Introduction
Breast cancer is the most common malignancy in women
and the most common cause of cancer-associated death among women
worldwide (1). Advances in early
detection and cancer therapy have led to a reduction in the
incidence of breast cancer, and breast cancer-associated deaths
have decreased by ~2% in the past decade (2). However, the prognosis of patients
varies greatly and is affected by numerous factors, including tumor
type, stage, treatment and geographical location; for example,
prognosis is better among patients in Western countries than among
those in developing countries (3).
Furthermore, a higher stage at diagnosis is associated with a
poorer prognosis; stage 0 ductal carcinoma in situ has an
excellent prognosis with a 10-year survival rate of ~98%, while
stage IV metastatic cancer has a poor prognosis with a 10-year
survival rate of <10% (4). This
discrepancy poses a substantial clinical challenge. Thus, it is
necessary to identify novel molecular targets and drugs that will
strengthen early intervention and effective therapeutic
strategies.
Several studies have indicated that patients with
type 2 diabetes mellitus (DM) have an increased risk for the
development of several types of cancer, including breast cancer
(5–11). Metformin is a first-line drug for
type 2 DM, and numerous studies have demonstrated that it is
associated with a lower risk of breast cancer in patients with type
2 DM (12–15). Metformin inhibits the proliferation
and colony formation of cancer cells by inducing cell cycle arrest
and apoptosis by modulating the expression levels of proteins that
regulate the G1-S cell cycle transition, including
cyclin D1, cyclin E1 and E2F transcription factor 1 (12,16–18). The
AMP kinase (AMPK) regulatory system is one of the main targets of
metformin therapy (19). The
activation of AMPK regulates tumor cell survival and tumor growth
through inhibition of the mTOR and fatty acid synthesis signaling
pathways, and it also stimulates the apoptotic pathway (p53/p21
axis) (14,20,21).
However, the precise molecular mechanisms of metformin in breast
cancer remain to be fully elucidated.
Cyclooxygenase-2 (COX-2) is expressed in numerous
types of solid tumor tissues and cells, and serves a key role in
the development of breast cancer (22,23).
COX-2 expression is upregulated in ~50% of breast cancer cases, and
high COX-2 expression is significantly associated with a poor
clinical outcome (24–27). COX-2 is associated with increased
proliferation and angiogenesis in human breast cancer (28,29).
Furthermore, COX-2 increases the production of prostaglandin E2
(PGE2), which stimulates breast cancer progression and bone
metastasis (30–32). Therefore, COX-2 inhibitors may have a
role in the prevention and treatment of breast cancer, and may have
value as novel biomarkers to stratify breast cancer risk in women
with atypia (33). Non-steroidal
anti-inflammatory drugs (NSAIDs), particularly the highly selective
COX-2 inhibitors, have been shown experimentally to stimulate
apoptosis and inhibit angiogenesis, two mechanisms that can
counteract tumor growth, progression and metastasis (34). In addition to NSAIDs, administration
of metformin can prevent the increase in COX-2 expression induced
by dehydroepiandrosterone and enhance the activation of
phosphorylated (p)-AMPKa expression in embryonic ovarian disorders
(35,36). While the aforementioned studies have
confirmed an association of high COX-2 expression with highly
aggressive tumors and have emphasized the anticancer action of
metformin in breast cancer, the underlying mechanisms remain
unclear.
The present study provided preliminary evidence that
metformin inhibited breast cancer cell proliferation via the well
known AMPK/mTOR signaling pathway and the novel AMPK/COX-2
signaling pathway.
Materials and methods
Clinical samples
Between January 2016 and December 2018, 63 patients
with invasive breast cancer that underwent curative surgical
resection at the Changhai Hospital (Shanghai, China) and Kunshan
Hospital (Jiangsu, China) were enrolled in the present study. Tumor
and adjacent normal tissues from patients with breast cancer were
obtained and the expression levels of AMPK/COX-2/mTOR signaling
proteins were analyzed. Non-tumor specimens were ≥1.5 cm from the
tumor margins. All 63 patients were female and aged between 38 and
78 years (mean age ± SD, 50.5±6.7 years). A total of 29 (46.0%) and
34 (54.0%) patients were aged <50 years and >50 years,
respectively. The tumors were staged following the
tumor-node-metastasis (TNM) staging system of the International
Union Against Cancer (37). A total
of 53 (84.1%) tumors were categorized as stage I–II and 10 (15.9%)
tumors as stages III–IV. None of the patients received preoperative
chemotherapy and/or radiation therapy. Freshly resected tissues
from patients with breast cancer were immediately snap-frozen in
liquid nitrogen. All the clinical specimens were obtained with
written informed consent, and the Institutional Review Boards of
the Affiliated Kunshan First People's Hospital of Jiangsu
University and Changhai Hospital approved the use of all tissues
and clinical information (approval no. CHEC2014-098).
Reagents and antibodies
Metformin (cat. no. 1115-70-4) was purchased from
Sangon Biotech Co., Ltd. Antibodies against p-AMPKα (Thr-172 or
D4D6D; cat. no. 50081), AMPKα (D5A2; cat. no. 5831), p-eukaryotic
translation initiation factor 4E-binding protein 1 (p-4E-BP1;
Thr-37/46; cat. no. 2855), 4E-BP1 (53H11; cat. no. 9644), COX-2
(D5H5; cat. no. 4842) and Cyclin D1 (E3P5S; cat. no. 55506) were
purchased from Cell Signaling Technology, Inc. Anti-proliferating
cell nuclear antigen (PCNA) antibody (EPR3821; cat. no. ab92552)
and anti-PGE2 antibody (cat. no. ab45295) were obtained from Abcam.
Anti-β-actin (C4; cat. no. sc-47778) was obtained from Santa Cruz
Biotechnology, Inc.
Cell culture
The breast cancer MCF-7 and 4T1 cell lines were
purchased from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. Both cell lines were grown in DMEM
(Invitrogen; Thermo Fisher Scientific, Inc.) with 10% FBS
(Invitrogen; Thermo Fisher Scientific, Inc.). The cell lines were
kept at 37°C in a humidified incubator with 5% CO2.
MTT assay
The MTT assay was used to assess the
anti-proliferative effects of metformin in MCF-7 and 4T1 cells. In
recent years of breast cancer research, the dosage range of
metformin has been 1–50 mM, and the highest dosage can reach 100 mM
(38,39). It has been reported that metformin
can induce the AMPK signaling pathway in the dose range of 10–20
mM, thereby increasing apoptosis (40). Therefore, metformin with the dose
range of 20–50 mM was selected to study its antitumor mechanism.
Exponentially growing cells were trypsinized, counted and plated at
5×103 cells/well in 96-well plates. After 24 h, cells
were treated with 0, 20 or 50 mM metformin for 24, 48, 96 or 144 h.
Thereafter, the medium was changed, and the cells were incubated
with 10 µl MTT (Sigma-Aldrich; Merck KGaA; 5 mg/ml) at 37°C for 4 h
before each test. The supernatant was then carefully discarded, and
150 µl DMSO (Sigma-Aldrich; Merck KGaA; cat. no. D2650) was added
to dissolve the precipitate. Absorbance at 490 nm (A570) for the
experimental group and DMSO (control) was measured using a
microplate reader (KHB ST-360; Shanghai Kehua Bio-Engineering Co.,
Ltd.). Finally, actual absorbance was calculated using the
following formula: Actual absorbance = absorbance of treated
cultures-absorbance of DMSO. To ensure consistency, the experiment
was repeated three times in all cases.
Colony formation assay
For each treatment, cells were plated at
1×103 cells/well in 6-well plates in triplicate. After
the cells were attached, the cells were treated with 0, 20 or 50 mM
metformin for 14 days and then fixed in −20°C precooled
methanol/acetone (1:1) solution for 10 min. After fixation, the
cells were stained using crystal violet (Sigma-Aldrich; Merck KGaA;
cat. no. C0775) for 1 min at room temperature. The colonies with
≥50 cells were manually counted under an inverted light microscope
(magnification, ×400; CX31; Olympus Corporation). The number of
colonies for breast cancer cells were calculated as:
Colonies/500×100.
Cell cycle analysis
The cells were seeded in 6-well plates
(2×105 cells/well) overnight and then exposed to 0, 20
and 50 mM metformin for another 48 h. Exponentially growing cells
were trypsinized and precipitated overnight with 70% ethanol at
4°C. Next, the cells were washed, resuspended in fresh PBS
containing propidium iodide with 0.1% Triton X-100 (Sigma-Aldrich;
Merck KGaA) and RNase A (Beyotime Institute of Biotechnology), and
then the reaction was further incubated for 30 min in the dark at
37°C. Cell cycle distribution was determined using the BD FACSAria™
III Cell Sorting system (Becton, Dickinson and Company) and
analyzed using the ModFit LT software (version 5.0; Becton,
Dickinson and Company).
Xenografts
Subcutaneous xenograft mouse models were used to
estimate the antitumor efficacy of metformin. A total of 10 female
athymic BALB/c nude mice (4-week-old; average weight, 20 g) were
kept under pathogen-free conditions. All animals were housed in
metabolic cages (dimensions, 500×360×200 mm), with 2 or 3 mice per
cage. During the experimental procedure, nude mice were kept at a
temperature of 23±2°C, humidity of 50±10% and light-controlled
environment (12/12-h light/dark cycle). The mice were free to drink
and eat. The litter was changed every other day. After completing
the experiment, the mice were sacrificed by cervical dislocation
after anesthesia with an intraperitoneal injection of 0.6% sodium
pentobarbital (40 mg/kg). Death was carefully verified by
monitoring cessation of corneal reflex, heartbeat and breathing.
4T1 cells (1×106 cells/mouse) in PBS were injected
subcutaneously into the right upper flank of the mice. When the
mean tumor diameter was ~4 mm, the animals were randomly assigned
to two groups (control group and metformin treatment group, n=5 in
each group). Metformin (250 mg/kg) was injected intraperitoneally
once daily. The control group was treated similarly but with
sterile saline solution instead of metformin (0.9% NaCl in
ultrapure water). Tumor long/short diameter was measured every 2–3
days. All animals were sacrificed after the end of the experiment
at day 14, and the tumors were harvested and weighed. The length of
tumor was measured by caliper, and tumor volume (mm3)
was estimated by the formula V = 0.52 × length × width × depth.
Humane endpoint criteria were defined as follows: i) body weight
loss persisted beyond 20% of predose weight; ii) tumor size
exceeding 3,000 mm3; and iii) anorexia or loss of
mobility. All animal experimental protocols were reviewed and
approved by the Animal Ethics Committee of the Second Military
Medical University (Shanghai, China).
Western blot analysis
Standard western blotting was performed as
previously described (41). In
short, whole-cell lysates were prepared from 4T1 and MCF7 cells at
0, 24, 48 and 72 h after the addition of metformin (0, 20 and 50
mM). Breast cancer cells were lysed using RIPA lysis buffer (Sangon
Biotech Co., Ltd.). Total protein concentration was quantified
using the BCA method. The protein samples (30 µg/lane) were
electrophoresed via 10% SDS-PAGE and then transferred to a
polyvinylidene fluoride membrane (Bio-Rad Laboratories, Inc.).
After being blocked with 1% BSA (Sigma-Aldrich; Merck KGaA) for at
least 2 h at room temperature, the membranes were incubated
overnight with the aforementioned primary antibodies for 10–12 h at
4°C (p-AMPKα, AMPKα, β-actin, cyclin D1, p-4E-BP1, 4E-BP1, PGE2 and
COX-2; all 1:1,000) and then incubated for 2 h at room temperature
with HRP-conjugated anti-rabbit (1:1,000; cat. no. ab205718; Abcam)
and anti-mouse (1:1,000; cat. no. ab205719; Abcam) secondary
antibodies. Finally, the membranes were washed three times and
immunoreactive proteins were detected using an enhanced
chemiluminescence kit (Amersham; Cytiva) and analyzed using
Quantity One software version 4.6 (Bio-Rad Laboratories, Inc.).
Hematoxylin and eosin (H&E)
staining and immunohistochemistry (IHC)
Tumor and adjacent normal tissues (confirmed as
normal by the pathology department) were fixed in 10% formalin for
24 h at room temperature, embedded in paraffin and cut into
4-µm-thick sections for IHC or H&E. IHC and H&E staining
were performed using the Histostain™ kit (Thermo Fisher Scientific,
Inc.) following the manufacturer's protocol. Briefly, the tissue
slides were deparaffinized in xylene (three washes for 5 min each)
and graded ethanol dilution series (100, 95, 70 and 50% ethanol,
each washed twice for 10 min each) and washed in deionized water
(two washes for 5 min each). The slides were then fixed in 10%
paraformaldehyde for 10 min at room temperature and subsequently
rinsed in PBS. Fixed samples were incubated with 3%
H2O2 solution at room temperature for 10 min
followed by washing with PBS three times. Antigen retrieval was
achieved by boiling slides in 0.01 M citrate buffer, pH 6.0, for 10
min, cooling to room temperature and rinsing three times with PBS.
Tissue sections were incubated with serum blocking solution
provided in the aforementioned kit for 10 min at room temperature,
followed by incubation with primary antibody against p-AMPKα
(1:100), COX-2 (1:100), cyclin D1 (1:100), PCNA (1:100) and
p-4E-BP1 (1:200) at room temperature for 1 h. After washing off the
primary antibody in PBS, sections were incubated with biotinylated
broad-spectrum secondary antibody (Histostain®-Plus 3rd
Gen IHC Detection kit; Invitrogen; Thermo Fisher Scientific, Inc.;
cat. no. 85-903) at room temperature for 10 min. At the end of
incubation, the sections were washed again in PBS, and then
incubated for a further 10 min at room temperature in the presence
of streptavidin-enzyme conjugate. After washing with PBS, sections
were incubated with substrate-chromogen mixture at room temperature
for 5 min and washed with PBS again. Hematoxylin was used to stain
slices for 1 min at room temperature, followed by a rinse with tap
water. Sections were finally mounted and dried until observation.
Tissue samples were evaluated and blindly scored by two independent
investigators using a light microscope (Olympus Corporation). The
criteria for scoring were as follows: 0, absence of positive
staining; 1, weak staining intensity; 2, moderate staining
intensity; and 3, strong staining intensity. The percentage of
positively stained cells was obtained in at least five different
visual fields at ×400 magnification for each section, and assigned
a value between 0 and 100%. The final score of immunoreactivity was
obtained by multiplying the staining intensity score by the
percentage of positive tumor cells in each case. The scores ranged
from 0 (0% of cells stained) to 3 (100% strong staining).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from cells was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). First, first-strand cDNA was reverse transcribed from 1 µg
total RNA using M-MLV reverse transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 10 min according to the
manufacturer's instructions of the PrimeScript RT reagent kit
(Takara Bio, Inc.). qPCR was performed on the 7500HT Fast Real-Time
PCR machine (Applied Biosystems; Thermo Fisher Scientific, Inc.)
using SYBR-Green Master mix (Takara Bio, Inc.). The thermocycling
conditions were as follows: Enzyme activation at 95°C for 30 sec;
30 cycles of denaturation and annealing at 95°C for 5 sec and 55°C
for 20 sec, respectively; hold at 4°C. Relative gene expression was
determined using the SYBR Green Master Mix (Takara Bio, Inc.).
GAPDH was used for internal expression normalization.
Relative expression levels were normalized to endogenous controls
and were expressed as 2−ΔΔCq (42). Each sample was analyzed in
triplicates for the target genes and internal control gene. The
primers for qPCR were as follows: COX-2 forward,
5′-ATCATTCACCAGGCAAATTGC-3′ and reverse,
5′-GGCTTCAGCATAAAGCGTTTG-3′; and GAPDH forward,
5′-TGTTGCCATCAATGACCCCTT-3′; and reverse,
5′-CTCCACGACGTACTCAGCG-3′.
Luciferase reporter assay
Breast cancer cells were seeded in 24-well plates
and grown to 70–80% confluence. The cells were transfected with the
human COX-2 promoter fragments (a generous gift from Professor Qi
Li, Shuguang Hospital, Shanghai, China) using Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C with 5%
CO2 for 6 h, which were inserted into the pGL3 plasmid
(Promega Corporation; cat. no. E1761) and monitor plasmid pRL-TK
(Promega Corporation; cat. no. E2241). These cells were treated
with 0, 20 or 50 mM metformin. After 48 h of treatment, cells were
collected, washed and harvested to measure the firefly and
Renilla luciferase activities using the Dual-Glo™ Luciferase
reporter assay system (Promega Corporation; cat. no. E2920)
according to the manufacturer's instructions. Luciferase activity
was normalized to that of the co-transfected pRL-TK plasmid. All
experiments were performed at least twice in triplicates.
Statistical analysis
All data were analyzed using either unpaired or
paired Student's t-test, Pearson's correlation, one-way ANOVA with
Tukey's post-hoc test or χ2 test. Data analysis were
applied using SPSS version 22.0 (IBM Corp.) and GraphPad Prism
version 6.0 (GraphPad Software, Inc.). For all tests, a two-sided
P<0.05 was considered to indicate a statistically significant
difference.
Results
Metformin inhibits proliferation and
colony formation in breast cancer cells
To investigate the effects of metformin on cell
proliferation, MCF7 and 4T1 cells were treated with various
concentrations (0, 20 and 50 mM) of metformin and cell viability
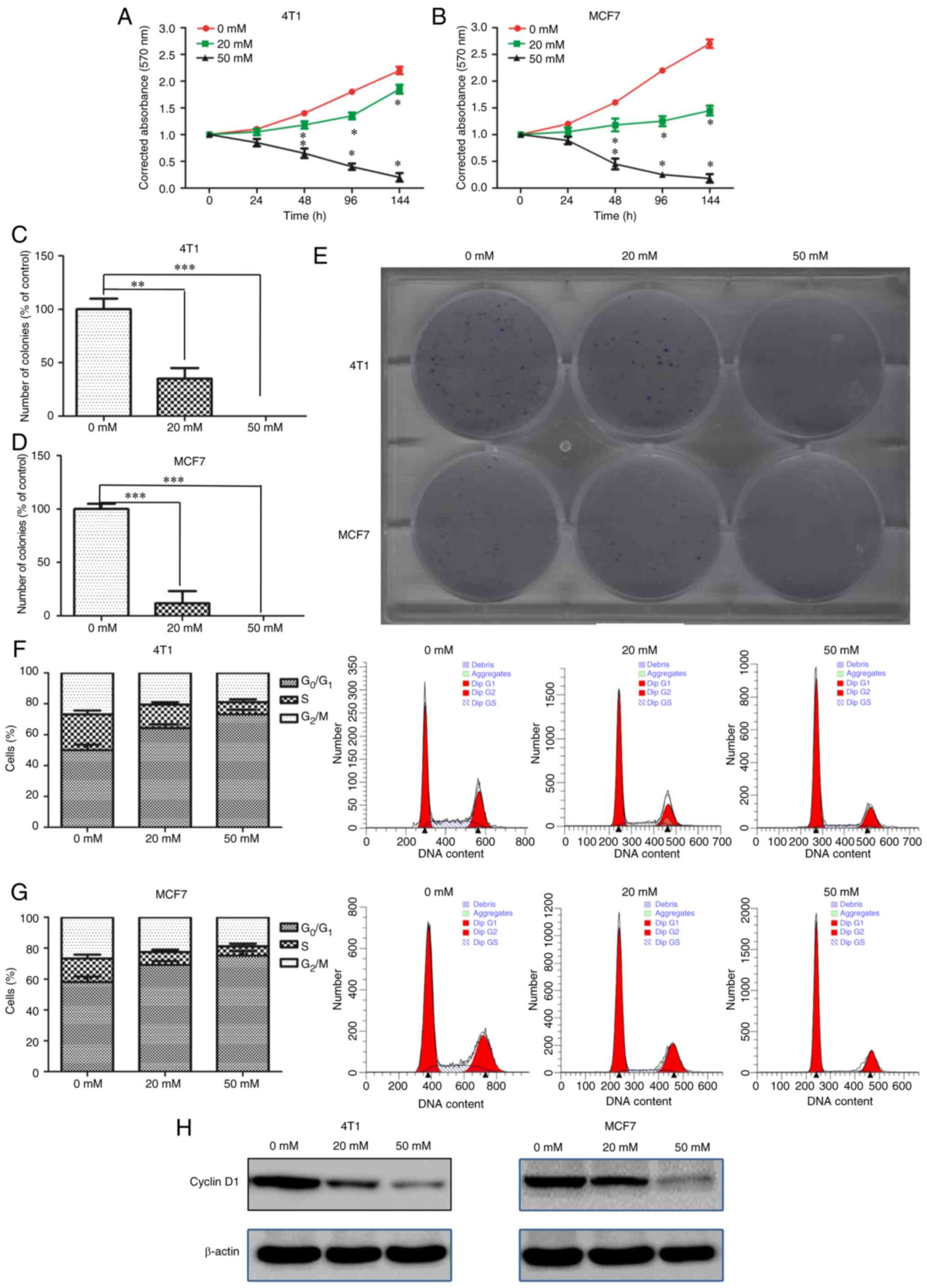
was measured at the indicated times (0–144 h). As shown in Fig. 1A and B, metformin led to a decrease
in cell proliferation in a dose- and time-dependent manner in the
two cell lines tested. At the maximum concentration of the drug (50
mM), the proliferation of the two cell lines was almost completely
blocked at 144 h. Furthermore, metformin significantly inhibited
the colony forming efficiency of both cell lines in a
dose-dependent manner (Fig. 1C-E).
At 20 mM concentration, metformin decreased the overall rate of
colony formation of MCF7 and 4T1 cells by >60% compared with
untreated cells, while at 50 mM, metformin inhibited colony
formation by 100% compared with untreated cells (Fig. 1C-E). These data indicated that
metformin effectively inhibited breast cancer cell proliferation
in vitro.
Metformin downregulates cyclin D1
expression and induces G0/G1 cell cycle
arrest in breast cancer cells
To further study the effect of metformin on cell
cycle progression, the percentage of cells in each respective cell
cycle phase was detected by flow cytometry. As shown in Fig. 1F and G, metformin markedly increased
the proportion of cells in G0/G1 phase by
>10% compared with untreated cells, whereas the number of cells
in the S phase was markedly decreased, especially in 4T1 cells.
Moreover, cyclin D1 protein expression, a key regular of the
G1/S transition, was markedly downregulated in a
dose-dependent manner in cells treated with metformin compared with
in control cells (Fig. 1H). Overall,
these results demonstrated that metformin exerted its antitumor
activity by inducing cell cycle arrest.
Metformin inhibits the growth of 4T1
cell xenografts in nude mice
To determine the inhibitory effect of metformin on
cell proliferation in vivo, nude mice were subcutaneously
injected with 4T1 cells, and treated daily with metformin (250
mg/kg) or saline when the diameter of xenograft tumors reached 4
mm. Mice were sacrificed after 2 weeks and tumors were excised. As
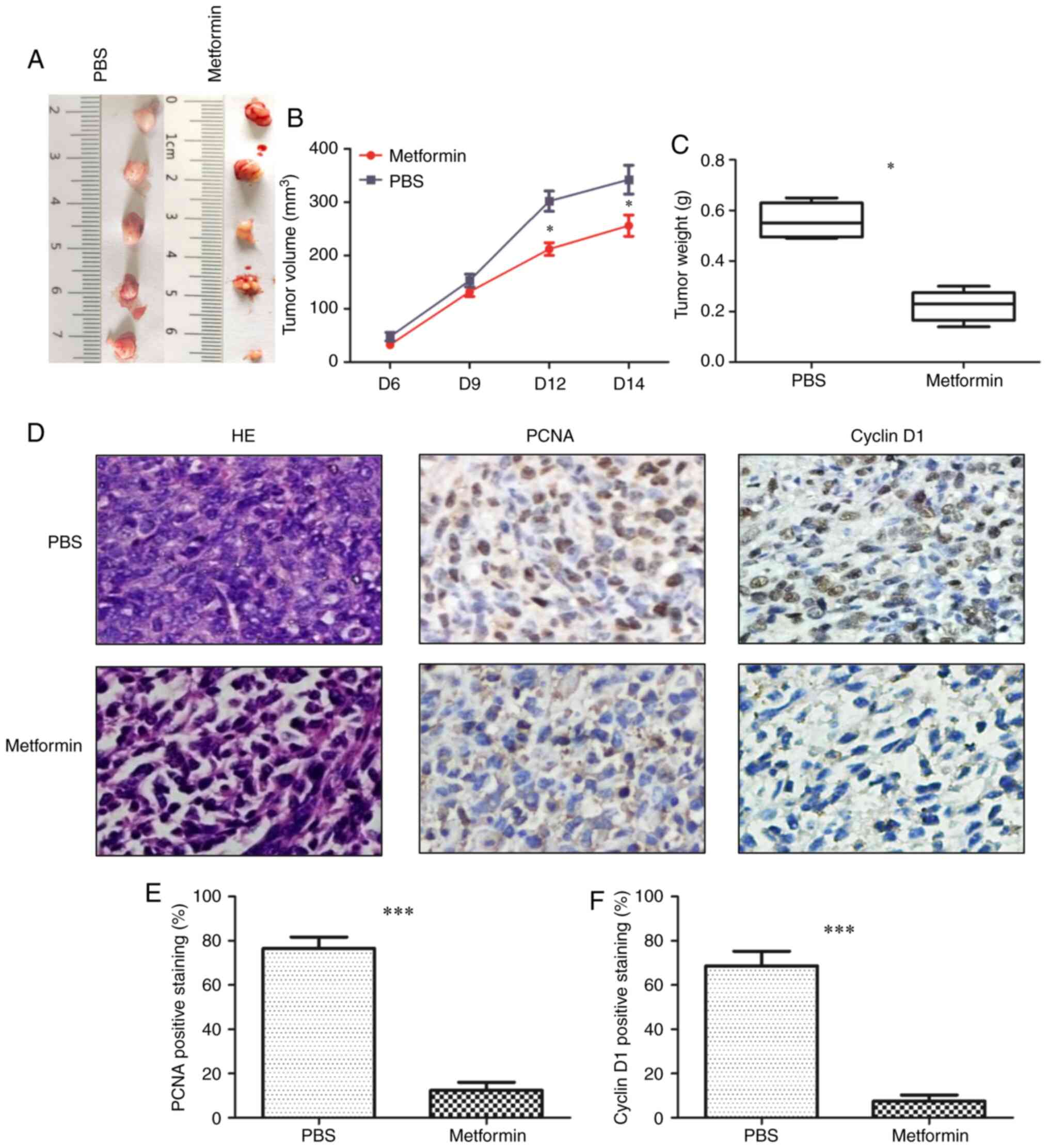
shown in Fig. 2A-C, the
administration of metformin alone significantly decreased the
growth of tumor-cell xenografts in vivo. The H&E-stained
slides of paraffin-embedded excised tumors indicated a decrease in
tumor cell volume and density in treated mice compared with in
untreated mice (Fig. 2D).
Furthermore, consistent with the aforementioned results in
vitro, a significant decrease in cyclin D1 and PCNA expression
was observed in metformin-treated tumors compared with in untreated
tumors (Fig. 2D-F). Thus, these
results demonstrated that metformin inhibited the growth of breast
cancer cell xenografts in nude mice.
Metformin activates AMPK and inhibits
COX-2 in vitro and in vivo
The anticancer effects of metformin are mediated via
the AMPK signaling pathway (43).
Therefore, the present study investigated whether metformin could
activate AMPK by measuring the levels of p-AMPK at Thr-172. As
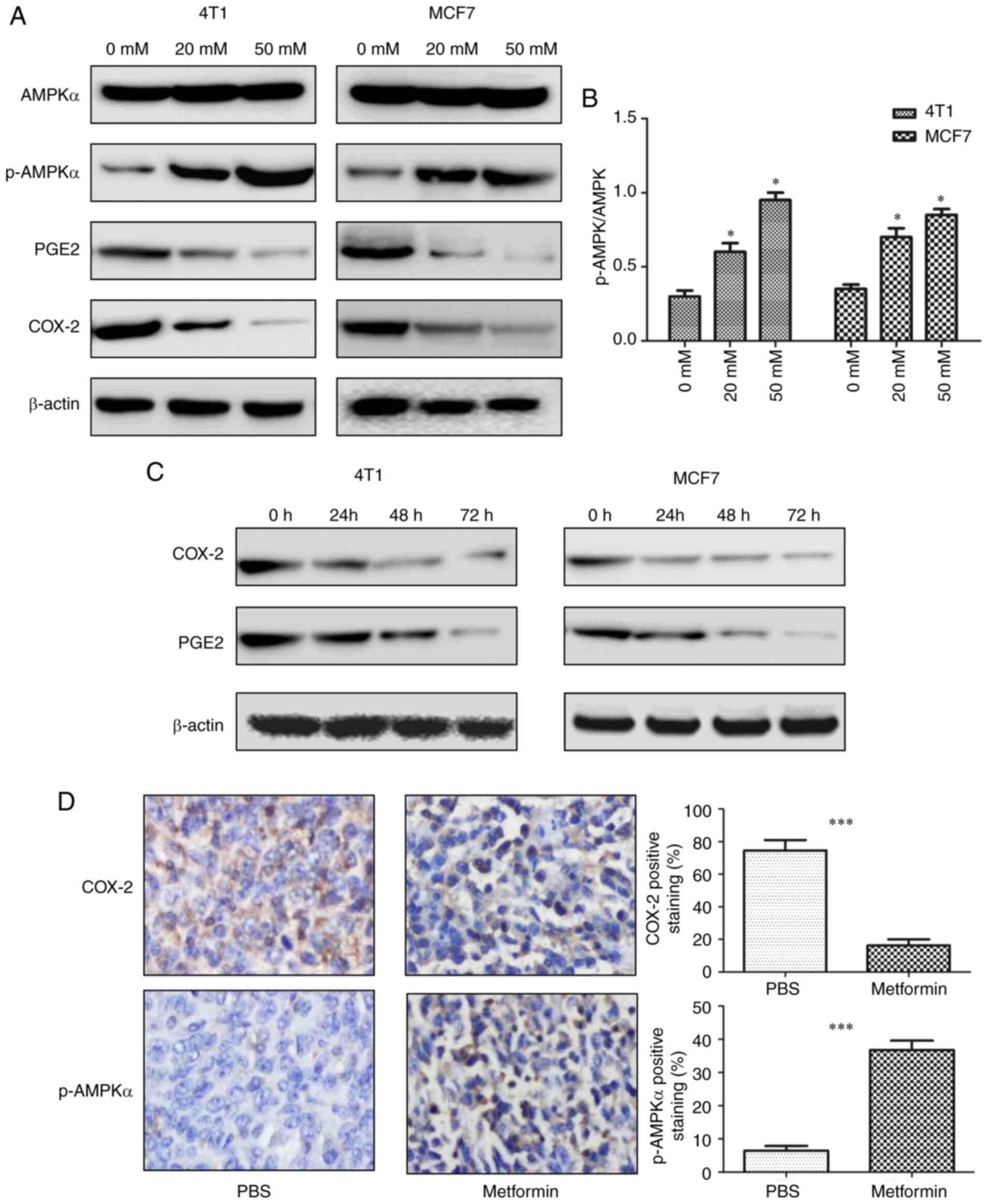
expected, treatment of 4T1 and MCF7 cells with metformin
significantly increased p-AMPK levels (Fig. 3A) and relative protein levels of
p-AMPK/AMPK (Fig. 3B) in a
dose-dependent manner. In vivo, significantly increased
levels of p-AMPKα were observed in xenograft tumor sections
obtained from mice after metformin treatment compared with in those
obtained from untreated mice (Fig.
3D). Previously, it has been shown that activation of the AMPK
signaling pathway with epigallocatechin-3-gallate (EGCG) abrogates
COX-2 expression and PGE2 production in colon cancer cells
(44). Thus, the present study
studied whether metformin could inhibit COX-2 expression and PGE2
production. The current results revealed that metformin inhibited
COX-2 and PGE2 expression in breast cancer cells in a dose- and
time-dependent manner (Fig. 3A and
C). This was also reflected in the percentage of
positively-stained cells in the tissues of mice treated with
metformin (Fig. 3D). In summary,
these findings strongly suggested that metformin may be a potent
inhibitor of the COX-2 signaling pathway and may modulate cell
cycle progression to restrain tumor growth.
Metformin inhibits the transcription
of COX-2
To determine whether metformin transcriptionally
regulates COX-2, a pGL3-COX-2 plasmid containing a 2,000-bp region
upstream of the human COX-2 promoter was used. The dual luciferase
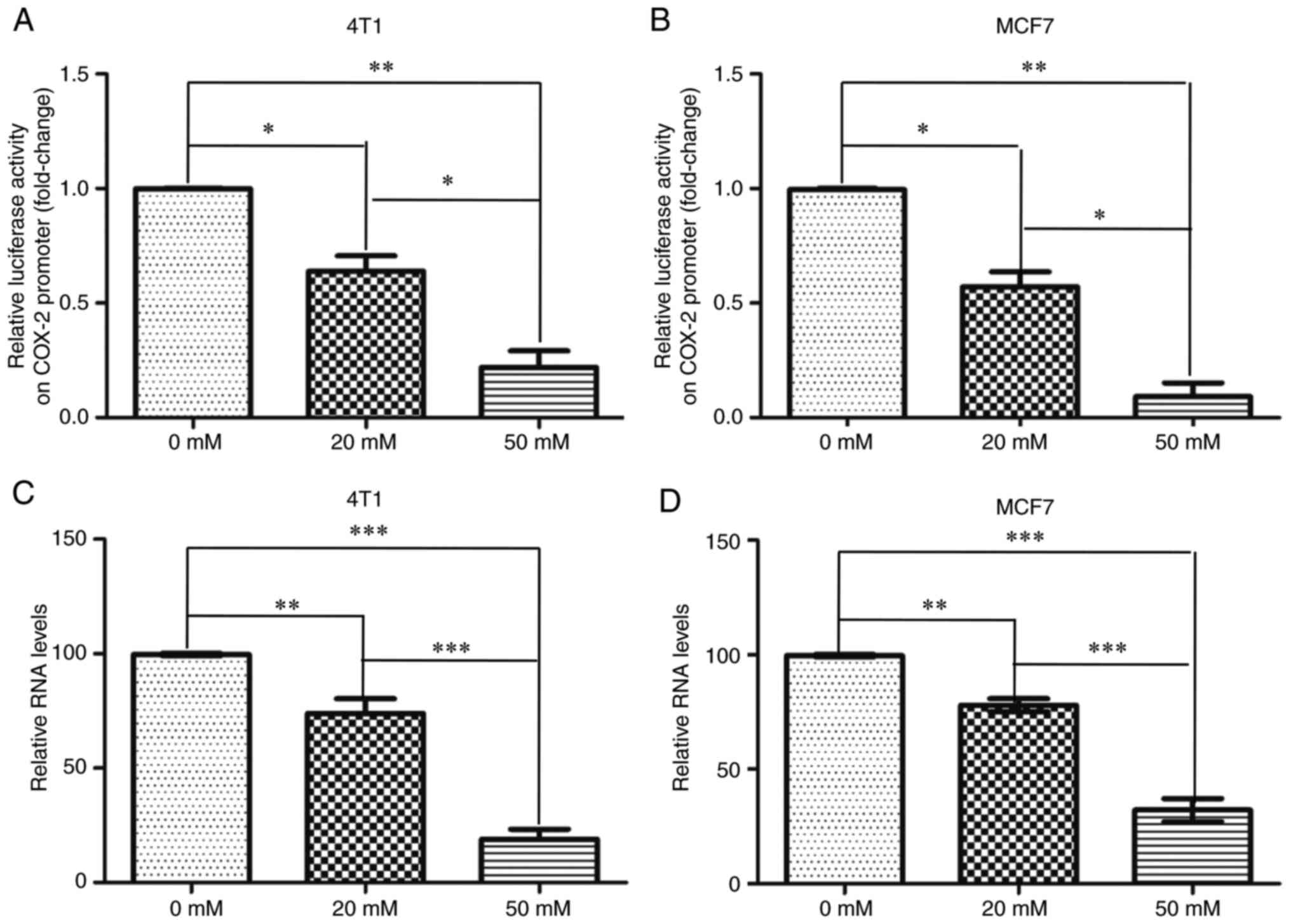
assay indicated that compared with no treatment, the relative COX-2
promoter activity was significantly decreased upon treatment of 4T1
and MCF7 cells with metformin (Fig. 4A
and B). Moreover, the mRNA expression levels of COX-2 were
significantly decreased upon treatment of both cells with metformin
(Fig. 4C and D). These data
indicated that metformin inhibited COX-2 transcription.
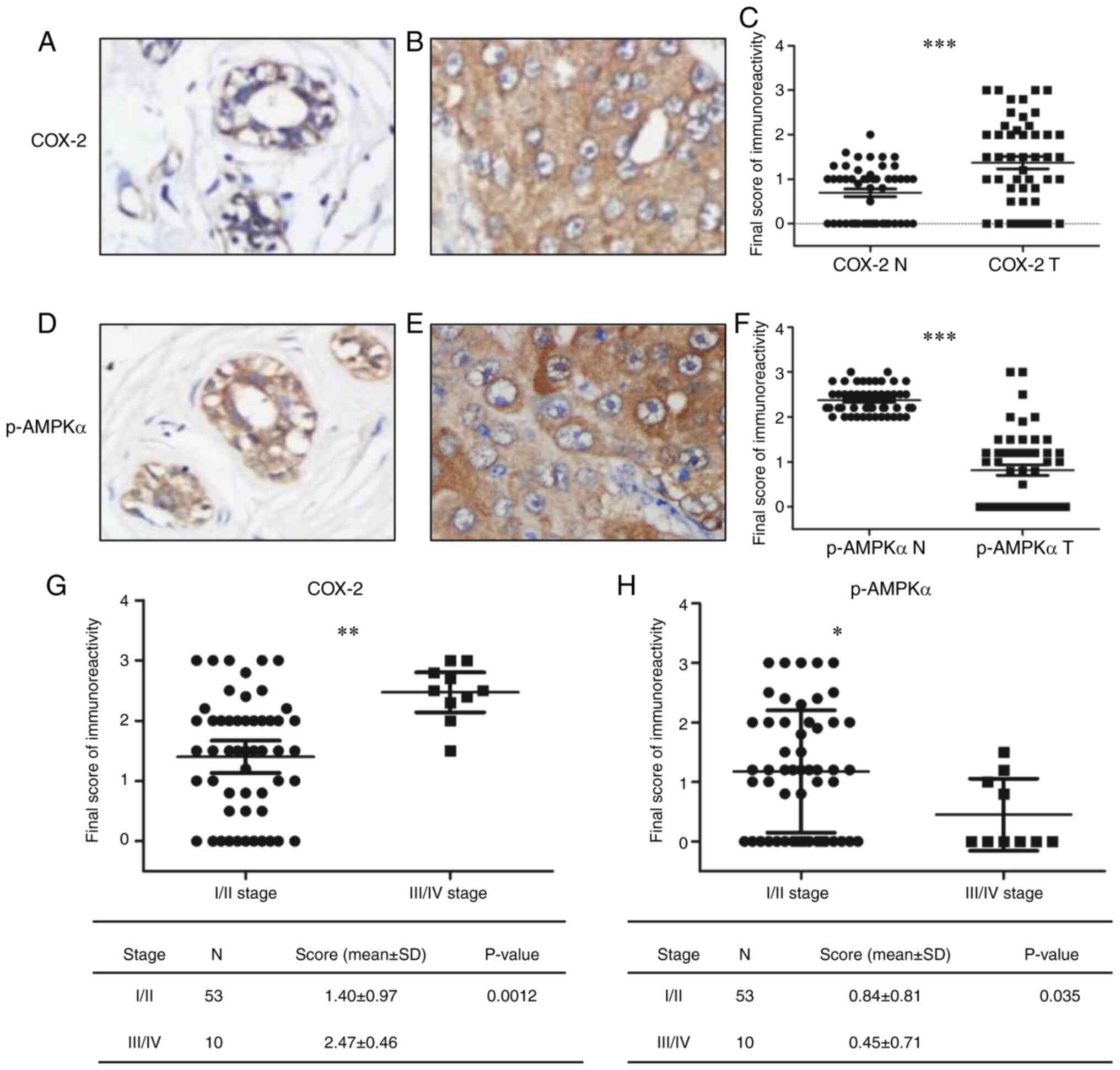
Expression profiles of COX-2 and
p-AMPKα in patients with breast cancer
Next, p-AMPKα and COX-2 protein expression was
measured in the tissues of 63 cases with invasive breast cancer.
p-AMPKα and COX-2 were primarily expressed in the cytoplasm of
cells obtained from normal breast and tumor tissues (Fig. 5). Furthermore, downregulation of
p-AMPKα and upregulation of COX-2 expression was observed in the
tumor tissues compared with in adjacent non-cancerous tissues
(Fig. 5A-F). The mean expression
values of COX-2 in tumor tissues were significantly increased
compared with in normal tissues (1.36±0.97 vs. 0.70±0.61,
respectively; Fig. 5E). By contrast,
the mean values of p-AMPKα in tumors (0.82±0.84) were significantly
lower than in normal tissues (2.37±0.31) (Fig. 5F).
The association of clinicopathological variables
with COX-2 and p-AMPKα expression is summarized in Table I. Increased levels of COX-2 were
significantly associated with lymphatic metastasis and TNM stage in
patients with breast cancer. COX-2 expression was more common in
breast tumors with lymphatic metastasis (81.0%) than in those
without lymphatic metastasis (42.9%) (P=0.004; Table I). Additionally, COX-2 expression was
more common in patients with stage III/IV (90.0%) than in those
with stage I/II disease (49.1%) (P=0.017; Table I). Moreover, COX-2 expression was
significantly lower in stage I/II tumors (1.40±0.97) compared with
in stage III/IV tumors (2.47±0.46) (Fig.
5G). No significant associations were identified between COX-2
expression and other clinicopathological factors, such as age, T
stage, and ER, PR, HER2 and p53 status (P>0.05; Table I). Compared with in non-cancerous
ductal tissues, p-AMPKα expression was significantly lower in
primary tumors (Fig. 5F), but there
was no significant association between p-AMPKα and any
clinicopathological variable (Table
I). However, p-AMPKα expression was significantly higher in
stage I/II tumors (0.84±0.81) compared with in stage III/IV tumors
(0.45±0.71) (Fig. 5H).
 | Table I.Association between COX-2, p-AMPKα
and p-4E-BP1 expression and clinicopathologic features in patients
with breast cancer (n=63). |
Table I.
Association between COX-2, p-AMPKα
and p-4E-BP1 expression and clinicopathologic features in patients
with breast cancer (n=63).
| Variables | N | COX-2, n (%) | P-value | p-AMPKα, n (%) | P-value | p-4E-BP1, n
(%) | P-value |
|---|
| Age, years |
|
|
|
|
|
|
|
|
<50 | 29 | 16 (55.2) | 0.955 | 15 (51.7) | 0.923 | 16 (55.2) | 0.596 |
|
≥50 | 34 | 19 (55.9) |
| 18 (52.9) |
| 21 (61.8) |
|
| pT |
|
|
|
|
|
|
|
|
pT1/2 | 51 | 28 (54.9) | 0.830 | 27 (52.9) | 0.854 | 31 (60.9) | 0.495 |
|
pT3/4 | 12 | 7
(58.3) |
| 6
(50.0) |
| 6
(50.0) |
|
| pN |
|
|
|
|
|
|
|
| No | 42 | 18 (42.9) | 0.004 | 23 (45.1) | 0.593 | 20 (47.6) | 0.011 |
|
Yes | 21 | 17 (81.0) |
| 10 (47.6) |
| 17 (81.0) |
|
| TNM stage |
|
|
|
|
|
|
|
|
I/II | 53 | 26 (49.1) | 0.017 | 30 (56.6) | 0.122 | 28 (52.8) | 0.029 |
|
III/IV | 10 | 9
(90.0) |
| 3
(30.0) |
| 9
(90.0) |
|
| ER |
|
|
|
|
|
|
|
| + | 34 | 16 (47.1) | 0.955 | 20 (58.8) | 0.268 | 16 (47.1) | 0.596 |
| − | 29 | 19 (65.5) |
| 13 (44.8) |
| 21 (72.4) |
|
| PR |
|
|
|
|
|
|
|
| + | 32 | 20 (62.5) | 0.260 | 18 (56.3) | 0.532 | 19 (59.4) | 0.685 |
| − | 31 | 15 (48.4) |
| 15 (48.4) |
| 18 (58.1) |
|
| Her2 |
|
|
|
|
|
|
|
| + | 20 | 14 (70.0) | 0.116 | 10 (50.0) | 0.796 | 15 (75.0) | 0.074 |
| − | 43 | 21 (48.8) |
| 23 (53.5) |
| 22 (51.2) |
|
| p53 |
|
|
|
|
|
|
|
| + | 26 | 18 (69.2) | 0.057 | 15 (57.7) | 0.479 | 19 (73.1) | 0.156 |
| − | 37 | 17 (45.9) |
| 18 (48.6) |
| 18 (48.6) |
|
| Total | 63 | 35 (55.6) |
| 33 (52.4) |
| 37 (58.7) |
|
Metformin inhibits phosphorylation of
4E-BP1 in vitro and in vivo
The activation of AMPK inhibits the mTOR signaling
pathway and decreases phosphorylation of S6 kinase (S6K) in breast
cancer (45). Thus, the specific
effects of metformin on 4E-BP1 (Thr-37/46), another downstream
target of mTOR, were examined in the present study. As shown in
Fig. 6A-D, the phosphorylation
levels of 4E-BP1 and relative protein levels of p-AMPK/AMPK
decreased in a dose- and time-dependent manner following metformin
treatment. The p-4E-BP1 levels were significantly decreased in
excised mouse tumors after treatment with metformin (Fig. 6E). The current results are consistent
with a previous study that has demonstrated that metformin inhibits
tumor growth mainly via the AMPK/mTOR signaling pathway (46).
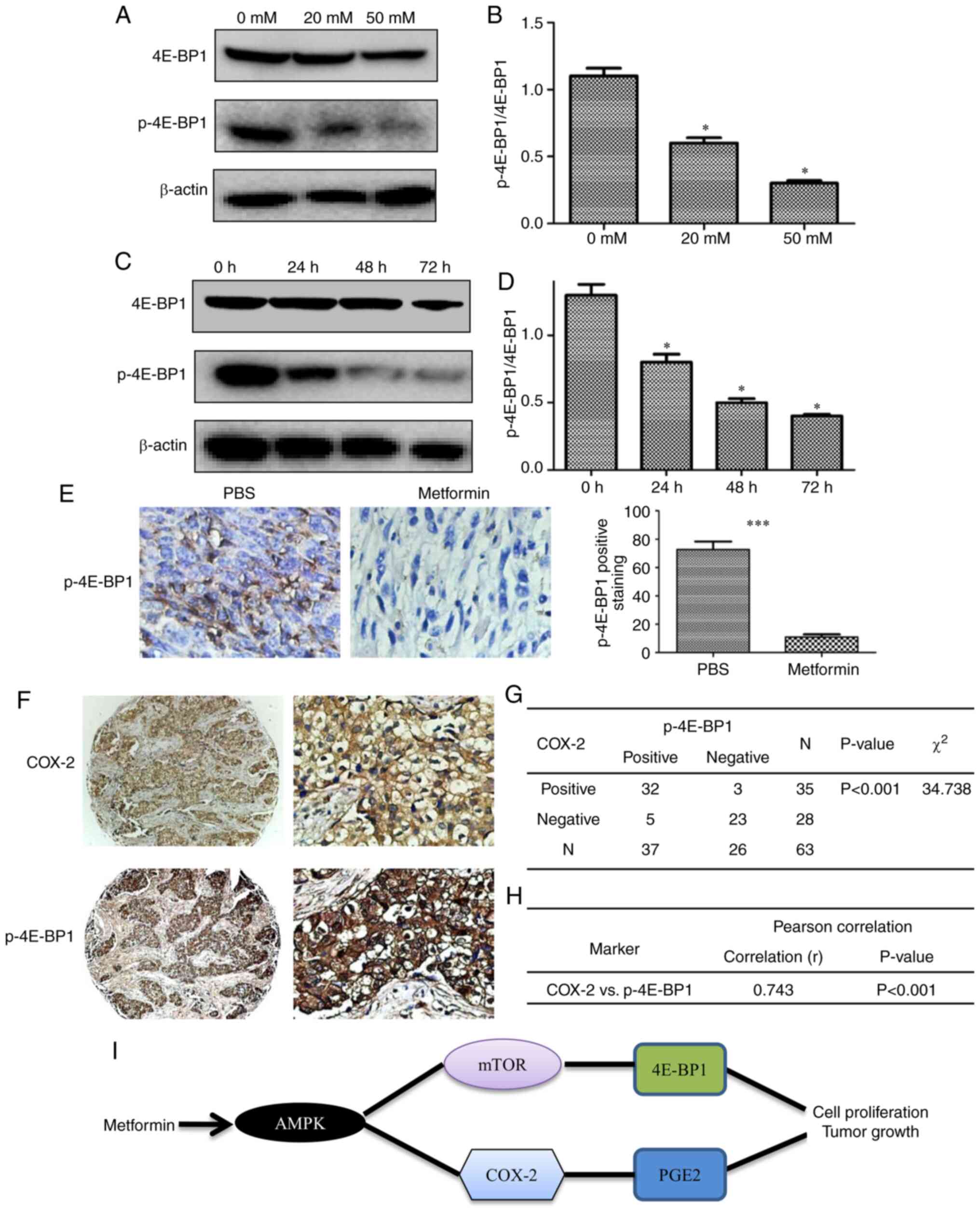 | Figure 6.Metformin treatment inhibits
phosphorylation of 4E-BP1. (A) Western blot analysis of 4E-BP1 and
p-4E-BP1 in 4T1 cells treated with metformin (0, 20 and 50 mM) for
48 h and (B) quantification of p-4E-BP1/4E-BP1 ratio. (C) Western
blot analysis of 4E-BP1 and p-4E-BP1 in 4T1 cells treated with
metformin (20 mM) for the indicated time periods and (D)
quantification of p-4E-BP1/4E-BP1 ratio. *P<0.05 vs. control
group. (E) Representative images (magnification, ×400) and
quantification of immunostaining for p-4E-BP1 in tumor sections
from metformin-treated and control mice. ***P<0.001. (F)
Representative staining of COX-2 and p-4E-BP1 in cancer tissues
from the same patient (left, ×10; right, ×400). (G) Association
between COX-2 and p-4E-BP1 staining and (H) correlation between
COX-2 and p-4E-BP1 expression. (I) Diagrammatic sketch showing that
activation of AMPK by metformin leads to inhibition of the mTOR
signaling pathway and COX-2 expression, which results in decreased
cell proliferation and tumor growth. COX-2, cyclooxyeganse-2; p,
phosphorylated; AMPK, AMP kinase; 4E-BP1, eukaryotic translation
initiation factor 4E-binding protein 1; PGE2, prostaglandin E2. |
Subsequently, the expression profile of p-4E-BP1 in
breast cancer specimens and the correlation between the levels of
p-4E-BP1 and COX-2 were evaluated. p-4E-BP1 staining was
preferentially localized to the cytoplasm and was markedly
increased in tumor tissues compared with in adjacent non-cancerous
breast epithelium (Fig. 6F). Of the
63 tumors, 37 (58.7%) exhibited high p-4E-BP1 levels, which were
significantly associated with lymphatic metastasis and TNM stage
(Table I). Co-expression of COX-2
and p-4E-BP1 was observed in 32 (50.8%) tumors, while 23 (36.5%)
showed no expression of either COX-2 or p-4E-BP1
(χ2=34.738; P<0.001; Fig.
6G). Pearson correlation analysis revealed that COX-2
expression was positively correlated with p-4E-BP1 expression among
the 63 breast tumors (r=0.743; P<0.001; Fig. 6H). Furthermore, COX-2 expression
co-localized with p-4E BP1 levels in the same specimens (Fig. 6F). Overall, the current data
suggested that activation of AMPK by metformin may lead to
inhibition of the mTOR signaling pathway and COX-2 expression,
resulting in decreased cell proliferation and tumor growth
(Fig. 6I).
Discussion
The present study aimed to explore a novel
anticancer mechanism of the anti-diabetic drug metformin. Metformin
is known to exert its antitumor effects by activating AMPK and
inhibiting mTOR-mediated phosphorylation of S6K1 and 4E-BP1 in the
AMPK/mTOR signaling pathway (47).
The current study described a novel role of metformin by
demonstrating that metformin significantly suppressed cell
proliferation in vitro and decreased tumor growth in
vivo by targeting the COX-2 and AMPK/mTOR signaling pathways.
Treatment with metformin alone significantly decreased the
expression levels of COX-2 and p-4E-BP1 in breast cancer cells.
Furthermore, COX-2 and p-4E-BP1 were frequently upregulated and
strongly associated with nodal metastasis and advanced disease
stage, implicating their dual-role as predictive biomarkers and
therapeutic targets in breast cancer.
Clinical and epidemiological studies have
demonstrated that compared with individuals without diabetes, the
relative risk of progression to breast cancer is increased in
patients with type 2 DM, and treatment with metformin decreases the
relative risk for breast cancer and cancer-associated mortality in
diabetic patients (48–50). Pre-clinical studies have indicated
that the majority of breast cancer cell lines show sensitivity to
metformin treatment (17,20). The present data further confirmed
that metformin was able to decrease cell viability in a dose- and
time-dependent manner, and inhibited the rate of colony formation
in breast cancer cells. The highest concentration of metformin (50
mM) completely blocked the proliferation rate of breast cancer
cells. These anticancer effects of metformin, as evidenced by the
increased proportion of cells in G0/G1 phase
and decreased cyclin D1 expression, were associated with cell cycle
arrest. Moreover, tumors excised from metformin-treated mice
exhibited significantly slower growth and lower tumor volume than
those from mice not treated with metformin. Moreover, metformin
treatment altered the morphology of cancer cells, as demonstrated
by significantly smaller cell types and larger intervals between
cells in the xenograft tissues of metformin-treated mice than those
in control mice. These changes may be due to a metabolic response
to metformin toxicity. Further immunohistochemical analysis
confirmed the low proliferation index (as indicated by PCNA) and
low levels of cyclin D1 in the metformin-treated mice compared with
in control mice (16,17).
Furthermore, consistent with previous studies
(51–53), the present study demonstrated that
metformin alone was sufficient to activate AMPK and inhibit
p-4E-BP1 and cyclin D1. Metformin decreases PGE2 synthesis by
activating AMPK (54), and COX-2 is
the key enzyme in PGE2 synthesis (55). Therefore, it was speculated that
metformin may serve an anticancer role through the AMPK-mediated
COX-2 signaling pathway. To the best of our knowledge, the present
study was the first to demonstrate that treatment with metformin
alone significantly decreased COX-2 protein expression in a dose-
and time-dependent manner in vitro and in vivo. COX-2
is undetectable in most normal tissues and accumulates in activated
macrophages and other cells at sites of inflammation (56). A previous study has indicated that
COX-2 expression is upregulated in various types of cancer,
including gastric, colorectal and lung cancer, and serves a crucial
role in tumorigenesis (57). AMPK
activation by selenium and EGCG abrogate COX-2 expression in colon
cancer cells (44). Similar results
were observed in the present study in breast cancer cells upon
treatment with another AMPK activator, metformin. The increase in
metformin concentrations significantly increased AMPK activity and
decreased COX-2 expression. Moreover, continued use of metformin
resulted in gradual reduction in COX-2 expression, including at the
mRNA level. Additionally, the inhibition of the activity of the
pGL3-COX-2-promoter suggested that AMPK activation abolished the
transactivation of COX-2. Overall, the current results suggested
that metformin regulated COX-2 production at both the
transcriptional and post-transcriptional levels, and identified a
potential association between AMPK activation by metformin and
inhibition of inflammatory events.
AMPK and COX-2 expression is well-studied in human
solid cancer tissues, such as gastric, colorectal and ovarian
cancer (58–61). p-AMPK expression is decreased in ~90%
of patients with breast cancer and is significantly associated with
higher histological grade and axillary node metastasis (62). In accordance with the aforementioned
studies, the present study demonstrated that p-AMPKα expression was
strong in normal breast epithelium and weak in primary breast
cancer tissues. The levels of p-AMPKα decreased with disease
progression, although no significant association was observed with
clinicopathological factors due to the relatively small cohort.
Given the association between AMPK and COX-2, it is plausible that
decreased p-AMPKα levels may be associated with increased COX-2
expression. The current data indicated that COX-2 expression was
increased in primary breast cancer specimens compared with in
normal breast epithelium, consistent with previous studies
(24,25). Additionally, COX-2 positivity was
significantly associated with high histological grade and lymph
node metastasis. Enhanced COX-2 expression was frequently
associated with decreased levels of p-AMPKα, although this
association was not significant due to the relatively small number
of cases in the present study. These data further suggest the
importance of the AMPK/COX-2 axis in breast cancer development and
progression.
Furthermore, a previous study has revealed that
metformin exerts anticancer effects by activating AMPK, which
results in inhibition of mTOR kinase and decreased S6K1 activity
(63). Consistent with a previous
study (18), the present study
indicated that metformin also suppressed another mTOR downstream
effector, 4E-BP1, thus making it a particularly attractive molecule
for investigation in breast cancer. The current data offer a novel
mechanistic insight into the potential use of metformin for
treatment of breast cancer. Previous studies have demonstrated that
p-4E-BP1 levels are associated with malignant progression and
adverse prognosis in breast cancer (64–66). The
current data revealed that p-4E-BP1 levels were significantly
elevated in the majority of breast cancer cases, and p-4E-BP1
positivity was common in cases with nodal metastasis and advanced
disease stage. The AMPK/mTOR axis is a well-known effective
therapeutic target in metabolic syndromes and cancer, while COX-2
is an established therapeutic target in inflammatory diseases and
cancer (67–70). Therefore, cross-talk between the
AMPK/mTOR and COX-2 signaling pathways can be expected in cancer
pathophysiology. The present results indicated that AMPK activation
by metformin decreased both p-4E-BP1 and COX-2 expression in breast
cancer cells, which supports the hypothesis that the two pathways
are connected. Moreover, immunostaining revealed a close
correlation between the levels of these proteins. Thus, p-4E-BP1
and COX-2 may have potential roles as predictive markers and
therapeutic targets in breast cancer.
To the best of our knowledge, the present study was
the first to demonstrate that metformin activated AMPK and
suppressed COX-2 expression to inhibit breast cancer cell
proliferation. The current findings are supported by clinical
research published by Jiralerspong et al (71), who reported a 3-fold greater complete
pathologic response in diabetic patients with breast tumors
receiving metformin and neoadjuvant chemotherapy than in those with
breast tumors not receiving metformin. Therefore, metformin
co-treatment with conventional therapy may serve as a successful
therapeutic strategy in the prevention of cancer recurrence and
improvement of long-term survival.
In conclusion, the present study revealed the novel
finding that metformin activated AMPK, which suppressed the
production of COX-2 and abrogated breast cancer cell proliferation.
Thus, metformin may serve as a potential therapeutic drug for the
treatment of patients with breast cancer, and further studies
should be performed to investigate how it may be used in cancer
therapy alone or in combination with other antitumor drugs.
Acknowledgements
The authors would like to thank Professor Yin Chen
(Changhai Hospital, Shanghai, China) for data processing and
helpful discussions.
Funding
The present study was partly sponsored by the
Guiding Program of Fujian (grant no. 2016Y9064) and the
Technological Innovation Foundation of Fujian (grant no.
2015J01408).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BS and WF conceived and designed the study. BS, XH,
HH and WF performed the experiments and prepared the manuscript, as
well as assessed and confirmed the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was performed according to the ethical
guidelines of the Declaration of Helsinki and was approved by the
Institutional Review Boards of the Kunshan First People's Hospital
affiliated with Jiangsu University and the First Affiliated
Hospital affiliated with the Second Military Medical University
(Changhai Hospital, Shanghai, China) (approval no. CHEC2014-098).
Written informed consent for the use of all the clinical specimens
was obtained from all patients. All animal experimental protocols
were reviewed and approved by the Animal Ethics Committee of the
Second Military Medical University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
American Cancer Society, . Breast Cancer
Facts and Figures, 2007–2008, May 2009. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2007-2008.pdfMay
21–2021
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller ME, Muhsen S, Olcese C, Patil S,
Morrow M and Van Zee KJ: Contralateral breast cancer risk in women
with ductal carcinoma in situ: Is it high enough to justify
bilateral mastectomy. Ann Surg Oncol. 24:2889–2897. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gandini S, Guerrieri-Gonzaga A, Puntoni M
and Decensi A: Metformin and breast cancer risk. J Clin Oncol.
31:973–974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahmadieh H and Azar ST: Type 2 diabetes
mellitus, oral diabetic medications, insulin therapy, and overall
breast cancer risk. ISRN Endocrinol. 2013:1812402013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brower V: Illuminating the diabetes-cancer
link. J Natl Cancer Inst. 104:1048–1050. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suh S and Kim KW: Diabetes and cancer: Is
diabetes causally related to cancer. Diabetes Metab J. 35:193–198.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scheen AJ, Beck E, De Flines J and Rorive
M: Obesity, insulin resistance and type 2 diabetes: Risk factors
for breast cancer. Rev Med Liege. 66:238–244. 2011.(In French).
PubMed/NCBI
|
|
10
|
Pandey A, Forte V, Abdallah M, Alickaj A,
Mahmud S, Asad S and McFarlane SI: Diabetes mellitus and the risk
of cancer. Minerva Endocrinol. 36:187–209. 2011.PubMed/NCBI
|
|
11
|
Vigneri P, Frasca F, Sciacca L, Pandini G
and Vigneri R: Diabetes and cancer. Endocr Relat Cancer.
16:1103–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen TW, Liang YN, Feng D, Tao LY, Qi K,
Zhang HY, Wang HX, Lin QS and Kong H: Metformin inhibits
proliferation and promotes apoptosis of HER2 positive breast cancer
cells by downregulating HSP90. J BUON. 18:51–56. 2013.PubMed/NCBI
|
|
13
|
Col NF, Ochs L, Springmann V, Aragaki AK
and Chlebowski RT: Metformin and breast cancer risk: A
meta-analysis and critical literature review. Breast Cancer Res
Treat. 135:639–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jalving M, Gietema JA, Lefrandt JD, de
Jong S, Reyners AK, Gans RO and de Vries EG: Metformin: Taking away
the candy for cancer. Eur J Cancer. 46:2369–2380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bodmer M, Meier C, Krähenbühl S, Jick SS
and Meier CR: Long-term metformin use is associated with decreased
risk of breast cancer. Diabetes Care. 33:1304–1308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu B, Fan Z, Edgerton SM, Deng XS,
Alimova IN, Lind SE and Thor AD: Metformin induces unique
biological and molecular responses in triple negative breast cancer
cells. Cell Cycle. 8:2031–2040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alimova IN, Liu B, Fan Z, Edgerton SM,
Dillon T, Lind SE and Thor AD: Metformin inhibits breast cancer
cell growth, colony formation and induces cell cycle arrest in
vitro. Cell Cycle. 8:909–915. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song CW, Lee H, Dings RP, Williams B,
Powers J, Santos TD, Choi BH and Park HJ: Metformin kills and
radiosensitizes cancer cells and preferentially kills cancer stem
cells. Sci Rep. 2:3622012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soga M, Ohashi A, Taniguchi M, Matsui T
and Tsuda T: The di-peptide Trp-His activates AMP-activated protein
kinase and enhances glucose uptake independently of insulin in L6
myotubes. FEBS Open Bio. 4:898–904. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in Metformin treated breast cancer cells involves activation
of AMPK, downregulation of cyclin D1, and requires p27Kip1 or
p21Cip1. J Mol Signal. 3:182008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hadad SM, Fleming S and Thompson AM:
Targeting AMPK: A new therapeutic opportunity in breast cancer.
Crit Rev Oncol Hematol. 67:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zong M, Fan DD, Lin S, Song YP, Wang ZY,
Ma XL, Qiu WH, Bai YH, Li L and Li S: Anti-cancer activity and
potential mechanism of a novel aspirin derivative. Eur J Pharmacol.
791:137–146. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aban M, Siddqui I, Saboor M, Pervez S and
Moatter T: Haplotypes of SNPs associated with COX-2 and their
comparison with histopathological features of breast cancer
patients. J Immuno Ther Cancer. 3:92015. View Article : Google Scholar
|
|
24
|
Dhakal HP, Naume B, Synnestvedt M, Borgen
E, Kaaresen R, Schlichting E, Wiedswang G, Bassarova A, Holm R,
Giercksky KE and Nesland JM: Expression of cyclooxygenase-2 in
invasive breast carcinomas and its prognostic impact. Histol
Histopathol. 27:1315–1325. 2012.PubMed/NCBI
|
|
25
|
Holmes MD, Chen WY, Schnitt SJ, Collins L,
Colditz GA, Hankinson SE and Tamimi RM: COX-2 expression predicts
worse breast cancer prognosis and does not modify the association
with aspirin. Breast Cancer Res Treat. 130:657–662. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Çiriş IM, Bozkurt KK, Başpinar S and
Kapucuoğlu FN: Immunohistochemical COX-2 overexpression correlates
with HER-2/neu overexpression in invasive breast carcinomas: A
pilot study. Pathol Res Pract. 207:182–187. 2011. View Article : Google Scholar
|
|
27
|
Kim HS, Moon HG, Han W, Yom CK, Kim WH,
Kim JH and Noh DY: COX2 overexpression is a prognostic marker for
Stage III breast cancer. Breast Cancer Res Treat. 132:51–59. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daneau G, Boidot R, Martinive P and Feron
O: Identification of cyclooxygenase-2 as a major actor of the
transcriptomic adaptation of endothelial and tumor cells to cyclic
hypoxia: Effect on angiogenesis and metastases. Clin Cancer Res.
16:410–419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lyons TR, Borges VF, Betts CB, Guo Q,
Kapoor P, Martinson HA, Jindal S and Schedin P:
Cyclooxygenase-2-dependent lymphangiogenesis promotes nodal
metastasis of postpartum breast cancer. J Clin Invest.
124:3901–3912. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Killian PH, Kronski E, Michalik KM,
Barbieri O, Astigiano S, Sommerhoff CP, Pfeffer U, Nerlich AG and
Bachmeier BE: Curcumin inhibits prostate cancer metastasis in vivo
by targeting the inflammatory cytokines CXCL1 and −2.
Carcinogenesis. 33:2507–2519. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karavitis J, Hix LM, Shi YH, Schultz RF,
Khazaie K and Zhang M: Regulation of COX2 expression in mouse
mammary tumor cells controls bone metastasis and PGE2-induction of
regulatory T cell migration. PLoS One. 7:e463422012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lucci A, Krishnamurthy S, Singh B,
Bedrosian I, Meric-Bernstam F, Reuben J, Broglio K, Mosalpuria K,
Lodhi A, Vincent L and Cristofanilli M: Cyclooxygenase-2 expression
in primary breast cancers predicts dissemination of cancer cells to
the bone marrow. Breast Cancer Res Treat. 117:61–68. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Visscher DW, Pankratz VS, Santisteban M,
Reynolds C, Ristimäki A, Vierkant RA, Lingle WL, Frost MH and
Hartmann LC: Association between cyclooxygenase-2 expression in
atypical hyperplasia and risk of breast cancer. J Natl Cancer Inst.
100:421–427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thun MJ, Henley SJ and Patrono C:
Nonsteroidal anti-inflammatory drugs as anticancer agents:
Mechanistic, pharmacologic, and clinical issues. J Natl Cancer
Inst. 94:252–266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luchetti CG, Mikó E, Szekeres-Bartho J,
Paz DA and Motta AB: Dehydroepiandrosterone and metformin modulate
progesterone-induced blocking factor (PIBF), cyclooxygenase 2
(COX2) and cytokines in early pregnant mice. J Steroid Biochem Mol
Biol. 111:200–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Elia E, Sander V, Luchetti CG, Solano ME,
Di Girolamo G, Gonzalez C and Motta AB: The mechanisms involved in
the action of metformin in regulating ovarian function in
hyperandrogenized mice. Mol Hum Reprod. 12:475–481. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sobin LH and Compton CC: TNM seventh
edition: What's new, what's changed: Communication from the
international union against cancer and the American Joint Committee
on Cancer. Cancer. 116:5336–5339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sabit H, Abdel-Ghany SE, M Said OA,
Mostafa MA and El-Zawahry M: Metformin reshapes the methylation
profile in breast and colorectal cancer cells. Asian Pac J Cancer
Prev. 19:2991–2999. 2018.PubMed/NCBI
|
|
39
|
Yue W, Zheng X, Lin Y, Yang CS, Xu Q,
Carpizo D, Huang H, DiPaola RS and Tan XL: Metformin combined with
aspirin significantly inhibit pancreatic cancer cell growth in
vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and
Bcl-2. Oncotarget. 6:21208–21224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hodges V, Tucci M and Benghuzzi H: The
effects of metformin and EGCG on PANC-1 cell survival. Biomed Sci
Instrum. 51:393–399. 2015.PubMed/NCBI
|
|
41
|
Liu K, Wang G, Ding H, Chen Y, Yu G and
Wang J: Downregulation of metastasis suppressor 1(MTSS1) is
associated with nodal metastasis and poor outcome in Chinese
patients with gastric cancer. BMC Cancer. 10:4282010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hwang JT, Ha J, Park IJ, Lee SK, Baik HW,
Kim YM and Park OJ: Apoptotic effect of EGCG in HT-29 colon cancer
cells via AMPK signal pathway. Cancer Lett. 247:115–121. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Okubo K, Isono M, Asano T and Sato A:
Metformin augments Panobinostat's Anti-bladder cancer activity by
activating AMP-activated protein kinase. Transl Oncol. 12:669–682.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lipscombe LL, Goodwin PJ, Zinman B,
McLaughlin JR and Hux JE: Diabetes mellitus and breast cancer: A
retrospective population-based cohort study. Breast Cancer Res
Treat. 98:349–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dowling RJ, Niraula S, Stambolic V and
Goodwin PJ: Metformin in cancer: Translational challenges. J Mol
Endocrinol. 48:R31–R43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen X, Li C, He T, Mao J, Li C, Lyu J and
Meng QH: Metformin inhibits prostate cancer cell proliferation,
migration, and tumor growth through upregulation of PEDF
expression. Cancer Biol Ther. 17:507–514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation-implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Iglesias DA, Yates MS, van der Hoeven D,
Rodkey TL, Zhang Q, Co NN, Burzawa J, Chigurupati S, Celestino J,
Bowser J, et al: Another surprise from Metformin: Novel mechanism
of action via K-Ras influences endometrial cancer response to
therapy. Mol Cancer Ther. 12:2847–2856. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sarfstein R, Friedman Y, Attias-Geva Z,
Fishman A, Bruchim I and Werner H: Metformin downregulates the
insulin/IGF-I signaling pathway and inhibits different uterine
serous carcinoma (USC) cells proliferation and migration in
p53-dependent or -independent manners. PLoS One. 8:e615372013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhou Y, Xu JN, Zeng C, Li X, Zhou YF, Qi Y
and Xue Q: Metformin suppresses prostaglandin E2-induced cytochrome
P450 aromatase gene expression and activity via stimulation of
AMP-activated protein kinase in human endometriotic stromal cells.
Reprod Sci. 22:1162–1170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bansal K, Narayana Y and Balaji KN:
Inhibition of TNF-alpha-induced cyclooxygenase-2 expression by
Mycobacterium bovis BCG in human alveolar epithelial A549 cells.
Scand J Immunol. 69:11–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zidar N, Odar K, Glavac D, Jerse M, Zupanc
T and Stajer D: Cyclooxygenase in normal human tissues-is COX-1
really a constitutive isoform, and COX-2 an inducible isoform. J
Cell Mol Med. 13:3753–3763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rodrigues S, Bruyneel E, Rodrigue CM,
Shahin E and Gespach C: Cyclooxygenase 2 and carcinogenesis. Bull
Cancer. 91:Spec No: S61-S76, 2004 (In French).
|
|
58
|
Jørgensen SB, Jensen TE and Richter EA:
Role of AMPK in skeletal muscle gene adaptation in relation to
exercise. Appl Physiol Nutr Metab. 32:904–911. 2007. View Article : Google Scholar
|
|
59
|
Röckl KS, Witczak CA and Goodyear LJ:
Signaling mechanisms in skeletal muscle: Acute responses and
chronic adaptations to exercise. IUBMB Life. 60:145–153. 2008.
View Article : Google Scholar
|
|
60
|
Huang SP, Wu MS, Shun CT, Wang HP, Hsieh
CY, Kuo ML and Lin JT: Cyclooxygenase-2 increases hypoxia-inducible
factor-1 and vascular endothelial growth factor to promote
angiogenesis in gastric carcinoma. J Biomed Sci. 12:229–241. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cianchi F, Cortesini C, Bechi P, Fantappiè
O, Messerini L, Vannacci A, Sardi I, Baroni G, Boddi V, Mazzanti R
and Masini E: Up-regulation of cyclooxygenase 2 gene expression
correlates with tumor angiogenesis in human colorectal cancer.
Gastroenterology. 121:1339–1347. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hadad SM, Baker L, Quinlan PR, Robertson
KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG,
et al: Histological evaluation of AMPK signalling in primary breast
cancer. BMC Cancer. 9:3072009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang T, Wang X, He D, Jin X and Guo P:
Metformin sensitizes human bladder cancer cells to TRAIL-induced
apoptosis through mTOR/S6K1-mediated downregulation of c-FLIP.
Anticancer Drugs. 25:887–897. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Takabatake M, Daino K, Imaoka T, Nishimura
M, Morioka T, Fukushi M and Shimada Y: Aberrant expression and
phosphorylation of 4E-BP1, a main target of mTOR signaling, in rat
mammary carcinomas: an association with etiology. In Vivo.
25:853–860. 2011.PubMed/NCBI
|
|
65
|
Coleman LJ, Peter MB, Teall TJ, Brannan
RA, Hanby AM, Honarpisheh H, Shaaban AM, Smith L, Speirs V,
Verghese ET, et al: Combined analysis of eIF4E and 4E-binding
protein expression predicts breast cancer survival and estimates
eIF4E activity. Br J Cancer. 100:1393–1399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Akcakanat A, Sahin A, Shaye AN, Velasco MA
and Meric-Bernstam F: Comparison of Akt/mTOR signaling in primary
breast tumors and matched distant metastases. Cancer.
112:2352–2358. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bort A, Quesada S, Ramos-Torres Á,
Gargantilla M, Priego EM, Raynal S, Lepifre F, Gasalla JM,
Rodriguez-Henche N, Castro A and Díaz-Laviada I: Identification of
a novel 2-oxindole fluorinated derivative as in vivo antitumor
agent for prostate cancer acting via AMPK activation. Sci Rep.
8:43702018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rehman G, Shehzad A, Khan AL and Hamayun
M: Role of AMP-activated protein kinase in cancer therapy. Arch
Pharm (Weinheim). 347:457–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lee MS, Han HJ, Han SY, Kim IY, Chae S,
Lee CS, Kim SE, Yoon SG, Park JW, Kim JH, et al: Loss of the E3
ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome
through AMPK activation. Nat Commun. 9:34042018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Janzen NR, Whitfield J and Hoffman NJ:
Interactive roles for AMPK and glycogen from cellular energy
sensing to exercise metabolism. Int J Mol Sci. 19:33442018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jiralerspong S, Palla SL, Giordano SH,
Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi
GN and Gonzalez-Angulo AM: Metformin and pathologic complete
responses to neoadjuvant chemotherapy in diabetic patients with
breast cancer. J Clin Oncol. 27:3297–3302. 2009. View Article : Google Scholar : PubMed/NCBI
|