Introduction
Colorectal cancer (CRC) is the third most common
cancer worldwide and the second most common cancer in Europe,
causing an estimated 9.4% of all cancer deaths in Europe. CRC is
also a serious societal problem in Slovakia, with an incidence rate
of 15,7% and a mortality rate of 15,6% [(1,2) GLOBOCAN
2020 data]. Many risk factors and causes are associated with the
likelihood of developing CRC, but the main reason is still not
fully understood. The considerable geographical variability
suggests that CRC is a complex polygenic disease caused by genetic
and environmental factors and their interactions. Age, sex,
lifestyle, and dietary habits (3,4),
including meat and alcohol consumption (5,6), tobacco
smoking (7–9), obesity and lack of physical activity
(4,10,11) play
a major role in the pathogenesis of CRC. Other well-known risk
factors may also be inflammatory bowel diseases, acromegaly, renal
transplantation with long-term immunosuppression, diabetes mellitus
and insulin resistance, cholecystectomy, or androgen deprivation
therapy (3,4).
Beside these, inherited susceptibility plays a
significant role in the etiology of CRC because it can be
responsible for about 35% of all cases of colorectal cancer.
However, high-penetrance germline variants in known genes (APC,
BRCA2, KRAS, NTS, SMAD4, POLE, BRAF, BMPR1A, POLD1, STK11,
MUTYH and DNA mismatch repair genes), which are associated with
severe hereditary syndromes, such as familial adenomatous polyposis
and Lynch syndrome (also called hereditary non-polyposis colorectal
cancer), account for only 5–7% of total CRC cases (4,12).
Therefore, the remaining unknown heritability is probably explained
by the interaction of common, low-penetrance variants identified
through genome-wide association studies (GWAS). GWAS in
ethnic/racial minority populations offers the opportunity to
uncover genetic susceptibility factors and discover new genomic
regions and loci that contribute risk for CRC development. Since
2007, more than 100 common risk variants have been successfully
identified in GWAS, which have helped to elucidate the etiology of
CRC (13–18).
Lynch syndrome (LS) is an autosomal dominant
hereditary cancer syndrome that accounts for approximately 3% of
all colorectal cancer cases (19).
From a clinical point of view, 10–82% (20) of LS cases are associated with a
lifetime risk of developing CRC, unless the risk is significantly
lower in other types of cancer (21,22). LS
is caused by pathogenic germline mutations in a class of genes
called DNA mismatch repair (MMR) genes, mainly MLH1, located
in 3p22.2 chromosome, and MSH2, located in 2p21 chromosome
(23), which represent 70–85% of
cases of LS (24). Mutations found
in MSH6 (2p16.3), PMS2 (7p22.1) (25) and MLH3 genes have lower
incidence (26). Molecular
investigations have also shown that MSH3 (27) and germline 3′deletions of the
EPCAM gene, which lead to epigenetic silencing of
MSH2 (28), are also
implicated in the pathogenesis of LS. As a consequence of MMR
pathway inactivation and loss of expression of MMR proteins, DNA
replication errors accumulate typically resulting in microsatellite
instability (MSI), which is generally detected in LS patients'
tumor tissues (29). The diagnosis
of LS involves three main steps, identification of patients and
their familial history that meet the Amsterdam or Bethesda
guidelines, presence of MSI in tumors and immunohistochemical
analysis (IHC) of MMR protein expression. A definitive diagnosis of
LS must be confirmed by detecting the germline mutations in MMR
genes (30).
Non-invasive prenatal testing (NIPT) based on
low-coverage massively parallel whole-genome sequencing of plasma
DNA from pregnant women generates a large amount of data that
provides the resources to investigate human genetic variations in
the population. In our previous studies, we described the re-use of
the data from NIPT for genome-scale population specific frequency
determination of small DNA variants (31) and CNVs (32). Since pregnant women represent a
relatively standard sample of the local female population, we
assumed this NIPT data could also be used in the population study
of CRC, the most common cancer in Slovakia. Some research
concerning on genomic analysis of plasma from NIPT has also
demonstrated NIPT data's efficiency and utility for viral genetic
studies (33), genetic profiling of
Vietnamese population (34) or
detection of CNV aberrations (32,35).
The main aim of our study was a detailed analysis of
common variants (MAF >0,05) that showed evidence of association
with CRC in GWAS datasets and characterization of population
variability from data generated by NIPT. We assumed that the
genetic factors, mainly the increased specific population frequency
of CRC and LS variants could be responsible for the high incidence
of CRC in Slovakia. To test this hypothesis, allele frequencies of
risk CRC variants identified in the Slovak population were compared
with allele frequencies of risk CRC variants in 6 worldwide
populations. As LS is among the most common hereditary CRC
syndromes, the aim of our study was also to analyze population
allele frequencies and describe clinical impacts of relevant
variants located in known LS predisposing genes. To our knowledge,
this was the first population study of CRC using NIPT data
conducted exclusively in the Slovak population.
Materials and methods
Data source
The laboratory procedure used, to generate the NIPT
data, were as follows: DNA from plasma of peripheral maternal blood
was isolated for NIPT analysis from 1,501 pregnant women after
obtaining a written informed consent consistent with the Helsinki
declaration from the subjects. The population cohort consisted from
women in reproductive age between 17–48 years with a median of 35
years. Genomic information from a sample consisted of maternal and
fetal DNA fragments. Each included individual agreed to use their
genomic data in an anonymized form for general biomedical research.
The NIPT study (study ID 35900_2015) was approved by the Ethical
Committee of the Bratislava Self-Governing Region (Sabinovska
ul.16, 820 05 Bratislava) on 30th April of 2015 under the decision
ID 03899_2015. Blood samples were collected to EDTA tubes and
plasma was separated in dual centrifugation procedure. DNA was
isolated from 700 µl of plasma using DNA Blood Mini kit (Qiagen)
according to standard protocol. Sequencing libraries were prepared
from each sample using TruSeq Nano kit HT (Illumina) following
standard protocol with omission of DNA fragmentation step. Each
sample was normalized to 4 nm library and the final concentration
of libraries was 2,8 pM. Individual barcode labelled libraries were
pooled and sequenced using low-coverage whole-genome sequencing on
an Illumina NextSeq500 platform (Illumina) by performing paired end
sequencing of 2×35 bases (36).
Data analysis
The datasets generated and/or analyzed during the
current study are available in the DSpace repository, https://dspace.uniba.sk/xmlui/handle/123456789/27
(31).
Analyses of common variants previously
reported to be risk variants for CRC
We combined genotype data from all previously
reported GWAS studies available online (https://www.gwascentral.org/) for the years 2007–2020,
specifically 66 GWAS studies of CRC risk variants that included
individuals with European, Asian and African American ancestry.
Using data from these GWAS datasets, we identified 116 risk
variants associated with CRC, which were then merged with our data
of identified variants from NIPT. Risk variants that were not found
in NIPT data were excluded from the analysis. All identified
variants in the Slovak population used for further analyses were
common (MAFs >0.05). Subsequently, allele frequencies of CRC
risk variants for each population (East Asian, South Asian,
African, American, Finnish European and non-Finnish European) were
extracted from the gnomAD database available online (v3.0,
downloaded from http://gnomad.broadinstitute.org/downloads) and
compared with our frequencies determined for the Slovak population
from NIPT data. Allele frequency in each population and allele
frequency differences were plotted using boxplots. Outliers of
boxplots that represent variants with highly different frequencies
between Slovak and non-Finnish populations were annotated via
published literature and studies [in dbSNP (https://www.ncbi.nlm.nih.gov/snp/) and GWAS
(https://www.gwascentral.org/)]. To
assess the relations between allele frequency of CRC risk variants
in each population, we also used Principal Component Analysis (PCA)
using matplotlib.pyplot library, which reduces the dimension of the
data to a graphically interpretable 2D or 3D dimension.
Consequently, we obtained information on which populations have
similar or different allele frequencies of the identified CRC risk
variants.
Analyses of variants located in genes
associated with LS
After analyzing variants associated with CRC, we
focused on the study of variants associated with LS. First, we
filtered out a group of variants located in 7 genes known to be
associated with LS (MLH1, PMS2, MSH6, MLH3, MSH2, TGFBR2,
EPCAM). The genomic locations of genes were determined by the
GeneCards database (https://www.genecards.org/). From the dataset of
identified variants in LS associated genes, we excluded variants in
low complexity genomic regions (soft masked in the reference FASTA
file), eliminating the variants that could represent sequencing
artifacts or repetitive regions. All variants that were used for
further analysis were annotated using Ensembl Variant Effect
Predictor (VEP, version 101_GRCh38). In our dataset, based on
ClinVar database annotation of the most common types of pathogenic
and likely pathogenic variants associated with LS (https://www.ncbi.nlm.nih.gov/clinvar),
we selected variants including frameshift, missense, nonsense,
splice site, non-coding and UTR variants. After this filtering,
allele frequencies for both groups of variants (all variants
identified in LS genes and selected types of variants) for each
population (East Asian, South Asian, African, American, Finnish
European and non-Finnish European) were extracted from the gnomAD
database (v3.0, downloaded from http://gnomad.broadinstitute.org/downloads) and
compared with our frequencies determined for the Slovak population
from NIPT data. Allele frequency in each population and allele
frequency differences were plotted using boxplots and PCA analysis
using matplotlib.pyplot library. Outliers of boxplots representing
variants with allele frequency differences more than 10% were
annotated via published literature and studies [in dbSNP
(https://www.ncbi.nlm.nih.gov/snp/)
and GWAS (https://www.gwascentral.org/].
Results
Analyses of common variants previously
reported to be risk variants for CRC
In the analysis of the 66 GWAS studies that included
all identified risk variants associated with colorectal
carcinogenesis from 2007–2020, we investigated 116 variants. There
were 25 independent CRC risk variants in Asian population, 62 risk
variants found in European population, 27 risk variants in both
European and Asian population and 2 risk variants located in
African American population that were previously reported in GWAS
(Table I).
 | Table I.Identification of 116 risk variants
associated with colorectal cancer from 66 genome-wide association
studies between 2007 and 2020. |
Table I.
Identification of 116 risk variants
associated with colorectal cancer from 66 genome-wide association
studies between 2007 and 2020.
| First author,
year | rs_ID | Chr | POS | POP | Gene | PMID | (Refs.) |
|---|
| Wang et al,
2017 | rs7252505 | 19q13 | 33,084,158 | AFR A | GPATCH1 | 28295283 | (46) |
| Wang et al,
2017 | rs56848936 | 19q13.3 | 46,321,507 | AFR A | SYMPK | 28295283 | (46) |
| Lu et al,
2019 | rs7542665 | 1p31.3 | 62,673,037 | ASN | L1TD1 | 30529582 | (13) |
| Law et al,
2019 | rs12143541 | 1p32.3 | 55,247,852 | ASN | TTC22 |
31089142 | (43) |
| Lu et al,
2019 | rs201395236 | 1q44 | 245,181,421 | ASN | EFCAB2 | 30529582 | (13) |
| Lu et al,
2019 | rs7606562 | 2p16.3 | 48,686,695 | ASN | PPP1R21 | 30529582 | (13) |
| Lu et al,
2019 | rs113569514 | 3q22.2 | 133,748,789 | ASN | SLCO2A1 | 30529582 | (13) |
| Lu et al,
2019 | rs12659017 | 5q23.2 | 125,988,175 | ASN | ALDH7A1, PHAX | 30529582 | (13) |
| Law et al,
2019 | rs639933 | 5q31.1 | 134,467,751 | ASN | C5orf66,
LOC105379188 | 31089142 | (43) |
| Jia et al,
2013 | rs647161 | 5q31.1 | 134,499,092 | ASN | PITX1 | 23263487 | (58) |
| Law et al,
2019 | rs6933790 | 6p21.1 | 41,672,769 | ASN | TFEB | 31089142 | (43) |
| Zeng et al,
2016 | rs4711689 | 6p21.1 | 41,692,812 | ASN | TFEB | 26965516 | (14) |
| Schmit et
al, 2018 | rs6906359 | 6p21.31 | 35,528,378 | ASN | FKBP5 | 29917119 | (18) |
| Lu et al,
2019 | rs3830041 | 6p21.32 | 32,191,339 | ASN | NOTCH4 | 30529582 | (13) |
| Lu et al,
2019 | rs6584283 | 10q24.2 | 101,290,301 | ASN | NKX2-3 | 30529582 | (13) |
| Lu et al,
2019 | rs77969132 | 12p11.21 | 31,594,813 | ASN | DENND5B | 30529582 | (13) |
| Zeng et al,
2016 | rs11064437 | 12p13.31 | 6,982,162 | ASN | SPSB2 | 26965516 | (14) |
| Lu et al,
2019 | rs2730985 | 12q12 | 43,130,624 | ASN | PRICKLE1 | 30529582 | (13) |
| Lu et al,
2019 | rs1886450 | 13q22.1 | 73,986,628 | ASN | KLF5, KLF12 | 30529582 | (13) |
| Lu et al,
2019 | rs4341754 | 16q23.2 | 80,039,621 | ASN | WWOX, MAF | 30529582 | (13) |
| Lu et al,
2019 | rs1078643 | 17p12 | 10,707,241 | ASN | PIRT | 30529582 | (13) |
| Law et al,
2019 | rs73975588 | 17p13.3 | 816,741 | ASN | NXN | 31089142 | (43) |
| Law et al,
2019 | rs9797885 | 19q13.2 | 41,873,001 | ASN | TMEM91 | 31089142 | (43) |
| Law et al,
2019 | rs6055286 | 20p12.3 | 7,718,045 | ASN | None | 31089142 | (43) |
| Jia et al,
2008 | rs2423279 | 20p12.3 | 7,812,350 | ASN | HAO1 | 23263487 | (58) |
| Law et al,
2019 | rs2179593 | 20q13.12 | 42,660,286 | ASN | TOX2 | 31089142 | (43) |
| Lu et al,
2019 | rs13831 | 20q13.32 | 57,475,191 | ASN | GNAS | 30529582 | (13) |
| Law et al,
2019 | rs61776719 | 1p34.3 | 38,461,319 | EUR | None | 31089142 | (43) |
| Peters et
al, 2013 | rs10911251 | 1q25.3 | 183,112,059 | EUR | LAMC1 | 23266556 | (59) |
| Whiffin et
al, 2014 | rs10911251 | 1q25.3 | 183,112,059 | EUR | LAMC1 | 24737748 | (60) |
| Houlston et
al, 2010 | rs6687758 | 1q41 | 222,164,948 | EUR | None | 20972440 | (61) |
| Houlston et
al, 2010 | rs6691170 | 1q41 | 222,045,446 | EUR | DUSP10 | 20972440 | (61) |
| Law et al,
2019 | rs11692435 | 2q11.2 | 98,275,354 | EUR | ACTR1B | 31089142 | (43) |
| Law et al,
2019 | rs11893063 | 2q33.1 | 199,601,925 | EUR | LOC105373831 | 31089142 | (43) |
| Law et al,
2019 | rs7593422 | 2q33.1 | 200,131,695 | EUR | None | 31089142 | (43) |
| Orlando et
al, 2016 | rs992157 | 2q35 | 219,154,781 | EUR | TMBIM1 | 27005424 | (62) |
| Law et al,
2019 | rs9831861 | 3p21.1 | 53,088,285 | EUR | None | 31089142 | (43) |
| Law et al,
2019 | rs12635946 | 3q13.2 | 112,916,918 | EUR | None | 31089142 | (43) |
| Houlston et
al, 2010 | rs10936599 | 3q26.2 | 169,774,313 | EUR | MYNN | 20972440 | (61) |
| Schmit et
al, 2018 | rs1370821 | 4q22.2 | 94,943,383 | EUR | None | 29917119 | (18) |
| Law et al,
2019 | rs17035289 | 4q24 | 106,048,291 | EUR | None | 31089142 | (43) |
| Law et al,
2019 | rs75686861 | 4q31.21 | 145,621,328 | EUR | HHIP | 31089142 | (43) |
| Schmit et
al, 2014 | rs35509282 | 4q32.2 | 163,333,405 | EUR | FSTL5 | 25023989 | (63) |
| Schmit et
al, 2018 | rs58791712 | 5p13.1 | 40,281,797 | EUR | PTGER4 | 29917119 | (18) |
| Schmit et
al, 2018 | rs2735940 | 5p15.33 | 1,296,486 | EUR | TERT | 29917119 | (18) |
| Peters et
al, 2012 | rs2853668 | 5p15.33 | 1299910 | EUR | TERT | 21761138 | (64) |
| Schmit et
al, 2018 | rs62404968 | 6p12.1 | 55,714,314 | EUR | BMP5 | 29917119 | (18) |
| Dunlop et
al, 2012 | rs1321311 | 6p21.2 | 36,622,900 | EUR | CDKN1A | 22634755 | (48) |
| Law et al,
2019 | rs9271770 | 6p21.32 | 32,594,248 | EUR | LOC107987449 | 31089142 | (43) |
| Law et al,
2019 | rs3131043 | 6p21.33 | 30,758,466 | EUR | HCG20 | 31089142 | (43) |
| Law et al,
2019 | rs2070699 | 6p24.1 | 12,292,772 | EUR | EDN1 | 31089142 | (43) |
| Law et al,
2019 | rs6928864 | 6q21 | 105,966,894 | EUR | None | 31089142 | (43) |
| Law et al,
2019 | rs10951878 | 7p12.3 | 46,926,695 | EUR | None | 31089142 | (43) |
| Law et al,
2019 | rs3801081 | 7p12.3 | 47,511,161 | EUR | TNS3 | 31089142 | (43) |
| Tomlinson et
al, 2008 | rs16892766 | 8q23.3 | 117,630,683 | EUR | EIF3H | 18372905 | (64) |
| Law et al,
2019 | rs1412834 | 9p21.3 | 22,110,131 | EUR | CDKN2B-AS1 | 31089142 | (43) |
| Schmit et
al, 2018 | rs10994860 | 10q11.23 | 52,645,424 | EUR | A1CF | 29917119 | (18) |
| Al Tassan et
al, 2015 | rs10904849 | 10p13 | 16,955,267 | EUR | CUBN | 25990418 | (15) |
| Law et al,
2019 | rs4450168 | 11p15.4 | 10,286,755 | EUR | SBF2 | 31089142 | (43) |
| Dunlop et
al, 2012 | rs3824999 | 11q13.4 | 74,345,550 | EUR | POLD3 | 22634755 | (48) |
| Tenesa et
al, 2008 | rs3802842 | 11q23.1 | 111,171,709 | EUR | COLCA2 | 18372901 | (66) |
| Houlston et
al, 2010 | rs7136702 | 12q13.12 | 50,880,216 | EUR | LARP4 | 20972440 | (61) |
| Law et al,
2019 | rs7398375 | 12q13.3 | 57,540,848 | EUR | LRP1 |
31089142 | (43) |
| Schmit et
al, 2018 | rs72013726 | 12q24.21 | 115,890,835 | EUR | MED13L | 29917119 | (18) |
| Schmit et
al, 2018 | rs10161980 | 13q13.2 | 34,093,518 | EUR | STARD13 | 29917119 | (18) |
| Law et al,
2019 | rs12427600 | 13q13.3 | 37,460,648 | EUR | SMAD9 | 31089142 | (43) |
| Law et al,
2019 | rs45597035 | 13q22.1 | 73,649,152 | EUR | KLF5 | 31089142 | (43) |
| Law et al,
2019 | rs1330889 | 13q22.3 | 78,609,615 | EUR | LINC00446 | 31089142 | (43) |
| Law et al,
2019 | rs7993934 | 13q34 | 111,074,915 | EUR | COL4A2 | 31089142 | (43) |
| Tomlinson et
al, 2011 | rs1957636 | 14q22.2 | 54,560,018 | EUR | BMP4 | 21655089 | (67) |
| Houlston et
al, 2008 | rs4444235 | 14q22.2 | 54,410,919 | EUR | BMP4 | 19011631 | (68) |
| Tomlinson et
al, 2011 | rs4444235 | 14q22.2 | 54,410,919 | EUR | BMP4 | 21655089 | (67) |
| Tomlinson et
al, 2008 | rs11632715 | 15q13.3 | 33,004,247 | EUR | None | 18372905 | (65) |
| Tomlinson et
al, 2008 | rs16969681 | 15q13.3 | 32,993,111 | EUR | SCG5 | 18372905 | (65) |
| Tomlinson et
al, 2011 | rs16969681 | 15q13.3 | 32,993,111 | EUR | SCG5 | 21655089 | (67) |
| Tomlinson et
al, 2008 | rs4779584 | 15q13.3 | 32,994,756 | EUR | CRAC1 | 18372905 | (65) |
| Law et al,
2019 | rs4776316 | 15q22.31 | 67,007,813 | EUR | SMAD6 | 31089142 | (43) |
| Law et al,
2019 | rs10152518 | 15q23 | 68,177,162 | EUR | None | 31089142 | (43) |
| Law et al,
2019 | rs7495132 | 15q26.1 | 91,172,901 | EUR | CRTC3 | 31089142 | (43) |
| Houlston et
al, 2008 | rs9929218 | 16q22.1 | 68,820,946 | EUR | CDH1 | 19011631 | (68) |
| Law et al,
2019 | rs61336918 | 16q23.2 | 80,007,266 | EUR | None | 31089142 | (43) |
| Schmit et
al, 2018 | rs2696839 | 16q24.1 | 86,340,448 | EUR | FOXF1 | 29917119 | (18) |
| Broderick et
al, 2007 | rs4939827 | 18q21 | 46,453,463 | EUR | SMAD7 | 17934461 | (69) |
| Tenesa et
al, 2008 | rs4939827 | 18q21 | 46,453,463 | EUR | SMAD7 | 18372901 | (66) |
| Law et al,
2019 | rs285245 | 19p13.11 | 16,420,817 | EUR | None | 31089142 | (43) |
| Houlston et
al, 2008 | rs10411210 | 19q13.11 | 33,532,300 | EUR | RHPN2 | 19011631 | (68) |
| Law et al,
2019 | rs12979278 | 19q13.33 | 49,218,602 | EUR | MAMSTR | 31089142 | (43) |
| Tomlinson et
al, 2011 | rs4813802 | 20p12.3 | 6,699,595 | EUR | BMP2 | 21655089 | (67) |
| Peters et
al, 2012 | rs4813802 | 20p12.3 | 6,699,595 | EUR | BMP2 | 21761138 | (64) |
| Houlston et
al, 2008 | rs961253 | 20p12.3 | 6,404,281 | EUR | BMP2 | 19011631 | (68) |
| Schmit et
al, 2018 | rs2295444 | 20q11.22 | 33,173,883 | EUR | PIGU | 29917119 | (18) |
| Schmit et
al, 2018 | rs1810502 | 20q13.13 | 49,057,488 | EUR | PTPN1 | 29917119 | (18) |
| Law et al,
2019 | rs3787089 | 20q13.33 | 62,316,630 | EUR | RTEL1 | 31089142 | (42) |
| Houlston et
al, 2010 | rs4925386 | 20q13.33 | 60,921,044 | EUR | LAMA5 | 20972440 | (61) |
| Schumacher et
al, 2015 | rs8124813 | 3p14.1 | 43,476,841 | EUR, ASN | LRIG1 | 26151821 | (70) |
| Schumacher et
al, 2015 | rs35360328 | 3p22.1 | 40,924,962 | EUR, ASN | CTNNB1 | 26151821 | (70) |
| Lu et al,
2019 | rs1476570 | 6p22.1 | 29,809,860 | EUR, ASN | HLA-G | 30529582 | (13) |
| Tomlinson et
al, 2008 | rs2450115 | 8q23.3 | 117,624,093 | EUR, ASN | EIF3H | 18372905 | (67) |
| Tomlinson et
al, 2008 | rs6469656 | 8q23.3 | 117,647,788 | EUR, ASN | EIF3H | 18372905 | (67) |
| Haiman et
al, 2007 | rs6983267 | 8q24.21 | 128,413,305 | EUR, ASN | POU5F1B | 17618282 | (71) |
| Tomlinson et
al, 2007 | rs6983267 | 8q24.21 | 128,413,305 | EUR, ASN | POU5F1B | 17618284 | (72) |
| Hutter et
al, 2010 | rs6983267 | 8q24.21 | 128,413,305 | EUR, ASN | POU5F1B | 21129217 | (73) |
| Cui et al,
2011 | rs6983267 | 8q24.21 | 128,413,305 | EUR, ASN | POU5F1B | 21242260 | (4) |
| Law et al,
2019 | rs12255141 | 10q25.2 | 114,294,892 | EUR, ASN | VTI1A |
31089142 | (43) |
| Zhang et al,
2014 | rs704017 | 10q22.3 | 80,819,132 | EUR, ASN | ZMIZ1 | 24836286 | (16) |
| Whiffin et
al, 2014 | rs1035209 | 10q24.2 | 101,345,366 | EUR, ASN | SLC25A28 | 24737748 | (60) |
| Zeng et al,
2016 | rs4919687 | 10q24.32 | 104,595,248 | EUR, ASN | CYP17A1 | 26965516 | (14) |
| Zhang et al,
2014 | rs11196172 | 10q25.2 | 114,726,843 | EUR, ASN | TCF7L2 | 24836286 | (16) |
| Wang et al,
2014 | rs12241008 | 10q25.2 | 114,280,702 | EUR, ASN | VTI1A | 25105248 | (75) |
| Zhang et al,
2014 | rs1535 | 11q12.2 | 61,597,972 | EUR, ASN | FADS2 | 24836286 | (16) |
| Zhang et al,
2014 | rs174550 | 11q12.2 | 61,571,478 | EUR, ASN | FADS1 | 24836286 | (16) |
| Zhang et al,
2014 | rs4246215 | 11q12.2 | 61,564,299 | EUR, ASN | FEN1 | 24836286 | (16) |
| Zhang et al,
2014 | rs174537 | 11q12.2 | 61,552,680 | EUR, ASN | MYRF | 24836286 | (16) |
| Law et al,
2019 | rs10849438 | 12p13.31 | 6,412,036 | EUR, ASN | None |
31089142 | (43) |
| Zhang et al,
2014 | rs10849432 | 12p13.31 | 6,385,727 | EUR, ASN | PLEKHG6 | 24836286 | (16) |
| Peters et
al, 2013 | rs3217810 | 12p13.32 | 4,388,271 | EUR, ASN | CCND2 | 23266556 | (59) |
| Whiffin et
al, 2014 | rs3217810 | 12p13.32 | 4,388,271 | EUR, ASN | CCND2 | 24737748 | (60) |
| Jia et al,
2013 | rs10774214 | 12p13.32 | 4,368,352 | EUR, ASN | CCND2 | 23263487 | (58) |
| Zhang et al,
2014 | rs12603526 | 17p13.3 | 800,593 | EUR, ASN | NXN | 24836286 | (16) |
| Zhang et al,
2014 | rs7229639 | 18q21.1 | 46,450,976 | EUR, ASN | SMAD7 | 24836286 | (16) |
| Zhang et al,
2014 | rs1800469 | 19q13.2 | 41,860,296 | EUR, ASN | TMEM91 | 24836286 | (16) |
| Zhang et al,
2014 | rs2241714 | 19q13.2 | 41,869,392 | EUR, ASN | B9D2 | 24836286 | (16) |
| Schumacher et
al, 2015 | rs606682520 | 20q13.13 | 897,353 | EUR, ASN | PREX1 | 26498495 | (70) |
| Schumacher et
al, 2015 | rs6066825 | 20q13.13 | 47,340,117 | EUR, ASN | PREX1 | 26151821 | (70) |
| Dunlop et
al, 2012 | rs5934683 | Xp22.2 | 9,751,474 | EUR, ASN | SHROOM2 | 22634755 | (48) |
After merging all identified variants from GWAS (116
risk variants) with our NIPT data, we identified 106 common risk
CRC variants (Table SI), while 10
risk variants that were not called in the Slovak population were
excluded from further analysis. The allele frequencies of 106
variants identified in our population sample (Slovak population)
and the allele frequencies of variants for 6 world populations
(East Asian, South Asian, African, American, Finnish European and
non-Finnish European) obtained by gnomAD database (Table SII) are shown in graphical
comparison by Boxplots (Fig. 1) and
principal component analysis (PCA) (Fig.
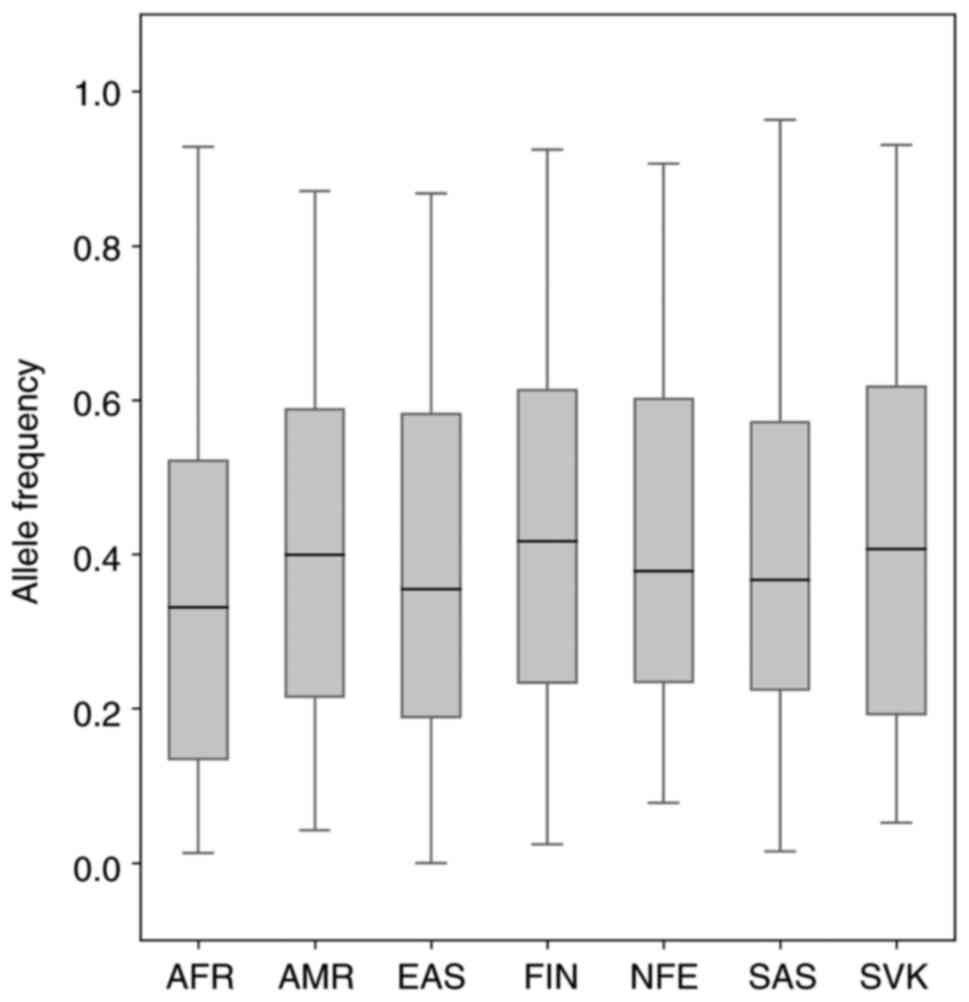
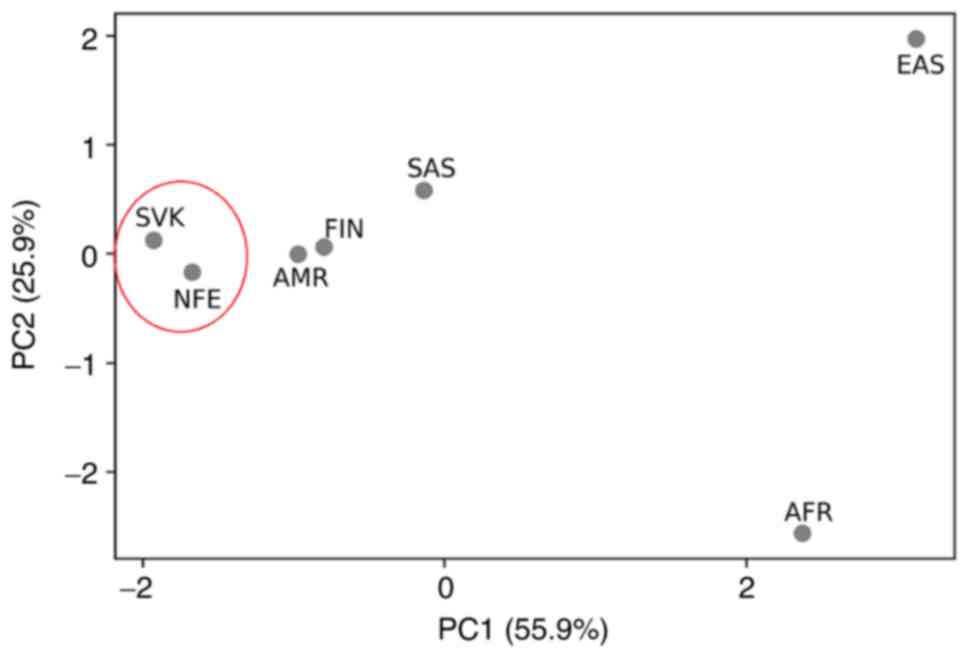
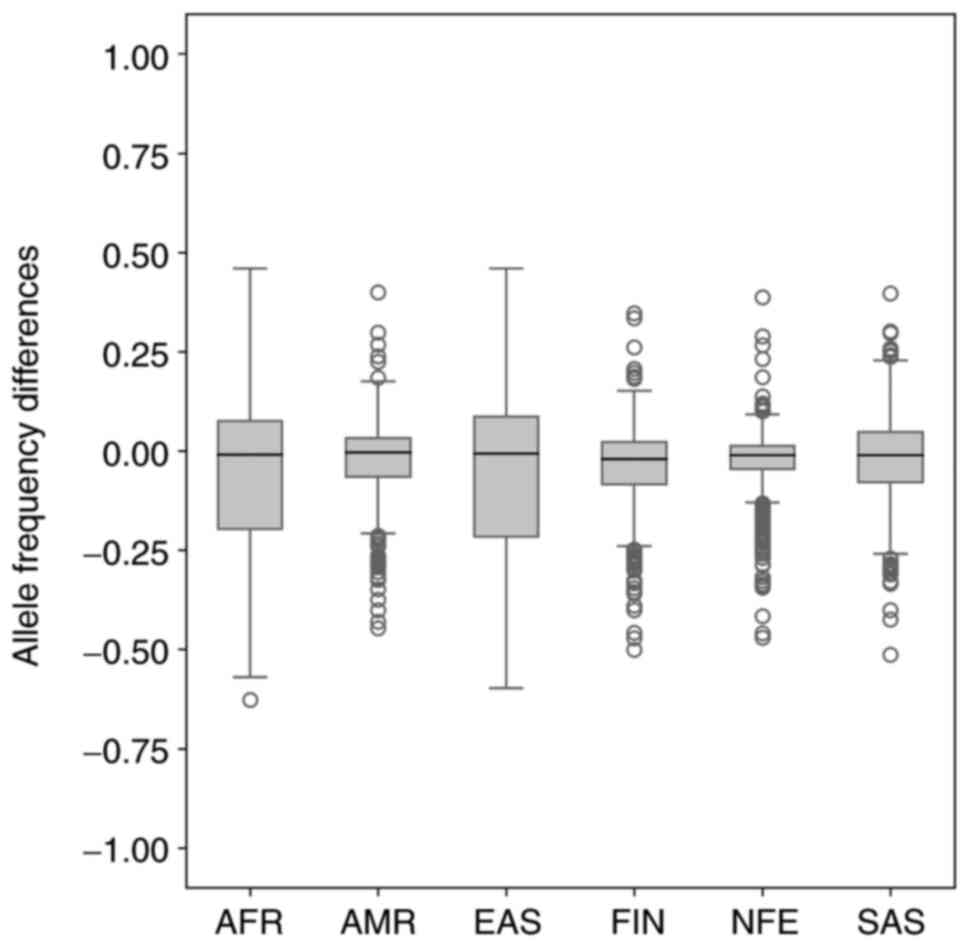
2). As shown in Fig. 1, the MAF
ranged from 0.0–0.963109 in 6 world populations that is comparable
with the Slovak population (0.0521–0.931). The median allele
frequency for the Slovak population reached the value of 0.4072,
which is closest to the value of the median of the American
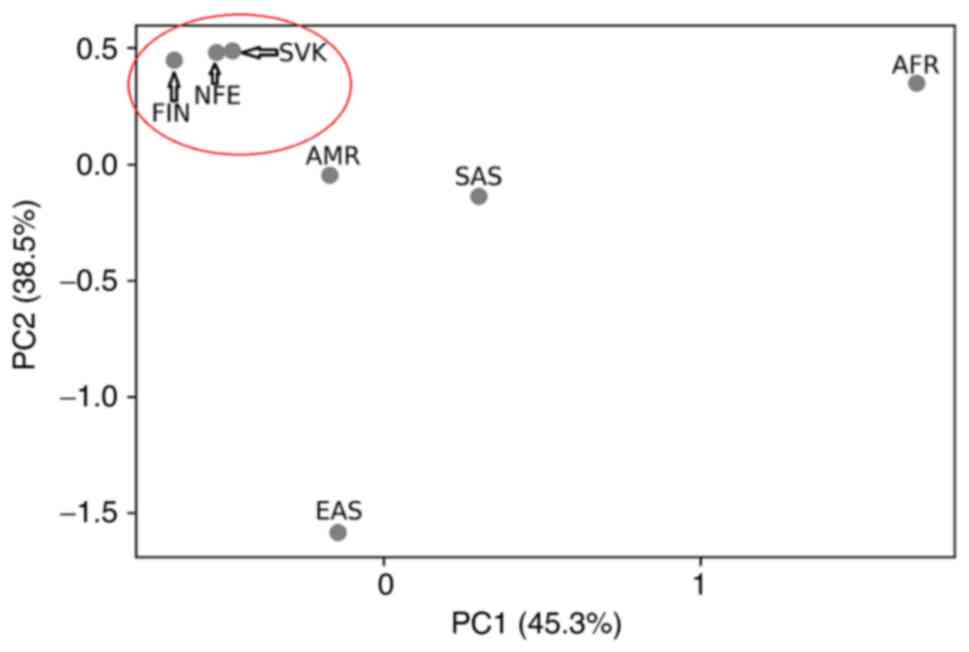
population (MED=0.3991) and Finnish population (MED=0.4166). PCA
placed our sample set most closely to the two gnomAD population
sample sets, i.e., to the Finnish and non-Finnish European
population.
Next, we compared known allele frequencies of 106
CRC risk variants in our sample set from the Slovak population to
allele frequencies of CRC variants in six world populations. The
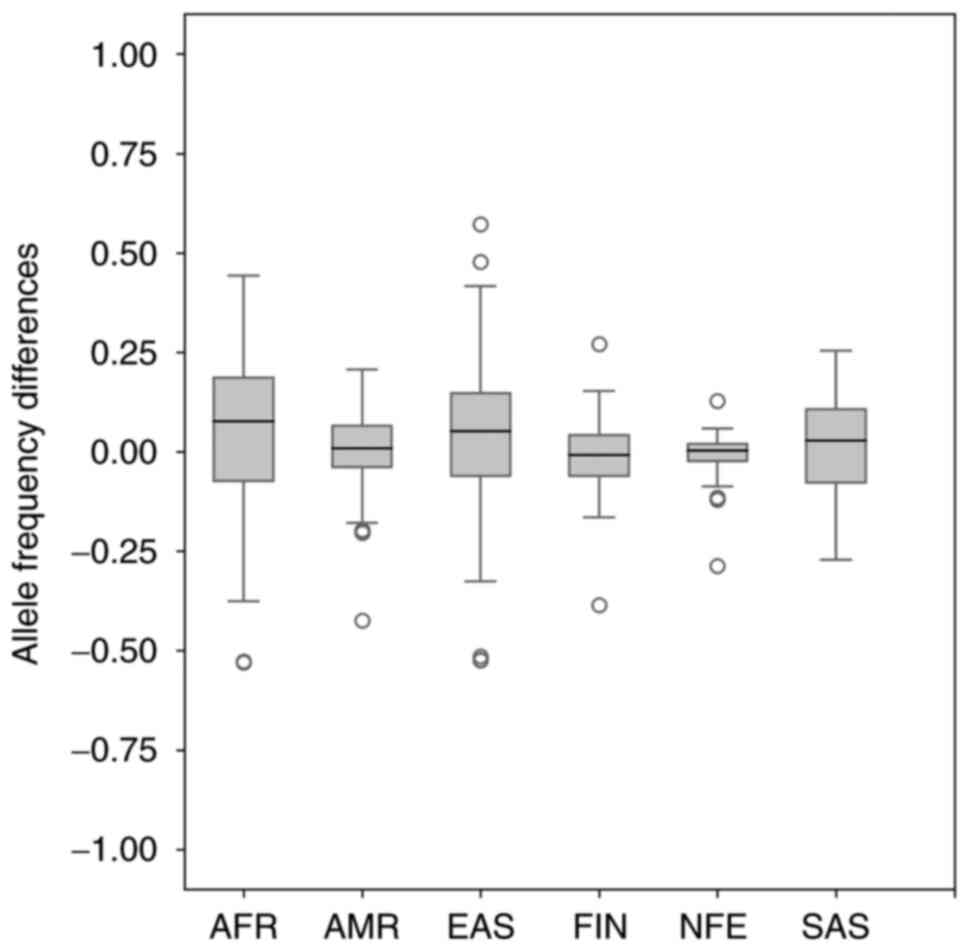
final findings of allele frequency differences are shown in
Fig. 3. The median allele frequency
for comparing the Slovak population and non-Finnish European
population reached the value of 0.002285. Together, we identified
14 outliers in Fig. 3 (3 of 14
variants reached a similar value and overlapped, so they are not
clearly visible in Fig. 3). Since
the same variant rs4246215 was identified in 4 different population
comparisons (Slovak-American, Slovak-Finnish European,
Slovak-non-Finnish European, and also identified in Slovak-East
Asian population comparison) and the rs3131043 variant in 2
population comparisons (Slovak-Finnish European and
Slovak-non-Finnish European population comparison), we identified a
total of 10 variants, whose difference in population allele
frequency was more than 10%. Table
II includes annotation information about these variants by
dbSNP NCBI, ClinVar database, and population comparison in which
they were identified.
| Table II.Outliers identified in boxplots that
show allele frequency differences of Slovak and the other six world
populations for 106 risk colorectal cancer variants identified from
genome-wide association studies. |
Table II.
Outliers identified in boxplots that
show allele frequency differences of Slovak and the other six world
populations for 106 risk colorectal cancer variants identified from
genome-wide association studies.
| rs_ID | Population
comparison | Chr | POS | Variant type | Gene | Consequence | Clinical
significance |
|---|
| rs5934683 | Slovak-East
Asian | chrX | 9783434 | SNV | GPR143 | Intron Variant | Not Reported in
ClinVar |
| rs7252505 | Slovak-African | chr19 | 33084158 | SNV | GPATCH1 | Intron Variant | Not Reported in
ClinVar |
| rs4779584 | Slovak-East
Asian | chr15 | 32702555 | SNV | None | None | Not Reported in
ClinVar |
| rs174550 |
Slovak-American | chr11 | 61804006 | SNV | FADS1 | Intron Variant | Not Reported in
ClinVar |
| rs4246215 |
Slovak-American | chr11 | 61796827 | SNV | FEN1 | 3′ UTR Variant | Not Reported in
ClinVar |
|
| Slovak-European
(Finnish) | chr11 | 61796827 | SNV | FEN1 | 3′ UTR Variant | Not Reported in
ClinVar |
|
| Slovak-European
(non-Finnish) | chr11 | 61796827 | SNV | FEN1 | 3′ UTR Variant | Not Reported in
ClinVar |
|
| Slovak-East
Asian | chr11 | 61796827 | SNV | FEN1 | 3′ UTR Variant | Not Reported in
ClinVar |
| rs10904849 | Slovak-European
(non-Finnish) | chr10 | 16955267 | SNV | CUBN | Intron Variant | Not Reported in
ClinVar |
| rs6928864 | Slovak-African | chr6 | 105519019 | SNV | None | None | Not Reported in
ClinVar |
| rs3131043 | Slovak-European
(non-Finnish) | chr6 | 30790689 | SNV | HCG20 | Intron Variant | Not Reported in
ClinVar |
|
| Slovak-European
(Finnish) | chr6 | 30790689 | SNV | HCG20 | Intron Variant | Not Reported in
ClinVar |
| rs12659017 | Slovak-East
Asian | chr5 | 126652483 | SNV | None | None | Not Reported in
ClinVar |
| rs397775554 | Slovak-European
(non-Finnish) | chr5 |
40281696-40281704 | Indel | None | None | Not Reported in
ClinVar |
Analyses of variants located in genes
associated with LS
In the analysis of LS, we identified 1212 variants
in our sample set from NIPT that were located in genes known to be
associated with LS, i.e., MLH1, PMS2, MSH6, TGFBR2, MLH3,
MSH2 and EPCAM. After excluding variants from low
complexity regions, we obtained 648 variants that were finally
annotated by VEP and used for further analysis. The allele
frequencies of 648 variants identified in our population sample
(Slovak population) and the allele frequencies of variants for 6
world populations (East Asian, South Asian, African, American,
Finnish European and non-Finnish European) obtained by gnomAD
database (Table SIII) are shown in
graphical comparison by Boxplots (Fig.
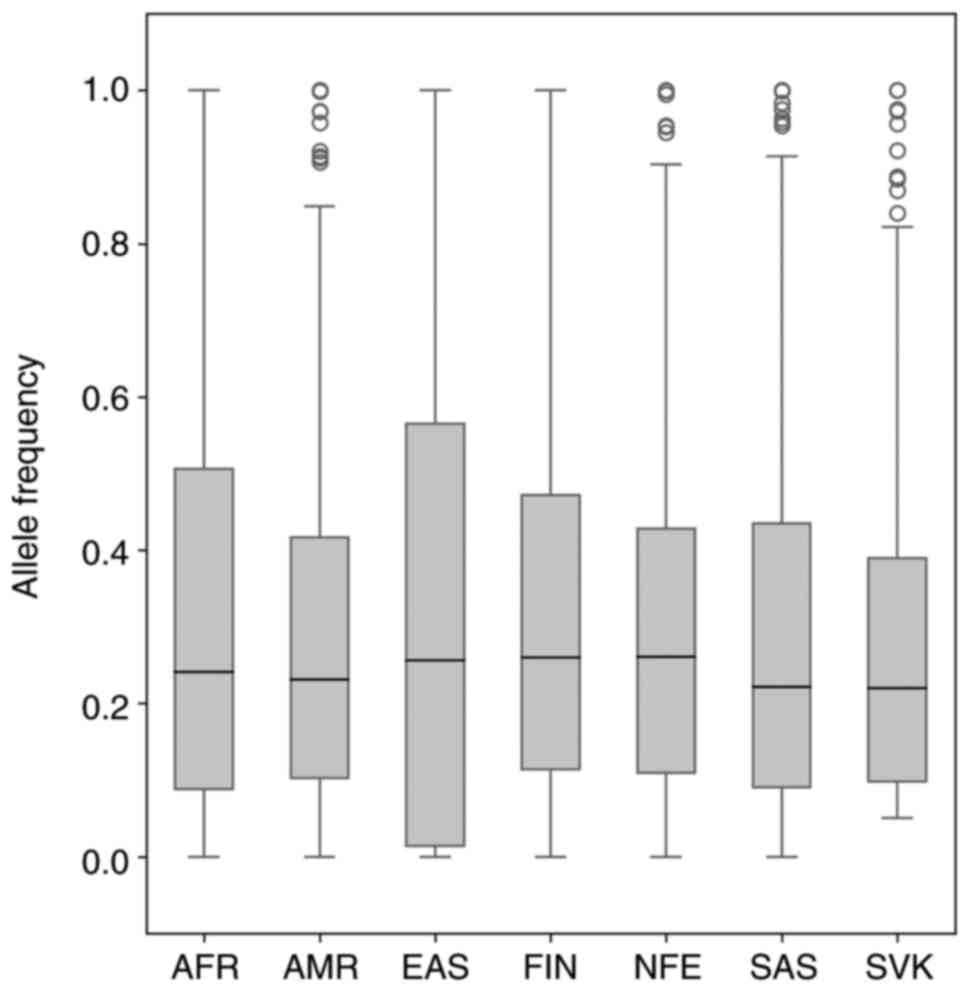
4) and principal component analysis (PCA) (Fig. 5). As shown in Fig. 4, the MAF ranged from 0.0–1.0 in 6
world populations. In the Slovak population, all variants were with
MAF>0.05 (0.0502–1.0). The median allele frequency for the
Slovak population reached the value of 0.2204, which is closest to
the value of the median of the South Asian population
(MED=0.221274). PCA placed our sample set most closely to the
non-Finnish European population (Fig.
5).
In the next step, to identify variants having
significantly different frequencies, we compared known allele
frequencies of 648 variants located in genes associated with LS
identified in our sample set from the Slovak population to allele
frequencies of these variants in six gnomAD world populations. The
final findings of allele frequency differences are shown in
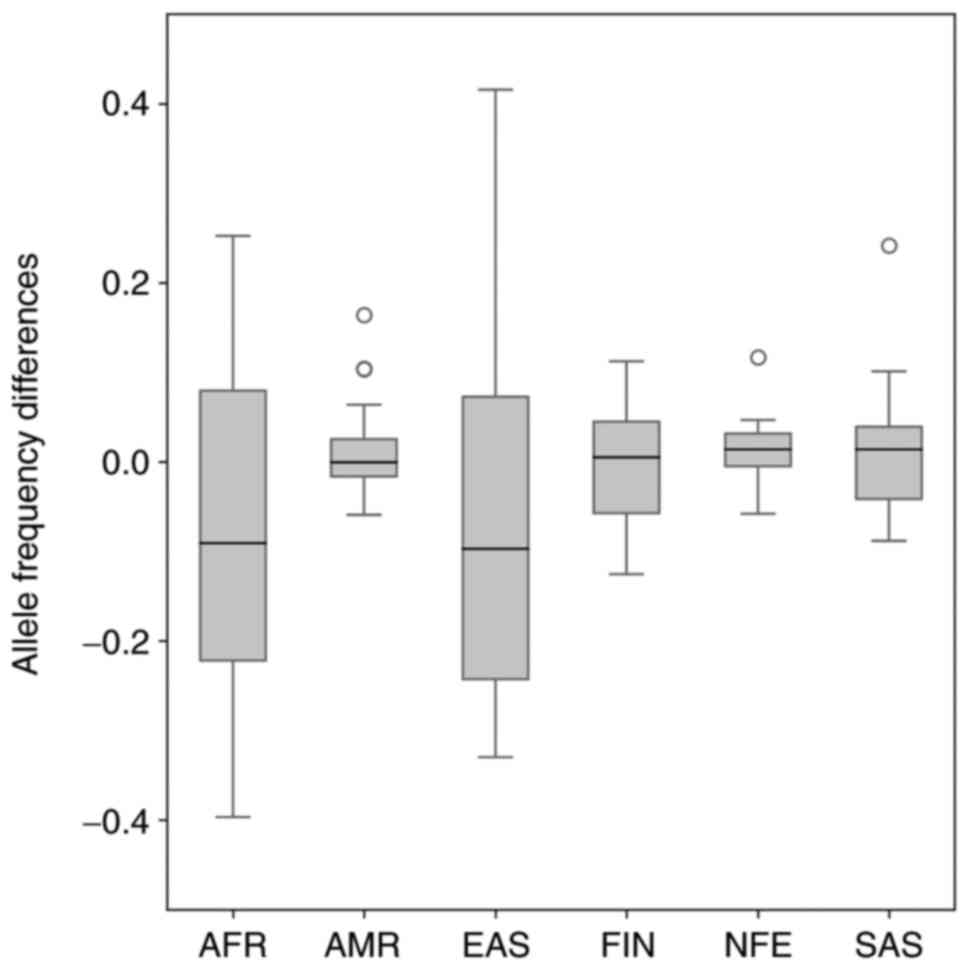
Fig. 6. The median allele frequency
for the comparison of the Slovak population and non-Finnish
European population reached the value of −0.01093. By comparing the
allele frequency of variants of the Slovak and non-Finnish
populations, we identified a total of 64 outliers. Most outliers
were found in the MSH2 gene, others in MSH6, TGFBR2,
PMS2, MLH1 and EPCAM. We did not identify any outlying
variant in the MLH3 gene. The variation type of all outliers
was ‘intronic variant’ and the clinical significance of all
outliers was not reported in ClinVar. All this annotation
information, also including chromosome position, variants ID,
reference and alternative allele, genes, is available in Table SIV.
Our analysis also included allele frequency
comparison of selected variants from 648 variants identified in our
sample set of NIPT data. We focused on frameshift, missense,
nonsense, splice site, non-coding and UTR variants, annotated in
the ClinVar database as the most common types of pathogenic and
likely pathogenic variants associated with LS. However, from these
selected types of variants, we found only UTR and non-coding
variants in our dataset of 648 variants. Other types of variants
(downstream, upstream, and intron) were excluded from further
analysis. Finally, we selected 18 variants, 10 UTR and 8 non-coding
variants (all selected variants with annotation information by VEP
and ClinVar are available in Table
III). We compared known allele frequencies of these 18 selected
variants identified in our population sample (Slovak population) to
the six gnomAD world populations. The final findings of allele
frequency differences are shown in Fig.
7. The median allele frequency for the comparison of the Slovak
population and non-Finnish European population reached the value of
0.014215, which is closest to the value of the median allele
frequency comparison of the South Asian and Slovak population
(MED=0.014186). By comparing the allele frequency of variants of
the Slovak and six gnomAD world population, we identified a total
of 4 outliers-rs10951973, rs10951972 (identified in Slovak-American
population comparison), rs6791557 (in Slovak-American and
Slovak-non-Finnish European population comparison) and rs9852378
(in Slovak-South Asian population comparison). All outliers were
non-coding variants, rs10951973 and rs10951972 located in the
PMS2 and rs6791557 located in the TGFBR2 were not
reported in ClinVar. The rs9852378 SNP, detected in the
MLH1, was reported as benign by ClinVar.
| Table III.Identification of 18 selected
variants (UTR and non-coding) from all 648 variants in genes
associated with Lynch syndrome from non-invasive prenatal testing
data in the Slovak population. |
Table III.
Identification of 18 selected
variants (UTR and non-coding) from all 648 variants in genes
associated with Lynch syndrome from non-invasive prenatal testing
data in the Slovak population.
| rs_ID | Chr | POS | Variant type | Gene | Consequence | Clinical
significance |
|---|
| rs11901645 | chr2 | 47510079 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs11891189 | chr2 | 47510259 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs876936 | chr2 | 47513059 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs72872839 | chr2 | 47565070 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs17036769 | chr2 | 47633503 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs2969774 | chr2 | 47661684 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs2952372 | chr2 | 47661919 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs2969773 | chr2 | 47662141 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs2705765 | chr2 | 47662389 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs12328344 | chr2 | 47662542 | SNV | MSH2 | 3′ UTR variant | Not reported in
ClinVar |
| rs10427209 | chr2 | 47709297 | SNV | MSH6 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs10427344 | chr2 | 47709476 | SNV | MSH6 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs3136240 | chr2 | 47784947 | SNV | MSH6 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs6791557 | chr3 | 30614676 | SNV | TGFBR2 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs1817338 | chr3 | 30631239 | SNV | TGFBR2 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs9852378 | chr3 | 36997280 | SNV | MLH1 | Non-coding
transcript exon variant | Benign in
ClinVar |
| rs10951972 | chr7 | 6002187 | SNV | PMS2 | Non-coding
transcript exon variant | Not reported in
ClinVar |
| rs10951973 | chr7 | 6002205 | SNV | PMS2 | Non-coding
transcript exon variant | Not reported in
ClinVar |
Finally, we analyzed allele frequencies of
pathogenic and likely pathogenic variants associated with Lynch
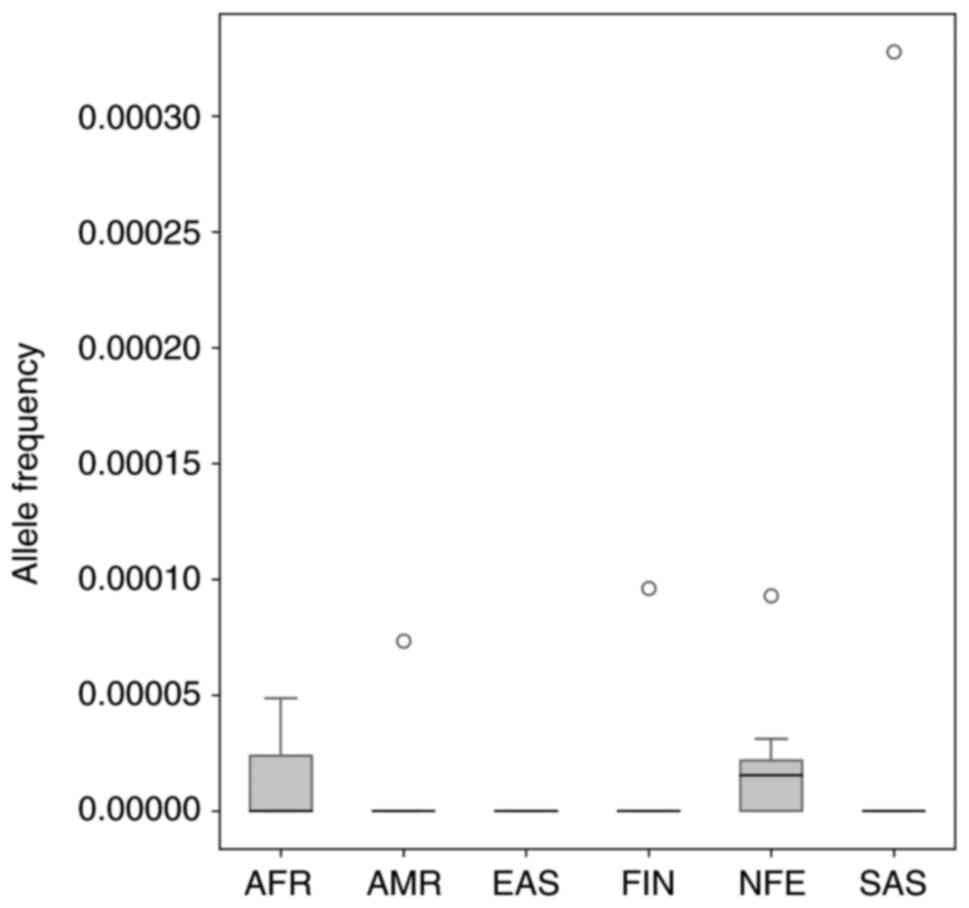
syndrome annotated in the ClinVar database. From 229 SNPs with
pathogenic and likely pathogenic clinical significance, only 15
have non-zero AF records in the gnomAD database. As shown in
Fig. 8, all found AF are
significantly below 5% (Table IV
and Fig. 8).
| Table IV.Identification of 15 pathogenic and
likely pathogenic variants associated with Lynch syndrome with
non-zero allele frequency in gnomAD database. |
Table IV.
Identification of 15 pathogenic and
likely pathogenic variants associated with Lynch syndrome with
non-zero allele frequency in gnomAD database.
|
|
|
|
|
| Allele frequency in
population |
|---|
|
|
|
|
|
|
|
|---|
| rs_ID | Chr | POS | REF Allele | ALT Allele | African | American | East Asian | European
(Finnish) | European
(non-Finnish) | South Asian |
|---|
| rs63750615 | 2 | 47403333 | G | T | 0 | 0 | 0 | 0 | 0 |
3.28×10−5 |
| rs1194793421 | 2 | 47414417 | AG | A | 0 | 0 | 0 | 0 |
2.75×10−5 | 0 |
| rs63750636 | 2 | 47476492 | C | T | 0 | 0 | 0 | 0 |
1.55×10−5 | 0 |
| rs63749873 | 2 | 47795903 | C | G | 0 | 0 | 0 | 0 |
3.10×10−5 | 0 |
| rs587783056 | 2 | 47799684 | GTT | G |
4.76×10−5 | 0 | 0 | 0 | 0 | 0 |
| rs63751017 | 2 | 47800714 | C | T | 0 | 0 | 0 | 0 |
3.10×10−5 | 0 |
| rs876660943 | 2 | 47806359 | G | T | 0 | 0 | 0 | 0 |
1.55×10−5 | 0 |
| rs63751221 | 3 | 37001045 | C | T |
2.38×10−5 | 0 | 0 | 0 | 0 | 0 |
| rs587779338 | 7 | 5977589 | G | A |
4.86×10−5 | 0 | 0 | 0 |
1.58×10−5 | 0 |
| rs267608161 | 7 | 5982885 | C | T |
4.80×10−5 | 0 | 0 |
9.61×10−5 |
1.55×10−5 | 0 |
| rs63751422 | 7 | 5986838 | G | A |
2.38×10−5 | 0 | 0 | 0 | 0 | 0 |
| rs63750250 | 7 | 5986933 | A | AT | 0 | 0 | 0 | 0 |
9.30×10−5 | 0 |
| rs200640585 | 7 | 5992018 | G | A | 0 | 0 | 0 | 0 |
1.55×10−5 | 0 |
| rs267608154 | 7 | 5995572 | ACTGT | A | 0 | 0 | 0 | 0 |
1.55×10−5 | 0 |
| rs63750871 | 7 | 6002590 | G | A | 0 |
7.33×10−5 | 0 | 0 | 0 | 0 |
Discussion
Population genetic studies currently have a huge
impact on the study of genomics (37). The detection of risk variants in a
population and identifying their genetic relationships have
advanced our understanding of the human genome's variability and
led to the elucidation of many factors that influence cancer risk.
In recent years, NGS technologies have played a key role in
colorectal cancer research and have become a useful tool for cancer
diagnostics and screening (38–41). Due
to the high incidence of colorectal cancer in the Slovak
population, it is crucial to determine the possible causes of the
high incidence of this disease in Slovakia.
Non-invasive prenatal testing of common fetal
chromosomal aberrations, using low-coverage massively parallel
whole-genome sequencing of maternal plasma cfDNA of pregnant women,
has become the fastest low-cost genomic DNA test that is rapidly
implemented in clinical practice. Currently, more than 3 million
NIPT tests are carried out worldwide each year, and the large
amount of data generated during NIPT provides the resources to
investigate human genetic variations in the population (31). In our study, we analyzed low-coverage
massively parallel whole-genome sequencing data of total plasma DNA
from pregnant women generated for NIPT screening to characterize
the variants in genes associated with CRC and LS in the Slovak
population. To our knowledge, the present study is the first
population analysis of CRC and LS variants worldwide and also in
the Slovak population using NIPT data. We illustrate the utility of
these genomic data for clinical genetics and population
studies.
Over the past two decades, GWAS offer the
opportunity to uncover genetic susceptibility factors for CRC and
provide insights into the biological basis of CRC etiology. These
studies have demonstrated that only a fraction of CRC heritability
is explained by known risk-conferring genetic variation, whereas
the remaining genetic risk of CRC may be accounted for by a
combination of high-prevalence and low-penetrance of common genetic
variants. To date, a large number of common genetic variants have
been identified by the GWAS approach, which has intimately
connected to the onset of CRC (13,18,42–46).
By pooling GWAS data of risk variants associated
with colorectal carcinogenesis from 2007–2020 and data variants in
our population sample from NIPT, we have identified 106 common risk
CRC variants. When we compared allele frequencies of these variants
to allele frequencies in six gnomAD world population, finally 13
common risk variants were found that showed statistically
significant differences in population allele frequencies-rs5934683,
rs7252505, rs4779584, rs1535, rs174550, rs4246215, rs11196172,
rs10904849, rs6928864, rs3131043, rs1476570, rs12659017,
rs397775554.
The SNP rs5934683 is located on chromosome Xp22.2
between two genes, GPR143 (G protein-coupled receptor 143),
which is expressed by melanocytes and retinal pigment epithelium
and SHROOM2 (shroom family member 2), a human homolog of the
Xenopus laevis APX gene that has important functions in cell
morphogenesis including endothelial and epithelial tissue
development (44). Missense
mutations in this gene have been detected in large-scale screens
for recurring mutations in cancer cell lines. Both GPR143
and SHROOM2 play a role in melanosome biogenesis and retinal
pigmentation. It is known that abnormal retinal pigmentation,
similar to the congenital hypertrophy of retinal pigment epithelium
lesions, are typical of the familial adenomatous polyposis syndrome
(FAP), one of the inherited syndromes of CRC (47). The relationship between Xp22.2 and
CRC risk represents the first evidence for the role of X-chromosome
variation in predisposition to non-sex-specific cancer (48).
The SNP rs7252505, located in the 19q13 risk locus,
is in an intron of the gene GPATCH1 (G-patch domain
containing 1). Although GPATCH1 is expressed in the colon,
little is known about its function other than the fact that it
contains a G-patch domain, a domain typically associated with RNA
processing. One study found that rs7252505 was associated with CRC
in African Americans (46,49).
Intergenic variant rs4779584 in chromosomal region
15q13.3 lies between SCG5 and GREM1, and the
association between this SNP to CRC has been identified in several
GWAS studies (13,50).
The rs4246215 polymorphism is located in the FEN1 in
the long arm of chromosome 11 (11q12.2). The association between
this SNP and the potential risk of different types of cancers,
including esophageal, lung, gastrointestinal, gallbladder, breast
cancer in Chinese and Iran populations, glioma and childhood
leukemia, has been previously studied. The rs4246215 variant was
also associated with colorectal cancer in East Asians and the
Chinese population (51,52).
To identify variants that may predispose to LS and
may cause the high incidence of CRC in Slovakia, we used NIPT data,
including variants with at least 5% AF and coverage at least 100
reads per variant. To verify the reliability of the found variants
using NIPT, we selected 15 variants with AF below 5% and validated
them using Sanger sequencing. For this reason, it is not possible
to find rare variants with AF under 5%. Initially, we selected gene
variants known to be associated with LS and we focused on their
population AF in gnomAD database and as well as on pathogenicity as
reported in public database ClinVar. No publications are available
for all variants showing statistically significant differences in
population allele frequencies and selected 18 variants. The
rs9852378 SNP was reported as benign by ClinVar, and other variants
were not reported in ClinVar.
Our study has several key shortcomings. None of the
variants identified in this study are pathogenic or likely
pathogenic due to their extremely low frequency in the general
population (Fig. 8). From the total
number of pathogenic or likely pathogenic variants annotated in the
ClinVar database, we could determine the population frequency of
only 15 variants even when using the gnomAD database (Table IV). Second, the sample size was
relatively small and it is strongly biased towards females. We
assume that even larger sample sets will also not offer
opportunities to detect such low frequencies of LS variants in the
population using NIPT data. Third, a substantial portion of
identified variants was removed from analyses due to technical
limitations, mainly because of their location in low complexity
regions. Although these could be technical artifacts (53), they could also be real variants
having biological effects that are yet generally hardly
determinable, but likely existing (54). Moreover, colorectal cancer is a
disease caused by a combination of multiple genes and environmental
factors. To assess the relationship between the variants identified
in population and CRC development, it is very important in future
research to study the interaction between genes and also the
environment on the colorectal cancer risk. Although suitable for
the determination of general population frequencies of independent
variants, NIPT data are unsuitable for calculations (such as
polygenic risk score determinations) based on exact combinations of
these variants in individuals, which may de facto determine
the risk of individuals to develop certain diseases.
The underlying mechanism for a high incidence of CRC
in the Slovak population is still unclear at the moment; however,
it is possible that genetic factors, like the most common inherited
syndrome-LS, play a crucial role in colorectal etiology. We have
performed a literature search in PubMed focused on population
studies of CRC and LS in Slovakia from 2010–2020 using
next-generation sequencing. In the Slovak population, only a few
population studies of risk variants have been conducted to
elucidate the etiology of CRC (55–57). In
general, little is known about risk variants associated with CRC or
LS in the Slovak population.
Identifying mutations associated with CRC in
populations with high mortality rate, such as the Slovak
population, is important to reduce the incidence of this
multifactor disorder. The findings from these studies suggest a
lack of understanding of the mechanism of many risk variants of
CRC. Due to study limitations, we could not identify any pathogenic
variants associated with LS in the Slovak population using NIPT
data. On the other hand, NIPT data is not a major obstacle to
better results, as pathogenic variants have extremely low
frequencies in the general population. Even in most cases, the
frequencies are not known. However, we identified several promising
common risk variants associated with CRC previously reported in
GWAS studies that represent variants with highly different
frequencies between Slovak and non-Finnish populations in boxplots.
Since NIPT expands rapidly to millions of individuals each year,
the reuse of these data reduces the cost of large-scale population
studies and likely provides an acceptable background for
information about genomic variation. Finally, future population
studies on larger sample sets with various types of mutations are
needed to reveal new mechanisms of pathogenicity and links to new
biological pathways, which may be useful in designing preventive
strategies and treatment of CRC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the PANGAIA
project H2020-MSCA-RISE-2019 (grant no. 872539) funded under
H2020-EU.1.3.3. Programme, the OP Integrated Infrastructure for the
project ‘Long term strategic research and development focused on
the occurrence of Lynch syndrome in the Slovak population and
possibilities of prevention of tumors associated with this
syndrome’ (grant no. 313011V578) co-financed by the European
Regional Development Fund (ERDF), the Operational Program
Integrated Infrastructure (grant no. 313011F988) co-financed by the
ERDF, and the Scientific Grant Agency of the Ministry of Education,
Science, Research and Sport of the Slovak Republic and the Slovak
Academy of Sciences (grant no. 1/0305/19).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the DSpace repository, https://dspace.uniba.sk/xmlui/handle/123456789/27.
Authors' contributions
NF and JG performed data analysis. NF was
responsible for the literature search and manuscript writing. JG,
JB and JR were responsible for designing the study and supervising
the work. TS conceived the idea of the project and TS, JB and JR
performed proofreading of the manuscript. JG and JB confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The non-invasive prenatal testing study (study ID.
35900_2015) was approved by the Ethical Committee of the Bratislava
Self-Governing Region (Sabinovska ul.16, 820 05 Bratislava) on 30th
April 2015 (approval no. 03899_2015).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Global Cancer Observatory, . International
Agency for Research on Cancer. (Lyon, France). 2020.https://gco.iarc.fr/today/data/factsheets/populations/703-slovakia-fact-sheets.pdfNovember
9–2020
|
|
3
|
Thanikachalam K and Khan G: Colorectal
cancer and nutrition. Nutrients. 11:1642019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rawla P, Sunkara T and Barsouk A:
Epidemiology of colorectal cancer: Incidence, mortality, survival,
and risk factors. Prz Gastroenterol. 14:89–103. 2019.PubMed/NCBI
|
|
5
|
Cai S, Li Y, Ding Y, Chen K and Jin M:
Alcohol drinking and the risk of colorectal cancer death: A
meta-analysis. Eur J Cancer Prev. 23:532–539. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dashti SG, Buchanan DD, Jayasekara H,
Ouakrim DA, Clendenning M, Rosty C, Winship IM, Macrae FA, Giles
GG, Parry S, et al: Alcohol consumption and the risk of colorectal
cancer for mismatch repair gene mutation carriers. Cancer Epidemiol
Biomarkers Prev. 26:366–375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Botteri E, Iodice S, Bagnardi V, Raimondi
S, Lowenfels AB and Maisonneuve P: Smoking and colorectal cancer: A
meta-analysis. JAMA. 300:2765–2778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Limsui D, Vierkant RA, Tillmans LS, Wang
AH, Weisenberger DJ, Laird PW, Lynch CF, Anderson KE, French AJ,
Haile RW, et al: Cigarette smoking and colorectal cancer risk by
molecularly defined subtypes. J Natl Cancer Inst. 102:1012–1022.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ordóñez-Mena JM, Walter V, Schöttker B,
Jenab M, O'Doherty MG, Kee F, Bueno-de-Mesquita B, Peeters PH,
Stricker BH, Ruiter R, et al: Impact of prediagnostic smoking and
smoking cessation on colorectal cancer prognosis: A meta-analysis
of individual patient data from cohorts within the CHANCES
consortium. Ann Oncol. 29:472–483. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thrift AP, Gong J, Peters U, Chang-Claude
J, Rudolph A, Slattery ML, Chan AT, Locke AE, Kahali B, Justice AE,
et al: Mendelian randomization study of body mass index and
colorectal cancer risk. Cancer Epidemiol Biomarkers Prev.
24:1024–1031. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gharahkhani P, Ong JS, An J, Law MH,
Whiteman DC, Neale RE and MacGregor S: Effect of increased body
mass index on risk of diagnosis or death from cancer. Br J Cancer.
120:565–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu Y, Kweon SS, Tanikawa C, Jia WH, Xiang
YB, Cai Q, Zeng C, Schmit SL, Shin A, Matsuo K, et al: Large-scale
genome-wide association study of east asians identifies loci
associated with risk for colorectal cancer. Gastroenterology.
156:1455–1466. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng C, Matsuda K, Jia WH, Chang J, Kweon
SS, Xiang YB, Shin A, Jee SH, Kim DH, Zhang B, et al:
Identification of susceptibility loci and genes for colorectal
cancer risk. Gastroenterology. 150:1633–1645. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Tassan NA, Whiffin N, Hosking FJ,
Palles C, Farrington SM, Dobbins SE, Harris R, Gorman M, Tenesa A,
Meyer BF, et al: A new GWAS and meta-analysis with 1000Genomes
imputation identifies novel risk variants for colorectal cancer.
Sci Rep. 5:104422015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang B, Jia WH, Matsuda K, Kweon SS,
Matsuo K, Xiang YB, Shin A, Jee SH, Kim DH, Cai Q, et al:
Large-scale genetic study in East Asians identifies six new loci
associated with colorectal cancer risk. Nat Genet. 46:533–542.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takahashi Y, Sugimachi K, Yamamoto K,
Niida A, Shimamura T, Sato T, Watanabe M, Tanaka J, Kudo S,
Sugihara K, et al: Japanese genome-wide association study
identifies a significant colorectal cancer susceptibility locus at
chromosome 10p14. Cancer Sci. 108:2239–2247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmit SL, Edlund CK, Schumacher FR, Gong
J, Harrison TA, Huyghe JR, Qu C, Melas M, Van Den Berg DJ, Wang H,
et al: Novel common genetic susceptibility loci for colorectal
cancer. J Natl Cancer Inst. 111:146–157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biller LH, Syngal S and Yurgelun MB:
Recent advances in lynch syndrome. Fam Cancer. 18:211–219. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yurgelun MB and Hampel H: Recent advances
in lynch syndrome: Diagnosis, treatment, and cancer prevention. Am
Soc Clin Oncol Educ Book. 38:101–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Møller P, Seppälä T, Bernstein I,
Holinski-Feder E, Sala P, Evans DG, Lindblom A, Macrae F, Blanco I,
Sijmons R, et al: Incidence of and survival after subsequent
cancers in carriers of pathogenic MMR variants with previous
cancer: A report from the prospective lynch syndrome database. Gut.
66:1657–1664. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Møller P, Seppälä TT, Bernstein I,
Holinski-Feder E, Sala P, Evans DG, Lindblom A, Macrae F, Blanco I,
Sijmons RH, et al: Cancer risk and survival in carriers by gene and
gender up to 75 years of age: A report from the prospective lynch
syndrome database. Gut. 67:1306–1316. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soares BL, Brant AC, Gomes R, Pastor T,
Schneider NB, Ribeiro-Dos-Santos Â, de Assumpção PP, Achatz MI,
Ashton-Prolla P and Moreira MA: Screening for germline mutations in
mismatch repair genes in patients with lynch syndrome by next
generation sequencing. Fam Cancer. 17:387–394. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cox VL, Bamashmos AA, Foo WC, Gupta S,
Yedururi S, Garg N and Kang HC: Lynch syndrome: Genomics update and
imaging review. Radiographics. 38:483–499. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Le S, Ansari U, Mumtaz A, Malik K, Patel
P, Doyle A and Khachemoune A: Lynch syndrome and muir-torre
syndrome: An update and review on the genetics, epidemiology, and
management of two related disorders. Dermatol Online J.
23:130302017.
|
|
26
|
Peltomäki P: Update on lynch syndrome
genomics. Fam Cancer. 15:385–393. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with Lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuiper RP, Vissers LELM, Venkatachalam R,
Bodmer D, Hoenselaar E, Goossens M, Haufe A, Kamping E, Niessen RC,
Hogervorst FB, et al: Recurrence and variability of germline EPCAM
deletions in Lynch syndrome. Hum Mutat. 32:407–414. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shah SN, Hile SE and Eckert KA: Defective
mismatch repair, microsatellite mutation bias, and variability in
clinical cancer phenotypes. Cancer Res. 70:431–435. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Martin-Morales L, Rofes P, Diaz-Rubio E,
Llovet P, Lorca V, Bando I, Perez-Segura P, de la Hoya M, Garre P,
Garcia-Barberan V and Caldes T: Novel genetic mutations detected by
multigene panel are associated with hereditary colorectal cancer
predisposition. PLoS One. 13:e02038852018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Budis J, Gazdarica J, Radvanszky J,
Harsanyova M, Gazdaricova I, Strieskova L, Frno R, Duris F, Minarik
G, Sekelska M, et al: Non-invasive prenatal testing as a valuable
source of population specific allelic frequencies. J Biotechnol.
299:72–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pös O, Budis J, Kubiritova Z, Kucharik M,
Duris F, Radvanszky J and Szemes T: Identification of structural
variation from NGS-Based non-invasive prenatal testing. Int J Mol
Sci. 20:44032019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu S, Huang S, Chen F, Zhao L, Yuan Y,
Francis SS, Fang L, Li Z, Lin L, Liu R, et al: Genomic analyses
from non-invasive prenatal testing reveal genetic associations,
patterns of viral infections, and Chinese population history. Cell.
175:347–359. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tran NH, Vo TB, Nguyen VT, Tran NT, Trinh
THN, Pham HAT, Dao THT, Nguyen NM, Van YLT, Tran VU, et al: Genetic
profiling of Vietnamese population from large-scale genomic
analysis of non-invasive prenatal testing data. Sci Rep.
10:191422020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pös O, Budiš J and Szemes T: Recent trends
in prenatal genetic screening and testing. F1000Res. 8:F10002019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minarik G, Repiska G, Hyblova M, Nagyova
E, Soltys K, Budis J, Duris F, Sysak R, Bujalkova MG, Vlkova-Izrael
B, et al: Utilization of benchtop next generation sequencing
platforms ion torrent PGM and MiSeq in noninvasive prenatal testing
for chromosome 21 trisomy and testing of impact of in silico and
physical size selection on its analytical performance. PLoS One.
10:e01448112015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Beyene J and Pare G: Statistical genetics
with application to population-based study design: A primer for
clinicians. Eur Heart J. 35:495–500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu L, Huang Y, Fang X, Liu C, Deng W,
Zhong C, Xu J, Xu D and Yuan Y: A novel and reliable method to
detect microsatellite instability in colorectal cancer by
next-generation sequencing. J Mol Diagn. 20:225–231. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yurgelun MB, Allen B, Kaldate RR, Bowles
KR, Judkins T, Kaushik P, Roa BB, Wenstrup RJ, Hartman AR and
Syngal S: Identification of a variety of mutations in cancer
predisposition genes in patients with suspected lynch syndrome.
Gastroenterology. 149:604–613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Valle L, de Voer RM, Goldberg Y, Sjursen
W, Försti A, Ruiz-Ponte C, Caldés T, Garré P, Olsen MF, Nordling M,
et al: Update on genetic predisposition to colorectal cancer and
polyposis. Mol Aspects Med. 69:10–26. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Budiš J, Kucharík M, Ďuriš F, Gazdarica J,
Zrubcová M, Ficek A, Szemes T, Brejová B and Radvanszky J: Dante:
Genotyping of known complex and expanded short tandem repeats.
Bioinformatics. 35:1310–1317. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jiao S, Peters U, Berndt S, Brenner H,
Butterbach K, Caan BJ, Carlson CS, Chan AT, Chang-Claude J, Chanock
S, et al: Estimating the heritability of colorectal cancer. Hum Mol
Genet. 23:3898–3905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Law PJ, Timofeeva M, Fernandez-Rozadilla
C, Broderick P, Studd J, Fernandez-Tajes J, Farrington S, Svinti V,
Palles C, Orlando G, et al: Association analyses identify 31 new
risk loci for colorectal cancer susceptibility. Nat Commun.
10:21542019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang K, Civan J, Mukherjee S, Patel F and
Yang H: Genetic variations in colorectal cancer risk and clinical
outcome. World J Gastroenterol. 20:4167–4177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hofer P, Hagmann M, Brezina S, Dolejsi E,
Mach K, Leeb G, Baierl A, Buch S, Sutterlüty-Fall H, Karner-Hanusch
J, et al: Bayesian and frequentist analysis of an Austrian
genome-wide association study of colorectal cancer and advanced
adenomas. Oncotarget. 8:98623–98634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang H, Schmit SL, Haiman CA, Keku TO,
Kato I, Palmer JR, van den Berg D, Wilkins LR, Burnett T, Conti DV,
et al: Novel colon cancer susceptibility variants identified from a
genome-wide association study in African Americans. Int J Cancer.
140:2728–2733. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Closa A, Cordero D, Sanz-Pamplona R, Solé
X, Crous-Bou M, Paré-Brunet L, Berenguer A, Guino E, Lopez-Doriga
A, Guardiola J, et al: Identification of candidate susceptibility
genes for colorectal cancer through eQTL analysis. Carcinogenesis.
35:2039–2046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dunlop MG, Dobbins SE, Farrington SM,
Jones AM, Palles C, Whiffin N, Tenesa A, Spain S, Broderick P, Ooi
LY, et al: Common variation near CDKN1A, POLD3 and SHROOM2
influences colorectal cancer risk. Nat Genet. 44:770–776. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang H, Haiman CA, Burnett T, Fortini BK,
Kolonel LN, Henderson BE, Signorello LB, Blot WJ, Keku TO, Berndt
SI, et al: Fine-mapping of genome-wide association study-identified
risk loci for colorectal cancer in African Americans. Hum Mol
Genet. 22:5048–5055. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hong SN, Park C, Kim JI, Kim DH, Kim HC,
Chang DK, Rhee PL, Kim JJ, Rhee JC, Son HJ and Kim YH: Colorectal
cancer-susceptibility single-nucleotide polymorphisms in Korean
population. J Gastroenterol Hepatol. 30:849–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moazeni-Roodi A, Ghavami S, Ansari H and
Hashemi M: Association between the flap endonuclease 1 gene
polymorphisms and cancer susceptibility: An updated meta-analysis.
J Cell Biochem. 120:13583–13597. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chou AK, Shen MY, Chen FY, Hsiao CL, Shih
LC, Chang WS, Tsai CW, Ying TH, Wu MH, Huang CY and Bau DT: The
association of flap endonuclease 1 genotypes with the
susceptibility of endometriosis. Cancer Genomics Proteomics.
14:455–460. 2017.PubMed/NCBI
|
|
53
|
Kubiritova Z, Gyuraszova M, Nagyova E,
Hyblova M, Harsanyova M, Budis J, Hekel R, Gazdarica J, Duris F,
Kadasi L, et al: On the critical evaluation and confirmation of
germline sequence variants identified using massively parallel
sequencing. J Biotechnol. 298:64–75. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Trost B, Engchuan W, Nguyen CM,
Thiruvahindrapuram B, Dolzhenko E, Backstrom I, Mirceta M, Mojarad
BA, Yin Y, Dov A, et al: Genome-wide detection of tandem DNA
repeats that are expanded in autism. Nature. 586:80–86. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mahmood S, Sivoňová M, Matáková T, Dobrota
D, Wsólová L, Dzian A, et al: Association of EGF and p53 gene
polymorphisms and colorectal cancer risk in the Slovak population.
Cent Eur J Med. 9:405–416. 2014.
|
|
56
|
Škereňová M, Halašová E, Matáková T,
Jesenská L, Jurečeková J, Šarlinová M, Čierny D and Dobrota D: Low
variability and stable frequency of common haplotypes of the tp53
gene region in colorectal cancer patients in a Slovak population.
Anticancer Res. 37:1901–1907. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kašubová I, Kalman M, Jašek K, Burjanivová
T, Malicherová B, Vaňochová A, Meršaková S, Lasabová Z and Plank L:
Stratification of patients with colorectal cancer without the
recorded family history. Oncol Lett. 17:3649–3656. 2019.PubMed/NCBI
|
|
58
|
Jia WH, Zhang B, Matsuo K, Shin A, Xiang
YB, Jee SH, Kim DH, Ren Z, Cai Q, Long J, et al: Genome-wide
association analyses in East Asians identify new susceptibility
loci for colorectal cancer. Nat Genet. 45:191–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Peters U, Jiao S, Schumacher FR, Hutter
CM, Aragaki AK, Baron JA, Berndt SI, Bézieau S, Brenner H,
Butterbach K, et al: Identification of genetic susceptibility loci
for colorectal tumors in a genome-wide meta-analysis.
Gastroenterology. 144:799–807. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Whiffin N, Hosking FJ, Farrington SM,
Palles C, Dobbins SE, Zgaga L, Lloyd A, Kinnersley B, Gorman M,
Tenes A, et al: Identification of susceptibility loci for
colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet.
23:4729–4737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Houlston RS, Cheadle J, Dobbins SE, Tenesa
A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E,
Farrington S, et al: Meta-analysis of three genome-wide association
studies identifies susceptibility loci for colorectal cancer at
1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 42:973–977. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Orlando G, Law PJ, Palin K, Tuupanen S,
Gylfe A, Hänninen UA, Cajuso T, Tanskanen T, Kondelin J, Kaasinen
E, et al: Variation at 2q35 (PNKD and TMBIM1) influences colorectal
cancer risk and identifies a pleiotropic effect with inflammatory
bowel disease. Hum Mol Genet. 25:2349–2359. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Schmit SL, Schumacher FR, Edlund CK, Conti
DV, Raskin L, Lejbkowicz F, Pinchev M, Rennert HS, Jenkins MA,
Hopper JL, et al: A novel colorectal cancer risk locus at 4q32.2
identified from an international genome-wide association study.
Carcinogenesis. 35:2512–2519. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Peters U, Hutter CM, Hsu L, Schumacher FR,
Conti DV, Carlson CS, Edlund CK, Haile RW, Gallinger S, Zanke BW,
et al: Meta-analysis of new genome-wide association studies of
colorectal cancer risk. Hum Genet. 131:217–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tomlinson IPM, Webb E, Carvajal-Carmona L,
Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A,
Sullivan K, et al: A genome-wide association study identifies
colorectal cancer susceptibility loci on chromosomes 10p14 and
8q23.3. Nat Genet. 40:623–630. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tenesa A, Farrington SM, Prendergast JGD,
Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E,
Cetnarskyj R, Cartwright N, et al: Genome-wide association scan
identifies a colorectal cancer susceptibility locus on 11q23 and
replicates risk loci at 8q24 and 18q21. Nat Genet. 40:631–637.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tomlinson IPM, Carvajal-Carmona LG,
Dobbins SE, Tenesa A, Jones AM, Howarth K, Palles C, Broderick P,
Jaeger EEM, Farrington S, et al: Multiple common susceptibility
variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part
of the missing heritability of colorectal cancer. PLoS Genet.
7:e10021052011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
COGENT Study, ; Houlston RS, Webb E,
Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I,
Vijayakrishnan J, Sullivan K, et al: Meta-analysis of genome-wide
association data identifies four new susceptibility loci for
colorectal cancer. Nat Genet. 40:1426–1435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Broderick P, Carvajal-Carmona L, Pittman
AM, Webb E, Howarth K, Rowan A, Lubbe S, Spain S, Sullivan K,
Fielding S, et al: A genome-wide association study shows that
common alleles of SMAD7 influence colorectal cancer risk. Nat
Genet. 39:1315–1317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Schumacher FR, Schmit SL, Jiao S, Edlund
CK, Wang H, Zhang B, Hsu L, Huang SC, Fischer CP, Harju JF, et al:
Genome-wide association study of colorectal cancer identifies six
new susceptibility loci. Nat Commun. 6:71382015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Haiman CA, Le Marchand L, Yamamato J,
Stram DO, Sheng X, Kolonel LN, Wu AH, Reich D and Henderson BE: A
common genetic risk factor for colorectal and prostate cancer. Nat
Genet. 39:954–956. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tomlinson I, Webb E, Carvajal-Carmona L,
Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood
W, et al: A genome-wide association scan of tag SNPs identifies a
susceptibility variant for colorectal cancer at 8q24.21. Nat Genet.
39:984–988. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hutter CM, Slattery ML, Duggan DJ,
Muehling J, Curtin K, Hsu L, Beresford SA, Rajkovic A, Sarto GE,
Marshall JR, et al: Characterization of the association between
8q24 and colon cancer: Gene-environment exploration and
meta-analysis. BMC Cancer. 10:6702010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cui R, Okada Y, Jang SG, Ku JL, Park JG,
Kamatani Y, Hosono N, Tsunoda T, Kumar V, Tanikawa C, et al: Common
variant in 6q26-q27 is associated with distal colon cancer in an
Asian population. Gut. 60:799–805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang H, Burnett T, Kono S, Haiman CA,
Iwasaki M, Wilkens LR, Loo LW, Van Den Berg D, Kolonel LN,
Henderson BE, et al: Trans-ethnic genome-wide association study of
colorectal cancer identifies a new susceptibility locus in VTI1A.
Nat Commun. 5:46132014. View Article : Google Scholar : PubMed/NCBI
|