Introduction
Lung cancer is the most frequently diagnosed
malignancy in the world. Out of patients over 50-years-old, ~1/5
are suffering from lung cancer in 2016 (1). It is also one of the most aggressive
malignancies, in which the mortality is predicted to increase by
2.53-fold by 2060 worldwide (2).
Smoking is the predominant risk factor for lung cancer development
(3). Non-small cell lung cancer
(NSCLC) accounts for 80–85% of lung cancer cases in the USA
(4). The 5-year survival rate of
early-stage NSCLC without metastasis is ~50%, whereas the rate in
advanced NSCLC with multiple metastases is only 1–2% in Asia
(5). Therefore, metastasis is a
notable factor in the poor prognosis of NSCLC.
The treatment of advanced, metastatic NSCLC remains
a challenge in the clinic. Due to the guidance of driver genes,
targeted therapies, such as EGFR-tyrosine kinase inhibitors (TKIs),
can extend the overall survival time (OS) of patients based on the
mutation status of oncogenes (6–9).
However, EGFR-TKI acquired resistance has emerged as a barrier to
effective clinical treatments (10).
To overcome EGFR-TKI resistance, several drug
resistance mechanisms have been studied, including secondary
mutations (T790M and C797S), aberrant downstream signaling pathways
(K-RAS mutations and loss of PTEN), activation of alternative
signaling pathways (hepatocyte growth factor receptor and
insulin-like growth factor 1 receptor) and the impairment of the
EGFR-TKIs-mediated apoptosis pathway (BCL2-like 11/BIM deletion
polymorphism) (11–20).
Tumor cells that are resistant to drugs also tend to
be more aggressive and have increased migration and invasion
capability compared with non-resistant parental cells. Also, a
majority of secondary tumors are more resistant to chemotherapy
drugs compared with primary tumors (21). Studies have shown that
platinum-resistant cells are more susceptible to the
epithelial-mesenchymal transition (EMT), which increases metastatic
potential (22–24). Tamoxifen promotes estrogen receptor
α36 expression and breast cancer metastasis by upregulating
aldehyde dehydrogenase family 1 member A1 (25). It has been reported that distant
metastases reduce the efficacy of TKIs in patients with mutant EGFR
who receive first-line treatment of EGFR-TKIs. Patient outcomes may
be worse if pathway genes are co-mutated in the PI3K catalytic
subunit α isoform or the PI3K/AKT/mTOR pathway, which contributes
to drug resistance and distant metastasis (26). Patients with multiple systemic
metastases often have multiple resistance-related mechanisms such
as HER2 amplification, EGFR-L858R mutation and EGFR-T790M mutation
(27). Studies suggest that patients
with EGFR-TKI resistance are more susceptible to metastasis
(28–30). However, the mechanisms of EGFR-TKI
resistance that are responsible for the promotion of metastasis
remain to be fully determined. Considering the challenges of
extending the OS of patients and delaying the relapse of drug
resistance, identifying the underlying molecular mechanisms
associated with metastasis after EGFR-TKI resistance are needed for
the development of novel EGFR-TKIs-based therapies and
EGFR-targeting agents.
Icotinib is an oral and safe first-generation TKI
that can significantly improve progression-free survival in
patients with advanced lung adenocarcinoma and EGFR mutations
(31). The present study established
icotinib-resistant (IcoR)-NSCLC cells and found that their
malignant abilities were significantly increased compared with
their respective parental cells. Furthermore, integrin α5 was
screened and shown to be a key molecule in promoting migration and
invasion rather than by contributing to icotinib resistance of
proliferation.
The present study showed that clinical treatments
could aim to block the integrin α5-FAK/STAT3/AKT signaling pathway
as a synergistic treatment to EGFR-TKIs. Inhibiting the expression
of integrin α5 may help to improve the prognosis and quality of
life of patients with EGFR-TKI resistance.
Materials and methods
Reagents and antibodies
Icotinib was gifted from Zhejiang Beta Pharma, Co.
Ltd. and was prepared in 5% dimethyl sulfoxide to obtain a stock
solution of 10 mM at −20°C. Stattic (cat. no. ab120952) was
obtained from Abcam, used in western blot as an inhibitor of STAT3.
Anti-AKT (1:1,000; cat. no. #9272S), anti-phosphorylated (p)-AKT
(1:500; cat. no. #9271L, Ser473), anti-p-ERK (1:1,000; cat. no.
#4370S, Thr202/Tyr204), anti-SRC (1:1,000; cat. no. #2110S),
anti-p-SRC (1:500; cat. no. #6943S), anti-FAK (1:1,000; cat. no.
#3285S), anti-p-FAK (1:250; cat. no. #3284, Y925), anti-STAT3
(1:1,000; cat. no. #132L), anti-p-STAT3 (1:1,000; cat. no. #9131L,
Tyr705) anti-EGFR (1:1,000; cat. no. #2646S), anti-p-EGFR (1:500;
cat. no. #2234S, Tyr1068) and anti-integrin α5 (1:500; cat. no.
#4705) were purchased from Cell Signaling Technology, Inc.
Anti-integrin β4 (1:1,000; cat. no. MAB4060) was purchased from
Novus Biologicals, Ltd. Anti-ERK (1:2,000; cat. no. sc292838),
anti-actin (1:1,000; cat. no. sc1616), horseradish peroxidase
(HRP)-conjugated secondary goat anti-rabbit (cat. no. sc-sc-2004,
targeting anti-AKT, p-AKT, p-ERK, p-SRC, FAK, p-FAK, STAT3,
p-STAT3, EGFR, p-EGFR, integrin α5 and ERK) and goat anti-mouse
antibodies (cat. no. sc-sc-2005, targeting anti-SRC, integrin β4
and actin) were purchased from Santa Cruz Biotechnology, Inc.
Cell culture and establishment of IcoR
cells
Human lung adenocarcinoma cells, PC9 and HCC827,
were obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences (Shanghai, China). The cells were
cultured in RPMI-1640 medium purchased from Thermo Fisher
Scientific, Inc. containing 10% heat-inactivated foetal bovine
serum (FBS; Biological Industries), penicillin (100 U/ml) and
streptomycin (100 mg/ml) at 37°C in an humidified atmosphere with
95% air and 5%CO2. Resistant cells were
established by exposing to 0.05 µM icotinib at the beginning and
the icotinib concentration was gradually increased in the totally
same incubator condition as previously mentioned. Finally, the
PC9/IcoR and 827/IcoR cells were maintained at 10 and 5 µM
icotinib, respectively.
MTT assay
The effect of icotinib on the cell viability was
evaluated using an MTT assay. PC9 and HCC827 cells (3,000 or 5,000
cells/well) in 96-well plate were treated with different
concentration of icotinib for 24 or 48 h. A total of 20 µl of the
MTT reagent (5 mg/l) was added per well and incubated for another 4
h. The supernatant was removed and 200 µl of dimethyl sulfoxide was
added. The absorbance was measured at 570 nm using an iMark
Absorbance Microplate (Bio-Rad Laboratories, Inc.).
Colony formation assay
A colony formation assay was used to estimate the
ability of cell proliferation. PC9, PC9/IcoR, HCC827 and 827/IcoR
cells at a density of 400 or 600/well in six-well plate were
treated with different concentrations of icotinib (0, 0.1, 1 and 10
µM) for 72 h at 37°C. Then, the supernatant of the medium was
discarded and the cells were cultured under the aforementioned
routine conditions for another 7 days. Subsequently, the colonies
were fixed with 95% ethanol for 5 min at room temperature, stained
with Wright-Giemsa (Wright staining for 1 min and Giemsa staining
for 25 min at room temperature). Only colonies containing >50
cells were counted were counted under an optical microscope (BX41;
Olympus Corporation) with white light by eye.
Small interfering (si)-RNA
transfections
To knockdown the expression of integrin α5, siRNAs
targeting integrin α5 (si-integrin α5) and a scrambled negative
control (NC) were transfected into PC9/IcoR and 827/IcoR cells were
transfected with 0.1 mM siRNAs using Lipofectamine® 2000
(Thermo Fisher Scientific, Inc.) reagent according to
manufacturer's instruction at 37°C. After 48 h, subsequent
experiment was performed. The coding strand of ITGA5 si1 was
5′-GUUUCACAGUGGAACUUCA-3′, and ITGA5 si2 was
5′-GCAGUGCUAUUCCCAGUAA-3′. The coding strand of NC was
5′-UUCUCCGAACGUGUCACGU-3′.
Wound healing assay
Cells (HCC827, PC9, 827/IcoR and PC9/IcoR) in
six-well plate were scratched with 200-µl pipet tips. After being
washed with PBS two or three times, 0 and 5 µmol/ml icotinib (for
HCC827 and 827/IcoR) or 10 µmol/ml icotinib (for PC9 and PC9/IcoR)
was added with RPMI-1640 medium containing 2% FBS onto the plate.
The scratches were observed using the CK40 inverted light
microscope (Olympus Corporation) and images were captured at 0 and
24 h and the wound width was measured using ImageJ version 1.52a
(National Institutes of Health). The healing rate was evaluated
relative to the starting wound width (0 h time point).
Transwell assay
The migration assay was performed using 8-µm
Transwell chambers (Corning, Inc.). PC9 or PC9/IcoR cells with a
density of 3×104 cells/200 µl and HCC827or 827/IcoR
cells with a density of 8×104 cells/200 µl were placed
into the upper chamber, and 500 µl of RPMI-1640 medium containing
2.5% FBS was added to the lower chamber with 37°C. After 24 h, the
remaining cells on the upper membrane were removed with a cotton
swab and cells that migrated to the bottom of the membranes were
stained with Wright-Giemsa staining (Wright staining for 1 min and
Giemsa staining for 25 min at room temperature). Next, four
randomly selected fields were counted using the BX41 microscope
with white lights and the average mean number of cells was
presented. Images were also captured. The invasion assay used the
same steps as the migration assay except for inserting 3% Matrigel
into the upper chamber before seeding 3×104 cells into
the culture system. The Matrigel was diluted to a 1:30 ratio at 4°C
and solidified at 37°C for 4 h, then the cells were seeded into the
upper chamber at 37°C for 24 h.
Western blotting
HCC827, 827/IcoR, PC9 and PC9/IcoR cells with or
without icotinib or Stattic or siRNA treatments were lysed in lysis
buffer, prepared with 1% Triton X-100,50 mM Tris-HCl pH 7.4, 150 mM
NaCl, 10 mM EDTA, 100 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 2 µg/ml
aprotinin. After quantification by Bradford assay, the protein
samples were mixed with 3×loading buffer. Equal amounts of protein
(20–40 µg/lane) were separated using 8% SDS-PAGE and transferred to
a PVDF membrane (EMD Millipore). The membranes were blocked using
5% skimmed milk for 1 h at room temperature and incubated with
primary antibodies overnight at 4°C. After exposure to appropriate
secondary antibodies for 30 min at room temperature, the proteins
were detected with chemiluminescence reagent (SuperSignal™ Western
Pico Chemiluminescent Substrate; Thermo Fisher Scientific, Inc.)
and analyzed using the Electrophoresis Gel Imaging Analysis system
(version 6.12, DNR Bio-Imaging Systems, Ltd.).
Bioinformatics analysis and whole
transcriptome resequencing
Total RNA of PC9 and PC9/IcoR cells were extracted
for gene expression microarray analysis using Illumina HumanHT12 v3
BeadChip (Shanghai Oui Biomedical Technology Co., Ltd.). The DEseq2
method was used to analyze the different gene expression levels
between the two groups with P<0.05 and a fold-change >2.0 as
the significance cut-off. Microarray data set GSE62504 were
downloaded from the Gene Expression Omnibus (GEO) database
(https://www.ncbi.nlm.nih.gov/gds/)
(32). GSE62504 contains the
expression profiling data of NSCLC sensitive EGFR-mutant cells
HCC827 and EGFR-TKIs resistant HCC827-BR cells, which were
established based on individual clones by maintenance of HCC827
cells in the presence of escalating concentrations of BIBW2992 of
up to 2 µM. GEO2R was used to screen differentially expressed genes
using a 2-fold-change cut-off. After downloading all the
differentially expressed genes, Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analysis was performed using the Database
for Annotation, Visualization and Integrated Discovery (33). The pathways of ‘focal adhesion’ and
‘extracellular matrix (ECM)-receptor interaction’ enriched from
expression profiling of PC9 and PC9/IcoR, and the pathway of ‘focal
adhesion’ from microarray data of HCC827 and HCC827/BR were
selected to screen potential target genes.
Statistical analysis
The experimental results were expressed as mean ±
standard deviation from at least three independent experiments and
analyzed using SPSS v16.0 software. The graphs were constructed
using GraphPad v6.0 Software. Unpaired Student's t-test was used to
analyze the differences between two independent groups. One-way
ANOVA was used to analyze the differences among multiple groups,
followed by Dunnett's and Sidak's multiple comparisons tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
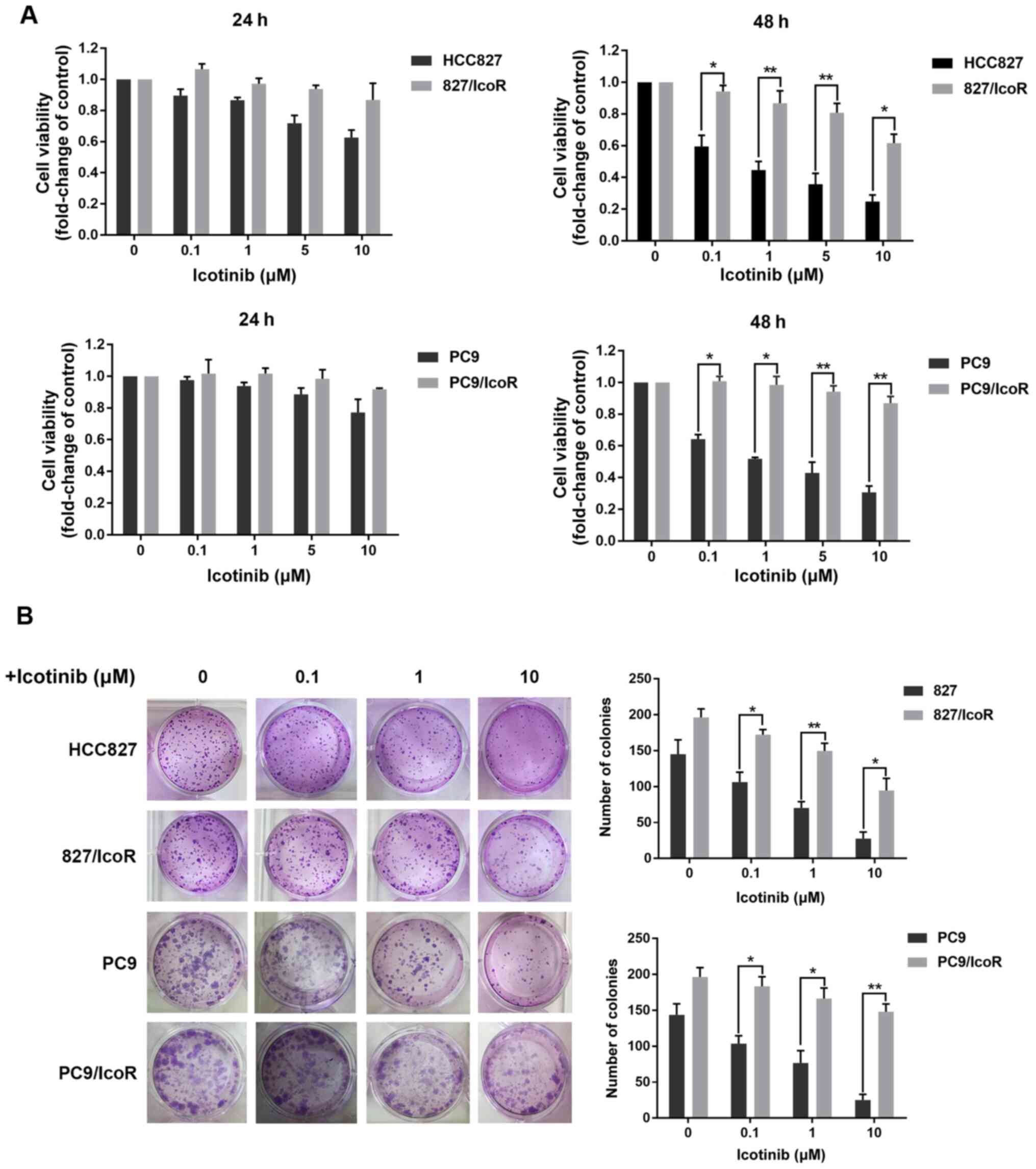
PC9/IcoR and 827/IcoR cells show
significant resistance to icotinib
The PC9/IcoR and 827/IcoR resistant cell lines were
established by gradually exposing PC9 and HCC827 parental cells to
icotinib up to 10 and 5 µM, respectively. As shown in the MTT
assays of 48 h, the resistant cell lines showed increased
resistance to icotinib compared with parental cells, which showed
dose-dependent inhibition of proliferation. Colony formation also
showed that icotinib failed to inhibit the formation of colonies
derived from PC9/IcoR and 827/IcoR compared with parental cells
(Fig. 1B). However, when treated
with icotinib for 24 h, there were no remarkable differences in the
inhibition of cell viability between parental and resistant cells
(Fig. 1A). Similar results were also
obtained when cells were exposed to gefitinib for 48 h (Fig. S1A). Together, these results
confirmed that the resistant cell lines are stable in the presence
of icotinib for long period (48 h), while there was no difference
between sensitive and resistant cell lines on the cell viability
inhibited by icotinib in a short period (24 h).
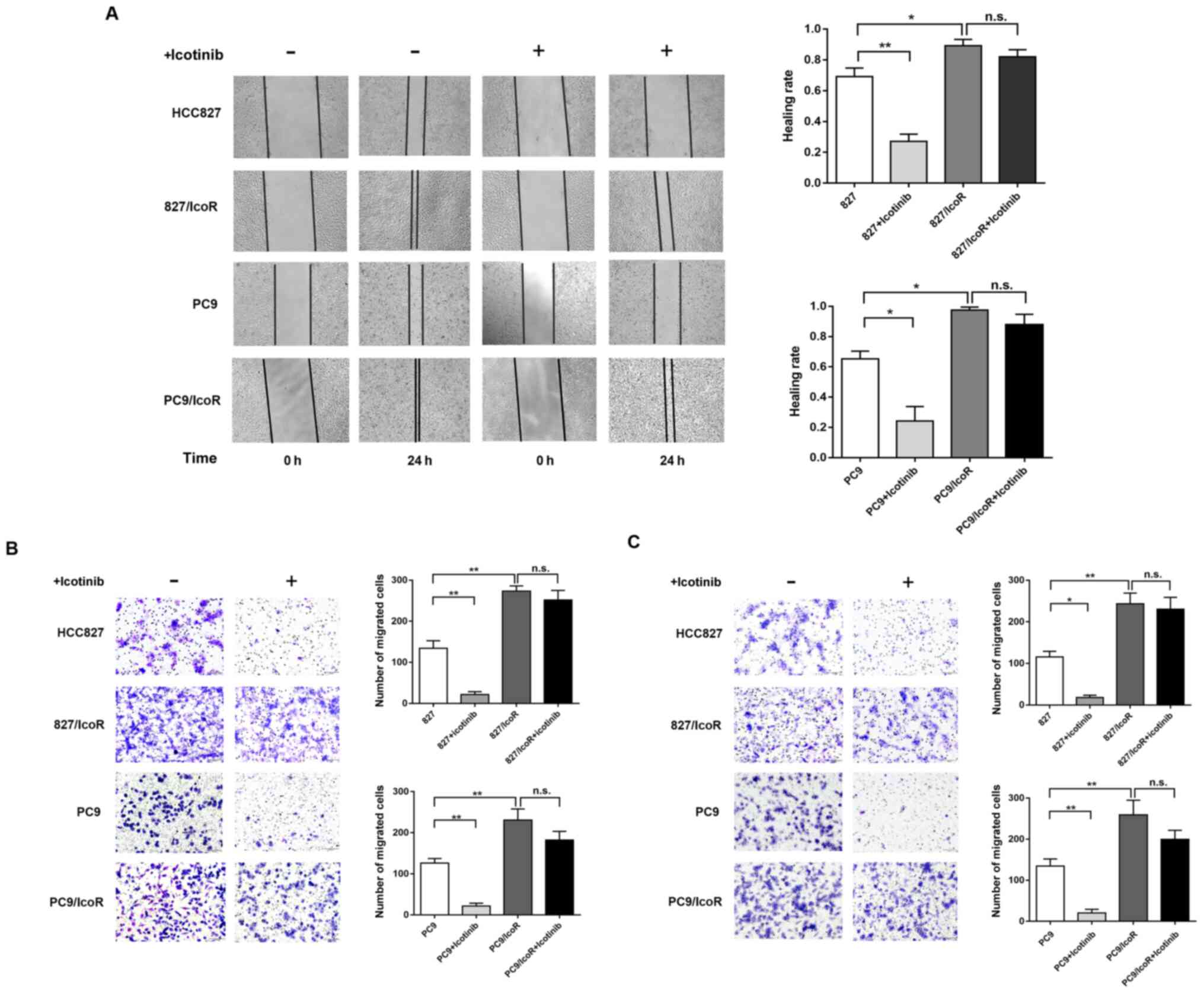
PC9/IcoR and 827/IcoR cell migration
and invasion ability is increased compared with parental cells
To further investigate the differences between the
resistant cells and parental cells, Transwell and wound healing
azssays were performed to determine the migration and invasive
capability of the cell lines. As shown in Fig. 2A, the healing rate of 827/IcoR and
PC9/IcoR cells was faster compared with the parental cells, in
which icotinib treatment decreased the healing rate. In addition,
results from the Transwell assay showed that the number of 827/IcoR
and PC9/IcoR cells that passed through the transwell membranes and
the Matrigel-coated membrane was higher compared with that of the
parental cells. Similarly, icotinib treatment reduced the migration
and invasion abilities of HCC827 and PC9 but had little impact on
the 827/IcoR and PC9/IcoR cells (Fig. 2B
and C). Collectively, these results showed that the migration
and invasion abilities of icotinib-resistant cells were
significantly enhanced compared with their parental cells.
Furthermore, icotinib could only inhibit the migration and invasion
capacities of parental cells but showed no effect on resistant
cells.
Integrin α5 is overexpressed in
icotinib-resistant cells
To determine the causes of enhanced migration and
invasion in icotinib-resistant cells, a gene microarray was used to
identify differentially expressed genes that were upregulated in
PC9/IcoR cells compared with parental cells. The top 20 pathways
were identified using KEGG enrichment analysis, among which the
‘focal adhesion’ pathway and the ‘ECM-receptor interaction pathway’
had higher scores, indicating that these significantly enriched
pathways were mostly associated with metastasis (Fig. 3A). The gene microarray data of HCC827
and HCC827/BR cell lines were downloaded in the GSE62504 microarray
dataset and the ‘focal adhesion’ pathway was identified using GEO2R
and KEGG analysis. To narrow the range and to obtain more precise
results, the intersection of the three pathways was identified, as
shown in Fig. 3B. Finally, four
genes were identified, including integrin α5 and integrin β4, as
members of the integrin family of proteins.
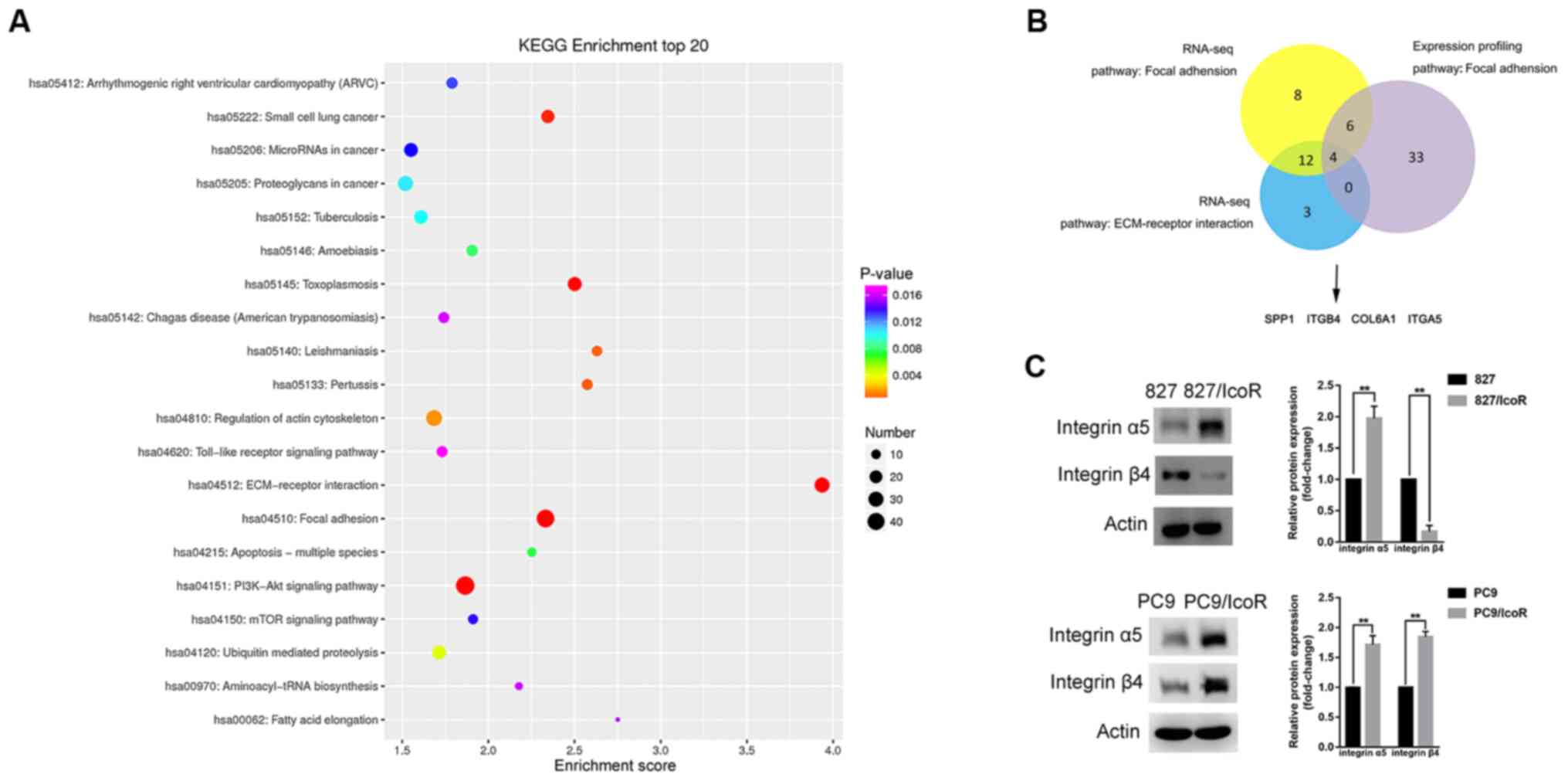 | Figure 3.Integrin α5 is highly expressed in
827/IcoR and PC9/IcoR cells. (A) Top 20 signaling pathways that
were upregulated in PC9/IcoR compared with PC9 were screened using
KEGG pathway enrichment analysis. (B) A total of four genes,
including integrin α5 and integrin β4, were obtained as the
intersection of the three pathways using Venn diagram. (C) Protein
expression of integrin α5 and integrin β4 of HCC827, 827/IcoR, PC9
and PC9/IcoR cells by western blotting. **P<0.01. IcoR,
icotinib-resistant; tRNA, transfer RNA; ECM, extracellular matrix;
SPP1, secreted phosphoprotein 1; ITGB4, integrin β4; COL6A1,
collagen alpha-1(VI) chain; ITGA5, integrin α5. |
To verify these results, western blotting analysis
was performed to identify the protein expression levels of integrin
α5 and integrin β4 in the cell lines (Fig. 3C). The data showed that the protein
expression levels of integrin α5 were higher in 827/IcoR and
PC9/IcoR cells compared with those in parental cells. The level of
integrin β4 expression was enhanced in PC9/IcoR cells but not in
827/IcoR cells compared with their respective parental cell lines.
Moreover, knockdown of integrin α5 did not change the expression
level of integrin β4, implying no association between them
(Fig. S1C). These data suggested
that it was integrin α5 instead of integrin β4 that played a key
role in icotinib resistance.
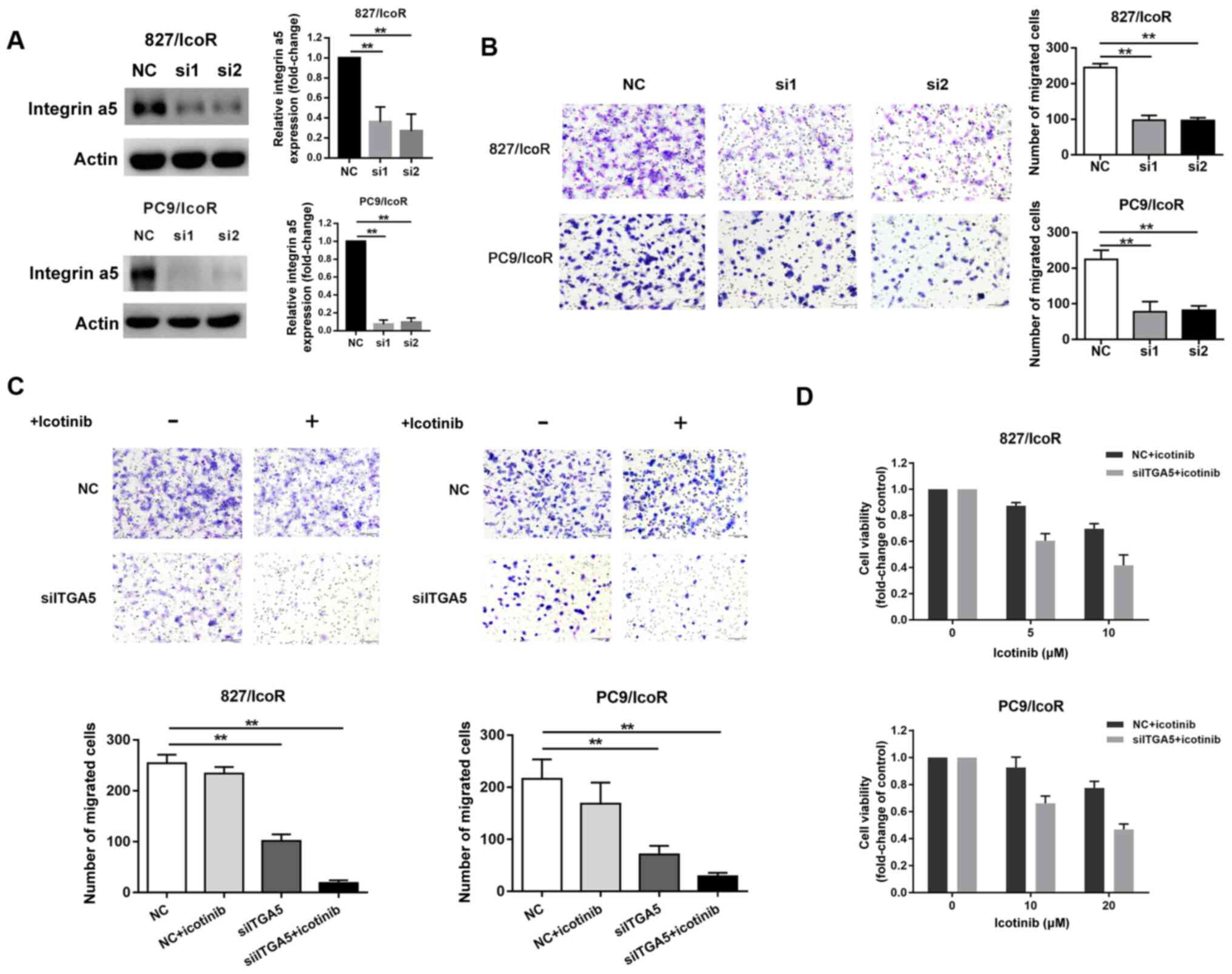
Integrin α5 promotes migration,
invasion and icotinib resistance in resistant cells
Compared with treatment with NC siRNA, treatment
with si-ITGA5 (si1 and si2) suppressed the expression of integrin
α5 protein in 827/IcoR and PC9/IcoR cells (Fig. 4A). Transwell assays showed that the
number of cells that had passed through the Transwell membranes
were significantly decreased after transfection with si1 and si2
(Fig. 4B). However, the migration
resistant cells decreased when treated with si-ITGA5 and icotinib
together (both P<0.001; Fig. 4C).
Moreover, the cell viability of 827/IcoR and PC9/IcoR cells were
partly decreased when treated with integrin α5 siRNA and icotinib
together (Fig. 4D). Therefore, the
sensitivity to icotinib of resistant cells was partly restored
after transfected with siITGA5.
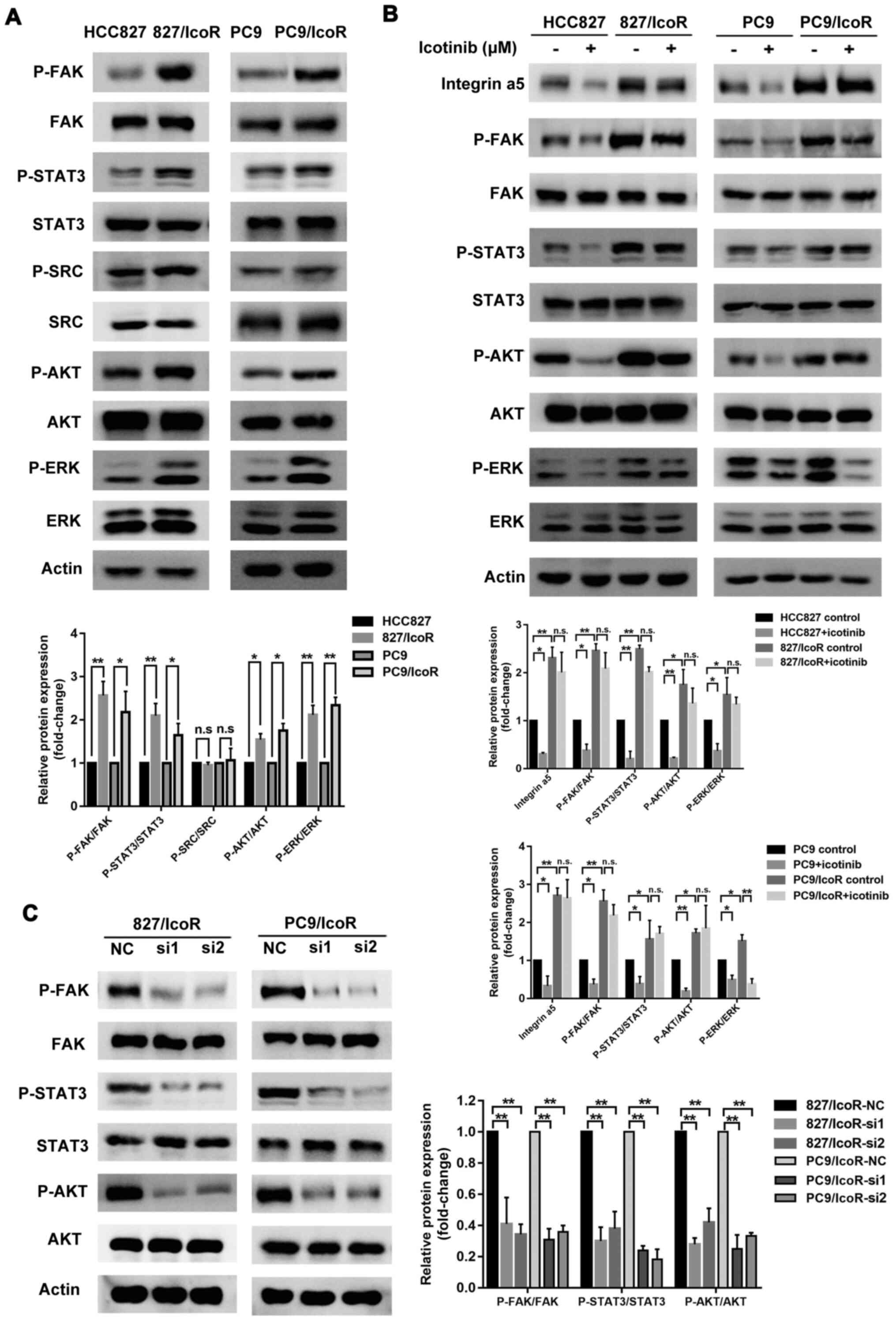
Integrin α5 increases icotinib
resistance-associated malignancy through the FAK-STAT3/AKT
signaling pathway
To further clarify the underlying mechanisms of
integrin α5 in icotinib resistance, western blotting was performed
to identify its related signaling effects. The data showed that the
expression levels of integrin α5 related signaling, such as p-FAK,
p-STAT3, p-AKT and p-ERK, were higher in the resistant cell lines
compared with the parental cell lines except p-SRC (Fig. 5A). The expression levels of p-FAK,
p-STAT3 and p-AKT were decreased in icotinib-treated sensitive
cells and did not change in icotinib-treated resistant cells except
p-ERK (Fig. 5B), which were
consistent with the results indicating that the migration and
invasion capacity of resistant cells were not affected by
icotinib.
The persistent activation of p-EGFR in PC9/IcoR and
827/IcoR may explain the resistance to gefitinib in
icotinib-resistant cells (Fig. S1A and
B). Furthermore, after knocking down integrin α5 using siRNAs
in the resistant cells, the protein expression levels of p-FAK,
p-STAT3 and p-AKT were notably decreased (Fig. 5C). When treated with STAT3 inhibitor,
Stattic, the phosphorylation level of AKT was significantly
decreased, whereas no change of FAK phosphorylation was observed
(Fig. S1D). These results suggested
that the integrin α5/FAK-STAT3/AKT signaling pathway played a
notable role in icotinib-resistant cells.
Discussion
The present study found that the malignant
properties of icotinib-resistant NSCLC cells were enhanced compared
with non-resistant cells. Further investigations showed that the
upregulation of integrin α5 significantly contributed to increased
invasion and migration ability and slightly increased icotinib
resistance through the FAK/STAT3/AKT signaling pathway.
To the best of our knowledge, the majority of
current research relating to the mechanism of resistance to
EGFR-TKIs has focused on increased proliferation and decreased
apoptosis with few studies considering impacts on metastasis
(34–36). However, clinical criteria for
acquired resistance to EGFR-TKIs are not limited to recurrence of
the primary tumor and metastasis into sites nearby, but also
include systemic multisite infiltration and distant metastasis
(37,38).
Few studies have focused on specific changes in the
malignant capacity of TKI-resistant cells and how changes in TKI
resistance-related genes affect malignancy. In order to find out
the mechanism for the change in malignancy, resistant cell lines
were established by gradually exposing PC9 and HCC827 parental
cells to icotinib, an effective TKI developed in China (31), and the resistant cell lines PC9/IcoR
and 827/IcoR were also found to be resistant to gefitinib. These
results were also similar to those presented in Chen et al
(39). Furthermore, the present
study found that capacity of migration and invasion was
significantly increased in icotinib-resistant cells, which provided
evidence to explain the frequently observed metastatic phenomenon
in patients diagnosed with EGFR-TKI-resistance. Furthermore, the
expression profiles of parental and drug-resistant cells were
screened using a combination of online analyses to demonstrate that
integrin α5 played a notable role in this process. These results
suggested that strategies targeting integrin may be effective in
the treatment of patients with TKI-resistant tumors.
Integrin family members are cell-surface
transmembrane protein receptors that consist of 18 different α
subunits and eight β subunits. Amongst these proteins, β1, β3, αv
and α5 are the main factors that affect metastasis and survival in
tumors (40). The integrin family
can provide a bridge for the mechanical adhesion of cells to the
ECM and increase the ability of tumor cells to metastasize and
invade (41,42). In addition, integrins transmit
signals to metastatic tumor cells in order to facilitate the
migration of these cells, which indicates that integrins are
involved in the signaling pathways that induce tumor metastasis
(43).
Previous studies have shown that integrin can
promote malignant behavior in multiple tumor types including
metastatic melanoma and breast, prostate, pancreatic and lung
cancer (44–47). In lung cancer, targeting TGF-β1 and
integrin β3 can significantly reduce lymph node metastasis
(48). The deletion of integrin β3
can also increase the sensitivity of lung cancer cells to
traditional chemotherapy drugs (49). Several reports have shown that high
expression levels of integrin β1 and β3 can increase the resistance
of lung cancer cells to EGFR-TKIs (50–52).
However, these studies only found that integrins are highly
expressed in TKIs-resistant NSCLC cells. After knockdown of
specific integrins such as integrin β1 and β3, cell proliferation
and apoptosis are impacted but the changes are not correlated with
alterations in metastatic capacity. Thus, the effect of integrin on
the migration and invasion of TKI-resistant cells remains
unclear.
Integrin α5 was selected as a key molecule that
could potentially affect the malignancy of drug-resistant cells in
the present study. The entire transcriptome of the drug-resistant
cell lines was sequenced and the data were compared with public
databases. Further investigation showed that knocking down integrin
α5 significantly reduced the migration capacity of TKI-resistant
cells, but the effect on proliferation ability was not obvious.
These results indicated that integrin α5 can promote cell migration
and also provided a basic mechanism for this in TKI-resistant tumor
cells. In addition, tumors with high expression levels of different
integrin subunits were characterized by different organ-specific
metastases in which integrin α5 targets the lung (41). Hoshino et al (41) showed that tumor cells releasing
integrin α5 exosomes are more prone to lung metastasis, whilst
cells producing β1 or β3 exosomes can metastasize to various organs
including bone, lung, liver and brain. Based on these
characteristics of integrin α5, the proportion of patients with
EGFR-TKI resistance alongside integrin α5-overexpression and the
metastatic burden of these patients merits further research.
In metastatic tumor cells, the regulation of
cellular activities is important after stable and firm adhesion to
the ECM (40). Ligand binding to
integrins induces survival signals to tumor cells to enhance cell
viability (42). However, tumor
cells that unstably adhere to the ECM undergo withdrawal of
survival signals and initiate apoptosis (43,53).
Thus, specific types of integrins are needed to help cells avoid
apoptosis, which is a key mechanism for successful metastasis. A
previous study reported that integrin signaling is associated with
several receptor tyrosine kinases that can enhance cancer cell
survival and proliferation, principally EGFR (54). Common integrin downstream signals are
also associated with cell survival, including the PI3K/AKT pathway,
which also leads to EGFR-TKI resistance. Integrins are non-receptor
kinase receptors, so the aforementioned signal transduction
pathways require kinase partners, such as FAK (55,56).
Integrin β1 has been shown to promote erlotinib resistance in lung
cancer by activating the SRC/AKT pathway (50). In the present study, although the
common integrin downstream signals FAK, SRC, AKT and ERK were
continuously activated in drug-resistant cells, there was no
difference in the activation of SRC between sensitive and
drug-resistant cells. Moreover, icotinib treatment significantly
decreased phosphorylation level of ERK in PC9/IcoR, suggesting that
the ERK pathway was not involved in integrin α5-mediated malignancy
and resistance. After knockdown of integrin α5, the FAK and AKT
signaling pathways in drug-resistant cells were inhibited. In
addition, our previous research found that TKIs induce the
activation of the STAT3 signaling pathway and reduce the
sensitivity of tumor cells to icotinib (57). The results of the present study
showed that knockdown of integrin α5 inhibited the activation of
the STAT3 signaling pathway. Therefore, we hypothesized that
integrin α5 affects the migration and invasion of TKIs-resistant
cells through the FAK-STAT3/AKT signaling pathway.
EMT is also regarded as one of the mechanisms of
acquired resistance to EGFR-TKIs with low E-cadherin (a
cell-adhesion protein) and high vimentin/fibronectin expression
levels. This is also upregulated by integrins in various tumors
including hepatocellular carcinoma, breast tumors and thyroid
carcinoma (58–60). However, to the best of our knowledge,
there is no evidence indicating the effect of integrins on the
metastatic capacity of TKIs-resistance by regulating the EMT
phenotype.
Evidence suggests that integrin αvβ5 may be a
potential therapeutic target in patients with lung adenocarcinoma
with brain metastases (61).
Integrin α5β1 can also be used as a biomarker to predict the
prognosis of early-stage NSCLC (62). As the integrins play notable roles in
tumor metastasis, integrin inhibitors may have potential value as
therapeutics. Currently, integrin inhibitors are divided into three
categories: Monoclonal antibodies, small molecule inhibitors and
small peptides. However, to the best of our knowledge, the majority
are in early-phase clinical trials (63). For example, Cilengitide is an
arginine-glycine-aspartate pentapeptide used as an αvβ3 and αvβ5
integrin inhibitor that has achieved good therapeutic effects in
phase II and III clinical trials of recurrent glioblastoma
(64). Volociximab, an integrin α5β1
inhibitor, is a chimeric human IgG4 antibody inhibitor that has
already been used in several phase II clinical trials including
metastatic melanoma, renal cell carcinoma and NSCLC (65). The present study identified integrin
α5 as a key molecule in mediating the malignant properties of
EGFR-TKIs resistant cells. Therefore, in future clinical
applications, the combination of EGFR-TKIs and integrin α5
inhibitors may help reduce drug resistance and the risk of
metastasis in patients with EGFR-TKI-resistant NSCLC. However,
there are limitations to the study, since no animal model or human
samples were included in the investigation of relationship between
integrin α5 and metastasis in vivo. These could be
considered in future study, but this approach still could provide a
basis for clinical applications and may help to improve prognosis
and quality of life for patients with lung cancer.
The present study demonstrated a promising
therapeutic approach using the interruption of the integrin
α5-FAK/STAT3/AKT signaling pathway as a synergistic combination
therapy with EGFR-TKI to overcome malignancy of cells after
development of EGFR-TKI resistance. Upregulated genes in
drug-resistant cells have a notable in promoting migration and may
have significant effects on proliferation or apoptosis. A
combination therapy to prevent metastasis in patients with TKI
resistance may be an effective treatment strategy, and may also
provide evidence to explore the underlying mechanisms of
TKI-resistance in the future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The study was funded by The National Key Research
and Development Program of China (grant no. 2017YF1309403), The
National Natural Science Foundation of China (grant nos. 81972197
and 81472193), The Key Research and Development Program of Liaoning
Province (grant no. 2018225060), The Natural Science Foundation of
Liaoning Province (grant no. 2019-ZD-777), The Technological
Special Project of Liaoning Province of China (grant no.
2019020176-JH1/103), The Science and Technology Plan Project of
Liaoning Province (grant no. 2013225585) and Science and Technology
Plan Project of Shenyang City (grant no. 19-112-4-099).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Gene Expression Omnibus
repository (https://www.ncbi.nlm.nih.gov/gds/). The accession
number is PRJNA690636.
Authors' contributions
YY, CZ, KH performed the experiments. YY, JW, XH and
YW drafted the manuscript. YW, YC and XC performed the online data
analysis. XC, CZ and KH revised the manuscript. XH, JZ and YL
conceptualized and designed the study. JW, YL, XH and YC
established the icotinib-resistant cell lines. YY and JZ confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mattiuzzi C and Lippi G: Current cancer
epidemiology. J Epidemiol Glob Health. 9:217–222. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mathers CD and Loncar D: Projections of
global mortality and burden of disease from 2002 to 2030. PLoS Med.
3:e4422006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bade BC and Dela Cruz CS: Lung cancer
2020: Epidemiology, etiology, and prevention. Clin Chest Med.
41:1–24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McGuire S: World Cancer Report 2014.
Geneva, Switzerland: World Health Organization, International
Agency for Research on Cancer, WHO Press, 2015. Adv Nutr.
7:418–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keedy VL, Temin S, Somerfield MR, Beasley
MB, Johnson DH, McShane LM, Milton DT, Strawn JR, Wakelee HA and
Giaccone G: American society of clinical oncology provisional
clinical opinion: Epidermal growth factor receptor (EGFR) Mutation
testing for patients with advanced non-small-cell lung cancer
considering first-line EGFR tyrosine kinase inhibitor therapy. J
Clin Oncol. 29:2121–2127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han SW, Kim TY, Hwang PG, Jeong S, Kim J,
Choi IS, Oh DY, Kim JH, Kim DW, Chung DH, et al: Predictive and
prognostic impact of epidermal growth factor receptor mutation in
non-small-cell lung cancer patients treated with gefitinib. J Clin
Oncol. 23:2493–2501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Somaiah N, Fidler MJ, Garrett-Mayer E,
Wahlquist A, Shirai K, Buckingham L, Hensing T, Bonomi P and Simon
GR: Epidermal growth factor receptor (EGFR) mutations are
exceptionally rare in thyroid transcription factor (TTF-1)-negative
adenocarcinomas of the lung. Oncoscience. 1:522–528. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirsch FR, Suda K, Wiens J and Bunn PA Jr:
New and emerging targeted treatments in advanced non-small-cell
lung cancer. Lancet. 388:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohashi K, Maruvka YE, Michor F and Pao W:
Epidermal growth factor receptor tyrosine kinase
inhibitor-resistant disease. J Clin Oncol. 31:1070–1080. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kuwano M, Sonoda K, Murakami Y, Watari K
and Ono M: Overcoming drug resistance to receptor tyrosine kinase
inhibitors: Learning from lung cancer. Pharmacol Ther. 161:97–110.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA and Janne PA: Mechanisms of
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small cell lung cancer. Clin Cancer Res.
14:2895–2899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Song Y and Liu D: EAI045: The
fourth-generation EGFR inhibitor overcoming T790M and C797S
resistance. Cancer Lett. 385:51–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morgillo F, Della Corte CM, Fasano M and
Ciardiello F: Mechanisms of resistance to EGFR-targeted drugs: Lung
cancer. ESMO Open. 1:e0000602016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan CS, Cho BC and Soo RA: Next-generation
epidermal growth factor receptor tyrosine kinase inhibitors in
epidermal growth factor receptor-mutant non-small cell lung cancer.
Lung Cancer. 93:59–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okamoto K, Okamoto I, Hatashita E, Kuwata
K, Yamaguchi H, Kita A, Yamanaka K, Ono M and Nakagawa K:
Overcoming erlotinib resistance in EGFR mutation-positive non-small
cell lung cancer cells by targeting survivin. Mol Cancer Ther.
11:204–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krawczyk P, Mlak R, Powrozek T, Nicos M,
Kowalski DM, Wojas-Krawczyk K and Milanowski J: Mechanisms of
resistance to reversible inhibitors of EGFR tyrosine kinase in
non-small cell lung cancer. Contemp Oncol (Pozn). 16:401–406.
2012.PubMed/NCBI
|
|
20
|
Yeo CD, Park KH, Park CK, Lee SH, Kim SJ,
Yoon HK, Lee YS, Lee EJ, Lee KY and Kim TJ: Expression of
insulin-like growth factor 1 receptor (IGF-1R) predicts poor
responses to epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitors in non-small cell lung cancer patients harboring
activating EGFR mutations. Lung Cancer. 87:311–317. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang Y, McDonnell S and Clynes M:
Examining the relationship between cancer invasion/metastasis and
drug resistance. Curr Cancer Drug Targets. 2:257–277. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brozovic A: The relationship between
platinum drug resistance and epithelial-mesenchymal transition.
Arch Toxicol. 91:605–619. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Niwa N, Tanaka N, Hongo H, Miyazaki Y,
Takamatsu K, Mizuno R, Kikuchi E, Mikami S, Kosaka T and Oya M:
TNFAIP2 expression induces epithelial-to-mesenchymal transition and
confers platinum resistance in urothelial cancer cells. Lab Invest.
99:1702–1713. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang L, Chen Y, Li F, Bao L and Liu W:
Atezolizumab and bevacizumab attenuate cisplatin resistant ovarian
cancer cells progression synergistically via suppressing
epithelial-mesenchymal transition. Front Immunol. 10:8672019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Q, Jiang J, Ying G, Xie XQ, Zhang X,
Xu W, Zhang X, Song E, Bu H, Ping YF, et al: Tamoxifen enhances
stemness and promotes metastasis of ERalpha36(+) breast cancer by
upregulating ALDH1A1 in cancer cells. Cell Res. 28:336–358. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng LL, Gao G, Deng HB, Wang F, Wang ZH
and Yang Y: Co-occurring genetic alterations predict distant
metastasis and poor efficacy of first-line EGFR-TKIs in EGFR-mutant
NSCLC. J Cancer Res Clin Oncol. 145:2613–2624. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lim SM, Syn NL, Cho BC and Soo RA:
Acquired resistance to EGFR targeted therapy in non-small cell lung
cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev.
65:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yufen X, Binbin S, Wenyu C, Jialiang L and
Xinmei Y: The role of EGFR-TKI for leptomeningeal metastases from
non-small cell lung cancer. Springerplus. 5:12442016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang JJ, Chen HJ, Yan HH, Zhang XC, Zhou
Q, Su J, Wang Z, Xu CR, Huang YS, Wang BC, et al: Clinical modes of
EGFR tyrosine kinase inhibitor failure and subsequent management in
advanced non-small cell lung cancer. Lung Cancer. 79:33–39. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu X, Chen L, Liu L and Niu X:
EMT-Mediated acquired EGFR-TKI resistance in NSCLC: Mechanisms and
strategies. Front Oncol. 9:10442019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi YK, Wang L, Han BH, Li W, Yu P, Liu
YP, Ding CM, Song X, Ma ZY, Ren XL, et al: First-line icotinib
versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for
patients with advanced EGFR mutation-positive lung adenocarcinoma
(CONVINCE): A phase 3, open-label, randomized study. Ann Oncol.
28:2443–2450. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kurppa KJ, Liu Y, To C, Zhang T, Fan M,
Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, et al:
Treatment-induced tumor dormancy through YAP-Mediated
transcriptional reprogramming of the apoptotic pathway. Cancer
Cell. 37:104–122.e12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tong X, Tanino R, Sun R, Tsubata Y,
Okimoto T, Takechi M and Isobe T: Protein tyrosine kinase 2: A
novel therapeutic target to overcome acquired EGFR-TKI resistance
in non-small cell lung cancer. Respir Res. 20:2702019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu YN, Tsai MF, Wu SG, Chang TH, Tsai TH,
Gow CH, Chang YL and Shih JY: Acquired resistance to EGFR tyrosine
kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL
signaling in lung cancer. Int J Cancer. 145:1609–1624. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Watanabe H, Okada M, Kaji Y, Satouchi M,
Sato Y, Yamabe Y, Onaya H, Endo M, Sone M and Arai Y: New response
evaluation criteria in solid tumours-revised RECIST guideline
(version 1.1). Gan To Kagaku Ryoho. 36:2495–2501. 2009.(In
Japanese). PubMed/NCBI
|
|
38
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Janne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Wu J, Yan HF, Cheng Y, Wang Y,
Yang Y, Deng M, Che X, Hou K, Qu X, et al: Lymecycline reverses
acquired EGFR-TKI resistance in non-small-cell lung cancer by
targeting GRB2. Pharmacol Res. 159:1050072020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hamidi H and Ivaska J: Every step of the
way: Integrins in cancer progression and metastasis. Nat Rev
Cancer. 18:533–548. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hoshino A, Costa-Silva B, Shen TL,
Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di
Giannatale A, Ceder S, et al: Tumour exosome integrins determine
organotropic metastasis. Nature. 527:329–335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kechagia JZ, Ivaska J and Roca-Cusachs P:
Integrins as biomechanical sensors of the microenvironment. Nat Rev
Mol Cell Biol. 20:457–473. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim YN, Koo KH, Sung JY, Yun UJ and Kim H:
Anoikis resistance: An essential prerequisite for tumor metastasis.
Int J Cell Biol. 2012:3068792012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nemlich Y, Baruch EN, Besser MJ, Shoshan
E, Bar-Eli M, Anafi L, Barshack I, Schachter J, Ortenberg R and
Markel G: ADAR1-mediated regulation of melanoma invasion. Nat
Commun. 9:21542018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo W, Pylayeva Y, Pepe A, Yoshioka T,
Muller WJ, Inghirami G and Giancotti FG: Beta 4 integrin amplifies
ErbB2 signaling to promote mammary tumorigenesis. Cell.
126:489–502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fromont G and Cussenot O: The integrin
signalling adaptor p130CAS is also a key player in prostate cancer.
Nat Rev Cancer. 11:2272011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu M, Zhang Y, Yang J, Cui X, Zhou Z,
Zhan H, Ding K, Tian X, Yang Z, Fung KA, et al: ZIP4 increases
expression of transcription factor ZEB1 to promote integrin α3β1
signaling and inhibit expression of the gemcitabine transporter
ENT1 in pancreatic cancer cells. Gastroenterology. 158:679–692.e1.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Salvo E, Garasa S, Dotor J, Morales X,
Pelaez R, Altevogt P and Rouzaut A: Combined targeting of TGF-β1
and integrin β3 impairs lymph node metastasis in a mouse model of
non-small-cell lung cancer. Mol Cancer. 13:1122014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hong SK, Lee H, Kwon OS, Song NY, Lee HJ,
Kang S, Kim JH, Kim M, Kim W and Cha HJ: Large-scale
pharmacogenomics based drug discovery for ITGB3 dependent
chemoresistance in mesenchymal lung cancer. Mol Cancer. 17:1752018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kanda R, Kawahara A, Watari K, Murakami Y,
Sonoda K, Maeda M, Fujita H, Kage M, Uramoto H, Costa C, et al:
Erlotinib resistance in lung cancer cells mediated by integrin
β1/Src/Akt-driven bypass signaling. Cancer Res. 73:6243–6253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Seguin L, Kato S, Franovic A, Camargo MF,
Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, et
al: An integrin β3-KRAS-RalB complex drives tumour
stemness and resistance to EGFR inhibition. Nat Cell Biol.
16:457–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ju L, Zhou C, Li W and Yan L: Integrin
beta1 over-expression associates with resistance to tyrosine kinase
inhibitor gefitinib in non-small cell lung cancer. J Cell Biochem.
111:1565–1574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vachon PH: Integrin signaling, cell
survival, and anoikis: Distinctions, differences, and
differentiation. J Signal Transduct. 2011:7381372011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Somanath PR, Malinin NL and Byzova TV:
Cooperation between integrin alphavbeta3 and VEGFR2 in
angiogenesis. Angiogenesis. 12:177–185. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Luo DY, Wazir R, Tian Y, Yue X, Wei TQ and
Wang KJ: Integrin αv mediates contractility whereas integrin α4
regulates proliferation of human bladder smooth muscle cells via
FAK pathway under physiological stretch. J Urol. 190:1421–1429.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mitra SK, Hanson DA and Schlaepfer DD:
Focal adhesion kinase: In command and control of cell motility. Nat
Rev Mol Cell Biol. 6:56–68. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang J, Wang Y, Zheng L, Hou K, Zhang T,
Qu X, Liu Y, Kang J, Hu X and Che X: Tyrosine kinase
inhibitor-induced IL-6/STAT3 activation decreases sensitivity of
EGFR-mutant non-small cell lung cancer to icotinib. Cell Biol Int.
42:1292–1299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang PF, Li KS, Shen YH, Gao PT, Dong ZR,
Cai JB, Zhang C, Huang XY, Tian MX, Hu ZQ, et al: Galectin-1
induces hepatocellular carcinoma EMT and sorafenib resistance by
activating FAK/PI3K/AKT signaling. Cell Death Dis. 7:e22012016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Akrida I, Nikou S, Gyftopoulos K, Argentou
M, Kounelis S, Zolota V, Bravou V and Papadaki H: Expression of EMT
inducers integrin-linked kinase (ILK) and ZEB1 in phyllodes breast
tumors is associated with aggressive phenotype. Histol Histopathol.
33:937–949. 2018.PubMed/NCBI
|
|
60
|
Guan Y, Bhandari A, Xia E, Kong L, Zhang X
and Wang O: Downregulating integrin subunit alpha 7 (ITGA7)
promotes proliferation, invasion, and migration of papillary
thyroid carcinoma cells through regulating
epithelial-to-mesenchymal transition. Acta Biochim Biophys Sin
(Shanghai). 52:116–124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schittenhelm J, Klein A, Tatagiba MS,
Meyermann R, Fend F, Goodman SL and Sipos B: Comparing the
expression of integrins αvβ3, αvβ5, αvβ6, αvβ8, fibronectin and
fibrinogen in human brain metastases and their corresponding
primary tumors. Int J Clin Exp Pathol. 6:2719–2732. 2013.PubMed/NCBI
|
|
62
|
Dingemans AM, van den Boogaart V, Vosse
BA, van Suylen RJ, Griffioen AW and Thijssen VL: Integrin
expression profiling identifies integrin alpha5 and beta1 as
prognostic factors in early stage non-small cell lung cancer. Mol
Cancer. 9:1522010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Stupp R, Hegi ME, Neyns B, Goldbrunner R,
Schlegel U, Clement PM, Grabenbauer GG, Ochsenbein AF, Simon M,
Dietrich PY, et al: Phase I/IIa study of cilengitide and
temozolomide with concomitant radiotherapy followed by cilengitide
and temozolomide maintenance therapy in patients with newly
diagnosed glioblastoma. J Clin Oncol. 28:2712–2718. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mas-Moruno C, Rechenmacher F and Kessler
H: Cilengitide: The first anti-angiogenic small molecule drug
candidate design, synthesis and clinical evaluation. Anticancer
Agents Med Chem. 10:753–768. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kuwada SK: Drug evaluation: Volociximab,
an angiogenesis-inhibiting chimeric monoclonal antibody. Curr Opin
Mol Ther. 9:92–98. 2007.PubMed/NCBI
|