Introduction
In recent years, the achievements of targeted therapy for cancer treatment have been self-evident (1,2). The difference between targeted therapy and conventional chemotherapy is that the cytotoxicity of normal cells is greatly reduced in targeted therapy due to its specific targeting (3,4). With the development of molecular biology and the gradual unfolding of mechanisms employed by tumor-associated factors, molecular targeted therapy will become the principal direction of antitumor treatment. However, the heterogeneity of drug resistance in tumor cells and the limited number of alternative targets are also clinical bottlenecks, which must be addressed (5). Therefore, it is urgent to identify and elucidate the abnormally activated or silenced signaling pathways in cancer cells, which are useful in exploring valuable therapeutic targets.
Tumorigenesis partly results from dysregulation of apoptosis, leading to apoptosis evasion of cells, which then become cancerous (6,7). There are three pathways to regulate apoptosis (death receptor cell death pathway, endoplasmic reticulum (ER) cell death pathway and mitochondrial cell death pathway), among which the mitochondrial cell death pathway is mainly regulated by the Bcl-2 family proteins (8). BAD, a member of the Bcl-2 family, acts as the main pro-apoptotic protein that regulates the cellular survival-apoptosis balance, and its phosphorylation may contribute to cancer progression (9). In addition, defender against apoptotic cell death 1 (DAD1), a subunit of oligosaccharyltransferase (OST) acting on N-glycosylation residing in the ER (10), is a negative regulator of programmed cell death associated with the ER cell death pathway (11). Increasing experimental evidence has indicated deep engagement of the two proteins in tumorigenesis, particularly in cellular apoptosis, invasion, chemosensitivity and diagnostic/prognostic judgment. Therefore, their key roles in signaling transduction pathways and their close association with cellular behavior may provide insights and novel alternative molecular agents for targeted therapy of cancer.
Physiological characteristics and cancerous activity of BAD
Bcl-2 family and overview of BAD
The Bcl-2 family, a group of cooperative proteins, exerts a great influence on the regulation of apoptosis via the mitochondrial cell death pathway (12,13). Bcl-2 homology domains (BH1-4) have been determined to be a collective characteristic of Bcl-2 family members, and two of the member proteins can form homo- or heterodimers as an essential functional unit to promote or suppress apoptosis (14). The effects of the Bcl-2 family are antagonistic, which means that some of the members, including BAD, Bax and BH3 interacting domain death agonist, serve a pro-apoptotic role in cellular modulation, while others, including Bcl-2, myeoid cell leukemin-1 (Mcl-1) and Bcl-xL, appear to suppress apoptosis (12). The mechanism underlying the regulation of the mitochondrial cell death pathway has been demonstrated to be the action of the Bcl-2 family proteins, which determine the permeability of transition pore embedded in the mitochondrial membrane (15,16). Upstream apoptosis signals make the non-selective pore an irreversible access point between the cytoplasm and mitochondrial matrix mediated by the Bcl-2 family of proteins (15,16). Mitochondrial matrix-deprived cytochrome c combines with apoptotic protease activating factor-1 and caspase-9 proenzyme to form apoptosomes in the cytoplasm, which then activate caspase-9, triggering a cascade reaction of apoptotic proteases to subsequently induce apoptosis (15,16).
BAD was first cloned from a mouse cDNA library, and the homologous human gene was cloned later (17). BAD comprises 168 amino acids, of which Ser112, Ser136 and Ser155 are the three known regulatory residues, which can be sequentially phosphorylated by several kinase proteins (17). Among them, ribosomal protein S6 kinase and cAMP dependent protein kinase (PKA) mediate the phosphorylation of Ser112, Akt mediates the phosphorylation of Ser136 (18), and PKA preferentially mediates the phosphorylation of Ser155, which is located in the center of the BH3 domain (19–21). Normal phosphorylation at the three residues helps to maintain cytoplasmic sequestration of BAD, and thus, apoptosis is attenuated (22). Phosphorylation of BAD at Ser26 by the IκB kinase complex inhibits the pro-apoptotic activity of BAD (23). A novel synthetic compound, N-cyclopentyl-3-((4-(2,3-dichlorophenyl)piperazin-1-yl) (2-hydroxyphenyl) methyl) benzamide, a specific inhibitor of BAD phosphorylation at Ser99, could suppress the vitality of cancer cells in vivo and in vitro (24). Normally, phosphorylated BAD (p-BAD) combines with the amphipathic groove of chaperone 14-3-3 (25). BAD is different from most Bcl-2 family members, as it has no C-terminal transmembrane domain that anchors the outer mitochondrial membrane and nuclear envelope (17). Therefore, BAD is sequestered in the cytoplasm, and apoptosis is inhibited (22). In the presence of survival signals, dephosphorylated BAD, which is generated by phosphatases, disassociates from 14-3-3 and begins to displace Bax in the Bcl-2/Bax or Bcl-xL/Bax heterodimer to form a Bcl-2/BAD or Bcl-xL/BAD heterodimer via the BH3 homologous domain in a concentration-dependent manner (17). Additionally, free Bax homodimerization is increased, and apoptosis is initiated by the Bax homodimer integrated into the outer mitochondrial membrane (17).
To simplify, subcellular relocation to the mitochondria of BAD that triggers apoptosis is tied to its phosphorylation status at the three amino acid residues. The switch between p-BAD and dephosphorylated BAD determines its role in the pathway, i.e., pro-apoptosis or pro-survival, wherein 14-3-3, several kinases and phosphatases are key regulators (26). In addition to apoptosis-related roles, BAD also has multiple non-apoptotic functions, such as regulation of the cell cycle (27–29), autophagy (30), immune engagement (31,32), glucose metabolism (15,33), and control of localized translation (34), all of which are closely associated with its cellular effects in cancer. Among all these pathways converging due to BAD, the phosphorylation status coordinates the multiple functions of BAD, and its BH3 domain is utilized. p-BAD manages cytoplasmic sequestration to prevent apoptosis, while other metabolic pathways, such as suppression of gluconeogenesis and activation of oxidative metabolism of glucose in the mitochondria of liver cells (33,35), are activated to promote survival according to different ligands matched with BH3. Therefore, the central status of BAD in several apoptosis-related and other metabolism-related signaling pathways may result in it being an appealing target in cancer.
Expression and function of BAD in cancer
BAD is usually expressed in the colon, stomach, prostate, kidney, brain and adipose tissues (36). The tumor-associated effects of abnormal levels of p-BAD are reflected in several aspects as described subsequently.
Cell proliferation, survival and apoptosis
Several types of cancer cells have been detected to exhibit higher levels of p-BAD than corresponding immortalized normal cells in vivo and in vitro, and cell apoptosis or survival is regulated by the BAD phosphorylation status (17,37). The effects of p-BAD on cell proliferation, survival and apoptosis are summarized in Table I.
 |
Table I.
Interaction of (p-)BAD with relevant genes and effect on cell proliferation, survival and apoptosis.
|
Table I.
Interaction of (p-)BAD with relevant genes and effect on cell proliferation, survival and apoptosis.
| First author/s, year |
Tissue/cell type |
Interaction of (p-)BAD with relevant genes |
Effect |
(Refs.) |
| Sastry et al, 2006 |
Prostate cancer |
Ras/MEK and Rac/PKA1 signaling pathways mediate the phosphorylation of BAD at Ser112 and Ser136, respectively. |
Prevents apoptosis |
(38) |
| Smith et al, 2009 |
|
Silencing of BAD by shRNA. |
Suppresses cell proliferation |
(46) |
| Kulik, 2019 |
|
Upregulation of p-BAD by the activation of the ADRB2/PKA signaling pathway. |
Inhibits apoptosis |
(48) |
| She et al, 2005 |
PTEN-deficient tumor cell lines |
Phosphorylation defect of BAD at Ser112 and Ser136 via EGFR/MEK/MAPK and PI3K/Akt signaling pathways, respectively. |
Promotes apoptosis |
(39) |
| Polzien et al, 2011 |
B-Raf-mutated cancer cell lines |
Raf kinase phosphorylates BAD at Ser134. |
Promotes proliferation |
(40) |
| Winter et al, 2014 |
JAK-depleted myeloproliferative neoplasms |
JAK2 phosphorylates BAD. |
Promotes survival |
(44) |
| Stickles et al, 2015 |
Colorectal adenocarcinoma, breast cancer, endometrial adenocarcinoma and ovarian cancer |
PP2C deletion induces higher levels of p-BAD at Ser155. |
Promotes growth |
(47) |
| Mann et al, 2019 |
Breast cancer |
Phosphorylation of BAD at Ser118 increases Ser99 phosphorylation, 14-3-3 binding and Akt activation. |
Promotes growth and survival |
(49) |
| |
|
BAD stimulates mitochondrial complex I activity. |
Facilitates growth and sensitizes cells to apoptosis in response to complex I blockade |
|
| Lu et al, 2019 |
Ovarian cancer |
Artificial fusion p53-BAD locates to the mitochondria. |
Promotes apoptosis |
(50) |
Sastry et al (38) determined that there are two signaling pathways that phosphorylate BAD to protect prostate cancer cells from apoptosis under the stimulus of epidermal growth factor. One signaling pathway induces phosphorylation at Ser112 via Ras/MEK. Another signaling pathway induces phosphorylation at Ser136 via Ras-related C3 botulinum toxin substrate (Rac)/P21-activated kinase 1 (PAK1) (38). However, She et al (39) reported that phosphorylation of Ser112 and Ser136 could also be mediated by the EGFR/MEK/MAPK and phosphatidylinositol 3-kinase/Akt signaling pathways, respectively. Furthermore, survival signaling-induced kinases, such as PAK1 and Raf, promote the proliferation of cancer cells in the presence of wild-type BAD (39). Polzien et al (40) determined that Raf kinases could phosphorylate BAD at Ser134 to promote cell proliferation in B-Raf-V600E-containing tumor cells. Furthermore, replacement of Ser134 with alanine leads to phosphorylation, suggesting that BAD phosphorylation at Ser134 is essential for sufficient cell proliferation (27). Myeloproliferative neoplasms (MPNs) may result in activating mutations of the Janus kinase (JAK) gene (41–43). A previous study has reported that the phosphorylation of BAD induced by JAK2 promotes cell survival in JAK-depleted MPN cells (44). Additionally, in cells sensitive to JAK inhibitor, treatment with JAK inhibitor results in dephosphorylation of BAD and affects its combination with Bcl-xL, initiating apoptosis (44). Huang et al (45) revealed that overexpression of BAD inhibits the proliferation of tumor cells in vitro and reduces tumor volume in vivo by promoting cell apoptosis and suppressing cell proliferation. Smith et al (46) observed contrasting results compared with Huang et al (45) in prostate cancer cells, wherein increased BAD expression could promote cell proliferation and silencing of BAD by short hairpin RNA suppressed cell proliferation. Stickles et al (47) demonstrated that protein phosphatase 2C (PP2C) deletion leads to higher levels of p-BAD at Ser155, which is beneficial for cell proliferation in vitro. Sastry et al (9) determined that the phosphorylation of BAD is indispensable for the survival of cancer stem cells (CSCs). Deficient expression of p-BAD induces apoptosis of CSCs, which could be reversed by the BH3 mimetic ABT-737, revealing that only the BH3 homologous domain is essential in BAD (9). Furthermore, the downregulation of BAD weakens the frequency and renewal capacity of CSCs (9). Kulik (48) reported that upregulated levels of p-BAD, together with Mcl-1 mediated by the activation of the adrenoceptor β2 (ADRB2)/PKA signaling pathway, are responsible for increased inhibition of apoptosis of prostate cancer cells. Furthermore, Mann et al (49) verified the two distinct mechanisms underlying the BAD-regulated increase in cell proliferation in breast cancer cells. Specifically, phosphorylation of BAD at Ser118 increases Ser99 phosphorylation, 14-3-3 binding and Akt activation, which promotes cell proliferation and survival. On the other hand, BAD stimulates mitochondrial oxygen consumption in a novel manner downstream of substrate entry into the mitochondria (49). BAD stimulates complex I activity that facilitates cell proliferation and sensitizes cells to apoptosis in response to complex I blockade, which may result in large but non-aggressive breast cancer (49). These results suggest that BAD-induced apoptosis may not only depend on mitochondrial membrane reactions between Bcl-2 family proteins, but it may also be associated with oxidative metabolism.
Lu et al (50) designed a novel chimeric gene fusion p53-BAD to overcome the dominant negative inhibition of wild-type p53 and multiple genetic aberrations in ovarian cancer (OVCA). By introducing Ser122A and Ser136A mutations to prevent phosphorylation at the two residues, p53-BAD constructs could always be located in the mitochondria. Furthermore, they observed that p53-BAD constructs exhibited higher pro-apoptotic activity, which was direct and rapid via the mitochondrial cell death pathway (50). This pro-apoptotic effect was consistent in several OVCA cell lines, regardless of the endogenous p53 status (50).
It is worth mentioning that Datta et al (32) explored the physiological significance of BAD phosphorylation for cell survival in vivo. They generated BAD3SA mutant mice, in which the three phosphoregulatory residues were shifted to alanine; thus, endogenous BAD was not responsive to survival signaling. They demonstrated that growth factor-mediated BAD phosphorylation is indispensable to prevent cells from undergoing apoptotic stimuli. Notably, they validated that the levels of BAD phosphorylation via growth factors could raise the threshold at which mitochondria release cytochrome c in response to apoptotic stimuli. In summary, the levels of BAD phosphorylation may be a sensor that determines the extent to which cells undergo apoptosis, and it is also one of the mechanisms employed by survival factors to block apoptosis (32).
Invasion and distant metastasis
A previous study revealed that the expression levels of BAD in hepatocellular carcinoma (HCC) are associated with vascular invasion (51). Cekanova et al (52) reported that the levels of BAD and p-BAD in clinical breast cancer tissues are lower than those in normal breast tissues. The expression levels of several proteins associated with invasiveness (c-Jun, Akt and signal transducer and activator of transcription proteins), epithelial-mesenchymal-transition (EMT; transcription factor Sp1 and β-catenin) and metastasis (vascular endothelial growth factor) are decreased by BAD in BAD-overexpressing breast cancer cells (52). The novel anti-invasion and EMT inhibition functions of BAD are distinct from its traditional role in prompting the mitochondrial cell death pathway (52). Furthermore, 33 out of 60 clinical salivary gland adenoid cystic carcinoma cases exhibited high expression levels of BAD, and the expression levels of BAD were associated with distant metastasis (53).
Clinical characteristics
Hu et al (51) reported that the expression levels of BAD are decreased in clinical HCC tissues compared with non-tumorous adjacent tissues. The expression levels of BAD are negatively associated with several clinical characteristics, including α-fetoprotein levels, clinical stage and tumor size. Furthermore, subsequent multivariate analyses revealed that BAD can act as an independent indicator of overall HCC survival. This study demonstrated that BAD may act as a potential biomarker for poor prognosis in clinical HCC (51). Furthermore, Yu et al (54) reported similar results in small cell lung carcinoma (SCLC), and notably decreased expression levels of BAD were detected in clinical SCLC specimens compared with in neighboring non-tumorous tissues. The downregulated levels of BAD were significantly associated with overall survival, disease-free survival and several clinical characteristics (e.g., tumor recurrence, tumor size and clinical stage) of patients with SCLC (54). Multivariate analyses further indicated that BAD can act as an independent indicator of overall survival in SCLC (54). Another study on triple-negative breast cancer (TNBC) constructed a BAD pathway gene expression signature score system derived from principal component analysis to evaluate the overall expression and activation of the BAD pathway, and the results demonstrated that BAD pathway expression was associated with triple-negative status and overall survival (55).
Chemosensitivity
Chon et al (56) revealed that BAD phosphorylation has an important cisplatin sensitivity in endometrial cancer (EC) cells. Since BAD can be phosphorylated by PP2C, they observed that higher levels of p-BAD resulted in lower chemosensitivity to cisplatin in PKA small interfering RNA (siRNA) EC cells. However, p-BAD presented higher chemosensitivity to cisplatin in EC cells when PKA dephosphorylation was knocked down (56).
Interestingly, Hayakawa et al (57) reported that treatment of both cisplatin-sensitive and cisplatin-resistant OVCA cell lines with cisplatin could result in the phosphorylation of BAD at both Ser122 and Ser136, which was later determined to be mediated by the ERK and Akt cascades, respectively. Furthermore, they determined that inhibition of either of the two cascades could render OVCA cells more sensitive to cisplatin (57). Marchion et al (58) observed that the parallel effect of p-BAD increased with cisplatin resistance both in OVCA cells and in primary patients. Apart from the evidence presented, there are several other kinases or phosphatases derived from the BAD apoptosis pathway that are associated with the evolution of cisplatin resistance by exerting influence on p-BAD status. For example, Bansal et al (59) selected CDK1 and PP2C to validate OVCA sensitivity to cisplatin. Lower expression levels of PP2C and higher expression levels of CDK1 increased cisplatin resistance. In addition, they revealed that downregulation of CDK1 by siRNA infection increased cisplatin sensitivity (59). Taken together, these results demonstrated that inhibition of p-BAD enhanced chemosensitivity in OVCA chemotherapy (57–59).
Yu et al (60) reported that Bcl-2(−)BAD(+) breast cancer cells exhibited higher chemosensitivity to four types of anticancer drugs (epirubicin, 5-fluorouracil, navelbine and cisplatin) than other breast cancer cells [Bcl-2(+)BAD(−) or Bcl-2(+)BAD(+)]. Therefore, the joint detection of Bcl-2 and BAD expression may help in chemotherapy drug selection. Boac et al (55) demonstrated that patients with TNBC express higher levels of p-BAD isoforms than patients with breast cancer that is not triple negative, and the levels of p-BAD-Ser136 are different, while the differences in p-BAD-Ser112 and p-BAD-Ser155 levels are not significant. Furthermore, the study demonstrated that targeted inhibition of kinases known to phosphorylate BAD results in increased sensitivity to nonspecific chemotherapeutic agents, such as cisplatin, in vitro (55). In a later report, BAD enhanced docetaxel sensitivity by facilitating longer mitotic arrest and activating cell death in mitosis in vivo and in vitro (61). Notably, death in mitosis has been observed to be an abnormal type of apoptosis, one that was dependent on Bcl-2 interaction and caspase activation; in fact, it was necroptosis (61). This type of BAD-enhanced docetaxel-mediated necroptotic cell death is dependent on reactive oxygen species, which indicates the chemosensitivity amplification effect of BAD in breast cancer (61).
In acute myeloid leukemia (AML), Yu et al (62) developed a system to quantify the chemosensitivity of dormant AML cells. The results revealed that two BAD mimetics, ABT-199 and ABT-737, were both able to effectively target dormant primary leukemia cells and decrease the dormant fraction of leucineaminopeptidase cells to 84 and 80%, respectively, revealing their good efficacy against cells protected by dormancy. Yiau et al (63) compared the alterations in the expression levels of CD34 and BAD in blood samples collected from patients with AML before and at day 3 after induction therapy. They observed that the average percentages of CD34 and p-BAD were higher in chemoresistant than chemosensitive samples, indicating potential CD34 signaling-associated chemotherapy resistance via p-BAD in AML (63).
Zhou et al (64) explored the relationship between low glucose levels and hypoxia-induced autophagy and chemoresistance in HCC cells. The study revealed that autophagy induced by low glucose and hypoxia in central solid tumors could reduce the protein expression levels of BAD and Bcl-2 interacting mediator of cell death (Bim), and elevated chemoresistance of HCC cells. Furthermore, they observed that chemotherapy-induced apoptosis could be reduced or promoted by RNA interference or upregulation, respectively. These results revealed that the downregulation of BAD and Bim is involved in the chemoresistance of HCC (64).
Constitutive engagement with other dominant molecules
Kim et al (65) reported that Epstein-Barr virus (EBV)-derived microRNA, microRNA-BART20-5p, inhibits BAD-mediated caspase-3-dependent apoptosis by targeting BAD in gastric carcinoma. The study demonstrated that BAD could act as a potential target of BAD in EBV-associated gastric carcinogenesis (65). Tang et al (66) revealed that downstream molecules, such as caspase-3, are influenced by BAD, together with cytokines affected by the NF-κB signaling pathway. Remodeling is performed when Akt is knocked out in primary liver cancer cells, which induces an altered inflammatory response and apoptosis. Additionally, they revealed that carnosic acid nanoparticles could activate the NF-κB signaling pathway and that the overexpression of caspase-3 can moderate inflammation, as well as promote apoptosis in Akt-knockout liver cancer cells (66). Zhao et al (67) observed that downregulation of prostate cancer associated transcript 1 in esophageal cancer cells results in upregulated BAD expression, inhibits cell proliferation, decreases migration and invasion, and enhances apoptosis. Liu et al (68) reported that inhibition of protein kinase AMP-activated catalytic subunit α1 (AMPK) with dorsomorphin (a specific AMPK inhibitor) in AMPK-driven hematological cancer types upregulates the expression levels of BAD to induce apoptosis.
Mansouri and Percival (69) observed that the anticancer effect of cranberry extract may result in a decrease in Akt induced in HL-60 cells, which leads to an increase in dephosphorylated BAD and subsequent activation of the intrinsic apoptosis pathway. Endo et al (70) demonstrated that the pro-apoptotic effect of curcumin is partly associated with BAD transfer from the cytoplasm to the mitochondrial membrane to trigger the mitochondrial cell death pathway by inhibiting the expression of 14-3-3 in the cytoplasm. An Akt-dependent manner was determined to be utilized to promote the dephosphorylation of BAD by curcumin (70). Furthermore, Gao et al (71) revealed that the JNK-p21/BAD signaling pathway may be involved in the process of cell proliferation inhibition and cisplatin resistance mediated by downregulation of cell death inducing DFFA like effector A protein in esophageal cancer.
Overview of DAD1 and its cancerous role
Biological characteristics and function of DAD1
DAD1 was originally cloned from a temperature-sensitive nephrogenic cell line (TSBN7) (11). Since a somatic mutation of DAD1 in a temperature-sensitive cell line was responsible for the induction of apoptosis when shifted to a non-permissive temperature, it was proposed that DAD1 may inhibit apoptosis and it was named based on this function (11). Subsequently, a number of studies have demonstrated another role of DAD1, acting as a subunit of OST, which participates in aspartic acid-mediated N-linked glycosylation (72,73). Human DAD1 gene mapping at chromosome 14q11-q12, encoding 113 amino acids (74), is widely expressed in thyroid, adrenal, kidney and lung cells (75). The molecular structure of DAD1 is conserved and stable during biological evolution, which indicates that it has an important function in cellular modulation (76).
Research on the function of DAD1 mainly focuses on two aspects: DAD1 as a crucial component of OST in catalyzing N-linked glycosylation and DAD1 as a pivotal negative regulator in programmed cell death. N-linked glycosylation is a type of co-translational or post-translational modification of proteins. The newly synthesized N-glycosylated chains are added to the asparagine residues of the peptide chains by the OST complex. However, to the best of our knowledge, the mechanism by which DAD1 regulates apoptosis remains unclear. One of the apoptotic mechanisms proposed is the DAD1 loss-induced N-linked glycosylation block (77). Researchers have demonstrated that deletion of DAD1 in hamster TSBN7 cells induces apoptosis (11). However, cycloheximide (a protein synthesis inhibitor) inhibits this process, while Bcl-2, a conventional anti-apoptotic molecule, does not (11). Notably, apoptosis induced by DAD1 deletion could not be rescued by Bcl-2, suggesting that DAD1 may serve a pivotal role in the ER pathway rather than the mitochondrial and death receptor pathways. Brewster et al (78) and Hong et al (79) have reported that DAD1 is necessary for development beyond the blastocyst stage, and its deletion promotes apoptosis in mouse embryos. However, in terms of T cell development and activation, DAD1 enhances T cell proliferation instead of preventing apoptosis in vivo (80).
Gene cloning and/or functional exploration of heterogenetic DAD1 has been performed and validated in other species, including Chlamydomonas (81), Hessian fly Mayetiola destructor (82) and bay scallop Argopecten irradians (83). Furthermore, enhanced expression levels of DAD1 have been suggested to be accountable for unanticipated stimulus in case of cell injury or apoptosis (83). Zhang et al (76) demonstrated that DAD1 in Drosophila melanogaster (DmDAD1) contributes to tissue enrichment, and upregulation of DmDAD1 facilitates N-linked glycosylation. Furthermore, feasible mechanisms have been proposed in terms of the deletion of DmDAD1, resulting in subsequent apoptosis (76). Loss of DmDAD1 leads to blocked N-linked glycosylation and the accumulation of unfolded or misfolded peptide chains, and enhancement of ER stress. Defects in DmDAD1, which employs the JNK pathway downstream to implement apoptosis, activate the protein kinase R-like endoplasmic reticulum kinase (Perk)/activating transcription factor 4 (Atf4) signaling pathway (76). On the other hand, compensatory proliferation of neighboring cells is driven by the Perk/Atf4 signaling pathway to sustain tissue homeostasis (76). Furthermore, Wang et al (84) cloned the homologous gene of DAD1 in Chlamys farreri (CfDAD1) and observed that suppression of CfDAD1 with specific dsRNA injection results in increased cell apoptosis. In addition, high mRNA expression levels of CfDAD1 are detected in the hepatopancreas and gill, which are regarded as immune battlefields, indicating its key role in the innate immunity of scallops (84). Another study revealed the interaction between DAD1 and Mcl-1, an anti-apoptotic member of the Bcl-2 family, and apoptosis triggered by DAD1 depletion could be inhibited by Mcl-1, indicating the feasible interaction between the two apoptosis pathways (85). Notably, the anti-apoptotic effect of DAD1 has also been determined in humans in vivo. The expression levels of DAD1 are increased in neutrophils from patients with sepsis after multiple traumas (86). Furthermore, increased expression levels of the DAD1 gene have been observed in thymocytes of enhancer Eα's downstream CTCF binding sites (EACBE)−/− mice (87). The increased DAD1 expression in EACBE-deleted CD4+CD8+ double-positive thymocytes can be explained by the increased interaction between enhancer Eα and DAD1 (87). EACBE is essential for the sub-topologically associating domains boundary, which separates the Tcra-Tcrd locus and the downstream region including the DAD1 gene (87). DAD1 has also been reported to be an adipokine candidate in adipose tissue (88).
Cancerous role of DAD1
Since DAD1 is a negative regulator of apoptosis, the anti-apoptotic function of DAD1 may pose potential advantages for tumor cells to allow them to infinitely proliferate, and research on this aspect highlights the role of DAD1 in cancer therapy. The aberrant expressive alterations of the DAD1 gene in different cancer types are summarized in Table II.
|
Table II.
Aberrant expressive alterations of DAD1 gene in diverse cancer cells.
|
Table II.
Aberrant expressive alterations of DAD1 gene in diverse cancer cells.
| First author/s, year |
Cancer type |
Protein or mRNA alterations of DAD1 gene |
(Refs.) |
| Tanaka et al, 2001 |
Hepatocellular carcinoma |
mRNA upregulated |
(89) |
| Bandres et al, 2004 |
Colorectal carcinoma |
Protein upregulated |
(90) |
| Kulke et al, 2008 |
Small bowel carcinoid tumor |
Protein upregulated |
(91) |
| Zhu et al, 2014 |
Solid pseudopapillary tumor of pancreas |
Protein downregulated |
(94) |
| Schnormeier et al, 2020 |
Chronic lymphocytic leukemia |
Protein upregulated |
(95) |
| Ayala et al, 2004; True et al, 2006; Bhasin, 2015 |
Prostate cancer |
Protein upregulated |
(96,97,100) |
| Wang et al, 2016 |
Invasive bladder cancer |
mRNA downregulated |
(98) |
| Yoon et al, 2010 |
Cisplatin-resistant ovarian cancer |
Protein and mRNA upregulated |
(99) |
Aberrant expression in cancer
Tanak et al (89) identified that DAD1 mRNA, and antisecretory factor-1, gp96 and CDC34, are highly expressed in HCC cells compared with adjacent non-tumorous liver tissues or normal liver tissues. Bandres et al (90) reported that DAD1 expression is upregulated in colorectal carcinoma with lymph node metastasis compared with that without lymph node metastasis, indicating the potential positive lymph node involvement. Kulke et al (91) reported that the expression levels of DAD1 in small bowel carcinoid tumor cells are higher than those in normal mucosa or the surrounding stroma. In addition, Wilson (92) conducted a meta-analysis to explore the genes involved in small ubiquitin-related modifier (SUMO) signaling pathways. In a meta-analysis, 10 out of 15 analyzed studies reported that DAD1 is co-expressed with SUMO1, which was the highest co-expression finding with SUMO1 (92). This result indicates that DAD1 might act as a member of the SUMO signaling pathway in cancer. Ter-Minassian et al (93) examined genetic associations with sporadic neuroendocrine tumor (NET) risk between patients with sporadic NET and healthy controls using a custom array containing 1,536 SNPs in 355 candidate genes. Ter-Minassian et al (93) demonstrated that DAD1 contained two of the SNPs found to be associated with NET risk, including in another independent duplication set, revealing that the DAD1-associated apoptosis pathway may participate in neuroendocrine tumorigenesis. Zhu et al (94) conducted a pioneering study on the proteomics of solid pseudopapillary tumor of the pancreas (SPTP), in which isobaric tags for relative and absolute quantitation technology integrated in liquid chromatography-tandem mass spectrometry analysis were utilized to determine differentially expressed proteins in SPTP samples compared with normal pancreatic tissues. Bioinformatics analysis resulted in 1,171 qualified proteins. Immunohistochemistry was performed to confirm the differential expression of six representative proteins and revealed the downregulation of DAD1 in SPTP specimens (94). This suggests that DAD1, together with other abnormally expressed proteins, may be a potential biomarker of SPTP in clinical therapy. High expression levels and high variability of DAD1 have also been determined in chronic lymphocytic luekemia, one of the non-solid tumors (95).
Invasion
Ayala et al (96) observed that the expression levels of NF-κB, and its downstream agents DAD1 and pim-2 proto-oncogene, were increased in perineural prostate cancer cells. Concurrent to the positive association between DAD1 expression and Gleason score in prostate adenocarcinoma, higher levels of DAD1 expression are associated with cancerous epithelium and perineural invasion (97). Wang et al (98) revealed that DAD1 is one of 21 differentially expressed genes in bladder cancer. Downregulation of the lncRNA NONHSAG045391 co-expressed with DAD1 has been observed in invasive bladder cancer, suggesting that NONHSAG045391 may contribute to enhanced invasiveness by targeting DAD1 (98). However, to the best of our knowledge, the concrete mechanism remains obscure.
Cisplatin resistance
Cisplatin treatment of a cell line derived from a clinical patient with cisplatin-resistant OVCA facilitated DAD1 expression at both the transcription and protein levels, indicating that cisplatin resistance might partly result from the upregulation of DAD1 (99).
Novel performance
The role of DAD1 in prostate cancer has been previously analyzed (100). First, increased DAD1 expression was detected in samples derived from clinical patients with prostate cancer compared with that in normal adjacent tissues. Furthermore, the study revealed that different TNM grades and Gleason grades were associated with prominent differences in DAD1 expression levels, which gradually increased with the progression of prostate cancer, underlying its diagnostic or prognostic role as a biomarker (100). Receiver operating characteristic curve analysis revealed that serum DAD1 exhibited improved specificity and sensitivity compared with prostate-specific antigen (PSA) in distinguishing low Gleason and high Gleason prostate cancer (100). Additionally, Bhasin (100) determined that ribophorin I (RPN1), another subunit of OST, is essential for DAD1 retention in the ER. DAD1 could be exocytosed with the downregulation of RPN1; thus, intervention with DAD1 antibody was implemented to check if DAD1 exocytosis was necessary. As a result, the DAD1 antibody exhibited markedly increased cytotoxicity compared with the control antibody in cancer cells and suppressed cancer cell survival (100). Furthermore, Bhasin (100) pointed out that this type of apoptosis was the result of extracellular DAD1 interacting with Fas protein. This research highlighted the potential of DAD1 in targeted therapy of cancer.
Feasible relation and interaction mechanism between BAD and DAD1
Table III summarizes the genetic alterations involved in pathways with BAD and DAD1 participation and their effects on cellular behavior.
|
Table III.
Genetic alterations associated with BAD and DAD1 signaling pathways.
|
Table III.
Genetic alterations associated with BAD and DAD1 signaling pathways.
| A, BAD |
|
| First author/s, year |
Cellular behavior |
Gene alteration and outcome |
(Refs.) |
| Sastry et al, 2006 |
Proliferation, survival and apoptosis |
Phosphorylation of BAD on Ser112 and Ser136 via Ras/MEK and Rac/PKA1 signaling pathways attenuates apoptosis. |
(38) |
| She et al, 2005 |
|
Phosphorylation defect of BAD on Ser112 and Ser136 via EGFR/MEK/MAPK and PI3K/Akt signaling pathways promotes apoptosis. |
(39) |
| Polzien et al, 2011 |
|
Phosphorylation of BAD on Ser134 by Raf kinases promotes. proliferation |
(40) |
| Winter et al, 2014 |
|
Phosphorylation of BAD induced by JAK2 promotes survival. |
(44) |
| Stickles et al, 2015 |
|
High levels of p-BAD induced by PP2C deletion promote cell proliferation. |
(47) |
| Kulik, 2019 |
|
Upregulated p-BAD co-expressed with Mcl-1 leads to increased apoptosis inhibition. |
(48) |
| Mann et al, 2019 |
|
Phosphorylation of BAD at Ser118 increases 14-3-3 binding and Akt activation to promote growth. |
(49) |
| Cekanova et al, 2015 |
Invasion |
Overexpressed BAD decreases gene expression to inhibit invasion (cyclin D1, MMP 10, c-Jun, Akt and STATs), metastasis (Snail) and EMT (Sp1, β-catenin, GSK-3β). |
(52) |
| Chon et al, 2012 |
Chemosensitivity |
PP2C siRNA leads to higher levels of p-BAD and cisplatin resistance. |
(56) |
| Hayakawa et al, 2000 |
|
Phosphorylation defect of BAD at Ser112 and Ser136 mediated by the ERK and Akt cascade, respectively, sensitizes ovarian cancer cells to cisplatin. |
(57) |
| Marchion et al, 2011 |
|
Cisplatin resistance is associated with BAD signaling pathway genes, including Bax, Bcl-xL and PP2C/PPM1A. |
(58) |
| Bansal et al, 2012 |
|
CDK1 (BAD signaling pathway gene) siRNA increases cisplatin sensitivity. |
(59) |
| Yiau et al, 2019 |
|
CD34 and p-BAD are increased in chemoresistant samples. |
(63) |
|
| B, DAD1 |
|
| First author/s, year |
Cellular behavior |
Gene alteration and outcome |
(Refs.) |
|
| Zhang et al, 2016 |
Proliferation, survival and apoptosis |
DAD1 defect-induced endoplasmic reticulum stress triggers Perk-Atf4 signaling pathway to induce apoptosis. |
(76) |
| |
|
Perk-Atf4 signaling pathway indirectly induces compensatory cell proliferation. |
|
| Bhasin, 2015 |
|
Extracellular DAD1 interacting with Fas results in apoptosis. |
(100) |
| Ayala et al, 2004 |
Invasion |
Increased expression levels of DAD1, PIM2 and NF-κB is associated with enhanced perineural invasion. |
(96) |
| Wang et al, 2016 |
|
Long non-coding RNA NONHSAG045391 is co-expressed with DAD1 to enhance invasion. |
(98) |
| Yoon et al, 2010 |
Chemosensitivity |
Overexpression of DAD1 is found in cisplatin-resistant ovarian cancer cells. |
(99) |
The apoptotic function of BAD has been greatly explored since its discovery, and the present review proposes a novel role of BAD. AI-Bazz et al (101) reported that BAD expression appeared to be nuclear in addition to its cytoplasmic location according to immunostaining in primary breast cancer. In addition, using proliferating breast cancer cell lines, Fernando et al (102) observed that endogenous BAD exists in both the cytoplasm and nucleus, whereas the levels of p-BAD in the nucleus are lower than those in the cytoplasm. Overexpression of BAD could augment the levels of p-BAD in the nucleus and inhibit the expression of cyclin D1 on the basis of phosphorylation at Ser75 and Ser99 and in combination with c-Jun (102). Using a chromatin immunoprecipitation assay, they further demonstrated that BAD could bind to the 12-O-tetradecanoylphorbol-13-acetate response element (TRE) and cAMP response element (CRE) in the promoter region of the natural cyclin D1 gene and possibly attenuate the transcriptional activity of c-Jun to suppress cyclin D1 expression (102). Activator protein 1 (AP1), a putative transcription factor, is a heterodimer of c-Jun and c-Fos that was also observed to be abolished by overexpression of BAD (102). These findings indicate that BAD may act as a DNA promoter binding protein and exert a transcription factor-like effect to downregulate the expression levels of targeted genes (102). Interestingly, the sequences of the DAD1 promoter region (~2.0 kb) were searched to predict the binding sites of transcription factors using PROMO (version 8.3; http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3). The final results are listed and shown in Table IV and Fig. 1. It was identified that there were two putative transcription factors, CRE element binding, DNA-binding transcriptional regulator (CREB) and AP1, which are able to bind CRE and TRE, respectively, in the DAD1 promoter region. In other words, BAD, as reported by Fernando et al (102), can bind with CRE and TRE in the cyclin D1 promoter region and may also bind with the CRE and TRE elements in the DAD1 promoter region. A previous study (103) revealed that overexpression of BAD in esophageal cancer cells could inhibit DAD1 expression, which could be restored when BAD expression is downregulated. Taken together, it was hypothesized that there is a negative regulatory relationship between BAD and DAD1 [Fig. 2; (104–106)]. BAD can act as a transcription factor by binding to the promoter of DAD1 to inhibit its expression. Our hypothesis suggests that BAD can act as a transcription factor-like protein to negatively regulate the expression of targeted genes and explains the relationship between BAD and DAD1 in apoptosis regulation and the crosstalk between two apoptotic signaling pathways: The mitochondrial cell death pathway and the ER cell death pathway. Further studies should be performed to support this hypothesis.
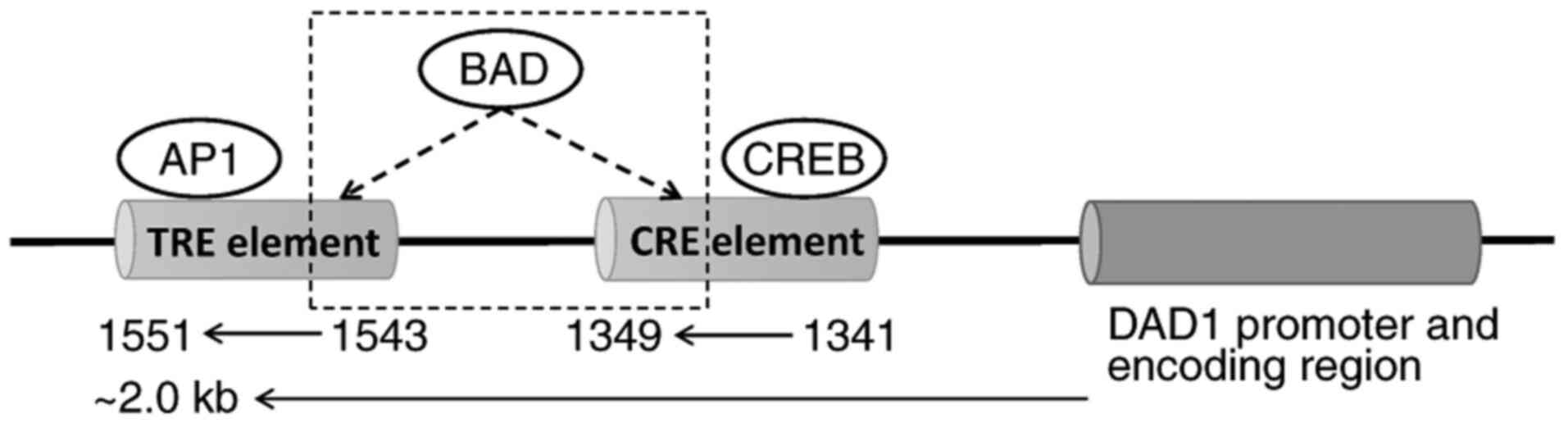 |
Figure 1.
Potential transcription factor binding sites in DAD1 promoter region. Two putative transcription factors, CREB and AP1, are predicted to bind with CRE and TRE, respectively. The possible binding relationship between BAD and CRE/TRE is highlighted with a dashed outline. AP1, activator protein 1; CRE, cAMP response element; CREB, CRE element binding, DNA-binding transcriptional regulator; DAD1, defender against apoptotic cell death 1; TRE, 12-O-tetradecanoylphorbol-13-acetate response element.
|
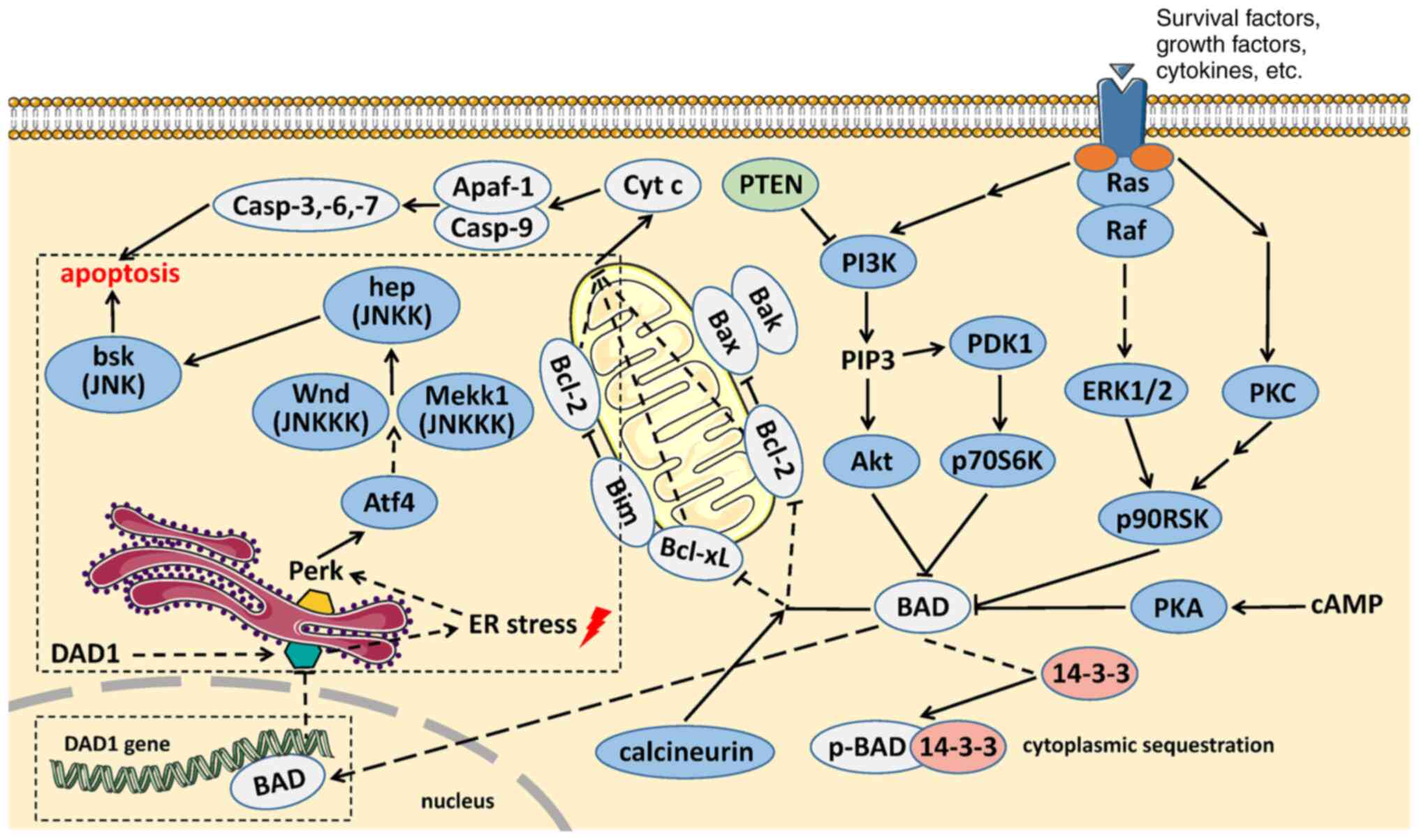 |
Figure 2.
Feasible interaction between BAD and DAD1 and sequential apoptosis. Our hypothesis is highlighted with a dashed outline. By exerting a transcription factor-like function, the combination between BAD and the promoter region of the DAD1 gene results in DAD1 knockdown. Loss of DAD1 function triggers ER stress and activates the Perk-Atf4 signaling pathway, which sequentially activates the JNK signaling pathway via MEKK1, together with Wnd, and eventually initiates apoptosis (76). The apoptosis signaling pathways in Fig. 2 were created with reference to signaling pathway figures on the Cell Signaling Technology, Inc. website (104–106). ER stress, endoplasmic reticulum stress.
|
|
Table IV.
PROMO online prediction of transcription factor binding sites in the defender against apoptotic cell death 1 promoter region.
|
Table IV.
PROMO online prediction of transcription factor binding sites in the defender against apoptotic cell death 1 promoter region.
| Transcription factor name |
Start position |
End position |
Dissimilarity |
String |
RE equally |
RE query |
| CREB [T00163] |
1341 |
1349 |
3.614755 |
ACAACGTCA |
0.10681 |
0.10409 |
| AP1 [T00029] |
1543 |
1551 |
14.681715 |
TGACTTGTT |
0.27466 |
0.28161 |
BAD and DAD1 as potential targets and biomarkers in cancer
Based on the studies presented, it can be clearly inferred that BAD and DAD1 serve an indispensable role in certain types of cancer development and progression, resulting from their key regulatory functions in apoptosis pathways and a number of other abilities affecting tumorigenesis (27–33,76,83,107). Therefore, it is possible to identify the two proteins as emerging useful targets and biomarkers in carcinogenesis and tumor therapies. Aberrant expression of both proteins in cancer cells is not always the same since their expression is influenced by cancer type and several unknown factors. However, this suggests that their ectopic expression indicates the disorder of apoptosis signaling pathways. Therefore, drugs that target BAD and DAD1 can be utilized to restore or suppress the activity of apoptosis signaling pathways. Furthermore, as summarized in the present review, the expression levels of BAD and DAD1 are associated with apoptosis (11,17,38–49,76–79), invasion enhancement and metastasis (51–53,96–98), and chemoresistance (55–64,99). Therefore, it is possible to predict the potential of apoptosis, invasion and chemoresistance by detecting the expression levels of BAD and DAD1 in pathological samples. Apart from their collective application value, there are several insights presented for the two proteins. As mentioned in the cell proliferation, survival and apoptosis subsection of the present review, the phosphorylation status of BAD determines the incline of the apoptosis-survival balance (17,27,37–44). Therefore, examination of the phosphorylation status of BAD may contribute to the efficacy evaluation in response to treatment. On the other hand, kinases and phosphatases associated with BAD phosphorylation status can also be exploited as potential targets due to their plausible interactions with p-BAD. Furthermore, as summarized in the clinical characteristics subsection of the present review, the expression levels of BAD are associated with a number of clinical pathological characteristics of cancer, such as overall survival, clinical stage and tumor size, and thus, BAD also acts as an independent biomarker of prognosis (51–55). In terms of DAD1, the favorable proliferation inhibition by treatment with DAD1 antibody against prostate cancer cells in vitro has inventively demonstrated that DAD1 could act as a potential target in tumor therapy, together with its improved sensitivity and specificity as a diagnostic/prognostic biomarker compared with PSA (100).
Conclusion
The present review summarizes insights on the functional roles of BAD and DAD1, particularly in cancer, and contributions to apoptosis, invasion and chemosensitivity are emphasized. However, the underlying molecular mechanisms involved require further exploration. It is gradually becoming clear that the two proteins are mainly involved in tumorigenic signaling regulation, whether as a constitutive molecule picked up by other dominant molecules or as central target of signaling pathways, affecting cellular behavior independently. Finally, a hypothesis was proposed to reveal the feasible interaction mechanism between BAD and DAD1. It was highlighted that decreased DAD1 expression results from BAD binding to the DAD1 gene promoter region, exerting a novel transcription factor-like function. BAD and DAD1, two emerging molecules acting as targets and biomarkers in tumorigenesis, their specific functional mechanisms and their exploitation value should be given more importance when considering the broader clinical therapeutic applications.
Acknowledgements
Not applicable.
Funding
This review was supported by Natural Science Foundation of Xinjiang Uygur Autonomous Region (grant no. 2020D01C171). The Key Discipline of the 13th Five-Year Plan (Plateau Discipline) of Xinjiang Uygur Autonomous Region also provided support to this study.
Availability of data and materials
Not applicable.
Authors' contributions
YL was the major contributor in writing of the manuscript, as well as in the preparation of the tables and figures. YW and HH searched and integrated all the supporting references, helped to refine the figures and tables and edited the manuscript. NY helped to improve the language and logic of the text in the present study. YC was responsible for the study design. Data authentication is not applicable. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
DAD1
|
defender against apoptotic cell death 1
|
|
OST
|
oligosaccharyltransferase
|
|
ER
|
endoplasmic reticulum
|
|
Mcl-1
|
myeoid cell leukemin-1
|
|
PKA
|
cAMP dependent protein kinase
|
|
p-BAD
|
phosphorylated BAD
|
|
PAK1
|
P21-activated kinase 1
|
|
MPNs
|
myeloproliferative neoplasms
|
|
JAK
|
Janus kinase
|
|
PP2C
|
protein phosphatase 2C
|
|
CSCs
|
cancer stem cells
|
|
OVCA
|
ovarian cancer
|
|
HCC
|
hepatocellular carcinoma
|
|
EMT
|
epithelial-mesenchymal-transition
|
|
SCLC
|
small cell lung carcinoma
|
|
TNBC
|
triple-negative breast cancer
|
|
EC
|
endometrial cancer
|
|
AML
|
acute myeloid leukemia
|
|
Bim
|
Bcl-2 interacting mediator of cell death protein
|
|
EBV
|
Epstein-Barr virus
|
|
AMPK
|
protein kinase AMP-activated catalytic subunit α1
|
|
TSBN7
|
temperature-sensitive nephrogenic cell line
|
|
DmDAD1
|
Drosophila melanogaster DAD1
|
|
Perk
|
protein kinase R-like endoplasmic reticulum kinase
|
|
Atf4
|
activating transcription factor 4
|
|
CfDAD1
|
Chlamys farreri DAD1
|
|
SUMO
|
small ubiquitin-related modifier
|
|
NET
|
neuroendocrine tumor
|
|
SPTP
|
solid pseudopapillary tumor of the pancreas
|
|
PSA
|
prostate-specific antigen
|
|
RPN1
|
ribophorin I
|
|
TRE
|
12-O-tetradecanoylphorbol-13-acetate response element
|
|
CRE
|
cAMP response element
|
|
AP1
|
activator protein 1
|
|
CREB
|
CRE element binding, DNA-binding transcriptional regulator
|
References
|
1
|
El Bali M, Bakkach J and Mechita MB: Colorectal cancer: From genetic landscape to targeted therapy. J Oncol. 2021:99181162021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan J, Shen X, Wang Y, Zhou HL, Liu G, Li YL and Xu ZX: Biomarkers for immune checkpoint therapy targeting programmed death 1 and programmed death ligand 1. Biomed Pharmacother. 130:1106212020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang M, Shen A, Ding J and Geng M: Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol Sci. 35:41–50. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Troxell ML, Higgins JP and Kambham N: Antineoplastic treatment and renal injury: An update on renal pathology due to cytotoxic and targeted therapies. Adv Anat Pathol. 23:310–329. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun M, Wang T, Li L, Li X, Zhai Y, Zhang J and Li W: The application of inorganic nanoparticles in molecular targeted cancer therapy: EGFR targeting. Front Pharmacol. 12:7024452021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eisenberg-Lerner A, Bialik S, Simon HU and Kimchi A: Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 16:966–975. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hanahan D and Weinberg RA: Hallmarks of cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blandino G and Strano S: BCL-2: The pendulum of the cell fate. J Exp Clin Cancer Res. 16:3–10. 1997.PubMed/NCBI
|
|
9
|
Sastry KS, Al-Muftah MA, Li P, Al-Kowari MK, Wang E, Chouchane AI, Kizhakayil D, Kulik G, Marincola FM, Haoudi A and Chouchane L: Targeting proapoptotic protein BAD inhibits survival and self-renewal of cancer stem cells. Cell Death Differ. 21:1936–1949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanjay A, Fu J and Kreibich G: DAD1 is required for the function and the structural integrity of the oligosaccharyltransferase complex. J Biol Chem. 273:26094–26099. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakashima T, Sekiguchi T, Kuraoka A, Fukushima K, Shibata Y, Komiyama S and Nishimoto T: Molecular cloning of a human cDNA encoding a novel protein, DAD1, whose defect causes apoptotic cell death in hamster BHK21 cells. Mol Cell Biol. 13:6367–6374. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Czabotar PE, Lessene G, Strasser A and Adams JM: Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat Rev Mol Cell Biol. 15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bhola PD and Letai A: Mitochondria-judges and executioners of cell death sentences. Mol Cell. 61:695–704. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsu SY, Kaipia A, Zhu L and Hsueh AJ: Interference of BAD (Bcl-xL/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14-3-3 isoforms and P11. Mol Endocrinol. 11:1858–1867. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Danial NN: BAD: Undertaker by night, candyman by day. Oncogene. 27 (Suppl 1):S53–S70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Polzien L, Baljuls A, Rennefahrt UEE, Fischer A, Schmitz W, Zahedi RP, Sickmann A, Metz R, Albert S, Benz R, et al: Identification of novel in vivo phosphorylation sites of the human proapoptotic protein BAD: Pore-forming activity of BAD is regulated by phosphorylation. J Biol Chem. 284:28004–28020. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang E, Zha J, Jockel J, Boise LH, Thompson CB and Korsmeyer SJ: Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 80:285–291. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
del Peso L, González-García M, Page C, Herrera R and Nuñez G: Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 278:687–689. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan Y, Demeter MR, Ruan H and Comb MJ: BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 275:25865–25869. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lizcano JM, Morrice N and Cohen P: Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J. 349:547–557. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou XM, Liu Y, Payne G, Lutz RJ and Chittenden T: Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on Ser155. J Biol Chem. 275:25046–25051. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Radisavljevic Z: AKT as locus of cancer angiogenic robustness and fragility. J Cell Physiol. 228:21–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan J, Xiang J, Lin Y, Ma J, Zhang J, Zhang H, Sun J, Danial NN, Liu J and Lin A: Inactivation of BAD by IKK inhibits TNFα-induced apoptosis independently of NF-κB activation. Cell. 152:304–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pandey V, Wang B, Mohan CD, Raquib AR, Rangappa S, Srinivasa V, Fuchs JE, Girish KS, Zhu T, Bender A, et al: Discovery of a small-molecule inhibitor of specific serine residue BAD phosphorylation. Proc Natl Acad Sci USA. 115:E10505–E10514. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang H, Masters SC, Wang H and Fu H: The proapoptotic protein Bad binds the amphipathic groove of 14-3-3zeta. Biochim Biophys Acta. 1547:313–319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hekman M, Albert S, Galmiche A, Rennefahrt UEE, Fueller J, Fischer A, Puehringer D, Wiese S and Rapp UR: Reversible membrane interaction of BAD requires two C-terminal lipid binding domains in conjunction with 14-3-3 protein binding. J Biol Chem. 281:17321–17336. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM and Yang E: Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 22:5459–5470. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Linette GP, Li Y, Roth K and Korsmeyer SJ: Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc Natl Acad Sci USA. 93:9545–9552. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chattopadhyay A, Chiang CW and Yang E: BAD/BCL-[X(L)] heterodimerization leads to bypass of G0/G1 arrest. Oncogene. 20:4507–4518. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al: Functional and physical interaction between Bcl-X(L) and a BH3-like domain in beclin-1. EMBO J. 26:2527–2539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME and Korsmeyer SJ: Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 100:9324–9329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ and Greenberg ME: Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell. 3:631–643. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al: BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 424:952–956. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Githaka JM, Tripathi N, Kirschenman R, Patel N, Pandya V, Kramer DA, Montpetit R, Zhu LF, Sonenberg N, Fahlman RP, et al: BAD regulates mammary gland morphogenesis by 4E-BP1-mediated control of localized translation in mouse and human models. Nat Commun. 12:29392021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Giménez-Cassina A, Garcia-Haro L, Choi CS, Osundiji MA, Lane EA, Huang H, Yildirim MA, Szlyk B, Fisher JK, Polak K, et al: Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 19:272–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
National Center for Biotechnology Information (NCBI), . BAD BCL2 associated agonist of cell death [Homo sapiens (human)]. NCBI; Bethesda MD: 2021, http://www.ncbi.nlm.nih.gov/gene/572September 2–2021
|
|
37
|
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sastry KS, Karpova Y and Kulik G: Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J Biol Chem. 281:27367–27377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J and Rosen N: The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 8:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Polzien L, Baljuls A, Albrecht M, Hekman M and Rapp UR: BAD contributes to RAF-mediated proliferation and cooperates with B-RAF-V600E in cancer signaling. J Biol Chem. 286:17934–17944. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, et al: Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 363:1117–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al: Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 7:387–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, et al: A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 434:1144–1148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A and Wood KC: RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal. 7:ra1222014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang N, Zhu J, Liu D, Li YL, Chen BJ, He YQ, Liu K, Mo XM and Li WM: Overexpression of Bcl-2-associated death inhibits A549 cell growth in vitro and in vivo. Cancer Biother Radiopharm. 27:164–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Smith AJ, Karpova Y, D'Agostino R Jr, Willingham M and Kulik G: Expression of the Bcl-2 protein BAD promotes prostate cancer growth. PLoS One. 4:e62242009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Stickles XB, Marchion DC, Bicaku E, Al Sawah E, Abbasi F, Xiong Y, Zgheib NB, Boac BM, Orr BC, Judson PL, et al: BAD-mediated apoptotic pathway is associated with human cancer development. Int J Mol Med. 35:1081–1087. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kulik G: ADRB2-Targeting therapies for prostate cancer. Cancers (Basel). 11:3582019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mann J, Githaka JM, Buckland TW, Yang N, Montpetit R, Patel N, Li L, Baksh S, Godbout R, Lemieux H and Goping IS: Non-canonical BAD activity regulates breast cancer cell and tumor growth via 14-3-3 binding and mitochondrial metabolism. Oncogene. 38:3325–3339. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lu P, Bowman KE, Brown SM, Joklik-Mcleod M, Mause ER, Nguyen HTN and Lim CS: p53-bad: A novel tumor suppressor/proapoptotic factor hybrid directed to the mitochondria for ovarian cancer gene therapy. Mol Pharm. 16:3386–3398. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hu W, Fu J, Lu SX, Liu LL, Luo RZ, Yun JP and Zhang CZ: Decrease of Bcl-xL/Bcl-2-associated death promoter in hepatocellular carcinoma indicates poor prognosis. Am J Cancer Res. 5:1805–1813. 2015.PubMed/NCBI
|
|
52
|
Cekanova M, Fernando RI, Siriwardhana N, Sukhthankar M, Parra C, Woraratphoka J, Malone C, Ström A, Baek SJ, Wade PA, et al: BCL-2 family protein, BAD is down-regulated in breast cancer and inhibits cell invasion. Exp Cell Res. 331:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhu X, Yu Y, Hou X, Xu J, Tan Z, Nie X, Ling Z and Ge M: Expression of PIM-1 in salivary gland adenoid cystic carcinoma: Association with tumor progression and patients' prognosis. Oncol Lett. 15:1149–1156. 2018.PubMed/NCBI
|
|
54
|
Yu Y, Zhong Z and Guan Y: The downregulation of Bcl-xL/Bcl-2-associated death promoter indicates worse outcomes in patients with small cell lung carcinoma. Int J Clin Exp Pathol. 8:13075–13082. 2015.PubMed/NCBI
|
|
55
|
Boac BM, Abbasi F, Ismail-Khan R, Xiong Y, Siddique A, Park H, Han M, Saeed-Vafa D, Soliman H, Henry B, et al: Expression of the BAD pathway is a marker of triple-negative status and poor outcome. Sci Rep. 9:174962019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chon HS, Marchion DC, Xiong Y, Chen N, Bicaku E, Stickles XB, Zgheib NB, Judson PL, Hakam A, Gonzalez-Bosquet J, et al: The BCL2 antagonist of cell death pathway influences endometrial cancer cell sensitivity to cisplatin. Gynecol Oncol. 124:119–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T and Murata Y: Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 60:5988–5994. 2000.PubMed/NCBI
|
|
58
|
Marchion DC, Cottrill HM, Xiong Y, Chen N, Bicaku E, Fulp WJ, Bansal N, Chon HS, Stickles XB, Kamath SG, et al: BAD phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin Cancer Res. 17:6356–6366. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bansal N, Marchion DC, Bicaku E, Xiong Y, Chen N, Stickles XB, Sawah EA, Wenham RM, Apte SM, Gonzalez-Bosquet J, et al: BCL2 antagonist of cell death kinases, phosphatases, and ovarian cancer sensitivity to cisplatin. J Gynecol Oncol. 23:35–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu B, Sun X, Shen HY, Gao F, Fan YM and Sun ZJ: Expression of the apoptosis-related genes BCL-2 and BAD in human breast carcinoma and their associated relationship with chemosensitivity. J Exp Clin Cancer Res. 29:1072010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mann J, Yang N, Montpetit R, Kirschenman R, Lemieux H and Goping IS: BAD sensitizes breast cancer cells to docetaxel with increased mitotic arrest and necroptosis. Sci Rep. 10:3552020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yu N, Seedhouse C, Russell N and Pallis M: Quantitative assessment of the sensitivity of dormant AML cells to the BAD mimetics ABT-199 and ABT-737. Leuk Lymphoma. 59:2447–2453. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yiau SK, Lee C, Tohit ER, Chang KM and Abdullah M: Potential CD34 signaling through phosphorylated-BAD in chemotherapy-resistant acute myeloid leukemia. J Recept Signal Transduct Res. 39:276–282. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong X and Wei L: Autophagy inhibits chemotherapy-induced apoptosis through downregulating Bad and Bim in hepatocellular carcinoma cells. Sci Rep. 4:53822014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim H, Choi H and Lee SK: Epstein-barr virus microRNA miR-BART20-5p suppresses lytic induction by inhibiting BAD-mediated caspase-3-dependent apoptosis. J Virol. 90:1359–1368. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tang B, Tang F, Wang Z, Qi G, Liang X, Li B, Yuan S, Liu J, Yu S and He S: Upregulation of Akt/NF-κB-regulated inflammation and Akt/Bad-related apoptosis signaling pathway involved in hepatic carcinoma process: Suppression by carnosic acid nanoparticle. Int J Nanomedicine. 11:6401–6420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhao X, Fan Y, Lu C, Li H, Zhou N, Sun G and Fan H: PCAT1 is a poor prognostic factor in endometrial carcinoma and associated with cancer cell proliferation, migration and invasion. Bosn J Basic Med Sci. 19:274–281. 2019.PubMed/NCBI
|
|
68
|
Liu Z, Zhang G, Huang S, Cheng J, Deng T, Lu X, Adeshakin FO, Chen Q and Wan X: Induction of apoptosis in hematological cancer cells by dorsomorphin correlates with BAD upregulation. Biochem Biophys Res Commun. 522:704–708. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mansouri RA and Percival SS: Cranberry extract initiates intrinsic apoptosis in HL-60 cells by increasing BAD activity through inhibition of AKT phosphorylation. BMC Complement Med Ther. 20:712020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Endo H, Inoue I, Masunaka K, Tanaka M and Yano M: Curcumin induces apoptosis in lung cancer cells by 14-3-3 protein-mediated activation of Bad. Biosci Biotechnol Biochem. 84:2440–2447. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Gao YP, Li L, Yan J, Hou XX, Jia YX, Chang ZW, Guan XY and Qin YR: Down-regulation of CIDEA promoted tumor growth and contributed to cisplatin resistance by regulating the JNK-p21/bad signaling pathways in esophageal squamous cell carcinoma. Front Oncol. 10:6278452020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kelleher DJ and Gilmore R: DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc Natl Acad Sci USA. 94:4994–4999. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Roboti P and High S: The oligosaccharyltransferase subunits OST48, DAD1 and KCP2 function as ubiquitous and selective modulators of mammalian N-glycosylation. J Cell Sci. 125:3474–3484. 2012.PubMed/NCBI
|
|
74
|
Apte SS, Mattei MG, Seldin MF and Olsen BR: The highly conserved defender against the death 1 (DAD1) gene maps to human chromosome 14q11-q12 and mouse chromosome 14 and has plant and nematode homologs. FEBS Lett. 363:304–306. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
National Center for Biotechnology Information (NCBI), . DAD1, defender against cell death 1 [Homo sapiens (human)]. Gene ID: 1603. NCBI; Bethesda MD: 2021, http://www.ncbi.nlm.nih.gov/gene/1603September 3–2021
|
|
76
|
Zhang Y, Cui C and Lai ZC: The defender against apoptotic cell death 1 gene is required for tissue growth and efficient N-glycosylation in Drosophila melanogaster. Dev Biol. 420:186–195. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Makishima T, Nakashima T, Nagata-Kuno K, Fukushima K, Iida H, Sakaguchi M, Ikehara Y, Komiyama S and Nishimoto T: The highly conserved DAD1 protein involved in apoptosis is required for N-linked glycosylation. Genes Cells. 2:129–141. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Brewster JL, Martin SL, Toms J, Goss D, Wang K, Zachrone K, Davis A, Carlson G, Hood L and Coffin JD: Deletion of Dad1 in mice induces an apoptosis-associated embryonic death. Genesis. 26:271–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hong NA, Flannery M, Hsieh SN, Cado D, Pedersen R and Winoto A: Mice lacking Dad1, the defender against apoptotic death-1, express abnormal N-linked glycoproteins and undergo increased embryonic apoptosis. Dev Biol. 220:76–84. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hong NA, Kabra NH, Hsieh SN, Cado D and Winoto A: In vivo overexpression of Dad1, the defender against apoptotic death-1, enhances T cell proliferation but does not protect against apoptosis. J Immunol. 163:1888–1893. 1999.PubMed/NCBI
|
|
81
|
Moharikar S, D'Souza JS and Rao BJ: A homologue of the defender against the apoptotic death gene (dad1) in UV-exposed Chlamydomonas cells is downregulated with the onset of programmed cell death. J Biosci. 32:261–270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mittapalli O and Shukle RH: Molecular characterization and responsive expression of a defender against apoptotic cell death homologue from the Hessian fly, Mayetiola destructor. Comp Biochem Physiol B Biochem Mol Biol. 149:517–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhu L, Song L, Zhang H, Zhao J, Li C and Xu W: Molecular cloning and responsive expression to injury stimulus of a defender against cell death 1 (DAD1) gene from bay scallops Argopecten irradians. Mol Biol Rep. 35:125–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang MQ, Wang BJ, Liu M, Jiang KY and Wang L: Molecular characterization of a defender against apoptotic cell death 1 gene (CfDAD1) from the mollusk Chlamys farreri. Invertebr Surviv J. 15:294–301. 2018.
|
|
85
|
Makishima T, Yoshimi M, Komiyama S, Hara N and Nishimoto T: A subunit of the mammalian oligosaccharyltransferase, DAD1, interacts with Mcl-1, one of the bcl-2 protein family. J Biochem. 128:399–405. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Paunel-Görgülü A, Kirichevska T, Lögters T, Windolf J and Flohé S: Molecular mechanisms underlying delayed apoptosis in neutrophils from multiple trauma patients with and without sepsis. Mol Med. 18:325–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhao H, Li Z, Zhu Y, Bian S, Zhang Y, Qin L, Naik AK, He J, Zhang Z, Krangel MS and Hao B: A role of the CTCF binding site at enhancer Eα in the dynamic chromatin organization of the Tcra-Tcrd locus. Nucleic Acids Res. 48:9621–9636. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhang Y, Yu M, Dong J, Wu Y and Tian W: Identification of novel Adipokines through proteomic profiling of small extracellular vesicles derived from adipose tissue. J Proteome Res. 19:3130–3142. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Tanaka K, Kondoh N, Shuda M, Matsubara O, Imazeki N, Ryo A, Wakatsuki T, Hada A, Goseki N, Igari T, et al: Enhanced expression of mRNAs of antisecretory factor-1, gp96, DAD1 and CDC34 in human hepatocellular carcinomas. Biochim Biophys Acta. 1536:1–12. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Bandres E, Catalan V, Sola I, Honorato B, Cubedo E, Cordeu L, Andion E, Escalada A, Zarate R, Salgado E, et al: Dysregulation of apoptosis is a major mechanism in the lymph node involvement in colorectal carcinoma. Oncol Rep. 12:287–292. 2004.PubMed/NCBI
|
|
91
|
Kulke MH, Freed E, Chiang DY, Philips J, Zahrieh D, Glickman JN and Shivdasani RA: High-resolution analysis of genetic alterations in small bowel carcinoid tumors reveals areas of recurrent amplification and loss. Genes Chromosomes Cancer. 47:591–603. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wilson BJ: Meta-analysis of SUMO1. BMC Res Notes. 1:602008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ter-Minassian M, Wang Z, Asomaning K, Wu MC, Liu CY, Paulus JK, Liu G, Bradbury PA, Zhai R, Su L, et al: Genetic associations with sporadic neuroendocrine tumor risk. Carcinogenesis. 32:1216–1222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhu Y, Xu H, Chen H, Xie J, Shi M, Shen B, Deng X, Liu C, Zhan X and Peng C: Proteomic analysis of solid pseudopapillary tumor of the pancreas reveals dysfunction of the endoplasmic reticulum protein processing pathway. Mol Cell Proteomics. 13:2593–2603. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Schnormeier AK, Pommerenke C, Kaufmann M, Drexler HG and Koeppel M: Genomic deregulation of PRMT5 supports growth and stress tolerance in chronic lymphocytic leukemia. Sci Rep. 10:97752020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ayala GE, Dai H, Ittmann M, Li R, Powell M, Frolov A, Wheeler TM, Thompson TC and Rowley D: Growth and survival mechanisms associated with perineural invasion in prostate cancer. Cancer Res. 64:6082–6090. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R, Beer TM, Gelmann E, Datta M, Mostaghel E, et al: A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci USA. 103:10991–10996. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang M, Xiao X, Zeng F, Xie F, Fan Y, Huang C, Jiang G and Wang L: Common and differentially expressed long noncoding RNAs for the characterization of high and low grade bladder cancer. Gene. 592:78–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yoon J, Kim ES, Lee SJ, Park CW, Cha HJ, Hong BH and Choi KY: Apoptosis-related mRNA expression profiles of ovarian cancer cell lines following cisplatin treatment. J Gynecol Oncol. 21:255–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Bhasin N: DAD1 as potential therapeutic target and biomarker in prostate cancer (unpublished PhD thesis). Tulane University; 2015
|
|
101
|
Al-Bazz YO, Underwood JC, Brown BL and Dobson PR: Prognostic significance of Akt, phospho-Akt and BAD expression in primary breast cancer. Eur J Cancer. 45:694–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Fernando R, Foster JS, Bible A, Ström A, Pestell RG, Rao M, Saxton A, Baek SJ, Yamaguchi K, Donnell R, et al: Breast cancer cell proliferation is inhibited by BAD: Regulation of cyclin D1. J Biol Chem. 282:28864–28873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Meiliwuerti D: The regulation mechanism study of Bad on Dad1 gene in esophageal squamous cells. MaD ThesisXinjiang Medical University, China2017.(In Chinese).
|
|
104
|
Cell Signaling Technology (CST), . Mitochondrial control of apoptosis. CST; Danvers, MA: 2021, https://www.cellsignal.com/learn-and-support/order-support?countryId=10036#collapse00July 1–2021
|
|
105
|
Cell Signaling Technology (CST), . Regulation of apoptosis. CST; Danvers, MA: 2021, http://www.cellsignal.com/pathways/regulation-of-apoptosis-pathwayJuly 1–2021
|
|
106
|
Cell Signaling Technology (CST), . Inhibition of apoptosis. CST; Danvers, MA: 2021, http://www.cellsignal.com/pathways/inhibition-of-apoptosis-pathwayJuly 1–2021
|
|
107
|
Bui NL, Pandey V, Zhu T, Ma L, Basappa and Lobie PE: Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 415:177–186. 2018. View Article : Google Scholar : PubMed/NCBI
|














