.
Acknowledgements
Not applicable.
Funding
The present review was supported by grants from the
Project Program of Neurosurgical Clinical Research Center of
Sichuan Province (grant no. 17082), the Project Program of
Luzhou-Southwest Medical University (grant nos. 2016LZXNYD-G03 and
2018LZYD-ZK50), the Sichuan Province Returnees' Science and
Technology Activities Project [grant no. 2019(76)-72] and the
Applied and basic research program of the Science and Technology
Department of Sichuan Province (grant no. 2018JY0403 and
2018JY0404).
Availability of data and materials
Not applicable.
Authors' contributions
ZZ, JT, WZ, WX, YM, LC and JZ conceived and designed
the present review. ZZ performed the literature review and drafted
the initial manuscript. JZ and LC reviewed the manuscript for
important intellectual content. Data authentication is not
applicable. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kang X, Zheng Y, Hong W, Chen X, Li H,
Huang B, Huang Z, Tang H and Geng W: Recent advances in immune cell
therapy for glioblastoma. Front Immunol. 11:5445632020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Upadhaya PG, Pulakkat S and Patravale VB:
Nose-to-brain delivery: Exploring newer domains for glioblastoma
multiforme management. Drug Deliv Transl Res. 10:1044–1056. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alexander BM and Cloughesy TF: Adult
glioblastoma. J Clin Oncol. 35:2402–2409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ebrahimi S, Makvandi M, Abbasi S,
Azadmanesh K and Teimoori A: Developing oncolytic Herpes simplex
virus type 1 through UL39 knockout by CRISPR-Cas9. Iran J Basic Med
Sci. 23:937–944. 2020.PubMed/NCBI
|
|
5
|
Taylor TJ, Brockman MA, McNamee EE and
Knipe DM: Herpes simplex virus. Front Biosci. 7:d752–764. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Watanabe D and Goshima F: Oncolytic
virotherapy by HSV. Adv Exp Med Biol. 1045:63–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldstein DJ and Weller SK: Factor(s)
present in herpes simplex virus type 1-infected cells can
compensate for the loss of the large subunit of the viral
ribonucleotide reductase: Characterization of an ICP6 deletion
mutant. Virology. 166:41–51. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilcox DR and Longnecker R: The herpes
simplex virus neurovirulence factor γ34.5: Revealing Virus-host
interactions. PLoS Pathog. 12:e10054492016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Varghese S and Rabkin SD: Oncolytic herpes
simplex virus vectors for cancer virotherapy. Cancer Gene Ther.
9:967–978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glorioso JC, Cohen JB, Goins WF, Hall B,
Jackson JW, Kohanbash G, Amankulor N, Kaur B, Caligiuri MA, Chiocca
EA, et al: Oncolytic HSV vectors and anti-tumor immunity. Curr
Issues Mol Biol. 41:381–468. 2020.PubMed/NCBI
|
|
11
|
Martuza RL, Malick A, Markert JM, Ruffner
KL and Coen DM: Experimental therapy of human glioma by means of a
genetically engineered virus mutant. Science. 252:854–856. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peters C, Paget M, Tshilenge KT, Saha D,
Antoszczyk S, Baars A, Frost T, Martuza RL, Wakimoto H and Rabkin
SD: Restriction of replication of oncolytic herpes simplex virus
with a deletion of γ34.5 in glioblastoma stem-like cells. J Virol.
92:e00246–18. 2018. View Article : Google Scholar
|
|
13
|
Mineta T, Rabkin SD, Yazaki T, Hunter WD
and Martuza RL: Attenuated multi-mutated herpes simplex virus-1 for
the treatment of malignant gliomas. Nat Med. 1:938–943. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ,
Martuza RL, Wakimoto H and Rabkin SD: Effect of gamma34.5 deletions
on oncolytic herpes simplex virus activity in brain tumors. J
Virol. 86:4420–4431. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kambara H, Okano H, Chiocca EA and Saeki
Y: An oncolytic HSV-1 mutant expressing ICP34.5 under control of a
nestin promoter increases survival of animals even when symptomatic
from a brain tumor. Cancer Res. 65:2832–2839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Connor JH, Weiser DC, Li S, Hallenbeck JM
and Shenolikar S: Growth arrest and DNA damage-inducible protein
GADD34 assembles a novel signaling complex containing protein
phosphatase 1 and inhibitor 1. Mol Cell Biol. 21:6841–6850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakashima H, Nguyen T, Kasai K, Passaro C,
Ito H, Goins WF, Shaikh I, Erdelyi R, Nishihara R, Nakano I, et al:
Toxicity and efficacy of a novel GADD34-expressing oncolytic HSV-1
for the treatment of experimental glioblastoma. Clin Cancer Res.
24:2574–2584. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Friedman GK, Nan L, Haas MC, Kelly VM,
Moore BP, Langford CP, Xu H, Han X, Beierle EA, Markert JM, et al:
γ134.5-deleted HSV-1-expressing human cytomegalovirus
IRS1 gene kills human glioblastoma cells as efficiently as
wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 22:348–355.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cassady KA: Human cytomegalovirus TRS1 and
IRS1 gene products block the double-stranded-RNA-activated host
protein shutoff response induced by herpes simplex virus type 1
infection. J Virol. 79:8707–8715. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cassady KA, Bauer DF, Roth J, Chambers MR,
Shoeb T, Coleman J, Prichard M, Gillespie GY and Markert JM:
Pre-clinical assessment of C134, a chimeric oncolytic herpes
simplex virus, in mice and non-human primates. Mol Ther Oncolytics.
5:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campadelli-Fiume G, Petrovic B, Leoni V,
Gianni T, Avitabile E, Casiraghi C and Gatta V: Retargeting
strategies for oncolytic herpes simplex viruses. Viruses. 8:632016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Campadelli-Fiume G, Menotti L, Avitabile E
and Gianni T: Viral and cellular contributions to herpes simplex
virus entry into the cell. Curr Opin Virol. 2:28–36. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang C, Burger MC, Jennewein L, Genssler
S, Schönfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M,
Tonn T, et al: ErbB2/HER2-specific NK cells for targeted therapy of
glioblastoma. J Natl Cancer Inst. Dec 6–2016.(Epub ahead of print).
doi: 10.1093/jnci/djv375. View Article : Google Scholar
|
|
24
|
Menotti L, Cerretani A, Hengel H and
Campadelli-Fiume G: Construction of a fully retargeted herpes
simplex virus 1 recombinant capable of entering cells solely via
human epidermal growth factor receptor 2. J Virol. 82:10153–10161.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gambini E, Reisoli E, Appolloni I, Gatta
V, Campadelli-Fiume G, Menotti L and Malatesta P:
Replication-competent herpes simplex virus retargeted to HER2 as
therapy for high-grade glioma. Mol Ther. 20:994–1001. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
An Z, Aksoy O, Zheng T, Fan QW and Weiss
WA: Epidermal growth factor receptor and EGFRvIII in glioblastoma:
Signaling pathways and targeted therapies. Oncogene. 37:1561–1575.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uchida H, Marzulli M, Nakano K, Goins WF,
Chan J, Hong CS, Mazzacurati L, Yoo JY, Haseley A, Nakashima H, et
al: Effective treatment of an orthotopic xenograft model of human
glioblastoma using an EGFR-retargeted oncolytic herpes simplex
virus. Mol Ther. 21:561–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mazzacurati L, Marzulli M, Reinhart B,
Miyagawa Y, Uchida H, Goins WF, Li A, Kaur B, Caligiuri M, Cripe T,
et al: Use of miRNA response sequences to block off-target
replication and increase the safety of an unattenuated,
glioblastoma-targeted oncolytic HSV. Mol Ther. 23:99–107. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Q, Han B, Meng X, Duan C, Yang C, Wu
Z, Magafurov D, Zhao S, Safin S, Jiang C and Cai J: Immunogenomic
analysis reveals LGALS1 contributes to the immune heterogeneity and
immunosuppression in glioma. Int J Cancer. 145:517–530. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Q and Liu F: Advances and potential
pitfalls of oncolytic viruses expressing immunomodulatory transgene
therapy for malignant gliomas. Cell Death Dis. 11:4852020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim CY, Jeong M, Mushiake H, Kim BM, Kim
WB, Ko JP, Kim MH, Kim M, Kim TH, Robbins PD, et al: Cancer gene
therapy using a novel secretable trimeric TRAIL. Gene Therapy.
13:330–338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Passaro C, Alayo Q, De Laura I, McNulty J,
Grauwet K, Ito H, Bhaskaran V, Mineo M, Lawler SE, Shah K, et al:
Arming an oncolytic herpes simplex virus type 1 with a Single-chain
fragment variable antibody against PD-1 for experimental
glioblastoma therapy. Clin Cancer Res. 25:290–299. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wirsching HG, Zhang H, Szulzewsky F, Arora
S, Grandi P, Cimino PJ, Amankulor N, Campbell JS, McFerrin L,
Pattwell SS, et al: Arming oHSV with ULBP3 drives abscopal immunity
in lymphocyte-depleted glioblastoma. JCI insight. 4:e1282172019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu B, Ma R, Russell L, Yoo JY, Han J, Cui
H, Yi P, Zhang J, Nakashima H, Dai H, et al: An oncolytic
herpesvirus expressing E-cadherin improves survival in mouse models
of glioblastoma. Nat Biotechnol. Nov 26–2018.(Epub ahead of print).
doi: 10.1038/nbt.4302.
|
|
35
|
Zhu G, Su W, Jin G, Xu F, Hao S, Guan F,
Jia W and Liu F: Glioma stem cells targeted by oncolytic virus
carrying endostatin-angiostatin fusion gene and the expression of
its exogenous gene in vitro. Brain Res. 1390:59–69. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kucerova L, Durinikova E, Toro L, Cihova
M, Miklikova S, Poturnajova M, Kozovska Z and Matuskova M: Targeted
antitumor therapy mediated by prodrug-activating mesenchymal
stromal cells. Cancer Lett. 408:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakamura H, Mullen JT, Chandrasekhar S,
Pawlik TM, Yoon SS and Tanabe KK: Multimodality therapy with a
replication-conditional herpes simplex virus 1 mutant that
expresses yeast cytosine deaminase for intratumoral conversion of
5-fluorocytosine to 5-fluorouracil. Cancer Res. 61:5447–5452.
2001.PubMed/NCBI
|
|
38
|
Yamada S, Kuroda T, Fuchs BC, He X, Supko
JG, Schmitt A, McGinn CM, Lanuti M and Tanabe KK: Oncolytic herpes
simplex virus expressing yeast cytosine deaminase: Relationship
between viral replication, transgene expression, prodrug
bioactivation. Cancer Gene Ther. 19:160–170. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sattiraju A, Sai KKS and Mintz A:
Glioblastoma stem cells and their microenvironment. Adv Exp Med
Biol. 1041:119–140. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kanai R, Rabkin SD, Yip S, Sgubin D, Zaupa
CM, Hirose Y, Louis DN, Wakimoto H and Martuza RL: Oncolytic
virus-mediated manipulation of DNA damage responses: Synergy with
chemotherapy in killing glioblastoma stem cells. J Natl Cancer
Inst. 104:42–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aghi M, Rabkin S and Martuza RL: Effect of
chemotherapy-induced DNA repair on oncolytic herpes simplex viral
replication. J Natl Cancer Inst. 98:38–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wade A, Robinson AE, Engler JR, Petritsch
C, James CD and Phillips JJ: Proteoglycans and their roles in brain
cancer. FEBS J. 280:2399–2417. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dmitrieva N, Yu L, Viapiano M, Cripe TP,
Chiocca EA, Glorioso JC and Kaur B: Chondroitinase ABC I-mediated
enhancement of oncolytic virus spread and antitumor efficacy. Clin
Cancer Res. 17:1362–1372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jaime-Ramirez AC, Dmitrieva N, Yoo JY,
Banasavadi-Siddegowda Y, Zhang J, Relation T, Bolyard C, Wojton J
and Kaur B: Humanized chondroitinase ABC sensitizes glioblastoma
cells to temozolomide. J Gene Med. 19:10.1002/jgm.2942. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saha D, Rabkin SD and Martuza RL:
Temozolomide antagonizes oncolytic immunovirotherapy in
glioblastoma. J Immunother Cancer. 8:e0003452020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Advani SJ, Mezhir JJ, Roizman B and
Weichselbaum RR: ReVOLT: Radiation-enhanced viral oncolytic
therapy. Int J Radiat Oncol Biol Phys. 66:637–646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kanai R, Wakimoto H, Martuza RL and Rabkin
SD: A novel oncolytic herpes simplex virus that synergizes with
phosphoinositide 3-kinase/Akt pathway inhibitors to target
glioblastoma stem cells. Clin Cancer Res. 17:3686–3696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cheema TA, Kanai R, Kim GW, Wakimoto H,
Passer B, Rabkin SD and Martuza RL: Enhanced antitumor efficacy of
low-dose Etoposide with oncolytic herpes simplex virus in human
glioblastoma stem cell xenografts. Clin Cancer Res. 17:7383–7393.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang W, Fulci G, Wakimoto H, Cheema TA,
Buhrman JS, Jeyaretna DS, Stemmer Rachamimov AO, Rabkin SD and
Martuza RL: Combination of oncolytic herpes simplex viruses armed
with angiostatin and IL-12 enhances antitumor efficacy in human
glioblastoma models. Neoplasia. 15:591–599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kurozumi K, Hardcastle J, Thakur R, Yang
M, Christoforidis G, Fulci G, Hochberg FH, Weissleder R, Carson W,
Chiocca EA and Kaur B: Effect of tumor microenvironment modulation
on the efficacy of oncolytic virus therapy. J Natl Cancer Inst.
99:1768–1781. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang W, Fulci G, Buhrman JS,
Stemmer-Rachamimov AO, Chen JW, Wojtkiewicz GR, Weissleder R,
Rabkin SD and Martuza RL: Bevacizumab with angiostatin-armed oHSV
increases antiangiogenesis and decreases bevacizumab-induced
invasion in U87 glioma. Mol Ther. 20:37–45. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yoo JY, Jaime-Ramirez AC, Bolyard C, Dai
H, Nallanagulagari T, Wojton J, Hurwitz BS, Relation T, Lee TJ,
Lotze MT, et al: Bortezomib treatment sensitizes oncolytic
HSV-1-treated tumors to NK cell immunotherapy. Clin Cancer Res.
22:5265–5276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yoo JY, Hurwitz BS, Bolyard C, Yu JG,
Zhang J, Selvendiran K, Rath KS, He S, Bailey Z, Eaves D, et al:
Bortezomib-induced unfolded protein response increases oncolytic
HSV-1 replication resulting in synergistic antitumor effects. Clin
Cancer Res. 20:3787–3798. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ning J, Wakimoto H, Peters C, Martuza RL
and Rabkin SD: Rad51 degradation: Role in oncolytic
Virus-Poly(ADP-Ribose) polymerase inhibitor combination therapy in
glioblastoma. J Natl Cancer Inst. 109:1–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carbonell WS, DeLay M, Jahangiri A, Park
CC and Aghi MK: β1 integrin targeting potentiates antiangiogenic
therapy and inhibits the growth of bevacizumab-resistant
glioblastoma. Cancer Res. 73:3145–3154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lee TJ, Nair M, Banasavadi-Siddegowda Y,
Liu J, Nallanagulagari T, Jaime-Ramirez AC, Guo JY, Quadri H, Zhang
J, Bockhorst KH, et al: Enhancing therapeutic efficacy of oncolytic
herpes simplex virus-1 with integrin β1 blocking antibody OS2966.
Mol Cancer Ther. 18:1127–1136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bazzoni R and Bentivegna A: Role of notch
signaling pathway in glioblastoma pathogenesis. Cancers (Basel).
11:2922019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yahyanejad S, King H, Iglesias VS, Granton
PV, Barbeau LM, van Hoof SJ, Groot AJ, Habets R, Prickaerts J,
Chalmers AJ, et al: NOTCH blockade combined with radiation therapy
and temozolomide prolongs survival of orthotopic glioblastoma.
Oncotarget. 7:41251–41264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kahlert UD, Cheng M, Koch K, Marchionni L,
Fan X, Raabe EH, Maciaczyk J, Glunde K and Eberhart CG: Alterations
in cellular metabolome after pharmacological inhibition of Notch in
glioblastoma cells. Int J Cancer. 138:1246–1255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu R, Li X, Tulpule A, Zhou Y, Scehnet
JS, Zhang S, Lee JS, Chaudhary PM, Jung J and Gill PS: KSHV-induced
notch components render endothelial and mural cell characteristics
and cell survival. Blood. 115:887–895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Otani Y, Yoo JY, Chao S, Liu J,
Jaime-Ramirez AC, Lee TJ, Hurwitz B, Yan Y, Dai H, Glorioso JC, et
al: Oncolytic HSV-infected glioma cells activate NOTCH in adjacent
tumor cells sensitizing tumors to gamma secretase inhibition. Clin
Cancer Res. 26:2381–2392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
de Gooijer MC, Zhang P, Weijer R, Buil
LCM, Beijnen JH and van Tellingen O: The impact of P-glycoprotein
and breast cancer resistance protein on the brain pharmacokinetics
and pharmacodynamics of a panel of MEK inhibitors. Int J Cancer.
142:381–391. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yoo JY, Swanner J, Otani Y, Nair M, Park
F, Banasavadi-Siddegowda Y, Liu J, Jaime-Ramirez AC, Hong B, Geng
F, et al: Oncolytic HSV therapy increases trametinib access to
brain tumors and sensitizes them in vivo. Neuro Oncol.
21:1131–1140. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nguyen T, Avci NG, Shin DH, Martinez-Velez
N and Jiang H: Tune up in situ autovaccination against solid tumors
with oncolytic viruses. Cancers (Basel). 10:1712018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yin J, Markert JM and Leavenworth JW:
Modulation of the intratumoral immune landscape by oncolytic herpes
simplex virus virotherapy. Front Oncol. 7:1362017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ma W, He H and Wang H: Oncolytic herpes
simplex virus and immunotherapy. BMC Immunol. 19:402018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Delwar ZM, Kuo Y, Wen YH, Rennie PS and
Jia W: Oncolytic virotherapy blockade by microglia and macrophages
requires STAT1/3. Cancer Res. 78:718–730. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ikeda K, Ichikawa T, Wakimoto H, Silver
JS, Deisboeck TS, Finkelstein D, Harsh GR IV, Louis DN, Bartus RT,
Hochberg FH and Chiocca EA: Oncolytic virus therapy of multiple
tumors in the brain requires suppression of innate and elicited
antiviral responses. Nat Med. 5:881–887. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Han J, Chen X, Chu J, Xu B, Meisen WH,
Chen L, Zhang L, Zhang J, He X, Wang QE, et al: TGFβ treatment
enhances glioblastoma virotherapy by inhibiting the innate immune
response. Cancer Res. 75:5273–5282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Thorne AH, Meisen WH, Russell L, Yoo JY,
Bolyard CM, Lathia JD, Rich J, Puduvalli VK, Mao H, Yu J, et al:
Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated
oncolytic herpes simplex virus clearance. Mol Ther. 22:1678–1687.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hua L and Wakimoto H: Oncolytic herpes
simplex virus therapy for malignant glioma: Current approaches to
successful clinical application. Expert Opin Biol Ther. 19:845–854.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Duebgen M, Martinez-Quintanilla J, Tamura
K, Hingtgen S, Redjal N, Wakimoto H and Shah K: Stem cells loaded
with multimechanistic oncolytic herpes simplex virus variants for
brain tumor therapy. J Natl Cancer Inst. 106:dju0902014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Simpson GR, Han Z, Liu B, Wang Y, Campbell
G and Coffin RS: Combination of a fusogenic glycoprotein, prodrug
activation, and oncolytic herpes simplex virus for enhanced local
tumor control. Cancer Res. 66:4835–4842. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Pan M, Zhang Y, Deng Z, Yan F and Hong G:
Noninvasive and local delivery of adenoviral-mediated herpes
simplex virus thymidine kinase to treat glioma through focused
ultrasound-induced blood-brain barrier opening in rats. J Biomed
Nanotechnol. 14:2031–2041. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Roy DG and Bell JC: Cell carriers for
oncolytic viruses: Current challenges and future directions.
Oncolytic Virother. 2:47–56. 2013.PubMed/NCBI
|
|
76
|
Coukos G, Makrigiannakis A, Kang EH,
Caparelli D, Benjamin I, Kaiser LR, Rubin SC, Albelda SM and
Molnar-Kimber KL: Use of carrier cells to deliver a
replication-selective herpes simplex virus-1 mutant for the
intraperitoneal therapy of epithelial ovarian cancer. Clin Cancer
Res. 5:1523–1537. 1999.PubMed/NCBI
|
|
77
|
Lin W, Huang L, Li Y, Fang B, Li G, Chen L
and Xu L: Mesenchymal stem cells and cancer: Clinical challenges
and opportunities. Biomed Res Int. 2019:28208532019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dührsen L, Hartfuss S, Hirsch D, Geiger S,
Maire CL, Sedlacik J, Guenther C, Westphal M, Lamszus K, Hermann FG
and Schmidt NO: Preclinical analysis of human mesenchymal stem
cells: Tumor tropism and therapeutic efficiency of local HSV-TK
suicide gene therapy in glioblastoma. Oncotarget. 10:6049–6061.
2019. View Article : Google Scholar
|
|
79
|
Dewhirst MW and Secomb TW: Transport of
drugs from blood vessels to tumour tissue. Nature reviews Cancer.
17:738–750. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ribeiro Franco PI, Rodrigues AP, de
Menezes LB and Pacheco Miguel M: Tumor microenvironment components:
Allies of cancer progression. Pathol Res Pract. 216:1527292020.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dickson PV, Hamner JB, Sims TL, Fraga CH,
Ng CY, Rajasekeran S, Hagedorn NL, McCarville MB, Stewart CF and
Davidoff AM: Bevacizumab-induced transient remodeling of the
vasculature in neuroblastoma xenografts results in improved
delivery and efficacy of systemically administered chemotherapy.
Clin Cancer Res. 13:3942–3950. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Miller A, Nace R, Ayala-Breton CC, Steele
M, Bailey K, Peng KW and Russell SJ: Perfusion pressure is a
critical determinant of the intratumoral extravasation of oncolytic
viruses. Mol Ther. 24:306–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hill C and Carlisle R: Achieving systemic
delivery of oncolytic viruses. Expert Opin Drug Deliv. 16:607–620.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Rampling R, Cruickshank G, Papanastassiou
V, Nicoll J, Hadley D, Brennan D, Petty R, MacLean A, Harland J,
McKie E, et al: Toxicity evaluation of replication-competent herpes
simplex virus (ICP 34.5 null mutant 1716) in patients with
recurrent malignant glioma. Gene Ther. 7:859–866. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Papanastassiou V, Rampling R, Fraser M,
Petty R, Hadley D, Nicoll J, Harland J, Mabbs R and Brown M: The
potential for efficacy of the modified (ICP 34.5(−)) herpes simplex
virus HSV1716 following intratumoural injection into human
malignant glioma: A proof of principle study. Gene Ther. 9:398–406.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Harrow S, Papanastassiou V, Harland J,
Mabbs R, Petty R, Fraser M, Hadley D, Patterson J, Brown SM and
Rampling R: HSV1716 injection into the brain adjacent to tumour
following surgical resection of high-grade glioma: Safety data and
long-term survival. Gene Ther. 11:1648–1658. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Markert JM, Medlock MD, Rabkin SD,
Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore
C, Tufaro F and Martuza RL: Conditionally replicating herpes
simplex virus mutant, G207 for the treatment of malignant glioma:
Results of a phase I trial. Gene Ther. 7:867–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Markert JM, Liechty PG, Wang W, Gaston S,
Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA,
et al: Phase Ib trial of mutant herpes simplex virus G207
inoculated Pre-and Post-tumor resection for recurrent GBM. Mol
Ther. 17:199–207. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Markert JM, Razdan SN, Kuo HC, Cantor A,
Knoll A, Karrasch M, Nabors LB, Markiewicz M, Agee BS, Coleman JM,
et al: A phase 1 trial of oncolytic HSV-1, G207, given in
combination with radiation for recurrent GBM demonstrates safety
and radiographic responses. Mol Ther. 22:1048–1055. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bernstock JD, Wright Z, Bag AK, Gessler F,
Gillespie GY, Markert JM, Friedman GK and Johnston JM: Stereotactic
placement of intratumoral catheters for continuous infusion
delivery of herpes simplex virus-1 G207 in pediatric malignant
supratentorial brain tumors. World Neurosurgery. 122:e1592–e1598.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Taguchi S, Fukuhara H and Todo T:
Oncolytic virus therapy in Japan: Progress in clinical trials and
future perspectives. Jpn J Clin Oncol. 49:201–209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Fukuhara H, Ino Y and Todo T: Oncolytic
virus therapy: A new era of cancer treatment at dawn. Cancer Sci.
107:1373–1379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Desjardins A, Gromeier M, Herndon JE II,
Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F,
Muscat AM, Nair S, et al: Recurrent glioblastoma treated with
recombinant poliovirus. N Engl J Med. 379:150–161. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jayawardena N, Poirier JT, Burga LN and
Bostina M: Virus-receptor interactions and virus neutralization:
Insights for oncolytic virus development. Oncolytic Virother.
9:1–15. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ning J and Wakimoto H: Oncolytic herpes
simplex virus-based strategies: Toward a breakthrough in
glioblastoma therapy. Front Microbiol. 5:3032014. View Article : Google Scholar : PubMed/NCBI
|