Ubiquitylation is a key post-translational
modification that plays a significant role in the stability of the
intracellular environment, cell proliferation and differentiation,
as well as various other cellular functions (1). An imbalance in ubiquitination-mediated
protein degradation can represent the molecular basis of certain
human diseases, such as cancers (prostate cancer, lung cancer and
liver cancer) and nervous system diseases (Parkinson's disease,
Alzheimer's disease and Huntington's disease) (1). Ubiquitin is activated in an
ATP-dependent reaction catalyzed by the ubiquitin-activating enzyme
E1 (2,3). Subsequently, under the action of the
ubiquitin-conjugating enzyme E2, activated ubiquitin is transferred
to a specific substrate along with E3 ubiquitin ligase (1). Substrate recognition for ubiquitin
ligation is determined by E3 ubiquitin ligase (2,3), thus E3
ubiquitin ligase is specific compared with E1 and E2 enzymes and
constitutes an important topic in medical research. One of the best
known E3 ligase family is cullin (CUL)-RING E3 ubiquitin ligase,
which consists of a molecular scaffold connecting a
substrate-specific adaptor protein to a catalytic component
comprising a RING finger domain and an E2 ubiquitin-conjugating
enzyme (4–8). The human genome contains eight CUL
family members, namely, CUL1, −2, −3, −4A, −4B, −5, −7 and −9, all
of which have an evolutionarily conserved CUL homology domain at
the C-terminus, which promotes the interaction of CUL with RING box
protein (RBX)1 or RBX2 (9–18). E3 adaptors of CUL3, such as
speckle-type protein (SPOP) (19–29),
Kelch repeat and BTB domain-containing protein 8 (KBTBD8) (30–32) and
Kelch-like ECH-associated protein 1 (KEAP1) (33–35), are
composed of a similar structure called broad-complex, tramtrack and
bric-a-brac (BTB) domain (15–18),
which combines the substrate receptor and adaptor functions into
the CUL3-RBX1 E3 ubiquitin ligase complex (36,37).
Recently, increased attention has been paid to
leucine zipper-like transcription regulator 1 (LZTR1), due to its
far-reaching implications in physiological and pathological
conditions of cells and human diseases, such as Noonan syndrome
(NS), glioblastoma (GBM), chronic myeloid leukemia (CML) and
schwannomatosis (38,39). LZTR1, also a member of the BTB-Kelch
superfamily proteins, is the substrate-specific adaptor for CUL3
ubiquitin ligase complex (38–40).
Given its weak homology to known members of the basic leucine
zipper-like family, LZTR1 was initially identified as a
transcriptional regulator (40).
Proteins of the BTB-Kelch superfamily often interact with actin
filaments and play important roles in fundamental cell functions,
such as transcriptional regulation and protein ubiquitination
(41–45). However, unlike other BTB-Kelch
proteins, LZTR1 shows no interaction with actin but is localized on
the Golgi complex (40), suggesting
a unique function and status. Mutations in the LZTR1 gene occur in
≤8% of patients with NS (46,47),
4.4% of those with GBM (39,48) and 24.4% of those with schwannomatosis
(Table I) (49–51). The
occurrence of these diseases is related to abnormal function of RAS
proteins, demonstrating a close relationship between LZTR1 and
proteins of the RAS superfamily (38,52–54).
Notably, almost all members of the RAS superfamily, including KRAS,
NRAS, RAS-like without CAAX1 (RIT1) and RAF1, are substrates of
LZTR1 (38,55,56).
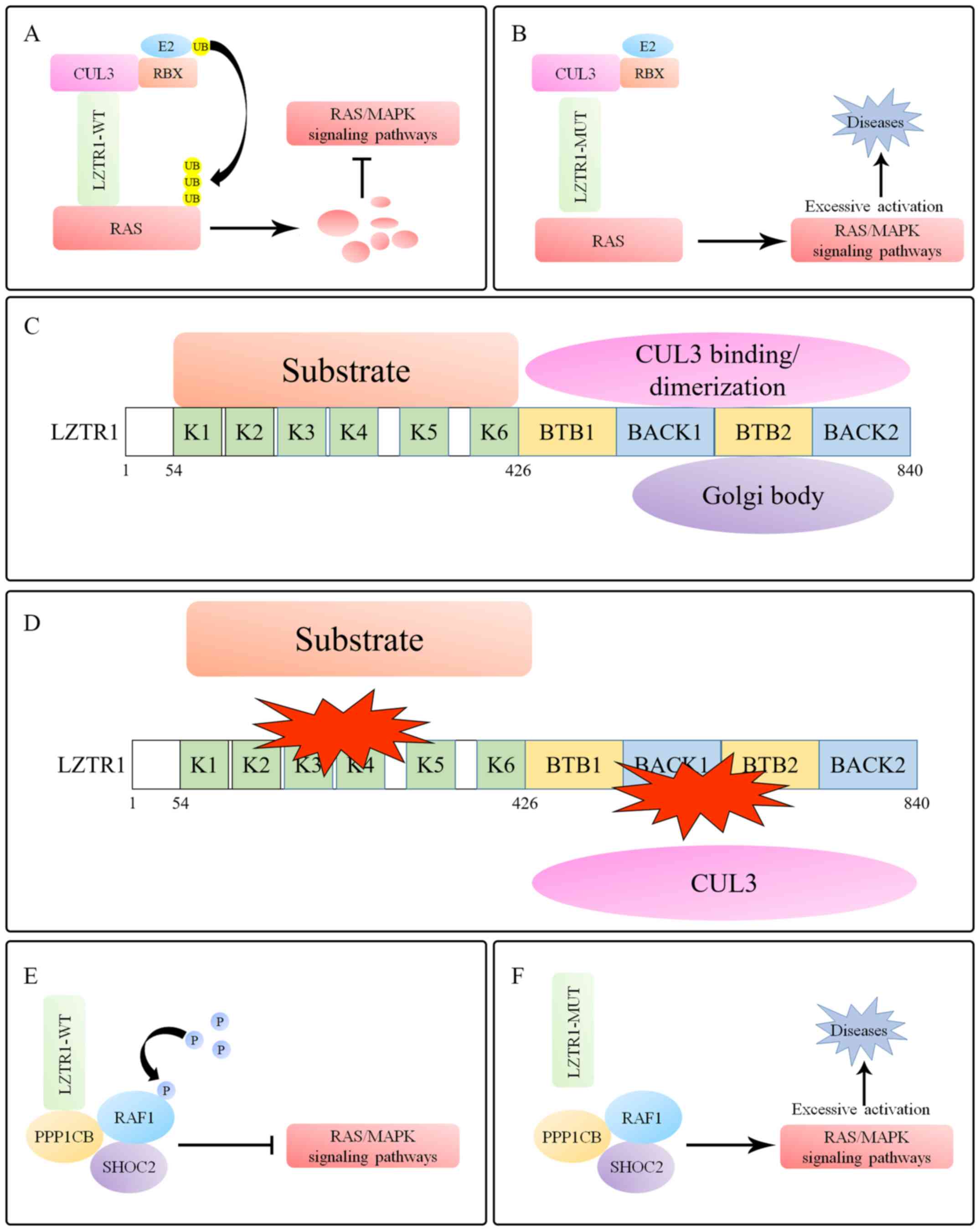
LZTR1 regulates the RAS/MAPK signaling pathway by inducing the
polyubiquitination and degradation of RAS-superfamily proteins
(KRAS, MRAS, NRAS and RAF1) (Fig.
1A) (57), whereas
disease-associated LZTR1 mutations lose this capability, leading to
excessive activation of RAS/MAPK signaling (Fig. 1B). The effect of LZTR1 on RAS/MAPK
signaling is specific as demonstrated by the failure of the other
two prominent CUL3 adaptors (KEAP1 and SPOP) in the process of RAS
ubiquitination (38).
The Kelch domains of the BTB-Kelch superfamily
proteins are located at the C-terminus of the BTB domains, and the
conserved BACK domains are present in almost all members (17). Different from the general
BTB-Kelch-superfamily proteins, the primary structure of LZTR1
includes six Kelch motifs at the N-terminus and two C-terminal
BTB-BACK domains (40). The Kelch
domains selectively recruit substrates, whereas the BACK domains
are considered to mediate dimerization and the binding to CUL3
(58). Furthermore, the second BTB
domain (from N to C) mediates the interaction between the Golgi
complex and LZTR1, suggesting that LZTR1 may be a novel Golgi
matrix-associated protein (Fig. 1C)
(40).
The majority of NS- and GBM-related LZTR1 mutations
are clustered in the Kelch domains (Table I), and these mutations are defective
in LZTR1-mediated substrate binding, thus preventing the formation
of substrate-LZTR1-CUL3 complexes and their efficient
ubiquitination and degradation, which results in excessive
activation of RAS/MAPK signaling in NS or GBM (39,55,58).
Furthermore, except for the Leu812Pro mutation, mutations in the
LZTR1 BACK domains exhibit reduced interaction with CUL3 and
although LZTR1-L812P retains the ability to bind to CUL3, it fails
to dimerize LZTR1 (38). The
endogenous and exogenous expression of wild-type LZTR1 displays
punctate endomembrane immunostaining, but diffuse and uniform
cytoplasmic staining when LZTR1 mutations occur in the BACK
domains, including in the Leu812Pro mutant (58). Interestingly, unlike NS- and
GBM-associated LZTR1 mutations, which are concentrated in the Kelch
domains, schwannomatosis-associated LZTR1 mutations are more evenly
distributed across all domains, and these mutations may result in
failure to bind with CUL3 (38,39,46–48). In
summary, disease-related LZTR1 mutations are defective in mediating
substrate ubiquitination by disrupting the formation of
substrate-LZTR1-CUL3 complexes (Fig.
1D; Table I).
RASopathies are defined as a class of inherited
diseases with germline mutations in genes encoding components of
the RAS/MAPK signaling pathway (58). RAS-superfamily proteins are a class
of evolutionarily conserved proteins with high affinity for GDP and
GTP (52,59,60). RAS
proteins, similar to molecular switches, are activated when
interacting with GTP and serve as positive regulators in the
physiological activities of cells, such as proliferation and cell
division (59,61–64).
RASopathy-associated RAS mutations located in the secondary locus
(mutations at these loci only slightly affect the physiological
function of RAS superfamily proteins) weakly affect GTPase activity
without causing embryonic-lethal death (53,54,65–69). NS,
one of the most common and typical RASopathies, is caused by
germline gain-of-function pathogenic RAS variants affecting the
RAS/MAPK signaling pathway (38).
The pathogenesis of NS depends on abnormal protein
degradation of RAS-superfamily proteins, including KRAS, NRAS, RIT1
and RAF1 (38,55,56,58).
LZTR1 has also been added to the list of genes causing NS, and
mutations in LZTR1 are present in ≤8% of patients with NS (46,70). NS
patients harboring LZTR1 mutations present typical NS facial
features, a webbed neck, cardiovascular defects and coagulation
dysfunction (71).
Previous studies have suggested that almost all
NS-related mutations in LZTR1 are dominant mutations that occur
mainly in the Kelch domains (38,58,70).
These mutations neither alter CUL3 binding nor affect subcellular
localization and stability of LZTR1 (72), although they may impair its ability
to specifically recognize substrates, such as KRAS, NRAS and HRAS,
ultimately contributing to the excessive activation of RAS/MAPK
signaling (58,72). However, a recent study reported
biallelic loss-of-function LZTR1 mutations in three patients NS
(Table I), suggesting recessive
inheritance of some NS cases (73).
Thus, considering the different effects of LZTR1 mutations,
determining the association between LZTR1 and the
autosomal-dominant and autosomal-recessive forms of NS may prove
useful.
RAF1 is the downstream substrate of most classical
RAS proteins, mediating the activation of the RAS/MAPK signaling
pathway. Protein phosphatase 1 (PP1), which interacts with the
leucine-rich repeat protein SHOC2 to form a complex encoded by an
NS-causing gene, dephosphorylates RAF1 to activate ERK (74,75). A
recent study on a case report identified eight LZTR1 variants,
including a de novo variant, in seven probands who were susceptible
to NS and one known de novo PP1 catalytic subunit β (PPP1CB)
variant in a patient with NS, indicating that LZTR1 and PPP1CB may
be closely related to the pathogenesis of NS (56). Moreover, co-immunoprecipitation
demonstrated that endogenous LZTR1 interacted with the RAF1-PPP1CB
complex and that RAF1 phosphorylation levels (Ser259), but not
ubiquitination, suggesting that LZTR1 may be involved in additional
pathways that mediate the phosphorylation of RAF1 to regulate its
activity (Fig. 1E and F) (56).
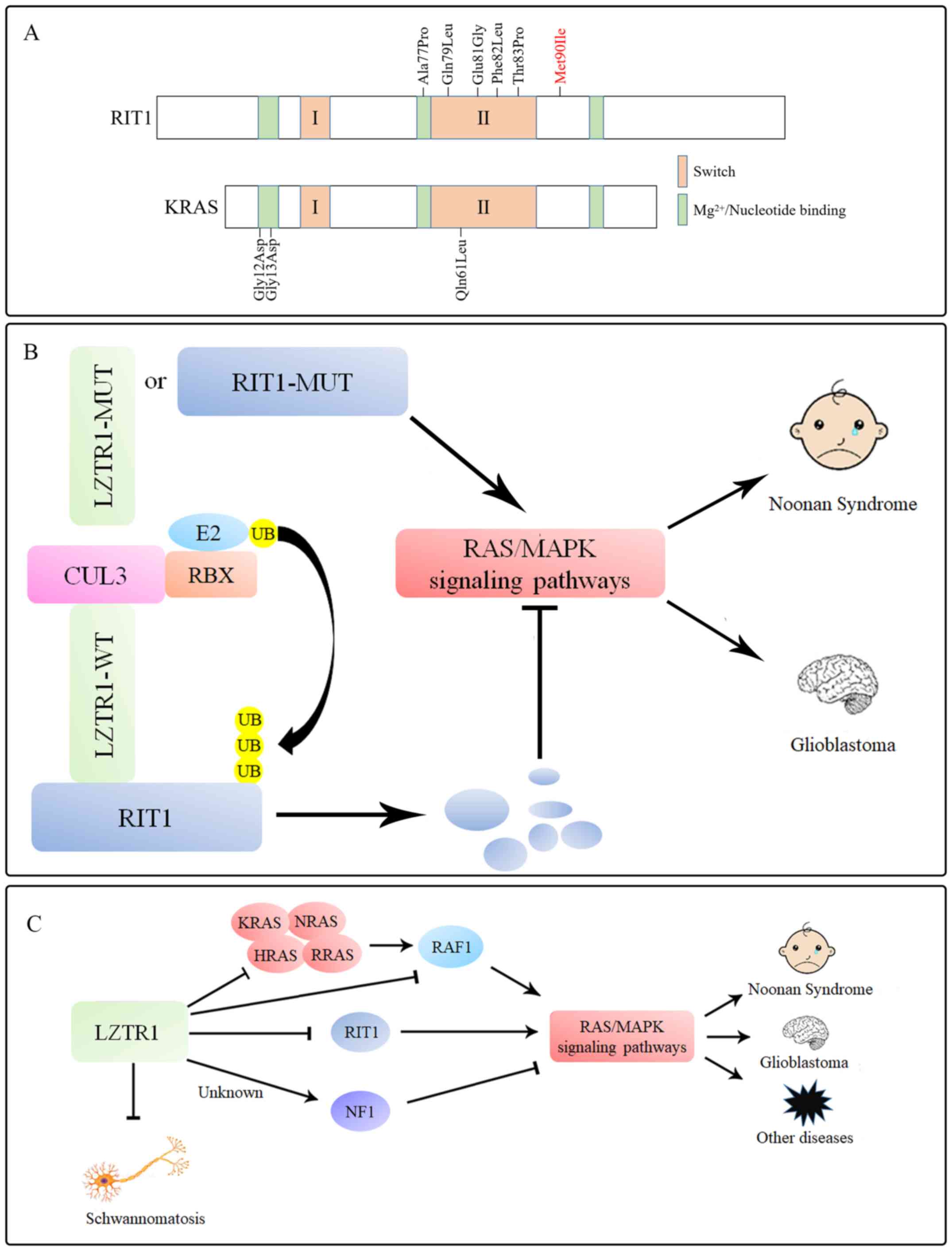
RIT1 also belongs to the RAS superfamily and is
highly similar to other members (K-, H-, R- and NRAS), regulating
the physiological activity of cells (76–79).
Germline mutations in RIT1 account for 5–9% of mutations in
patients with NS (54,80). The NS-associated mutation Met90Ile in
RIT1 (Fig. 2A), which is located in
an atypical site around the switch II region, contributes to the
symptoms of typical NS, suggesting a strong association between
RIT1 and NS (55). LZTR1 acts a
negative regulator in the control of RIT1 activity by inducing RIT1
ubiquitination via K48 (Lys48)-linked ubiquitination and
degradation (Fig. 2B). The exogenous
overexpression of LZTR1 in 293T cells leads to a reduction in RIT1
protein levels, which is reversed by the proteasomal inhibitor
bortezomib and by the CUL3 inhibitor MLN4924, but not by the
lysosomal inhibitor bafilomycin (81,82).
Notably, almost all NS-associated RIT1 mutants are
defective in their interaction with endogenous LZTR1 compared with
the wild-type RIT1, thus preventing the proteolysis and
ubiquitination of RIT1 mutants (39,54,55,77).
Similarly, a group of NS-associated LZTR1 mutations are associated
with impaired RIT1 degradation, thus revealing the close
relationship between LZTR1 and RIT1 (49,55).
Under physiological conditions, RIT1 is ubiquitinated by the
LZTR-CUL3 complex, then degraded by the ubiquitin-proteasome system
(UPS). Moreover, under pathological conditions, RIT1 escapes from
UPS-mediated degradation, thus leading to significant enhancement
of MAPK signaling in NS (Fig. 2B).
This suggests that LZTR1 serves a negative role in MAPK signaling,
whereas mutations in LZTR1 or members of the RAS superfamily affect
MAPK signaling and contribute to NS.
RAS is considered the most closely related
oncoprotein to human cancer (such as pancreatic, lung and colon
cancer, as well as hematological malignancies) and is commonly
activated in tumor cells (47,59,64,83).
Given their vital effects on GTPase activity, cancer-related RAS
mutations are often lethal in embryos, avoiding the transmission of
germline (60,84–86).
Brain cancer is one of the most common tumors in
adults; among the different types of brain cancer, GBM is the most
common (48%) and deadliest (median overall survival of 12–14
months) given its high tumor heterogeneity and poor survival
(87–93). Numerous genetic mutations, including
those in the LZTR1 gene, have been identified in GBM using
whole-exome sequencing. LZTR1 mutations in GBM include 4.4%
non-synonymous mutations and 22.4% focal deletions in the coding
sequence (94). RIT1 is regarded as
an important pathogenic factor of GBM, which participates in the
activation of the MAPK/ERK signaling pathway and plays crucial
roles in various physiological processes, including cell survival,
proliferation and differentiation (95–99).
Moreover, LZTR1 can interact with endogenous RIT1, which is
considered a tumor promoter in GBM (39,94).
Frattini et al (94) demonstrated
that 9 out of 10 LZTR1 mutations occur in the Kelch domains and
greatly impair the RIT1-LZTR1 interaction. Thus, LZTR1 suppresses
the MAPK/ERK signaling pathway by degrading RIT1 to inhibit
proliferation and migration of GBM cells; however, GBM-associated
LZTR1 mutations impair this function (Fig. 2B) (39,77,94).
Moreover, LZTR1 decreases tumor size by inducing glioma spheres,
enhancing glioma cell adhesion and reducing cell cycle
progression-related proteins (such as cyclin A, PLK1 and p107),
whereas mutations or deletions in LZTR1 impair glioma sphere
formation and promote the self-renewal ability of GBM cells
(39,77). These findings suggest that
GBM-associated LZTR1 mutations in human GBM drive self-renewal and
the growth of glioma spheres (39,55,77).
Moreover, LZTR1 simultaneously found in GBM with mutations and
deletions confirms the two-hit hypothesis of cancer (the two
alleles of tumor suppressor genes are required to be mutated into
‘loss of function’ in one cell (100). When a cell in a person is
heterozygous for a germline mutation (‘one hit’), it is required to
undergo a second, somatic event (‘second hit’) that inactivates the
other allele to initiate cancers (100), further indicating that LZTR1 acts
as a tumor suppressor in GBM (89–91,94).
Given the significance of LZTR1-RIT1 signaling in GBM, LZTR1 and
RIT1 may represent promising therapeutic targets for GBM.
The activation of key cellular pathways, including
the RAS/MAPK signaling pathway, affects the survival and
proliferation of BCR-ABL+ CML cells (101–103).
The occurrence of tyrosine kinase inhibitor (TKI) resistance is
also associated with dysregulation of RAS/MAPK signaling (38). In recent studies, the inactivation of
endogenous or exogenous LZTR1 increased the phosphorylation of MAPK
kinase 1 and −2, as well as ERK1 and −2 in CML cells (indicating
enhanced RAS/MAPK signaling activation) and promoted the resistance
of these cells to TKIs (104,105).
This phenotype may depend on the failed formation of the CUL3
ligase complex, as similar phenotypic changes were also observed
when CUL3 was genetically silenced (38). Thus, there is limited knowledge of
the link between LZTR1 inactivation and TKI resistance, and further
study of this molecular mechanism may help alleviate TKI resistance
in CML.
Schwannomatosis is a hereditary disease
characterized by schwannomas in the spinal and peripheral nerves
and predisposition to benign tumors throughout the nervous system
(106–108). Local or diffuse chronic pain is the
most common symptom reported by patients with schwannomatosis
(49). Germline mutations in the
LZTR1 gene occur in 41 out of 168 sporadic patients with
schwannomatosis (24.4%), highlighting the complex heterogeneity of
schwannomatosis (106,108). Interestingly,
schwannomatosis-associated LZTR1 mutations are uniformly located in
almost every domain (Table I). Thus,
in contrast with the mutations in NS and GBM, there is no
positional preference for LZTR1 mutations in schwannomatosis
(50). Notably, the occurrence of
schwannomatosis-associated LZTR1 mutations at the same genetic
locus as NS-associated ones and the coexistence of NS and
schwannomatosis in some patients have also been reported in
previous studies (108,109). According to the two-hit hypothesis,
biallelic loss of a tumor suppressor gene can lead to
tumorigenesis; hence, according to the two-hit hypothesis, the NS
patients carrying LZTR1 germline mutations are more likely to have
schwannomatosis (50,109,110).
However, the pathogenesis of this disease and the signaling
pathways involving LZTR1 have rarely been rarely studied.
SWI/SNF-related, matrix-associated, actin-dependent
regulator of chromatin subfamily B member 1 (SMARCB1) was
identified as the first predisposing gene for schwannomatosis
(111). Both SMARCB1 and LZTR1 are
located at the 22q centromere, and a previous study identified
germline variants of LZTR1 in 24 patients with SMARCB1-negative
schwannomatosis from 22 unrelated families (112). Notably, recent studies reported the
occurrence of GBM in patients with schwannomatosis and germline
LZTR1 mutations or SMARCB1 mutations (48,113,114).
Given the lack of evidence of protein interaction between SMARCB1
and LZTR1 or their participation in specific signaling pathways,
further research on LZTR1 and SMARCB1 may clarify the molecular
basis of schwannomatosis and GBM.
LZTR1 acts as a negative factor that suppresses RAS
function and MAPK signaling; mutations in this protein may
dysregulate RAS ubiquitination and lead to impaired protein
degradation of RAS superfamily proteins, leading to NS, GBM,
schwannomatosis and CML cell resistance to TKIs (Fig. 2C) (38,39,58).
Notably, NS- and GBM-related LZTR1 mutations are concentrated in
the Kelch domains, whereas schwannomatosis-associated LZTR1
mutations are uniformly located in almost every domain
(38,50.58.106,107,113-115). The distribution of these mutations is
associated with the severity of these diseases. Indeed, LZTR1
mutations located in Kelch domains directly affect the substrate
binding of the LZTR1 (40,58), which has the greatest effect on the
degradation level of the substrate, thus leading to more serious
diseases, such NS and GBM. However, schwannomatosis-associated
LZTR1 mutations occur in sites that only slightly affect the
function of LZTR1 and to some extent, substrate-LZTR1-CUL3
complexes still work. This hypothesis may be tested by studying
LZTR1 mutations in patients with concurrent GBM and
schwannomatosis.
For several genetic diseases, such as NS, early
detection may be more important than treatment. LZTR1 has also been
added to the list of genes causing NS and is present in ≤8% of NS
patients (46,70). Thus, prenatal screening for LZTR1
mutations and prophylactic use of RAS inhibitors may be a possible
way to avoid the occurrence of NS after birth. Moreover, for
patients with LZTR1 mutations in GBM and other cancer types, RAS
pathway inhibitors may be a good choice for treatment. Thus,
comprehensive understanding of other mechanisms of RAS activation
may benefit patients carrying LZTR1 mutations and offer new
therapeutic strategies (38,58).
LZTR1 is also closely related to the central nervous
system. LZTR1 interacts with CUL3 and neurofibromin 1 (NF1) to
regulate night-time sleep by increasing GABA receptor signaling and
has been associated with RAS-related neurological diseases created
by Nf1 deficiency (116). NF1 is a
negative growth factor of the RAS/MAPK signaling pathway, which,
together with LZTR1, may inhibit RAS/MAPK signaling pathways
(117). However, the molecular
mechanism of the interaction between LZTR1 and NF1 remains unknown.
The functions of LZTR1 in the nervous system are not restricted to
those already described and may be closely related to
neurodegenerative diseases, such as Alzheimer's disease and
Parkinson's disease (117).
Notably, the frequency of LZTR1 mutations in schwannomatosis are as
high as 25% (49), yet the specific
molecular mechanism of the disease has not been explored.
Characterizing LZTR1-associated functional changes in
schwannomatosis will provide additional insights into the disease
and design of new therapeutic strategies, thus benefitting patients
carrying LZTR1 mutations.
LZTR1 has previously been considered as a
ubiquitin-ligase enzyme, rather than phosphokinase; however, it
also regulates the phosphorylation but not the ubiquitination of
RAF1 (56). LZTR1 could mediate the
phosphorylation of RAF1 by inducing ubiquitination of PPP1CB in
complexes, and LZTR1 itself may also phosphorylate substrates.
However, such phenomenon has not been confirmed. Phosphorylation
and ubiquitination are common post-translational protein
modifications that play crucial roles in the functions of
substrates. Thus, the molecular basis of determining whether LZTR1
is capable of phosphorylating substrates directly is worth
exploring in the future.
Additionally, studying alternative mechanisms of RAS
regulation will also assist with the development of new treatments
for RAS-driven diseases. Notably, although KRAS is the most
frequently mutated member of the RAS family and is closely related
to human malignant tumors, it has long been considered to be
untargetable (23,53,118–125).
LZTR1 is the most specific and potent regulator of KRAS (38). Thus, further research on LZTR1 may
help in the successful targeting of KRAS in the future.
In conclusion, LZTR1 might become an important focus
of biomedical research. Further studies are required for improved
understanding of the biological function of LZTR1 and its role in
the occurrence of diseases, as well as the development of disease
treatments, such as rational design of LZTR1 promoters for patients
harboring loss-of-function LZTR1 mutations.
We would like to thank Dr Yuqi Wang (West Lake
University, China) for the kind help and good advice provided.
This work was supported by The Natural Science
Foundation of Zhejiang Province (grant no. LY20C070001), The
National Natural Science Foundation of China (grant no. 31801165),
The Natural Science Foundation of Ningbo (grant no. 2018A610213),
The National Undergraduate Training Program for Innovation and
Entrepreneurship (grant no. 202011646030), The Program of Xinmiao
(Potential) Talents in Zhejiang Province (grant nos. 2019R405061,
2019R405011 and 2020R405039), The Student Research and Innovation
Program of Ningbo University (grant nos. 2020SRIP1923,
2020SRIP1919, 2020SRIP1902 and 2020SRIP1901) and The K.C. Wong
Magna Fund in Ningbo University.
Not applicable.
XJ conceived the present study. HZ and XC drafted
the manuscript. QL, JW and YZ made substantial contributions to the
interpretation, drafting the study and revising it critically for
important intellectual content. XJ, HZ and XC were major
contributors in the manuscript. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mani RS: The emerging role of speckle-type
POZ protein (SPOP) in cancer development. Drug Discov Today.
19:1498–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zou T and Zhang J: Diverse and pivotal
roles of neddylation in metabolism and immunity. FEBS J. Oct
6;s2020doi: 10.1111/febs.15584.
|
|
4
|
Petroski MD and Deshaies RJ: Function and
regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol.
6:9–20. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Asmar AJ, Beck DB and Werner A: Control of
craniofacial and brain development by Cullin3-RING ubiquitin
ligases: Lessons from human disease genetics. Exp Cell Res.
396:1123002020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Achim W, Regina B, Nia T, Kaya DU and
Michael R: Multisite dependency of an E3 ligase controls
monoubiquitylation-dependent cell fate decisions. Elife.
7:e354072018. View Article : Google Scholar
|
|
7
|
Senft D, Qi J and Ronai ZA: Ubiquitin
ligases in oncogenic transformation and cancer therapy. Nat Rev
Cancer. 18:69–88. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rape M: Ubiquitylation at the crossroads
of development and disease. Nat Rev Mol Cell Biol. 19:59–70. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng N, Schulman BA, Song L, Miller JJ,
Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al:
Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase
complex. Nature. 416:703–709. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Enchev RI, Schulman BA and Peter M:
Protein neddylation: Beyond cullin-RING ligases. Nat Rev Mol Cell
Biol. 16:30–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Z, Liu P, Inuzuka H and Wei W: Roles
of F-box proteins in cancer. Nat Rev Cancer. 14:233–247. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Teixeira LK and Reed SI: Ubiquitin ligases
and cell cycle control. Annu Rev Biochem. 82:387–414. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lipkowitz S and Weissman AM: RINGs of good
and evil: RING finger ubiquitin ligases at the crossroads of tumour
suppression and oncogenesis. Nat Rev Cancer. 11:629–643. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cornelius RJ, Ferdaus MZ, Nelson JW and
McCormick JA: Cullin-Ring ubiquitin ligases in kidney health and
disease. Curr Opin Nephrol Hypertens. 28:490–497. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen RH: Cullin 3 and its role in
tumorigenesis. Adv Exp Med Biol. 1217:187–210. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, McCormick JA and Sigmund CD:
Cullin-3: Renal and vascular mechanisms regulating blood pressure.
Curr Hypertens Rep. 22:612020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang P, Song J and Ye D: CRL3s: The
BTB-CUL3-RING E3 ubiquitin Ligases. Adv Exp Med Biol. 1217:211–223.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cornelius RJ, Yang CL and Ellison DH:
Hypertension-causing cullin 3 mutations disrupt COP9 signalosome
binding. Am J Physiol Renal Physiol. 318:F204–F208. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin X, Shi Q, Li Q, Zhou L, Wang J, Jiang
L, Zhao X, Feng K, Lin T, Lin Z, et al: CRL3-SPOP ubiquitin ligase
complex suppresses the growth of diffuse large B-cell lymphoma by
negatively regulating the MyD88/NF-κB signaling. Leukemia.
34:1305–1314. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet.
44:685–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Le Gallo M, O'Hara AJ, Rudd ML, Urick ME,
Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et
al NIH Intramural Sequencing Center (NISC) Comparative Sequencing
Program, : Exome sequencing of serous endometrial tumors identifies
recurrent somatic mutations in chromatin-remodeling and ubiquitin
ligase complex genes. Nat Genet. 44:1310–1315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin X, Wang J, Gao K, Zhang P, Yao L, Tang
Y, Tang L, Ma J, Xiao J, Zhang E, et al: Dysregulation of
INF2-mediated mitochondrial fission in SPOP-mutated prostate
cancer. PLoS Genet. 13:e10067482017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei X, Fried J, Li Y, Hu L, Gao M, Zhang S
and Xu B: Functional roles of Speckle-Type Poz (SPOP) protein in
genomic stability. J Cancer. 9:3257–3262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cuneo MJ and Mittag T: The ubiquitin
ligase adaptor SPOP in cancer. FEBS J. 286:3946–3958. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin X, Wang J, Li Q, Zhuang H, Yang J, Lin
Z, Lin T, Lv Z, Shen L, Yan C, et al: SPOP targets oncogenic
protein ZBTB3 for destruction to suppress endometrial cancer. Am J
Cancer Res. 9:2797–2812. 2019.PubMed/NCBI
|
|
26
|
Wang Z, Song Y, Ye M, Dai X, Zhu X and Wei
W: The diverse roles of SPOP in prostate cancer and kidney cancer.
Nat Rev Urol. 17:339–350. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song Y, Xu Y, Pan C, Yan L, Wang ZW and
Zhu X: The emerging role of SPOP protein in tumorigenesis and
cancer therapy. Mol Cancer. 19:22020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Clark A and Burleson M: SPOP and cancer: A
systematic review. Am J Cancer Res. 10:704–726. 2020.PubMed/NCBI
|
|
29
|
Maekawa M and Higashiyama S: The roles of
SPOP in DNA damage response and DNA replication. Int J Mol Sci.
21:72932020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Werner A, Iwasaki S, McGourty CA,
Medina-Ruiz S, Teerikorpi N, Fedrigo I, Ingolia NT and Rape M:
Cell-fate determination by ubiquitin-dependent regulation of
translation. Nature. 525:523–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li YR, Peng RR, Gao WY, Liu P, Chen LJ,
Zhang XL, Zhang NN, Wang Y, Du L, Zhu FY, et al: The ubiquitin
ligase KBTBD8 regulates PKM1 levels via Erk1/2 and Aurora A to
ensure oocyte quality. Aging (Albany NY). 11:1110–1128. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lührig S, Kolb S, Mellies N and Nolte J:
The novel BTB-kelch protein, KBTBD8, is located in the Golgi
apparatus and translocates to the spindle apparatus during mitosis.
Cell Div. 8:32013. View Article : Google Scholar
|
|
33
|
Jiang T, Sánchez-Rivera F, Soto-Feliciano
Y, Yang Q, Song CQ, Bhuatkar A, Haynes CM, Hemann MT and Xue W:
Targeting de novo purine synthesis pathway via ADSL depletion
impairs liver cancer growth by perturbing mitochondrial function.
Hepatology. Dec 17–2020.(Epub ahead of print). doi:
10.1002/hep.31685. View Article : Google Scholar
|
|
34
|
Madden S and Itzhaki L: Structural and
mechanistic insights into the Keap1-Nrf2 system as a route to drug
discovery. Biochim Biophys Acta Proteins Proteom. 1868:1404052020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dhamodharan U, Ponjayanthi B, Sireesh D,
Bhakkiyalakshmi E and Ramkumar KM: Association of single-nucleotide
polymorphisms of the KEAP1 gene with the risk of various human
diseases and its functional impact using in silico analysis.
Pharmacol Res. 137:205–218. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pintard L, Kurz T, Glaser S, Willis JH,
Peter M and Bowerman B: Neddylation and deneddylation of CUL-3 is
required to target MEI-1/Katanin for degradation at the
meiosis-to-mitosis transition in C. elegans. Curr Biol. 13:911–921.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gray WM, Hellmann H, Dharmasiri S and
Estelle M: Role of the Arabidopsis RING-H2 protein RBX1 in RUB
modification and SCF function. Plant Cell. 14:2137–2144. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bigenzahn JW, Collu GM, Kartnig F, Pieraks
M, Vladimer GI, Heinz LX, Sedlyarov V, Schischlik F, Fauster A and
Rebsamen M: LZTR1 is a regulator of RAS ubiquitination and
signaling. Science. 362:1171–1177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Zhang J, Zhang P, Zhao Z, Huang Q,
Yun D, Chen J, Chen H, Wang C and Lu D: LZTR1 inactivation promotes
MAPK/ERK pathway activation in glioblastoma by stabilizing
oncoprotein RIT1. bioRxiv. Mar 15–2020.(Epub ahead of print).
|
|
40
|
Nacak TG, Leptien K, Fellner D, Augustin
HG and Kroll J: The BTB-kelch protein LZTR-1 is a novel Golgi
protein that is degraded upon induction of apoptosis. J Biol Chem.
281:5065–5071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Adams J, Kelso R and Cooley L: The kelch
repeat superfamily of proteins: Propellers of cell function. Trends
Cell Biol. 10:17–24. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Z, Wasney GA, Picaud S,
Filippakopoulos P, Vedadi M, D'Angiolella V and Bullock AN:
Identification of a PGXPP degron motif in dishevelled and
structural basis for its binding to the E3 ligase KLHL12. Open
Biol. 10:2000412020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Heng LZ, Kennedy J, Smithson S,
Newbury-Ecob R and Churchill A: New macular findings in individuals
with biallelic KLHL7 gene mutation. BMJ Open Ophthalmol.
4:e0002342019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Narahara S, Sakai E, Kadowaki T, Yamaguchi
Y, Narahara H, Okamoto K, Asahina I and Tsukuba T: KBTBD11, a novel
BTB-Kelch protein, is a negative regulator of osteoclastogenesis
through controlling Cullin3-mediated ubiquitination of NFATc1. Sci
Rep. 9:35232019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gao C, Pallett MA, Croll TI, Smith GL and
Graham SC: Molecular basis of cullin-3 (Cul3) ubiquitin ligase
subversion by vaccinia virus protein A55. J Biol Chem.
294:6416–6429. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nakaguma M, Jorge AA and Arnhold IJ:
Noonan syndrome associated with growth hormone deficiency with
biallelic LZTR1 variants. Genet Med:. 21:2602019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jacquinet A, Bonnard A, Capri Y, Martin D,
Sadzot B, Bianchi E, Servais L, Sacré JP, Cavé H and Verloes A:
Oligo-astrocytoma in LZTR1-related Noonan syndrome. Eur J Med
Genet. 63:1036172020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Deiller C, Van-Gils J, Zordan C, Tinat J,
Loiseau H, Fabre T, Delleci C, Cohen J, Vidaud M, Parfait B, et al:
Coexistence of schwannomatosis and glioblastoma in two families.
Eur J Med Genet. 62:1036802019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Merker VL, Esparza S, Smith MJ,
Stemmer-Rachamimov A and Plotkin SR: Clinical features of
schwannomatosis: A retrospective analysis of 87 patients.
Oncologist. 17:1317–1322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kehrer-Sawatzki H, Farschtschi S, Mautner
VF and Cooper DN: The molecular pathogenesis of schwannomatosis, a
paradigm for the co-involvement of multiple tumour suppressor genes
in tumorigenesis. Hum Genet. 136:129–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mansouri S, Suppiah S, Mamatjan Y,
Paganini I, Liu JC, Karimi S, Patil V, Nassiri F, Singh O,
Sundaravadanam Y, et al: Epigenomic, genomic, and transcriptomic
landscape of schwannomatosis. Acta Neuropathol. 141:101–116. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Simanshu DK, Nissley DV and McCormick F:
RAS proteins and their regulators in human disease. Cell.
170:17–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schubbert S, Zenker M, Rowe SL, Böll S,
Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner
LE, et al: Germline KRAS mutations cause Noonan syndrome. Nat
Genet. 38:331–336. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aoki Y, Niihori T, Banjo T, Okamoto N,
Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M, Ando T, et al:
Gain-of-function mutations in RIT1 cause Noonan syndrome, a
RAS/MAPK pathway syndrome. Am J Hum Genet. 93:173–180. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Castel P, Cheng A, Cuevas-Navarro A,
Everman DB, Papageorge AG, Simanshu DK, Tankka A, Galeas J, Urisman
A and McCormick F: RIT1 oncoproteins escape LZTR1-mediated
proteolysis. Science. 363:1226–1230. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Umeki I, Niihori T, Abe T, Kanno SI,
Okamoto N, Mizuno S, Kurosawa K, Nagasaki K, Yoshida M, Ohashi H,
et al: Delineation of LZTR1 mutation-positive patients with Noonan
syndrome and identification of LZTR1 binding to RAF1-PPP1CB
complexes. Hum Genet. 138:21–35. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Abe T, Umeki I, Kanno SI, Inoue SI,
Niihori T and Aoki Y: LZTR1 facilitates polyubiquitination and
degradation of RAS-GTPases. Cell Death Differ. 27:1023–1035. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Steklov M, Pandolf S, Baietti MF, Batiuk
A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, et al:
Mutations in LZTR1 drive human disease by dysregulating RAS
ubiquitination. Science. 362:1177–1182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zinatizadeh MR, Momeni SA, Zarandi PK,
Chalbatani GM, Dana H, Mirzaei HR, Akbari ME and Miri SR: The role
and function of Ras-association domain family in cancer: A Review.
Genes Dis. 6:378–384. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tidyman WE and Rauen KA: Pathogenetics of
the RASopathies. Hum Mol Genet. 25:R123–R132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Malaquias AC and Jorge AAL: Activation of
the MAPK pathway (RASopathies) and partial growth hormone
insensitivity. Mol Cell Endocrinol. 519:1110402021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Humphries B, Wang Z and Yang C: Rho
GTPases: Big Players in Breast Cancer Initiation, Metastasis and
Therapeutic Responses. Cells. 9:21672020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lavoie H, Gagnon J and Therrien M: ERK
signalling: A master regulator of cell behaviour, life and fate.
Nat Rev Mol Cell Biol. 21:607–632. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Moore AR, Rosenberg SC, McCormick F and
Malek S: RAS-targeted therapies: Is the undruggable drugged? Nat
Rev Drug Discov. 19:533–552. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cirstea IC, Kutsche K, Dvorsky R, Gremer
L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, König
R, et al: A restricted spectrum of NRAS mutations causes Noonan
syndrome. Nat Genet. 42:27–29. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
66
|
Higgins EM, Bos JM, Mason-Suares H, Tester
DJ, Ackerman JP, MacRae CA, Sol-Church K, Gripp KW, Urrutia R and
Ackerman MJ: Elucidation of MRAS-mediated Noonan syndrome with
cardiac hypertrophy. JCI Insight. 2:e912252017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Flex E, Jaiswal M, Pantaleoni F,
Martinelli S, Strullu M, Fansa EK, Caye A, De Luca A, Lepri F,
Dvorsky R, et al: Activating mutations in RRAS underlie a phenotype
within the RASopathy spectrum and contribute to leukaemogenesis.
Hum Mol Genet. 23:4315–4327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ratner N and Miller SJ: A RASopathy gene
commonly mutated in cancer: The neurofibromatosis type 1 tumour
suppressor. Nat Rev Cancer. 15:290–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dunnett-Kane V, Burkitt-Wright E,
Blackhall FH, Malliri A, Evans DG and Lindsay CR: Germline and
sporadic cancers driven by the RAS pathway: Parallels and
contrasts. Ann Oncol. 31:873–883. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Johnston JJ, van der Smagt JJ, Rosenfeld
JA, Pagnamenta AT, Alswaid A, Baker EH, Blair E, Borck G, Brinkmann
J, Craigen W, et al Members of the Undiagnosed Diseases Network, :
Autosomal recessive Noonan syndrome associated with biallelic LZTR1
variants. Genet Med. 20:1175–1185. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Aoki Y, Niihori T, Inoue S and Matsubara
Y: Recent advances in RASopathies. J Hum Genet. 61:33–39. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Motta M, Fidan M, Bellacchio E, Pantaleoni
F, Schneider-Heieck K, Coppola S, Borck G, Salviati L, Zenker M,
Cirstea IC, et al: Dominant Noonan syndrome-causing LZTR1 mutations
specifically affect the Kelch domain substrate-recognition surface
and enhance RAS-MAPK signaling. Hum Mol Genet. 28:1007–1022. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pagnamenta AT, Kaisaki PJ, Bennett F,
Burkitt-Wright E, Martin HC, Ferla MP, Taylor JM, Gompertz L,
Lahiri N, Tatton-Brown K, et al DDD Study, : Delineation of
dominant and recessive forms of LZTR1-associated Noonan syndrome.
Clin Genet. 95:693–703. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Rodriguez-Viciana P, Oses-Prieto J,
Burlingame A, Fried M and McCormick F: A phosphatase holoenzyme
comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions
as an M-Ras effector to modulate Raf activity. Mol Cell.
22:217–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Young LC, Hartig N, Muñoz-Alegre M,
Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M,
Gewinner C, Henderson S, et al: An MRAS, SHOC2, and SCRIB complex
coordinates ERK pathway activation with polarity and tumorigenic
growth. Mol Cell. 52:679–692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Shi GX, Cai W and Andres DA: Rit subfamily
small GTPases: Regulators in neuronal differentiation and survival.
Cell Signal. 25:2060–2068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Khalil A and Nemer G: The potential
oncogenic role of the RAS-like GTP-binding gene RIT1 in
glioblastoma. Cancer Biomark. 29:509–519. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Van R, Cuevas-Navarro A, Castel P and
McCormick F: The molecular functions of RIT1 and its contribution
to human disease. Biochem J. 477:2755–2770. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Song Z, Liu T, Chen J, Ge C, Zhao F, Zhu
M, Chen T, Cui Y, Tian H, Yao M, et al: HIF-1α-induced RIT1
promotes liver cancer growth and metastasis and its deficiency
increases sensitivity to sorafenib. Cancer Lett. 460:96–107. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Venugopal V and Romero CJ: Endocrine
complications of Noonan syndrome beyond short stature. Pediatr
Endocrinol Rev. 16 (Suppl 2):465–470. 2019.PubMed/NCBI
|
|
81
|
Soucy TA, Smith PG, Milhollen MA, Berger
AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP,
Critchley S, et al: An inhibitor of NEDD8-activating enzyme as a
new approach to treat cancer. Nature. 458:732–736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jin J, Ang XL, Shirogane T and Wade Harper
J: Identification of substrates for F-box proteins. Methods
Enzymol. 399:287–309. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li S, Balmain A and Counter CM: A model
for RAS mutation patterns in cancers: Finding the sweet spot. Nat
Rev Cancer. 18:767–777. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Pierpont ME, Brueckner M, Chung WK, Garg
V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et
al American Heart Association Council on Cardiovascular Disease in
the Young; Council on Cardiovascular and Stroke Nursing; and
Council on Genomic and Precision Medicine, : Genetic Basis for
Congenital Heart Disease: Revisited: A scientific statement from
the American Heart Association. Circulation. 138:e653–e711. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tajan M, Paccoud R, Branka S, Edouard T
and Yart A: The RASopathy family: Consequences of germline
activation of the RAS/MAPK pathway. Endocr Rev. 39:676–700. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kamihara J, Bourdeaut F, Foulkes WD,
Molenaar JJ, Mossé YP, Nakagawara A, Parareda A, Scollon SR,
Schneider KW, Skalet AH, et al: Retinoblastoma and neuroblastoma
predisposition and surveillance. Clin Cancer Res. 23:e98–e106.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: Primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14 (Suppl 5):v1–v49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Dunn GP, Rinne ML, Wykosky J, Genovese G,
Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et
al: Emerging insights into the molecular and cellular basis of
glioblastoma. Genes Dev. 26:756–784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lee E, Yong RL, Paddison P and Zhu J:
Comparison of glioblastoma (GBM) molecular classification methods.
Semin Cancer Biol. 53:201–211. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liang Y, Diehn M, Watson N, Bollen AW,
Aldape KD, Nicholas MK, Lamborn KR, Berger MS, Botstein D, Brown
PO, et al: Gene expression profiling reveals molecularly and
clinically distinct subtypes of glioblastoma multiforme. Proc Natl
Acad Sci USA. 102:5814–5819. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mischel PS, Nelson SF and Cloughesy TF:
Molecular analysis of glioblastoma: Pathway profiling and its
implications for patient therapy. Cancer Biol Ther. 2:242–247.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Diehn M, Nardini C, Wang DS, McGovern S,
Jayaraman M, Liang Y, Aldape K, Cha S and Kuo MD: Identification of
noninvasive imaging surrogates for brain tumor gene-expression
modules. Proc Natl Acad Sci USA. 105:5213–5218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Phillips HS, Kharbanda S, Chen R, Forrest
WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et
al: Molecular subclasses of high-grade glioma predict prognosis,
delineate a pattern of disease progression, and resemble stages in
neurogenesis. Cancer Cell. 9:157–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Frattini V, Trifonov V, Chan JM, Castano
A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al: The
integrated landscape of driver genomic alterations in glioblastoma.
Nat Genet. 45:1141–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lein PJ, Guo X, Shi GX, Moholt-Siebert M,
Bruun D and Andres DA: The novel GTPase Rit differentially
regulates axonal and dendritic growth. J Neurosci. 27:4725–4736.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Cai W, Rudolph JL, Harrison SM, Jin L,
Frantz AL, Harrison DA and Andres DA: An evolutionarily conserved
Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress
resistance. Mol Biol Cell. 22:3231–3241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Shi GX and Andres DA: Rit contributes to
nerve growth factor-induced neuronal differentiation via activation
of B-Raf-extracellular signal-regulated kinase and p38
mitogen-activated protein kinase cascades. Mol Cell Biol.
25:830–846. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shi GX, Han J and Andres DA: Rin GTPase
couples nerve growth factor signaling to p38 and b-Raf/ERK pathways
to promote neuronal differentiation. J Biol Chem. 280:37599–37609.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Rusyn EV, Reynolds ER, Shao H, Grana TM,
Chan TO, Andres DA and Cox AD: Rit, a non-lipid-modified
Ras-related protein, transforms NIH3T3 cells without activating the
ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene. 19:4685–4694.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ren R: Mechanisms of BCR-ABL in the
pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer.
5:172–183. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Garcia-Horton A and Lipton JH: Treatment
outcomes in chronic myeloid leukemia: Does one size fit all? J Natl
Compr Canc Netw. 18:1421–1428. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Crisà E, Nicolosi M, Ferri V, Favini C,
Gaidano G and Patriarca A: Atypical chronic myeloid leukemia: Where
are we now? Int J Mol Sci. 21:68622020. View Article : Google Scholar
|
|
104
|
Braun TP, Eide CA and Druker BJ: Response
and resistance to BCR-ABL1-targeted therapies. Cancer Cell.
37:530–542. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Vetrie D, Helgason GV and Copland M: The
leukaemia stem cell: Similarities, differences and clinical
prospects in CML and AML. Nat Rev Cancer. 20:158–173. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Evans DG, Bowers NL, Tobi S, Hartley C,
Wallace AJ, King AT, Lloyd SK, Rutherford SA, Hammerbeck-Ward C,
Pathmanaban ON, et al: Schwannomatosis: A genetic and
epidemiological study. J Neurol Neurosurg Psychiatry. 89:1215–1219.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kehrer-Sawatzki H, Farschtschi S, Mautner
VF and Cooper DN: The molecular pathogenesis of schwannomatosis, a
paradigm for the co-involvement of multiple tumour suppressor genes
in tumorigenesis. Hum Genet. 136:129–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Smith MJ, Isidor B, Beetz C, Williams SG,
Bhaskar SS, Richer W, O'Sullivan J, Anderson B, Daly SB, Urquhart
JE, et al: Mutations in LZTR1 add to the complex heterogeneity of
schwannomatosis. Neurology. 84:141–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yamamoto GL, Aguena M, Gos M, Hung C,
Pilch J, Fahiminiya S, Abramowicz A, Cristian I, Buscarilli M,
Naslavsky MS, et al: Rare variants in SOS2 and LZTR1 are associated
with Noonan syndrome. J Med Genet. 52:413–421. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lamlum H, Ilyas M, Rowan A, Clark S,
Johnson V, Bell J, Frayling I, Efstathiou J, Pack K, Payne S, et
al: The type of somatic mutation at APC in familial adenomatous
polyposis is determined by the site of the germline mutation: A new
facet to Knudson's ‘two-hit’ hypothesis. Nat Med. 5:1071–1075.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hulsebos TJ, Plomp AS, Wolterman RA,
Robanus-Maandag EC, Baas F and Wesseling P: Germline mutation of
INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet.
80:805–810. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
112
|
Paganini I, Chang VY, Capone GL, Vitte J,
Benelli M, Barbetti L, Sestini R, Trevisson E, Hulsebos TJ,
Giovannini M, et al: Expanding the mutational spectrum of LZTR1 in
schwannomatosis. Eur J Hum Genet. 23:963–968. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Smith MJ, Pathmanaban ON, Coope DJ, King
AT and Evans DG: Comment on: SMARCB1 gene mutation predisposes to
earlier development of glioblastoma: A case report of familial GBM.
J Neuropathol Exp Neurol. 80:289–290. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Fonkem E, Peng S, Berens M and Mukherjee
S: Authors' reply: SMARCB1 gene mutation predisposes to earlier
development of glioblastoma: A case report of familial GBM. J
Neuropathol Exp Neurol. 80:290–291. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Louvrier C, Pasmant E, Briand-Suleau A,
Cohen J, Nitschké P, Nectoux J, Orhant L, Zordan C, Goizet C,
Goutagny S, et al: Targeted next-generation sequencing for
differential diagnosis of neurofibromatosis type 2,
schwannomatosis, and meningiomatosis. Neuro Oncol. 20:917–929.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Maurer GW, Malita A, Nagy S, Koyama T,
Werge TM, Halberg KA, Texada MJ and Rewitz K: Analysis of genes
within the schizophrenia-linked 22q11.2 deletion identifies
interaction of night owl/LZTR1 and NF1 in GABAergic sleep control.
PLoS Genet. 16:e10087272020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ballester R, Marchuk D, Boguski M, Saulino
A, Letcher R, Wigler M and Collins F: The NF1 locus encodes a
protein functionally related to mammalian GAP and yeast IRA
proteins. Cell. 63:851–859. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu P, Wang Y and Li X: Targeting the
untargetable KRAS in cancer therapy. Acta Pharm Sin B. 9:871–879.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Buscail L, Bournet B and Cordelier P: Role
of oncogenic KRAS in the diagnosis, prognosis and treatment of
pancreatic cancer. Nat Rev Gastroenterol Hepatol. 17:153–168. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Krastev DB and Buchholz F: Ribosome
biogenesis and p53: Who is regulating whom? Cell Cycle.
10:3417–3418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Weiss RA: A perspective on the early days
of RAS research. Cancer Metastasis Rev. 39:1023–1028. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Uprety D and Adjei AA: KRAS: From
undruggable to a druggable cancer target. Cancer Treat Rev.
89:1020702020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Chen H and Zhao J: KRAS oncogene may be
another target conquered in non-small cell lung cancer (NSCLC).
Thorac Cancer. 11:3425–3435. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Goulding RE, Chenoweth M, Carter GC, Boye
ME, Sheffield KM, John WJ, Leusch MS, Muehlenbein CE, Li L, Jen MH,
et al: KRAS mutation as a prognostic factor and predictive factor
in advanced/metastatic non-small cell lung cancer: A systematic
literature review and meta-analysis. Cancer Treat Res Commun.
24:1002002020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Passiglia F, Malapelle U, Del Re M, Righi
L, Pagni F, Furlan D, Danesi R, Troncone G and Novello S: KRAS
inhibition in non-small cell lung cancer: Past failures, new
findings and upcoming challenges. Eur J Cancer. 137:57–68. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Matthew B, Juliati R and Field SJ: GOLPH3
links the Golgi, DNA damage, and cancer. Cancer Res. 75:624–627.
2015. View Article : Google Scholar : PubMed/NCBI
|