Introduction
The tumor protein (TP) 53, a tumor
suppressor gene, has been reported to be inactivated mainly by
missense mutation in approximately 50% of various advanced human
cancers (1,2). Even in cancers carrying wild-type (wt)
TP53, p53 is often suppressed by upregulated murine double
minute homolog 2 (MDM2) and MDM4 (3). MDM2 inhibits the p53 transcriptional
activity and ubiquitinates p53 leading to its degradation (4). MDM4 forms a complex with MDM2 that
participates in p53 degradation. Furthermore, MDM4 can block
p53-mediated transcription through direct binding to the
transactivation domain of p53. MDM4 is also a target of
MDM2-mediated ubiquitination and subsequent degradation (5,6). In
normal cells, an autoregulatory feedback loop formed with MDM2,
MDM4, and p53 ensures a dynamic equilibrium between these molecules
(4,7). In cancer cells, the regulatory
relationship between these three proteins is disrupted by
TP53 mutations or MDM2 and MDM4
overexpression, contributing to carcinogenesis and tumor
progression.
It has been expected that synthetic small
interfering RNAs (siRNAs) are promising therapeutics to be applied
to cancer therapy. They can be designed to specifically target
cancer-driver genes in a sequence-specific manner, enabling more
precise and personalized treatments (8). We previously reported that the MDM2
inhibitor nutlin-3 or siRNAs with DNA-substituted seed arms
targeting MDM2 (chiMDM2) inhibited tumor cell growth and
viability by inducing G1 arrest and apoptosis in colon and gastric
cancer cells carrying wt TP53 (9–11).
Furthermore, we revealed that MDM4 knockdown using chiMDM4 could
greatly enhance the antitumor effects of nutlin-3 and chiMDM2 in
those cancer cells via augmented p53 activation (9,10).
The interaction between the p53 pathway and the
RAS-RAF-mitogen-activated protein kinase kinase (MEK)-extracellular
signal-regulated kinase (ERK) cascade has been previously reported
(12,13). ERK1/2 upregulates phosphorylation of
MDM2 at Ser-166 and promotes MDM2-mediated p53 degradation. Dual
targeting of the p53 pathway and the RAS-RAF-MEK-ERK cascade may be
a rational therapeutic strategy for wt TP53 expressing
colorectal and gastric cancers with activated epidermal growth
factor receptor pathways. Further, this strategy might be
particularly important for tumors carrying mutant-type (mt)
RAS and RAF, exhibiting resistance to antibodies
targeted against these receptors. Recently, the synergism of MDM2
and MEK inhibitors was demonstrated in colorectal and non-small
cell lung cancer cells harboring mt KRAS (14). However, the precise mechanism of
action remains unclear.
In this study, we aimed to analyze whether MDM4
knockdown using chiMDM4 synergistically augments the antitumor
effect of the combination of MDM2 knockdown using chiMDM2 and
trametinib, a MEK1/2 inhibitor, or that of nutlin-3 and trametinib.
In addition, we investigated the molecular mechanism of the
antitumor effects induced by MDM4/MDM2 dual knockdown and
trametinib treatment in colon and gastric cancer cells with wt
TP53 and mt KRAS.
Materials and methods
Cell culture
HCT116 colon cancer cell line was purchased from
Horizon Discovery (Cambridge, UK). LoVo colon and SNU-1 gastric
cancer cell lines were purchased from the American Type Culture
Collection. HCT116 and SNU-1 cell lines were cultured in the
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich; Merck
KGaA). LoVo cell line was cultured in Ham's F-12 nutrient mixture
medium (Sigma-Aldrich; Merck KGaA) containing 10% FBS. Trametinib
was purchased from Cayman Chemical (Ann Arbor). Nutlin-3 was
purchased from Calbiochem.
siRNAs and transfection
Control siRNA and MDM2- and MDM4-targeting
DNA-modified siRNA sequences used in this study were adopted from a
previous report (10). Reverse siRNA
transfection was performed using lipofectamine RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.), in accordance with the
manufacturer's instructions. Transfection of the SNU-1 cell line
was performed as described previously (9). Transfection effects of chiMDM2 and
chiMDM4 are shown in Fig. S1.
Cell viability and combination
index
Cell viability was determined using the WST-8
colorimetric assay with the Cell Count Reagent SF (Nacalai Tesque)
as previously described (9). The
combination index (CI) was determined by the Chou-Talalay method
using the CalcuSyn software (Biosoft) (15). The CI values of <0.9, ≥0.9 and
<1.1, and ≥1.1 were defined as the synergistic effect, additive
effect, and antagonistic effect, respectively.
Immunoblot analysis and cell cycle
assay
Protein extraction, sodium dodecyl sulfate
polyacrylamide gel electrophoresis, and immunoblot analyses were
performed as previously described (9). Twenty micrograms of protein samples
were applied to gels. The primary antibodies used in this study are
shown in Table SI. Cell cycle assay
was performed using a Cycletest Plus DNA Reagent Kit (BD
Biosciences) as previously described (16).
Lentivirus production and
transduction
Human BCL2 cDNA was isolated from the pSVBT
plasmid (a kind gift from Dr Tsujimoto) (17) by XhoI digestion and was cloned
into pENTR1A (Invitrogen; Thermo Fisher Scientific, Inc.) at the
SalI-XhoI sites. Then, BCL2 cDNA was subcloned
into a lentivirus expression plasmid (pLenti6.3/V5-DEST,
Invitrogen) with LR Clonase II (Invitrogen; Thermo Fisher
Scientific, Inc.), and the resultant lentivirus plasmids were
designated as BCL2/pLenti6.3. Lentiviruses were produced by
the transfection of 293FT cells (Invitrogen; Thermo Fisher
Scientific, Inc.) with BCL2/pLenti6.3 or EGFP/pLenti6.3
along with pLP1, pLP2, and pLP/VSVG (Invitrogen; Thermo Fisher
Scientific, Inc.) mixtures using the Lipofectamine 2000
(Invitrogen) reagent according to the manufacturer's instructions.
Cells were infected with lentivirus at a multiplicity of infection
of five in the presence of 10 µg/ml of polybrene (Sigma-Aldrich;
Merck KGaA) overnight and replaced with fresh media. After 48 h,
the infected cells were selected using 10 µg/ml of blasticidin
(Invitrogen; Thermo Fisher Scientific, Inc.).
Statistical analysis
Each experiment was performed in triplicate, and all
data were shown as mean ± standard deviation. The Shapiro-Wilk test
was used to evaluate whether data were normal distribution or not.
The significance among three different groups was evaluated by
one-way ANOVA. Then, for post hoc pairwise multiple comparisons,
Tukey's test was used if Levene's test showed homogeneity of
variance, and Games-Howell's test was used if not. P-value of
<0.05 was considered a statistically significant difference. All
statistical analyses were performed using the SPSS version 27.0
(SPSS, Inc.).
Results
Antitumor activity
We examined whether chiMDM4 could enhance the
antitumor effects of chiMDM2 and trametinib in colon (HCT116 and
LoVo) and gastric (SNU-1) cancer cells harboring wt TP53 and
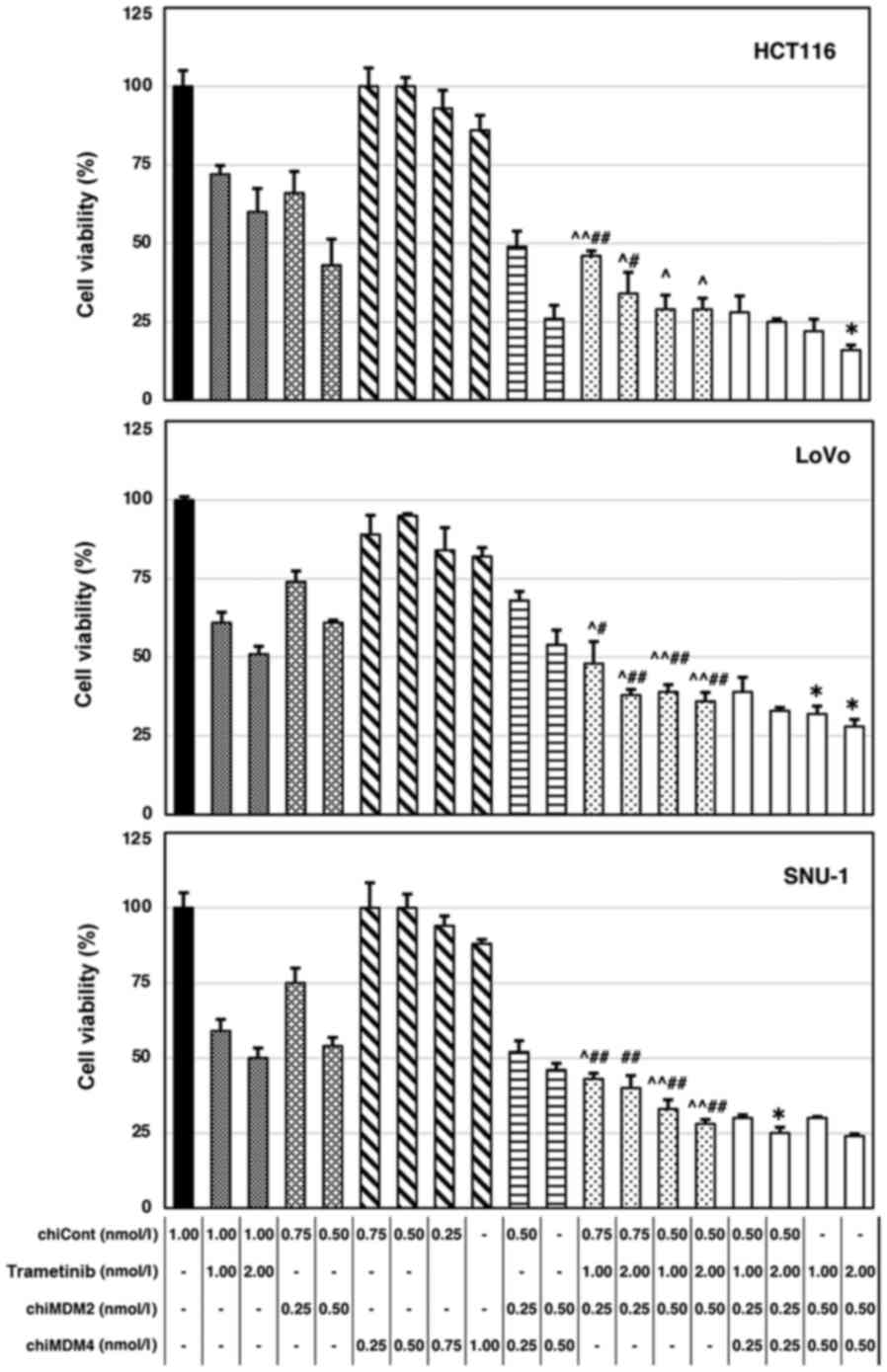
mt KRAS (wt/G13D). As shown in Fig. 1, trametinib alone and chiMDM2 alone
decreased cell viability in a dose-dependent manner. Further, the
combination of chiMDM2 and trametinib induced synergistic antitumor
effects. The CIs of chiMDM4, chiMDM2, and trametinib were
calculated and summarized (Table I).
The addition of chiMDM4 augmented the antitumor effects of the
combination of chiMDM2 and trametinib, which suggested that the
simultaneous inhibition of MDM2 and MDM4 might have an advantage
over the inhibition of MDM2 alone.
 | Table I.Combination index of chiMDM2,
trametinib and chiMDM4 in colon and gastric cancer cells. |
Table I.
Combination index of chiMDM2,
trametinib and chiMDM4 in colon and gastric cancer cells.
| A, HCT116 cell
line |
|---|
|
|---|
| chiMDM2
(nmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|---|
| 0.25 | 1.00 | – | 0.61 | 0.58 |
| 0.25 | 2.00 | – | 0.66 | 0.60 |
| 0.50 | 1.00 | – | 0.71 | 0.72 |
| 0.50 | 2.00 | – | 0.70 | 0.81 |
| 0.25 | 1.00 | 0.25 | 0.72 | 0.54 |
| 0.50 | 2.00 | 0.50 | 0.84 | 0.40 |
|
| B, LoVo cell
line |
|
| chiMDM2
(nmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|
| 0.25 | 1.00 | – | 0.52 | 0.68 |
| 0.25 | 2.00 | – | 0.62 | 0.57 |
| 0.50 | 1.00 | – | 0.61 | 0.58 |
| 0.50 | 2.00 | – | 0.64 | 0.66 |
| 0.25 | 1.00 | 0.25 | 0.61 | 0.70 |
| 0.50 | 2.00 | 0.50 | 0.72 | 0.76 |
|
| C, SNU-1 cell
line |
|
| chiMDM2
(nmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|
| 0.25 | 1.00 | – | 0.57 | 0.65 |
| 0.25 | 2.00 | – | 0.60 | 0.78 |
| 0.50 | 1.00 | – | 0.67 | 0.65 |
| 0.50 | 2.00 | – | 0.72 | 0.61 |
| 0.25 | 1.00 | 0.25 | 0.70 | 0.63 |
| 0.50 | 2.00 | 0.50 | 0.76 | 0.96 |
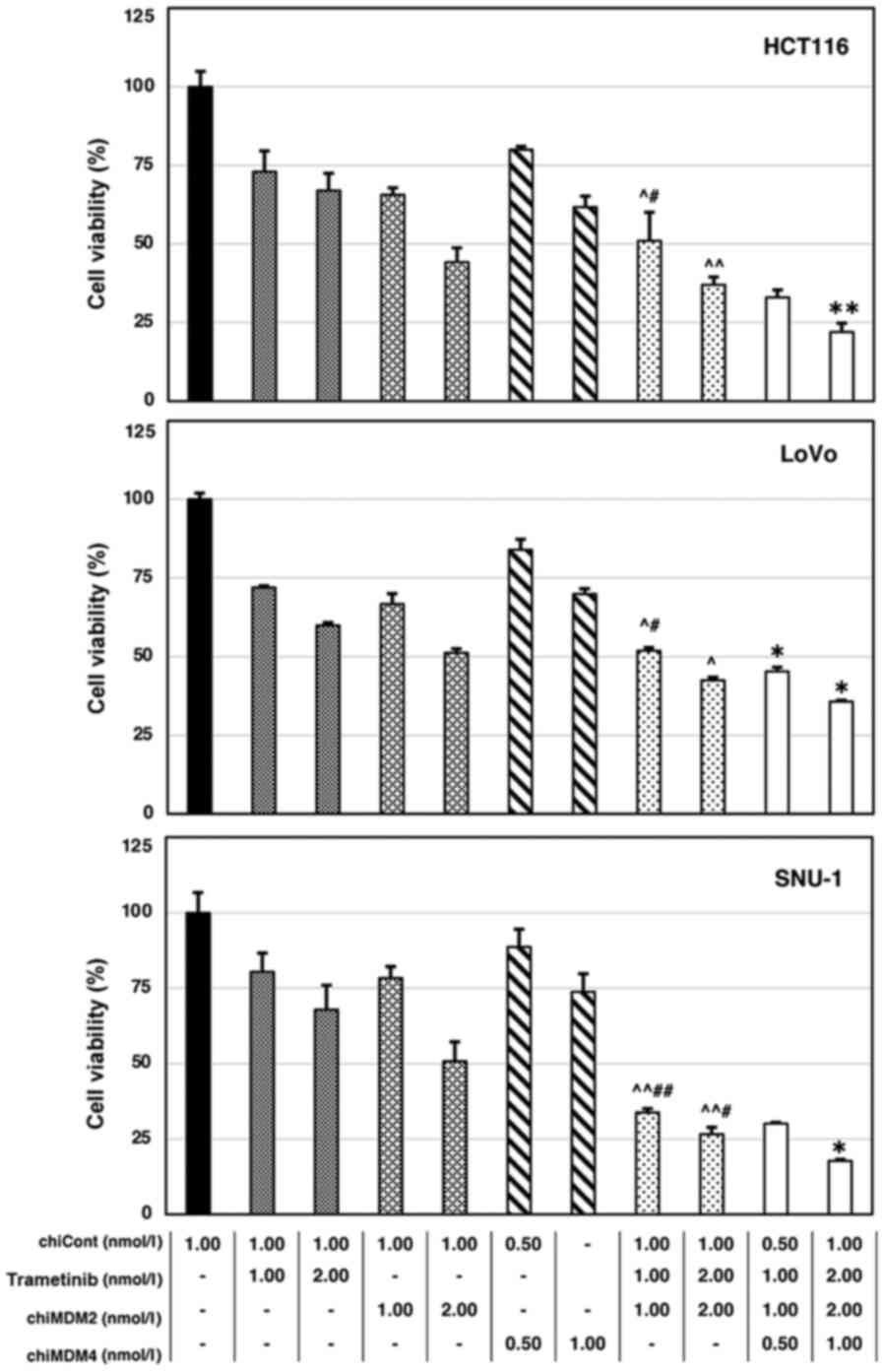
Similarly, we examined the cell viability after the
treatment of cell lines with chiMDM4, nutlin-3, and trametinib
(Fig. 2). The CI values of chiMDM4,
nutlin-3, and trametinib are listed in Table II. The addition of chiMDM4
synergistically enhanced nutlin-3 and trametinib mediated growth
suppression in the tumor cell lines.
| Table II.Combination index of nutlin-3,
trametinib and chiMDM4 in colon and gastric cancer cells. |
Table II.
Combination index of nutlin-3,
trametinib and chiMDM4 in colon and gastric cancer cells.
| A, HCT116 cell
line |
|---|
|
|---|
| Nutlin-3
(µmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|---|
| 1.00 | 1.00 | – | 0.49 | 0.72 |
| 2.00 | 2.00 | – | 0.63 | 0.84 |
| 1.00 | 1.00 | 0.50 | 0.67 | 0.56 |
| 2.00 | 2.00 | 1.00 | 0.78 | 0.59 |
|
| B, LoVo cell
line |
|
| Nutlin-3
(µmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|
| 1.00 | 1.00 | – | 0.48 | 0.84 |
| 2.00 | 2.00 | – | 0.58 | 1.08 |
| 1.00 | 1.00 | 0.50 | 0.55 | 0.82 |
| 2.00 | 2.00 | 1.00 | 0.64 | 0.89 |
|
| C, SNU-1 cell
line |
|
| Nutlin-3
(µmol/l) | Trametinib
(nmol/l) | chiMDM4
(nmol/l) | Effect | Combination
index |
|
| 1.00 | 1.00 | – | 0.66 | 0.45 |
| 2.00 | 2.00 | – | 0.73 | 0.72 |
| 1.00 | 1.00 | 0.50 | 0.70 | 0.85 |
| 2.00 | 2.00 | 1.00 | 0.83 | 0.53 |
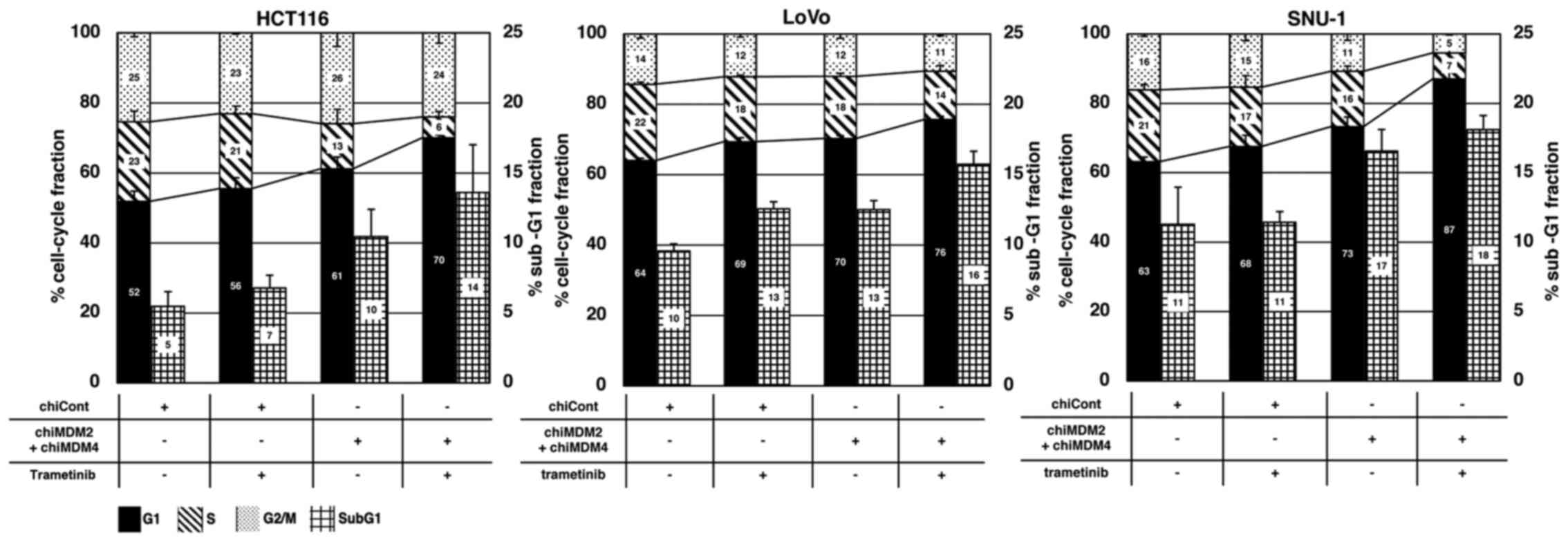
Cell cycle distribution and
apoptosis
To explore the mechanism by which trametinib
treatment and dual inhibition of MDM4/MDM2 exerted an enhanced
antitumor activity, their effects on cell cycle distribution and
apoptosis were analyzed using flow cytometry (Fig. S2). As shown in Fig. 3, trametinib alone and chiMDM4/chiMDM2
alone increased the cell fractions in the G1 phase, whereas it
reduced those in the S phases in all cell lines in a similar
manner, which indicated an induced G1 arrest. Simultaneous exposure
to trametinib and chiMDM4/chiMDM2 induced a profound G1 arrest.
Trametinib alone did not or only slightly increased the sub-G1
population, which is representative of cells undergoing apoptotic
cell death, whereas chiMDM4/chiMDM2 increased the sub-G1 population
moderately in HCT116 and SNU-1 cells and slightly in LoVo cells.
The combination of trametinib and chiMDM4/chiMDM2 further increased
the sub-G1 population in all three cell lines. These results
suggested that trametinib enhanced chiMDM4/chiMDM2-induced G1
arrest and apoptosis.
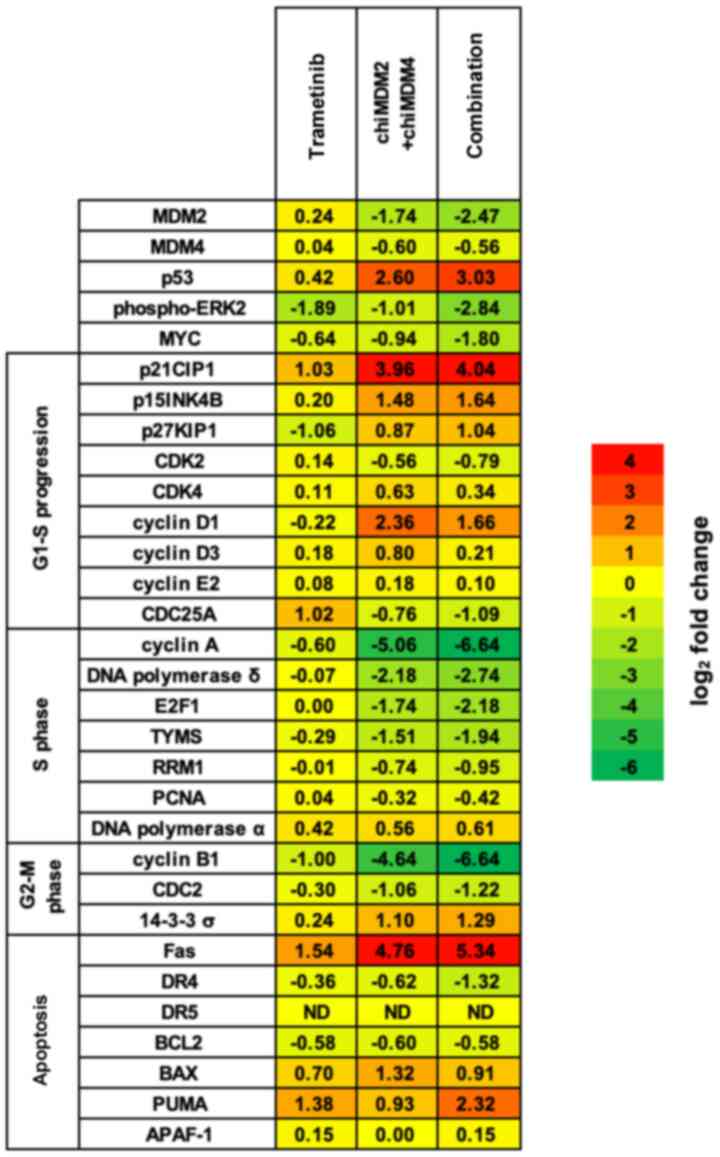
Alterations of cell cycle- and
apoptosis-regulating proteins
The combination of trametinib and chiMDM4/chiMDM2
treatment induced the synergistic antitumor activity by enhancing
G1 arrest and apoptosis in all cells with mt KRAS. To
explore the mechanisms, proteins regulating cell cycle and
apoptosis were examined in HCT116 cells using immunoblot analysis.
Results were summarized using a heatmap (Fig. 4). If the upregulation or
downregulation of the proteins by the combination of trametinib and
chiMDM4/chiMDM2 was two-fold or more than the control and each
agent alone, then it was considered as an important synergistic
antitumor effect of the combination treatment. The proteins such as
p53, p15, p27, and p53-activated proteins [p21, Fas, p53
upregulated modulator of apoptosis (PUMA), and 14-3-3 σ] were
upregulated. On the other hand, the downregulated proteins included
MDM2, p-ERK2, MYC, E2F1, and E2F1-activated proteins (cyclin A,
cyclin B1, DNA polymerase δ, TYMS, CDC2, and CDC25A).
Effects on p-ERK2, p53, p21, RB, Fas
and PUMA
We also analyzed the expression of those proteins in
LoVo and SNU-1 cell lines that were greatly upregulated or
downregulated in HCT116 cells (Fig.
5). Fold change values of the proteins in all three cell lines
are summarized in Table III.
Notably, p-ERK2, which functions as a positive regulator of
retinoblastoma (RB) phosphorylation and an inhibitor of cleavage of
both caspase-8 and caspase-9, was suppressed by the combination of
trametinib and chiMDM4/chiMDM2 treatment in all three cell lines
(4.0–5.3-fold) compared to controls. With respect to the changes in
protein expressions related to G1 arrest, the combination treatment
markedly reduced phosphorylated RB (a master regulator of
E2F-mediated transcription), cyclin A (one of the most efficiently
activated proteins by E2F1), and MYC (a direct target of ERK1/2) in
all three cell lines. ChiMDM4/chiMDM2 induced p53 and p21 in all
three cell lines (Table III). In
SNU-1 cells, as was observed in HCT116 cell, chiMDM4/chiMDM2
induced p53 and p21, which was enhanced by addition of trametinib.
In LoVo cells, chiMDM4/chiMDM2 similarly induced p53 and p21.
However, trametinib did not enhanced chiMDM4/chiMDM2-mediated p53
accumulation, or even decreased p21 accumulation. With regard to
the changes in protein expressions related to apoptosis,
chiMDM4/chiMDM2 induced moderate to high levels of Fas in HCT116
and SNU-1 cells, and a low level of Fas in LoVo cells (Table III). The addition of trametinib
increased Fas levels in HCT116, LoVo, and SNU-1 cells and markedly
increased cleaved caspase-8 in HCT116 cells, and slightly increased
it in LoVo cells; however, no increase of cleaved caspase-8 was
detected in SNU-1 cells until twice the amount of protein samples
was used in immunoblotting. Even so, trametinib slightly increased
cleaved caspase-8 in chiMDM4/chiMDM2-treated SNU-1 cells (Fig. S3). ChiMDM4/chiMDM2 induced PUMA
expression in HCT116 and LoVo cells, but not in SNU-1 cells.
Trametinib further enhanced the expression of PUMA that was induced
by chiMDM4/chiMDM2 in HCT116 and LoVo cells (Table III). This combination treatment
caused caspase-9 cleavage in HCT116 and LoVo cells, but not in
SNU-1 cells.
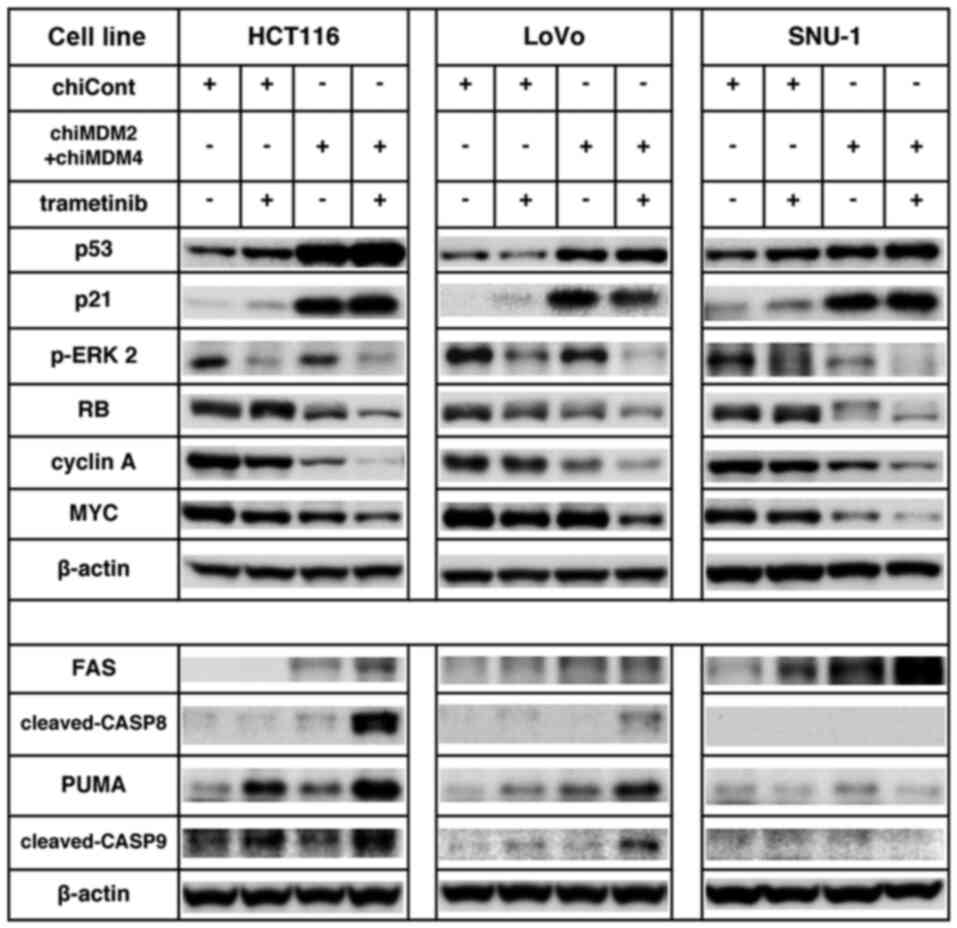 | Figure 5.ChiMDM4/chiMDM2 and trametinib
modulated protein levels of cell cycle progression and apoptosis
regulation in colon and gastric cancer cells. For immunoblotting,
20 µg proteins were loaded per lane. Effects of chiMDM4/chiMDM2 and
trametinib on p-ERK2, RB, MYC, cyclin A, PUMA and Fas levels were
analyzed using immunoblotting in colon (HCT116 and LoVo) and
gastric (SNU-1) cancer cells. Cells transfected with chiCont (1.0
nmol/l) or chiMDM4/chiMDM2 (0.5 nmol/l each) and trametinib (2.0
nmol/l) were analyzed via immunoblotting. β-actin was set as the
internal control. chiMDM, DNA-chimera small interfering RNA against
MDM; Cont, control; p, phosphorylated; ERK2, ERK, extracellular
signal-regulated kinase 2; RB, retinoblastoma; PUMA, p53
upregulated modulator of apoptosis. |
| Table III.Changes in phosphorylated-ERK, cyclin
A, Fas and PUMA levels induced by chiMDM4/chiMDM2 and trametinib in
colon and gastric cancer cells. |
Table III.
Changes in phosphorylated-ERK, cyclin
A, Fas and PUMA levels induced by chiMDM4/chiMDM2 and trametinib in
colon and gastric cancer cells.
| A, HCT116 cell
line |
|---|
|
|---|
|
| Fold change
relative to control |
|---|
|
|
|
|---|
| Up/downregulated
cell cycle apoptosis regulating protein | Trametinib | chiMDM2 +
chiMDM4 | Combination |
|---|
| Upregulated |
|
|
|
|
p53 | 1.6 | 7.7 | 11.3 |
|
p21CIP1 | 2.0 | 19.5 | 21.8 |
|
Fas | 2.9 | 27.1 | 40.4 |
|
PUMA | 2.6 | 1.9 | 5.0 |
| Downregulated |
|
|
|
|
Phospho-ERK2 | 2.2 | 1.0 | 5.0 |
|
MYC | 1.6 | 2.9 | 3.5 |
| Cyclin
A | 1.4 | 4.6 | 16.7 |
|
| B, LoVo cell
line |
|
|
| Fold change
relative to control |
|
|
|
| Up/downregulated
cell cycle apoptosis regulating protein |
Trametinib | chiMDM2 +
chiMDM4 |
Combination |
|
| Upregulated |
|
|
|
|
p53 | 1.3 | 3.1 | 3.1 |
|
p21CIP1 | 3.0 | 20.4 | 13.4 |
|
Fas | 1.1 | 1.2 | 1.3 |
|
PUMA | 1.9 | 2.1 | 4.2 |
| Downregulated |
|
|
|
|
Phospho-ERK2 | 2.1 | 1.4 | 5.3 |
|
MYC | 1.5 | 1.5 | 4.4 |
| Cyclin
A | 1.3 | 2.3 | 4.2 |
|
| C, SNU-1 cell
line |
|
|
| Fold change
relative to control |
|
|
|
| Up/downregulated
cell cycle apoptosis regulating protein |
Trametinib | chiMDM2 +
chiMDM4 |
Combination |
|
| Upregulated |
|
|
|
|
p53 | 1.4 | 2.0 | 2.9 |
|
p21CIP1 | 3.7 | 14.2 | 18.0 |
|
Fas | 2.1 | 3.3 | 5.1 |
|
PUMA | 1.1 | 2.2 | 1.7 |
| Downregulated |
|
|
|
|
Phospho-ERK2 | 1.8 | 1.1 | 4.0 |
|
MYC | 1.5 | 3.3 | 6.7 |
| Cyclin
A | 1.4 | 2.3 | 5.6 |
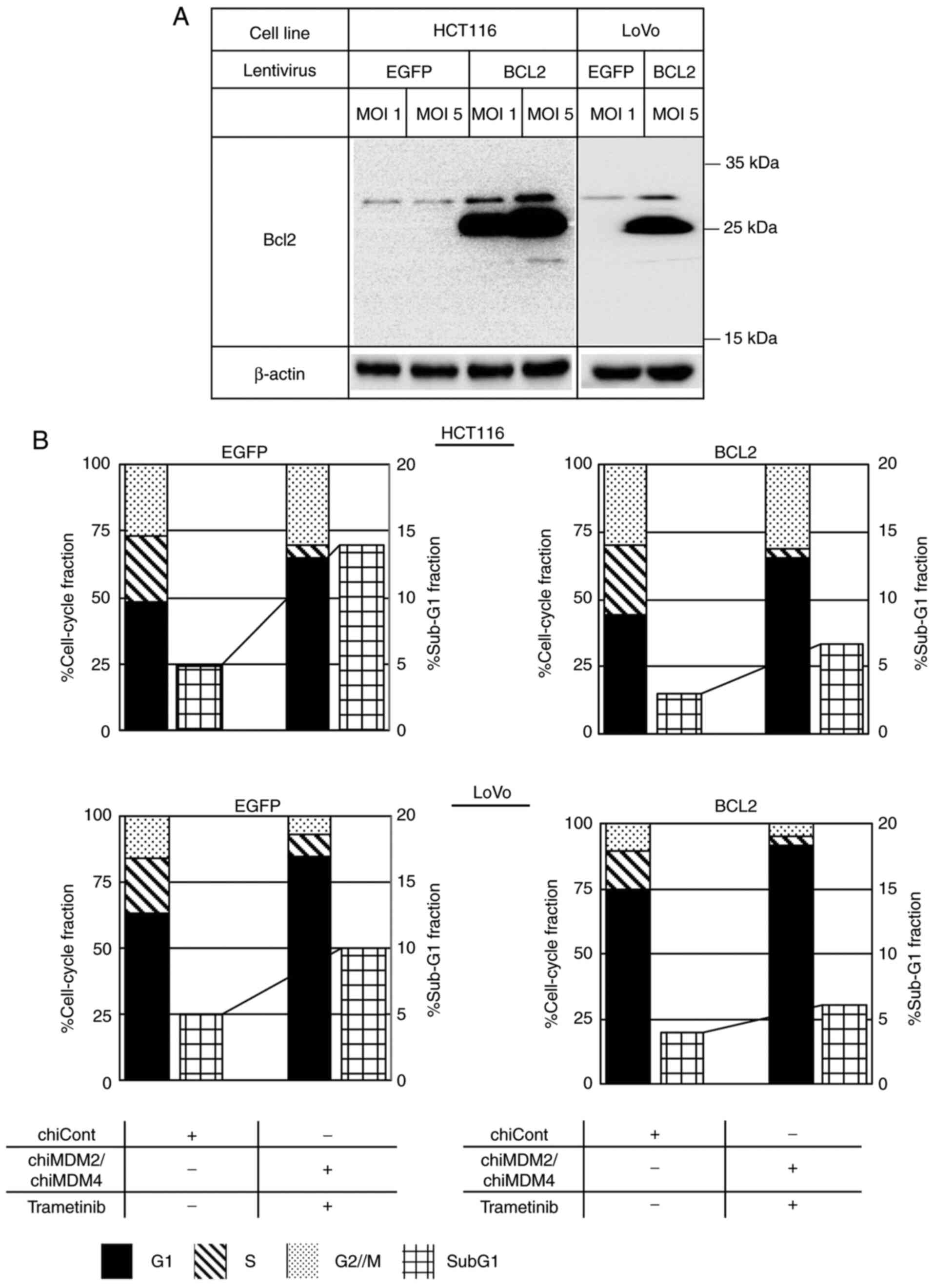
BCL2 induction in HCT116 and LoVo
cells
Bcl2 is an antiapoptotic protein of Bcl2 family,
regulating the mitochondria-mediated apoptosis in cells.
Overexpression of Bcl2 antagonizes proapoptotic Bcl2 family
members, such as PUMA, and represses caspase-9 activation by
reducing the translocation of proapoptotic Bax and cytochrome C
release, then inhibits the mitochondria-mediated apoptosis
(18,19). To investigate whether activation of
caspase-8 and caspase-9 was involved in the apoptosis caused by
combination treatment, the effect of Bcl2 expression on apoptosis
was analyzed in HCT116 and LoVo cells. BCL2 and a control
EGFP were stably transduced in HCT116 and LoVo cells using
lentiviruses (Fig. 6A). As shown in
Figs. 6B and S4, the combined treatment using
chiMDM4/chiMDM2 and trametinib increased the sub-G1 population from
5 to 14% in the control HCT116 cells, whereas this combination
increased the sub-G1 fraction from 3 to 7% in
BCL2-transduced HCT116 cells. This suggests that Bcl2
overexpression partially blocks apoptosis induction in HCT116 cells
via the combination treatment strategy. The combined treatment
increased the sub-G1 fraction from 5 to 10% in the control LoVo
cells and from 4 to 6% in BCL2-transduced LoVo cells,
suggesting that Bcl2 overexpression strongly blocked apoptosis in
LoVo cells via the combination treatment strategy.
Discussion
In this study, we confirmed the synergistic
antitumor effect of MEK and MDM2 inhibition in colon and gastric
cancer cells using the combination of trametinib and nutlin-3 and
that of trametinib and chiMDM2 as a previous study (14). More importantly, we also showed that
this synergistic antitumor effect was augmented by MDM4 knockdown.
In our previous study, chiMDM4 strongly enhanced p53 activation
mediated by nutlin-3 and chiMDM2 (9,10).
Therefore, concurrent inhibition of MDM4 may greatly benefit this
combination therapy in cancer cells with wt TP53 harboring
mt KRAS.
We demonstrated that enhanced induction of G1 arrest
and apoptosis was the mechanism by which chiMDM4/chiMDM2 and
trametinib exerted the synergistic antitumor effects in wt
TP53 colon and gastric cancer cells with KRAS
mutations. This is schematized in Fig.
7. The extensive analysis of protein expressions in HCT116
cells revealed that chiMDM4/chiMDM2 intensely accumulated p53 and
p21, which were associated with the downregulation of
E2F1-activated proteins (cyclin A, cyclin B1, DNA polymerase δ,
E2F1, and TYMS) and upregulation of pro-apoptotic proteins (Fas and
PUMA). Although trametinib alone induced only subtle changes in
these protein levels other than p-ERK2, cyclin B1, and p21, it
enhanced the alterations caused by chiMDM4/chiMDM2. These
synergistic antitumor effects of chiMDM4/chiMDM2 and trametinib
might involve the interaction between ERK1/2 and MDM2. Because
activated ERK1/2 upregulates MDM2 at the transcriptional and
post-translational levels (12,13),
trametinib and chiMDM2 may cooperatively suppress MDM2 expression
at various levels of transcription, post-transcription, and
post-translation, leading to further accumulation of p53.
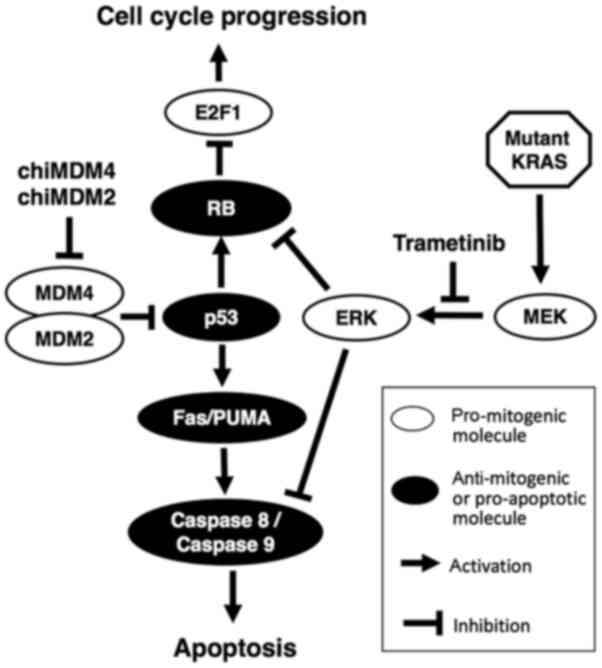 | Figure 7.Molecular mechanisms involved in
chiMDM4, chiMDM2 and trametinib-mediated cell cycle arrest and
apoptosis. ChiMDM4 and chiMDM2 reactivates p53, which decreases
phosphorylated RB levels. The MEK inhibitor, trametinib, inhibits
the downstream signaling pathway of mutant KRAS. RB phosphorylation
is also reduced by ERK inhibition. As a result, E2F1 is inhibited
by both pathways. Simultaneously, activated p53 induces
pro-apoptotic proteins, and inhibition of ERK by trametinib
promotes the same apoptosis pathway. chiMDM, DNA-chimera small
interfering RNA against MDM; RB, retinoblastoma; MEK,
mitogen-activated protein kinase kinase; ERK, extracellular
signal-regulated kinase; MDM2/4, murine double minute homolog
2/4. |
The combination of chiMDM4/chiMDM2 and trametinib
altered the expression levels of phosphorylated RB and
E2F-activated molecules more intensely than the p53 and CDK
inhibitor (p21, p15 and p27) levels in HCT116 cells (Figs. 4, 5
and Table III). Trametinib
enhanced neither chiMDM4/chiMDM2-induced accumulation of p53 nor
p21 in LoVo cells. These results suggest that activation of
p53-p21-RB pathway may not be a sole mechanism of synergistic
growth suppression; it might be regulated by some other pathways.
ERK1/2 directly participates in the regulation of RB
phosphorylation (20).
Hypo-phosphorylated RB binds to lamin A, forms a complex with E2F
and E2F-regulated promoters, and inhibits E2F-transcriptional
activity. Phosphorylated ERK1/2 enters the nucleus and competes
with RB at the same binding site of lamin A and thereby releases
RB. Free RB is rapidly phosphorylated and consequently promotes
cell cycle progression by E2F1-mediated gene expression (20). Suppression of ERK1/2 phosphorylation
by trametinib might coordinately inhibit RB phosphorylation with
p53 activation induced by chiMDM4/chiMDM2.
Trametinib enhanced chiMDM4/chiMDM2-induced
apoptosis in all the cell lines used in this study. It has been
reported that MDM2 and MEK inhibition increased the levels of
Bcl2-like protein 11 and PUMA and attributed the induction of
apoptosis as a reason for their accumulation in some cell lines
(14). We found that our combination
treatment similarly stimulated the intrinsic pathway involving
PUMA-caspase-9 in HCT116 and LoVo cells and also the extrinsic
apoptotic pathway involving Fas-caspase-8 in HCT116 cells and to a
lesser extent in LoVo and SNU-1 cells. The weaker cleaved caspase-8
induction by chiMDM4/chiMDM2 and trametinib, which is a little
inconsistent with certainly induced apoptosis in cell cycle assay
in SNU-1 cells, suggests that chiMDM4/chiMDM2 and trametinib may
induce caspase dependent as well as caspase independent apoptosis
in SNU-1 cells (21–23). BCL2 transduction inhibited
apoptosis more efficiently in LoVo cells than HCT116 cells,
suggesting that PUMA-caspase-9 pathway might be the major signaling
in the combination-induced apoptosis in LoVo cells whereas the
combination-induced apoptosis in HCT116 cells might involve both
Fas-caspase-8 and PUMA-caspase-9 pathways. These some differences
observed in the caspase-8 and caspase-9 activation among three cell
lines could be attributed to the different inducibility or
expressed levels of pro- and anti-apoptotic proteins regulating
apoptosis. Because ERK1/2 inhibits pro-caspase-8 and pro-caspase-9
cleavage by phosphorylating residues at the S387 (24) and T125 (25) sites, respectively, trametinib can
enhance apoptosis via both caspase-8- and caspase-9-mediated
routes.
This study has two methodological limitations.
First, all cell lines used in our study harbored mt KRAS.
Hence, it may be difficult to reach a definitive conclusion as to
whether the KRAS mutation status affects the synergistic
effect of the chiMDM4/chiMDM2 and trametinib combination treatment.
Second, the three cancer cell lines with mt KRAS harbored
PIK3CA or PIK3CB mutations. PIK3CA mutation
(H1047R) of HCT116 cells and PIK3CB mutation (E1051K) of
LoVo cells are pathogenic. We did not examine the interaction
between the PI3K-PTEN-Akt and p53 pathways as this might also
affect the synergistic effects observed in this study. There
remains another issue about toxicities of this combination
treatment. To resolve these issues, further studies are
warranted.
In conclusion, enhanced p53 activation by MDM4/MDM2
knockdown and trametinib treatment exerted the synergistic
antitumor activity through G1 arrest and apoptosis in wt
TP53 gastrointestinal cancers with aberrant KRAS signaling.
This simultaneous MDM2, MDM4, and MEK-ERK inhibition may be a
promising therapy for gastrointestinal cancers.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the Japanese Society for
the Promotion of Science (JSPS KAKENHI; grant no. JP18K07288).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW, YY, YN, TM, KY, and IH conceived and designed
the study. XW, MI, XZ, MS, AS, MH, SE and KY performed the
experiments. YY, KY, and IH confirm the authenticity of all the raw
data. XW and YY performed the statistical analyses. XW, YY, and IH
wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
chiCont
|
control chimeric siRNA
|
|
chiMDM2
|
chimeric siRNA with DNA-substituted
seed arms targeting MDM2
|
|
chiMDM4
|
chimeric siRNA with DNA-substituted
seed arms targeting MDM4
|
|
CI
|
combination index
|
|
wt
|
wild-type
|
|
mt
|
mutant-type
|
References
|
1
|
Sionov RV and Haupt Y: The cellular
response to p53: The decision between life and death. Oncogene.
18:6145–6157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vousden KH and Lu X: Live or let die: The
cell's response to p53. Nat Rev Cancer. 2:594–604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levine AJ: The tumor suppressor genes.
Annu Rev Biochem. 62:623–651. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moll UM and Petrenko O: The MDM2-p53
interaction. Mol Cancer Res. 1:1001–1008. 2003.PubMed/NCBI
|
|
5
|
Linares LK, Hengstermann A, Ciechanover A,
Müller S and Scheffner M: HdmX stimulates Hdm2-mediated
ubiquitination and degradation of p53. Proc Natl Acad Sci USA.
100:12009–12014. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shvarts A, Steegenga WT, Riteco N, van
Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt
W, Hateboer G, van der Eb AJ and Jochemsen AG: MDMX: A novel
p53-binding protein with some functional properties of MDM2. EMBO
J. 15:5349–5357. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu X, Bayle JH, Olson D and Levine AJ: The
p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7:1126–1132.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mahmoodi Chalbatani G, Dana H,
Gharagouzloo E, Grijalvo S, Eritja R, Logsdon CD, Memari F, Miri
SR, Rad MR and Marmari V: Small interfering RNAs (siRNAs) in cancer
therapy: A nano-based approach. Int J Nanomedicine. 14:3111–3128.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Imanishi M, Yamamoto Y, Wang X, Sugaya A,
Hirose M, Endo S, Natori Y, Yamato K and Hyodo I: Augmented
antitumor activity of 5-fluorouracil by double knockdown of MDM4
and MDM2 in colon and gastric cancer cells. Cancer Sci.
110:639–649. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirose M, Yamato K, Endo S, Saito R, Ueno
T, Hirai S, Suzuki H, Abei M, Natori Y and Hyodo I: MDM4 expression
as an indicator of TP53 reactivation by combined targeting of MDM2
and MDM4 in cancer cells without TP53 mutation. Oncoscience.
1:830–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Endo S, Yamato K, Hirai S, Moriwaki T,
Fukuda K, Suzuki H, Abei M, Nakagawa I and Hyodo I: Potent in vitro
and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric
cancer cells. Cancer Sci. 102:605–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Malmlöf M, Roudier E, Högberg J and
Stenius U: MEK-ERK-mediated phosphorylation of Mdm2 at Ser-166 in
hepatocytes. Mdm2 is activated in response to inhibited Akt
signaling. J Biol Chem. 282:2288–2296. 2007. View Article : Google Scholar
|
|
13
|
Ries S, Biederer C, Woods D, Shifman O,
Shirasawa S, Sasazuki T, McMahon M, Oren M and McCormick F:
Opposing effects of Ras on p53: Transcriptional activation of mdm2
and induction of p19ARF. Cell. 103:321–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hata AN, Rowley S, Archibald HL,
Gomez-Caraballo M, Siddiqui FM, Ji F, Jung J, Light M, Lee JS,
Debussche L, et al: Synergistic activity and heterogeneous acquired
resistance of combined MDM2 and MEK inhibition in KRAS mutant
cancers. Oncogene. 36:6581–6591. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bronkhorst AJ, Wentzel JF, Aucamp J, van
Dyk E, du Plessis L and Pretorius PJ: Characterization of the
cell-free DNA released by cultured cancer cells. Biochim Biophys
Acta. 1863:157–165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsujimoto Y and Croce CM: Analysis of the
structure, transcripts, and protein products of bcl-2, the gene
involved in human follicular lymphoma. Proc Natl Acad Sci USA.
83:5214–5218. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reshi L, Wang HV, Hui CF, Su YC and Hong
JR: Anti-apoptotic genes Bcl-2 and Bcl-xL overexpression can block
iridovirus serine/threonine kinase-induced
Bax/mitochondria-mediated cell death in GF-1 cells. Fish Shellfish
Immunol. 61:120–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rodríguez J, Calvo F, González JM, Casar
B, Andrés V and Crespo P: ERK1/2 MAP kinases promote cell cycle
entry by rapid, kinase-independent disruption of
retinoblastoma-lamin A complexes. J Cell Biol. 191:967–979. 2010.
View Article : Google Scholar
|
|
21
|
Bidère N and Senik A: Caspase-independent
apoptotic pathways in T lymphocytes: A minireview. Apoptosis.
6:371–375. 2001. View Article : Google Scholar
|
|
22
|
Wu M, Xu LG, Li X, Zhai Z and Shu HB:
AMID, an apoptosis-inducing factor-homologous
mitochondrion-associated protein, induces caspase-independent
apoptosis. J Biol Chem. 277:25617–25623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee TJ, Kim EJ, Kim S, Jung EM, Park JW,
Jeong SH, Park SE, Yoo YH and Kwon TK: Caspase-dependent and
caspase-independent apoptosis induced by evodiamine in human
leukemic U937 cells. Mol Cancer Ther. 5:2398–2407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mandal R, Raab M, Matthess Y, Becker S,
Knecht R and Strebhardt K: pERK 1/2 inhibit caspase-8 induced
apoptosis in cancer cells by phosphorylating it in a cell cycle
specific manner. Mol Oncol. 8:232–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Allan LA, Morrice N, Brady S, Magee G,
Pathak S and Clarke PR: Inhibition of caspase-9 through
phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 5:647–654.
2003. View Article : Google Scholar : PubMed/NCBI
|