Introduction
Melanomas are highly malignant tumors that readily metastasize and have poor prognosis, with a 5-year overall survival (OS) rate of 12–28% (1,2). Melanoma has several biologically distinct subtypes, each with the mutation profile of melanoma cells, such as B-Raf, NRAS and c-Kit (3,4). The B-Raf inhibitor vemurafenib provides a clinical benefit to B-Raf-mutated melanoma (5), while the MEK inhibitor MEK-162 prolongs OS and progression free survival in NRAS-mutated melanomas (6). This approach of using targeted therapies against these mutations has been successful for melanoma treatment.
The c-Kit proto-oncogene is hyperactivated in 5–10% of melanomas, by either somatic mutation or amplification (7). It is a receptor tyrosine kinase associated with malignant cancer, which can regulate proliferation, migration, invasion and metastasis (8). c-Kit knockdown is reported to inhibit melanoma cell proliferation, invasion and migration (9,10). Although clinical benefit has been achieved in melanoma cases with c-Kit aberration, results from molecular therapies targeting c-Kit in melanoma have been disappointing.
Sorafenib is a multikinase inhibitor that has been approved for the treatment of thyroid cancer, renal cell carcinoma and hepatocellular carcinoma (11). Sorafenib has been reported to inhibit B-RAF, c-RAF, platelet derived growth factor receptor (PDGFR), VEGFR and c-Kit (12). We hypothesized that sorafenib may be useful for treatment of metastatic melanoma with c-Kit aberration. Thus, the present study examined the effect of sorafenib in metastatic melanoma with c-Kit aberration in vitro and in vivo.
Materials and methods
Materials
For the in vitro studies, sorafenib (Funakoshi, Co., Ltd.) was dissolved in DMSO to a concentration of 20 mM (stock solution at −20°). The solution was diluted in PBS before use in the subsequent experiments at room temperature.
For the animal studies, sorafenib was made up as a 1.25 mg/ml (10 mg/kg of body weight), 3.75 mg/ml (30 mg/kg of body weight) and 6.25 mg/ml (50 mg/kg of body weight) solution in PBS containing 0.1% DMSO.
Cells and culture conditions
Murine melanoma cell lines, B16F1 and B16BL6, were kindly provided by Dr Inufusa of Kindai University (Osaka, Japan). B16F1 cells were established from lung metastasis lesions from the intravenous injection of B16 melanoma cells into a syngeneic C57BL/6J mouse, and B16F10 cells were selected by using 10 successive lung metastasis lesions. B16BL6 cells were derived from B16F10 cells that have invaded the mouse bladder membrane (13). MeWo cells were obtained from the Japanese Cancer Research Resources Bank. These cells were maintained in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 2 mM L-glutamine (FUJIFILM Wako Pure Chemical Corporation), 25 mM HEPES (FUJIFILM Wako Pure Chemical Corporation) and 100 µg/ml penicillin and streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2.
Mice
Female C57BL/6 mice (age, 6 weeks; weight, 21±3 g; n=116) were purchased from Shimizu Laboratory Supplies Co., Ltd. Mice were housed in an animal care facility with a 12-h light/dark cycle and had free access to water and commercially available chow. The mice were housed in a controlled environment with an ambient temperature of 22–25°C and relative humidity of 40–60%. All animal experiments were approved by the Animal Care and Use Committee of Kindai University (project identification code KAPS-27-021; April 1, 2015). Animal experimental procedures and ethical procedures were carried out strictly in accordance with the Recommendations for Handling of Laboratory Animals for Biomedical Research compiled by the Committee on Safety and Ethical Handling Regulations for Laboratory Animal Experiments, Kindai University and the United Kingdom Coordinating Committee for Cancer Research guidelines (14).
Cell viability
B16BL6, MeWo and B16F1 cells were plated in 96-well plates at 2×103 cells in a volume of 100 µl medium and cultured for 24 h at 37°C with 5% CO2. Then, sorafenib (0.1, 0.5, 1, 5, 10, 25 and 50 µM) was added to the well and cultured at 37°C with 5% CO2. After, 1, 3 and 5 days, the cells were stained with 0.4% trypan blue at room temperature and immediately counted using light microscopy (×100 magnification).
Migration assay
The in vitro migration was measured using 8.0-μm pore size Falcon cell culture inserts (Becton-Dickinson and Company). B16BL6 and MeWo cells (2×104) suspended in 200 µl serum-free medium were added in the upper chamber compartment, and incubated in the absence or presence of sorafenib (0.5, 1, 5 and 10 µM) at 37°C with 5% CO2. The lower chamber was filled with 500 µl medium supplemented with 10% FBS. After 24 h, the lower layer was fixed with 95% ethanol for 10 min at room temperature and stained with hematoxylin for 5 min at room temperature. The migrated cells were counted using light microscopy.
Matrigel invasion assay
The in vitro invasion was measured using 8.0-μm pore size Falcon cell culture inserts with Matrigel (Becton, Dickinson and Company). Cell culture inserts were precoated with Matrigel (50 µl) and incubated for 10 min at 37°C. Next, B16BL6 and MeWo cells (2×104) suspended in 200 µl medium containing 0.5% FBS were added in the upper chamber compartment, and incubated in the absence or presence of sorafenib (0.5, 1, 5 and 10 µM) at 37°C with 5% CO2. The lower chamber was filled with 500 µl medium supplemented with 10% FBS. After 24 h, the lower layer was fixed with 95% ethanol for 10 min at room temperature and stained with hematoxylin for 5 min at room temperature. The invaded cells were counted using light microscopy.
Western blot analysis
Western blot analysis was performed as described in our previous study (15). B16BL6 cells and the harvested tumor were lysed in lysis buffer [20 mM Tris-HCl (pH 7.5), 10 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 µM pepstatin, 1 µM leupeptin, 2 mM sodiumorthovanadate, 1 µM calpain inhibitor, phosphatase inhibitor cocktail I/II and 1 mM phenylmethylsulfonyl fluoride]. The protein concentrations were examined using the BCA protein assay kit (Thermo Fisher Scientific, Inc.). Then, equal amounts of protein (20 µg) were separated using 10% SDS-PAGE, and transferred to PVDF membranes (Cytiva). The proteins were detected using following specific antibodies overnight at 4°C: Phosphorylated (p)-c-Kit (cat. no. 3391), c-Kit (cat. no. 3074), p-PDGFR (cat. no. 3124), PDGFR (cat. no. 3175), p-VEGFR (cat. no. 2471), VEGFR (cat. no. 2469), p-B-Raf (cat. no. 2696), B-Raf (cat. no. 9433), p-c-Raf (cat. no. 9427), c-Raf (cat. no. 9422), p-ERK1/2 (cat. no. 9101), ERK1/2 (cat. no. 9102), p-Akt (cat. no. 9271), Akt (cat. no. 9272), p-STAT3 (cat. no. 9131), STAT3 (cat. no. 9132), p-NF-κB p65 (cat. no. 3033), NF-κB (cat. no. 4764), p-p38 (cat. no. 9215), p38 (cat. no. 9212), very late antigen (VLA)-1 (cat. no. 71747), VLA-4 (cat. no. 8440), VLA-5 (cat. no. 4705), VLA-6 (cat. no. 3750) (all from Cell Signaling Technology, Inc.; 1:3,000), β-actin (1:3,000; cat. no. A5316; Sigma-Aldrich; Merck KGaA), VLA-2 (1:1,000; cat. no. sc-6586), VLA-3 (1:1,000; cat. no. sc-6586) (all from Santa Cruz Biotechnology, Inc.) and MMP-14 (1:3,000; cat. no. MAB3328; Calbiochem; Merck KGaA). Next, the membranes were incubated with HRP-conjugated anti-rabbit secondary antibody (cat. no. 7074) or anti-mouse secondary antibody (cat. no. 7076) (both Cell Signaling Technology, Inc.) (both 1:5,000) at room temperature for 2 h, and the HRP activity was visualized using a Luminata Forte Western HRP Substrate (MilliporeSigma). The quantities of reactive proteins were determined based on densitometric measurements using a CS analyzer (ATTO Corporation).
Reverse transcription-quantitative (RT-q)PCR
Total RNA was isolated using the RNAiso Plus reagent (Takara Bio, Inc.), according to the manufacturer's protocol. Then, 1 µg purified total RNA was used for cDNA synthesis using the PrimeScript™ RT reagent kit (Takara Bio, Inc.), according to the manufacturer's protocol, and subjected to RT-qPCR, which was performed with the Thermal Cycler Dice Real Time system (Takara Bio, Inc.) using SYBR Premix Ex Taq (Takara Bio, Inc.). The following PCR conditions were used: Initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 94°C for 30 sec, annealing at 50°C for 30 sec and extension at 72°C for 30 sec. The following primers were used: MMP-1 forward, 5′-CGACTCTAGAAACACAAGAGCAAGA-3′ and reverse, 5′-AAGGTTAGCTTACTGTCACACGCTT-3′; MMP-2 forward, 5′-TGTGTCTTCCCCTTCACTTT-3′ and reverse, 5′-GATCTGAGCGATGCCATCAA-3′; MMP-9 forward, 5′-AGGCCTCTACAGAGTCTTTG-3′ and reverse, 5′-CAGTCCAACAAGAAAGGACG-3′; MMP-14 forward, 5′-ACACCCTTTGATGGTGAAGG-3′ and reverse, 5′-TCGGAGGGATCGTTAGAATG-3′; VLA-1 forward, 5′-GTCTTCATGCTCCCAACAGC-3′ and reverse, 5′-ACTTCTGACGTGATTACAGGAAGC-3′; VLA-2 forward, 5′-TCTGCGTGTGGACATCAGTTTGGA-3′ and reverse, 5′-GATAACCCCTGTCGGTACTTCTGC-3′; VLA-3 forward, 5′-ATTGACTCAGAGCTGGTGGAGGAG-3′ and reverse, 5′-TACTTGGGCATAATCCGGTAGTAG-3′; VLA-4 forward, 5′-GTCTTCATACTCCCAACAGC-3′ and reverse, 5′-ACTTCTGACGTGATTACAGGAAGC-3′; VLA-5 forward, 5′-CTGCAGCTGCATTTCCGAGTCTGG-3′ and reverse, 5′-GAAGCCGAGCTTGTAGAGGACGTA-3′; VLA-6 forward, 5′-GAGGAATATTCCAAACTGAACTAC-3′ and reverse, 5′-GGAATGCTGTCATCGTACCTAGAG-3′; and GAPDH forward, 5′-ACTTTGTCAAGCTCATTT-3′ and reverse, 5′-TGCAGCGAACTTTATTG-3′. The expression levels were normalized to the internal control GAPDH, and fold change of expression levels were calculated using the 2−ΔΔCq method (16).
Collagenase activity assay
MMP-1, MMP-2 and MMP-9 activity was measured using a type I collagenase assay kit (cat. no. AK37; Cosmo Bio Co., Ltd.) and a gelatinase activity assay kit (cat. no. CBA003; Merck KGaA) following the protocol of the manufacturer. MMP-1 activity from tumor lysates was estimated at 37°C for 2 h using a type I collagenase activity assay kit containing FITC. MMP-2 and MMP-9 activity from tumor lysates were estimated at 42°C for 2 h using a type IV collagenase activity assay kit containing FITC. MMPs activities were measured using a spectrophotofluorometer at an excitation wavelength of 495 nm and an emission wavelength of 540 nm.
Metastasis mouse model
B16BL6 cells (1×105 cells in 50 µl PBS) were inoculated into the right hind footpad of C57BL/6 mice (6-week-old female). The tumor in the footpad was excised surgically under anesthesia with 2% isoflurane (FUJIFILM Wako Pure Chemical Corporation). The mice were randomly separated (n=10 for each group), and orally treated with 0.1% DMSO (control) or sorafenib (10, 30 and 50 mg/kg of body weight) once a day. The number of metastatic nodules in the lungs were counted after the amputation.
Subcutaneous tumor growth studies
B16BL6 cells (1×105 cells in 50 µl PBS) were inoculated into the right hind footpad of C57BL/6 mice (6-week-old female). Then, the mice were randomly separated (n=9 for each group), and orally treated with 0.1% DMSO (control) or sorafenib (10, 30 and 50 mg/kg of body weight) once a day from the day of inoculation. Tumor volume was measured daily using calipers and calculated as (A × B2)/2, where A is the long axis and B is the short axis of the tumor. After 21 days, the mice were sacrificed by asphyxiation with CO2, and the right footpads were removed immediately stored at −80°C until assayed. During euthanasia, the mice were kept in a 5 liters cage, then 100% CO2 was introduced at a flow rate of 1 l/min. The mortality of the mice was confirmed by checking breathing.
Survival studies
B16BL6 cells (1×105 cells in 50 µl PBS) were injected into tail vain of C57BL/6 mice (6-week-old female). Then, the mice were randomly separated (n=10 for each group), and orally treated with 0.1% DMSO (control) or sorafenib (10, 30 and 50 mg/kg of body weight) once a day from the day of inoculation. Animals were monitored daily, and sacrificed via CO2 inhalation when they met the following humane endpoint criteria: Posture, gait or mobility that interfere with feeding behaviour, such as hind limb paralysis, consistent foot dragging or spinal curvature. During euthanasia, the mice were kept in a 5 liters cage, then 100% CO2 was introduced at a flow rate of 1 l/min. The mortality of the mice was confirmed by checking breathing.
Statistical analysis
Data are presented as the mean ± SD of two or three independent experiments. All analyses were conducted using SPSS version 21.0 software (IBM Corp.). Statistical analysis was performed using one-way analysis of variance (ANOVA) with Dunnett's test. P<0.05 was considered to indicate a statistically significant difference. For survival date, Kaplan-Meier curves were plotted using the log-rank test.
Results
Sorafenib inhibits the viability, migration and invasion of melanoma cells with c-Kit aberration
The present study used B16BL6 cells to investigate the effect of sorafenib on melanoma cells with c-Kit aberration, as B16BL6 cells expressed higher levels of p-c-Kit and c-Kit compared with B16F1 cells (Fig. S1). To clarify whether sorafenib exhibits cytotoxicity in melanoma cells with c-Kit aberration, the viability of B16BL6, MeWo and B16F1 cells was examined. It was found that B16BL6 cells treated with 0.1–5 µM sorafenib did not show inhibited viability (Fig. 1A). However, 10–50 µM sorafenib reduced B16BL6 cell viability compared with untreated cells. MeWo cells showed results similar to those observed with B16BL6 cells (Fig. 1A). In the same experiment, it was identified that B16F1 cells were less sensitive to sorafenib than B16BL6 and MeWo cells (Fig. S2). The IC50 values for the treatment of sorafenib at 72 h were 15.0 µM (B16BL6 cells), 16.6 µM (MeWo cells) and 33.7 µM (B16F1 cells), respectively.
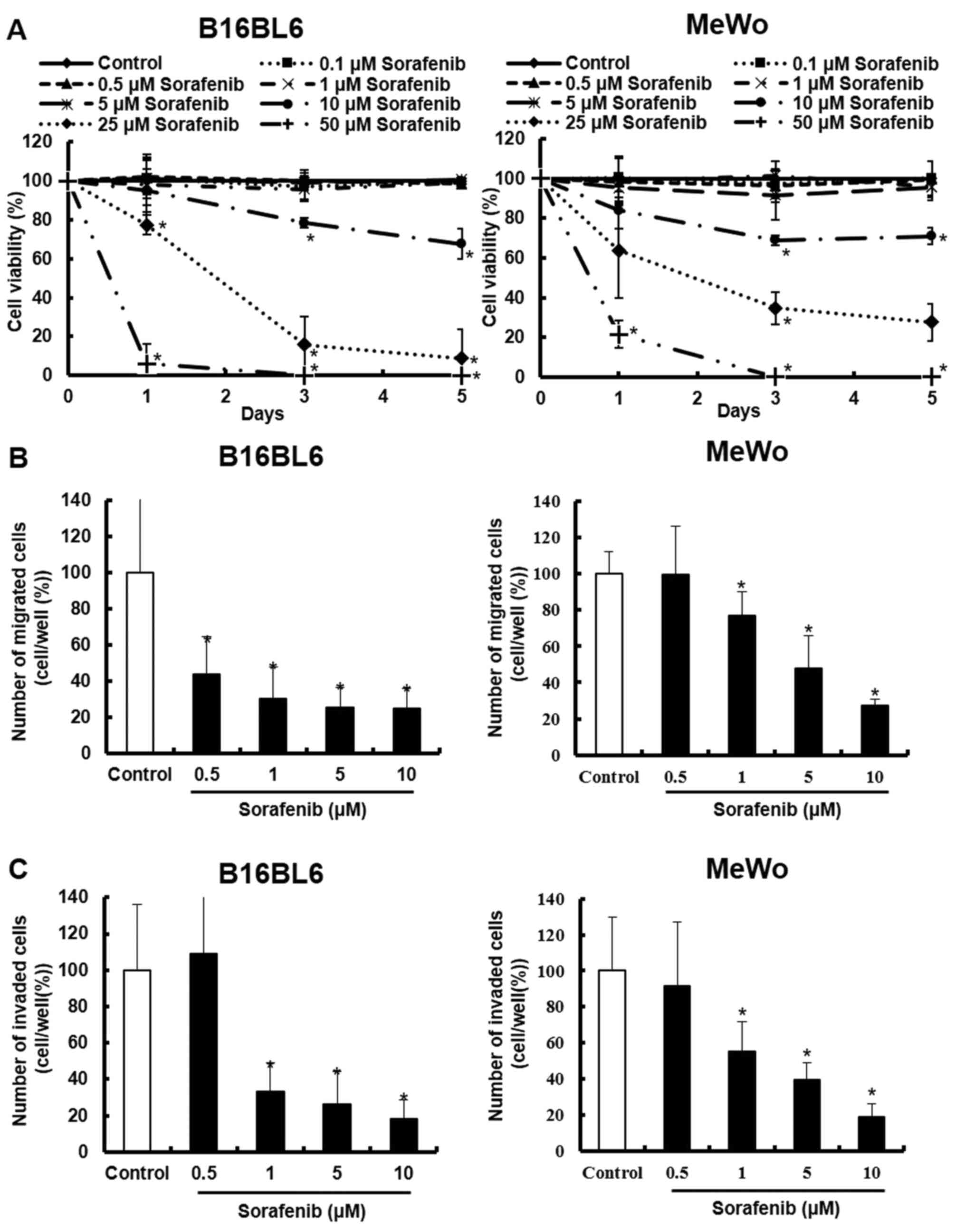 |
Figure 1.
Sorafenib inhibits the viability, migration and invasion of melanoma cells with c-Kit aberration. (A) B16BL6 and MeWo cells were treated with sorafenib (0.1, 0.5, 1, 5, 10, 25 and 50 µM), and cell viability was determined via trypan blue. The number of viable cells (unstained) and dead cells (stained) was counted using a haemocytometer. (B) B16BL6 and MeWo cells were treated with sorafenib (0.5, 1, 5 and 10 µM), and the migrated cells were measured in Falcon cell culture inserts. (C) B16BL6 and MeWo cells were treated with sorafenib (0.5, 1, 5 and 10 µM), and the invaded cells were measured in Falcon cell culture inserts with Matrigel. Data are presented as the mean ± SD of three independent experiments. *P<0.05 vs. control.
|
Next, the anti-migration and anti-invasion effects of sorafenib on B16BL6 and MeWo cells were examined. The results demonstrated that sorafenib inhibited the migration and invasion of B16BL6 and MeWo cells in a dose-dependent manner (Figs. 1B and C, and S3). These results suggest that sorafenib decreased melanoma cell viability, migration and invasion in a concentration-dependent manner.
Sorafenib inhibits c-Kit, PDGFR, VEGFR, B-Raf and c-Raf signaling pathways in melanoma cells with c-Kit aberration
Next, the molecular mechanism of the anti-proliferative, anti-migratory and anti-invasive effects of sorafenib were determined. Sorafenib has been shown to inhibit c-Kit, RAF, PDGFR and VEGFR activation (11). Therefore, the expression levels of the p- state of c-Kit, PDGFR, VEGFR, B-Raf, c-Raf, downstream ERK, Akt, STAT3, p38 and NF-κB protein were detected via western blotting. It was found that sorafenib suppressed the phosphorylation levels of c-Kit, PDGFR, VEGFR, B-Raf, c-Raf, ERK, Akt and STAT3 (Figs. 2 and S4). However, there were no significant changes in the levels of p38 and NF-κB phosphorylation. These results suggest that sorafenib suppresses the viability, migration and invasion of melanoma cells with c-Kit aberration via the inhibition of c-Kit, PDGFR, VEGFR, B-Raf and c-Raf signaling pathways.
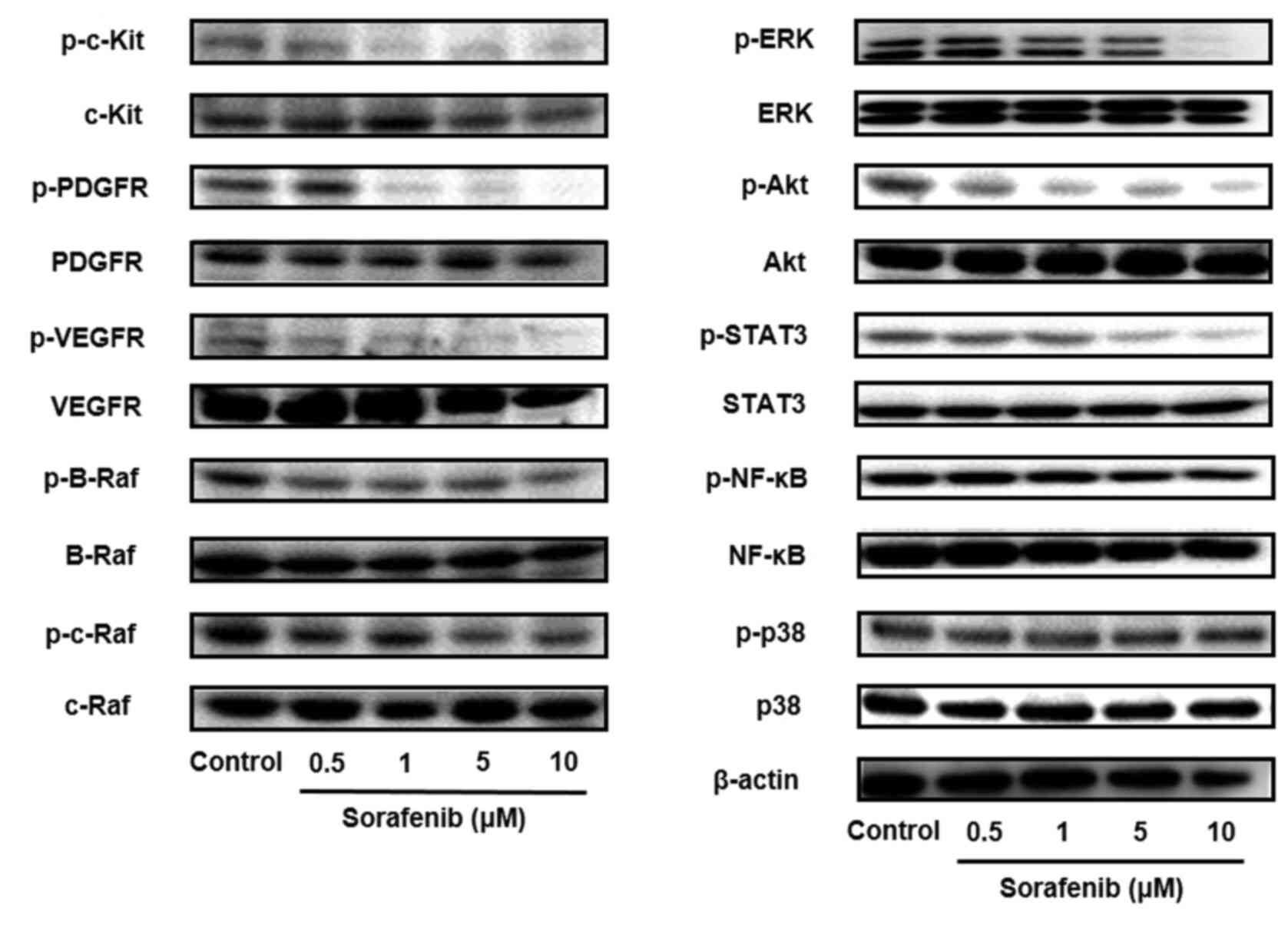 |
Figure 2.
Sorafenib inhibits c-Kit, PDGFR, VEGFR, B-Raf and c-Raf signaling pathways of melanoma cells with c-Kit aberration. B16BL6 cells were treated with sorafenib (0.5, 1, 5 and 10 µM), and the expression levels of p- and total c-Kit, PDGFR, VEGFR, B-Raf, c-Raf, ERK, Akt, STAT3, NF-κB and p38 proteins were analyzed via western blotting. β-actin was used as internal control. All determinations made in western blotting were from three different experiments. p-, phosphorylated; PDGFR, platelet derived growth factor receptor.
|
Sorafenib decreases the mRNA expression and enzymatic activities of MMPs, and the mRNA and protein expression levels of VLAs in melanoma cells with c-Kit aberration
MMPs and VLAs are responsible for the metastasis and invasion of malignant tumors (15,17). Therefore, the current study investigated the mRNA expression and enzymatic activities of MMPs, and the mRNA and protein expression levels of VLAs in melanoma cells with c-Kit aberration. Sorafenib decreased the mRNA expression and enzymatic activities of MMP-1, MMP-2, MMP-9 and MMP-14 in a concentration-dependent manner (Figs. 3 and S5A). In addition, sorafenib reduced the mRNA expression and protein expression levels of VLA-1, VLA-2, VLA-4, VLA-5 and VLA-6 (Figs. 4 and S5B). However, no change was observed in the expression of VLA-3. These data suggest that sorafenib decreased the mRNA expression and enzymatic activities of MMPs, and the mRNA and protein expression levels of VLAs in melanoma cells with c-Kit aberration.
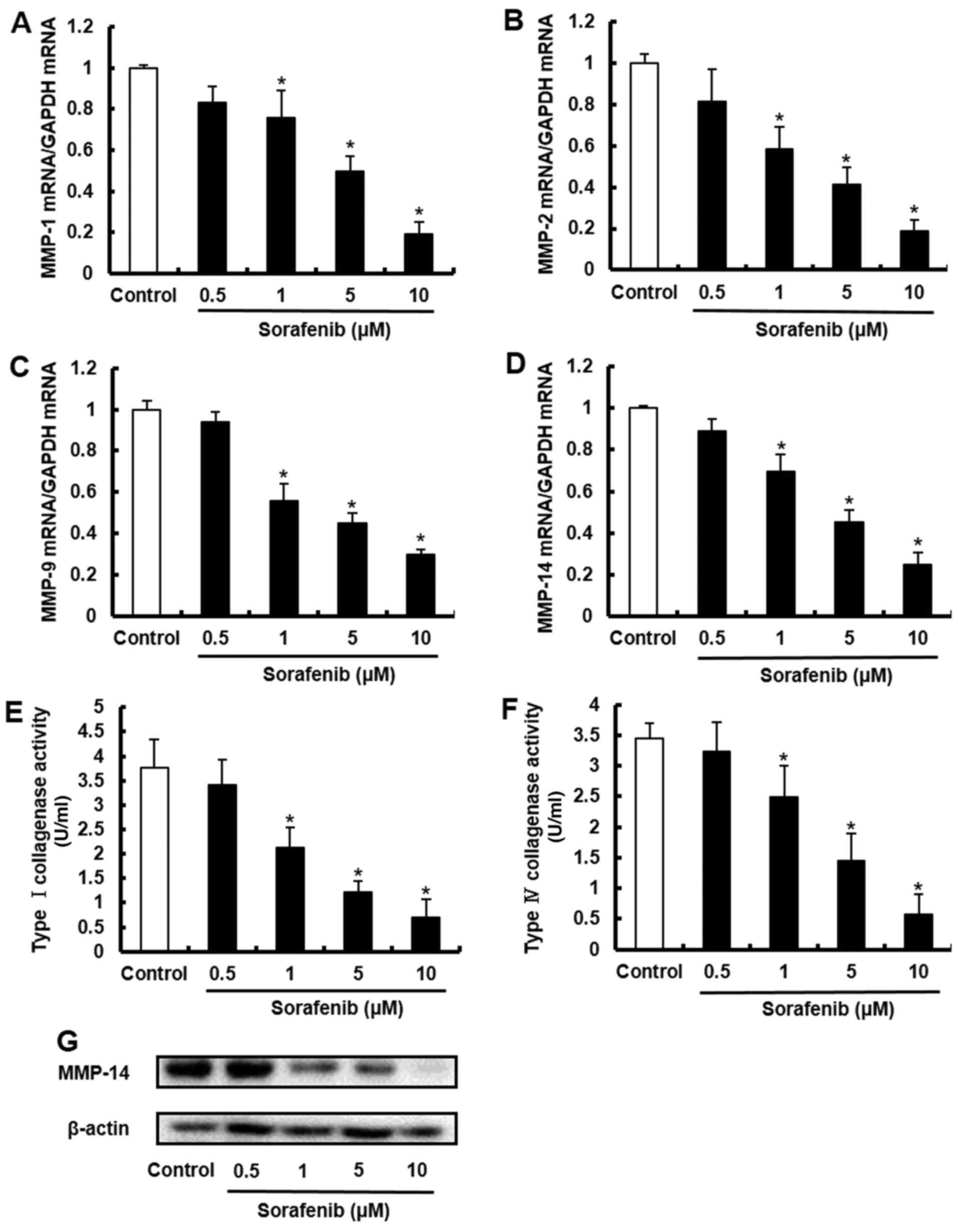 |
Figure 3.
Sorafenib decreases the mRNA expression and enzymatic activities of MMPs of melanoma cells with c-Kit aberration. B16BL6 cells were treated with sorafenib (0.5, 1, 5 and 10 µM). mRNA expression levels of (A) MMP-1, (B) MMP-2, (C) MMP-9 and (D) MMP-14 were measured via reverse transcription-quantitative PCR. (E and F) MMP-1, MMP-2 and MMP-9 activity was measured using assays for collagenase activity. (G) Expression level of MMP-14 was analyzed via western blotting. β-actin was used as internal control. All determinations made in western blotting were from three different experiments. Data are presented as the mean ± SD of three independent experiments. *P<0.05 vs. control.
|
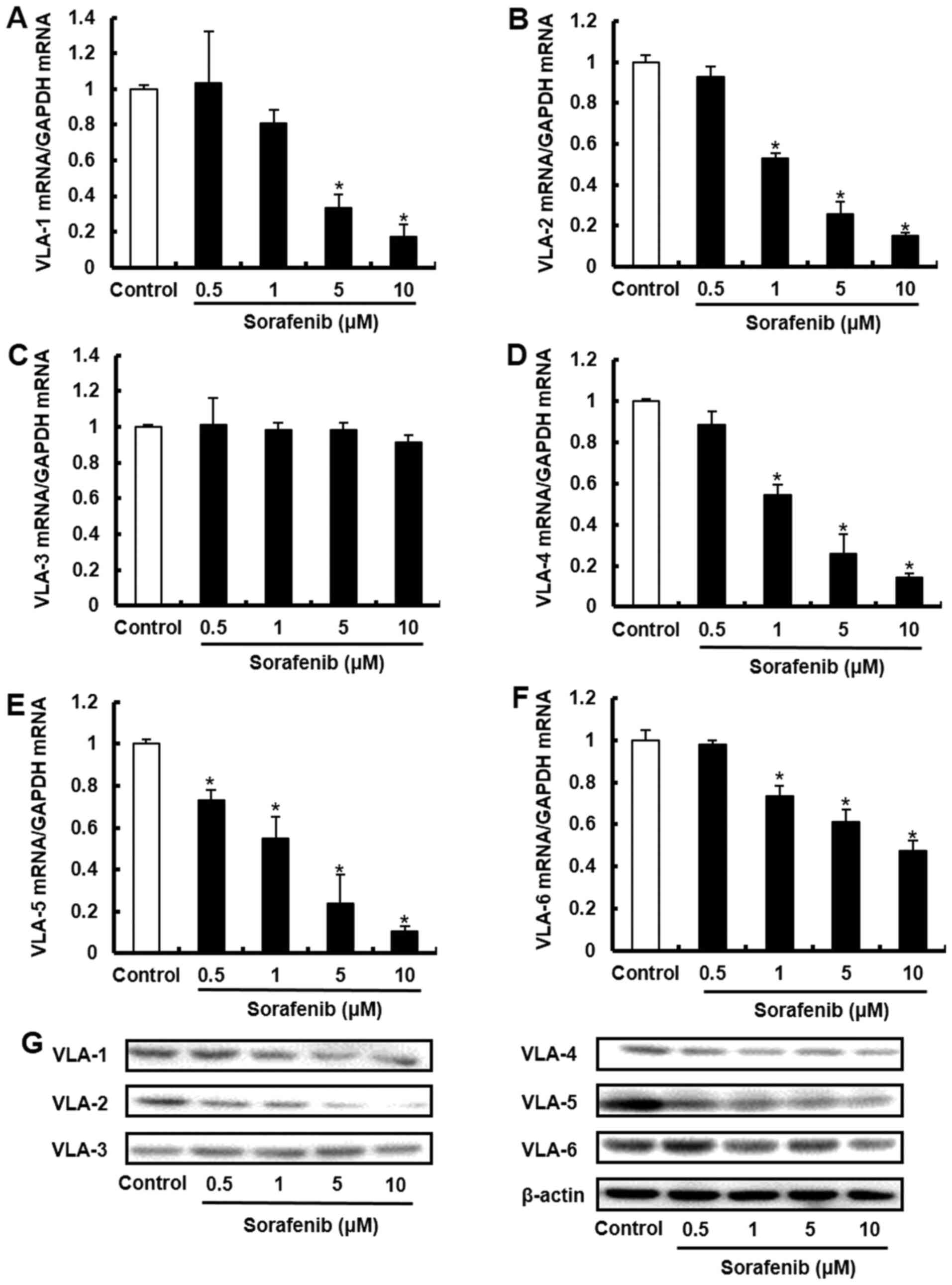 |
Figure 4.
Sorafenib decreases the mRNA and protein expression levels of VLAs in melanoma cells with c-Kit aberration. B16BL6 cells were treated with sorafenib (0.5, 1, 5 and 10 µM). mRNA expression levels of (A) VLA-1, (B) VLA-2, (C) VLA-3, (D) VLA-4, (E) VLA-5 and (F) VLA-6 were measured via reverse transcription-quantitative PCR. Data are presented as the mean ± SD of three independent experiments. *P<0.05 vs. control. (G) Protein expression levels of VLA-1, VLA-2, VLA-3, VLA-4, VLA-5 and VLA-6 were analyzed via western blotting. β-actin was used as internal control. All determinations were made in western blotting from three different experiments. VLA, very late antigen.
|
Sorafenib treatment has a significant effect on OS in mice by inhibiting metastasis and tumor growth in vivo
To clarify the anti-metastatic activity of sorafenib, the number of the lung metastatic nodules formed in a spontaneous metastasis model was counted. It was identified that sorafenib reduced the number of lung metastatic nodules in the sorafenib treated groups (Fig. 5A). The maximum tumor volumes of control, 10 mg/kg sorafenib, 30 mg/kg sorafenib, and 50 mg/kg sorafenib groups were 471.3 mm3 (11.9 mm is the long axis and 8.9 is the short axis), 285.9 mm3 (9.4 mm is the long axis and 7.8 is the short axis), 233 mm3 (9.0 mm is the long axis and 7.2 is the short axis) and 160.1 mm3 (8.5 mm is the long axis and 6.0 is the short axis), respectively. No significant change in body weight or side effects were observed in the sorafenib treated groups (data not shown).
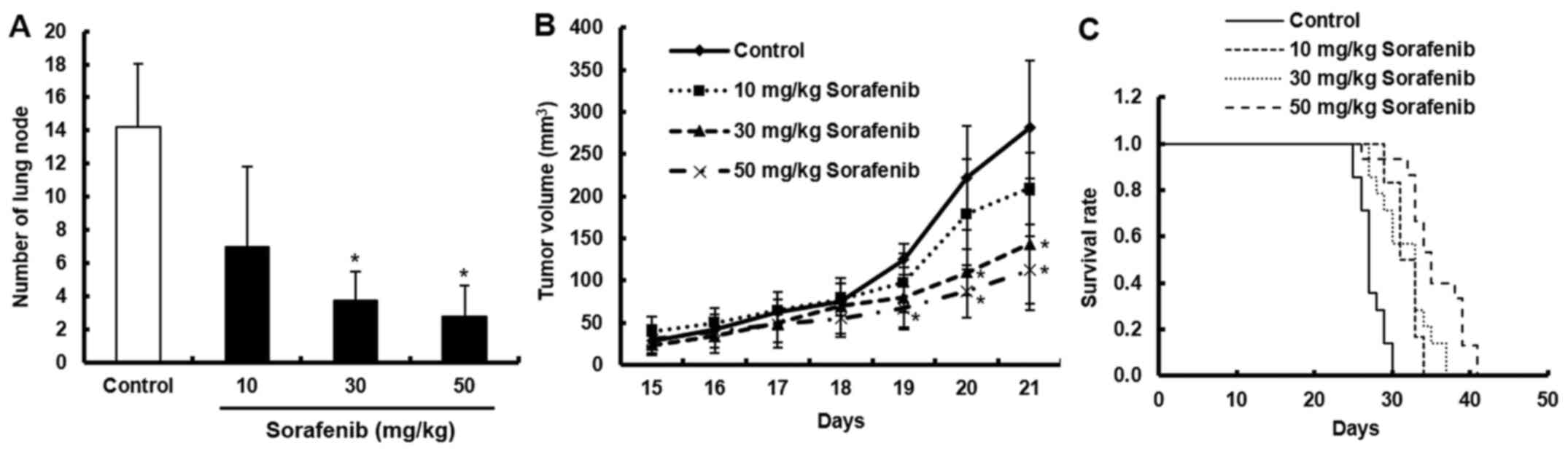 |
Figure 5.
Sorafenib treatment has a significant effect on overall survival in mice by inhibiting metastasis and tumor growth in vivo. (A) B16BL6 cells were inoculated into the right hind footpad of mice. The tumor in the footpad was excised surgically and treated with sorafenib (10, 30 and 50 mg/kg). The number of metastatic nodules in the lungs were counted. Data are presented as the mean ± SD of two independent experiments (n=10). (B) B16BL6 cells were inoculated into the right hind footpad of mice and treated with sorafenib (10, 30 and 50 mg/kg). Tumor volume was measured daily using calipers. Data are presented as the mean ± SD of two independent experiments (n=9). *P<0.05 vs. control. (C) B16BL6 cells were injected into tail vain of mice and treated with sorafenib (10, 30 and 50 mg/kg). Data are presented as the mean ± SD of two independent experiments (n=10; P<0.05, log-rank test).
|
In addition, the anti-tumor effect of sorafenib was evaluated in the in vivo model by inoculating the footpad of mice with B16BL6 cells. It was found that sorafenib inhibited tumor growth in the sorafenib treated group in a dose-dependent manner (Fig. 5B). The effect of sorafenib on overall mice survival time were also examined in the experimental metastasis model. There was a significant prolonged effect of sorafenib treatment on OS (Fig. 5C). The median survival times of the control, 10 mg/kg sorafenib, 30 mg/kg sorafenib and 50 mg/kg sorafenib-treated groups were 27.4, 31.8, 31.9 and 35.5 days, respectively. These results indicated that sorafenib treatment had a significant effect on OS in mice by inhibiting metastasis and tumor growth in vivo.
Sorafenib inhibits the expression of p-c-Kit, p-PDGFR, p-VEGFR, p-B-Raf and p-c-Raf in vivo
Next, the effect of c-Kit, PDGFR, VEGFR, B-Raf and c-Raf inhibition by sorafenib treatment in tumor lysates was examined via western blotting. Sorafenib inhibited c-Kit, PDGFR, VEGFR, B-Raf and c-Raf phosphorylation in a dose-dependent manner (Figs. 6 and S6). Consistent with the results of the in vitro studies, sorafenib abolished the phosphorylation of c-Kit, PDGFR, VEGFR, B-Raf and c-Raf. These data support the notion that sorafenib inhibits metastasis and tumor growth via the inhibition of c-Kit, PDGFR, VEGFR, B-Raf and c-Raf pathways.
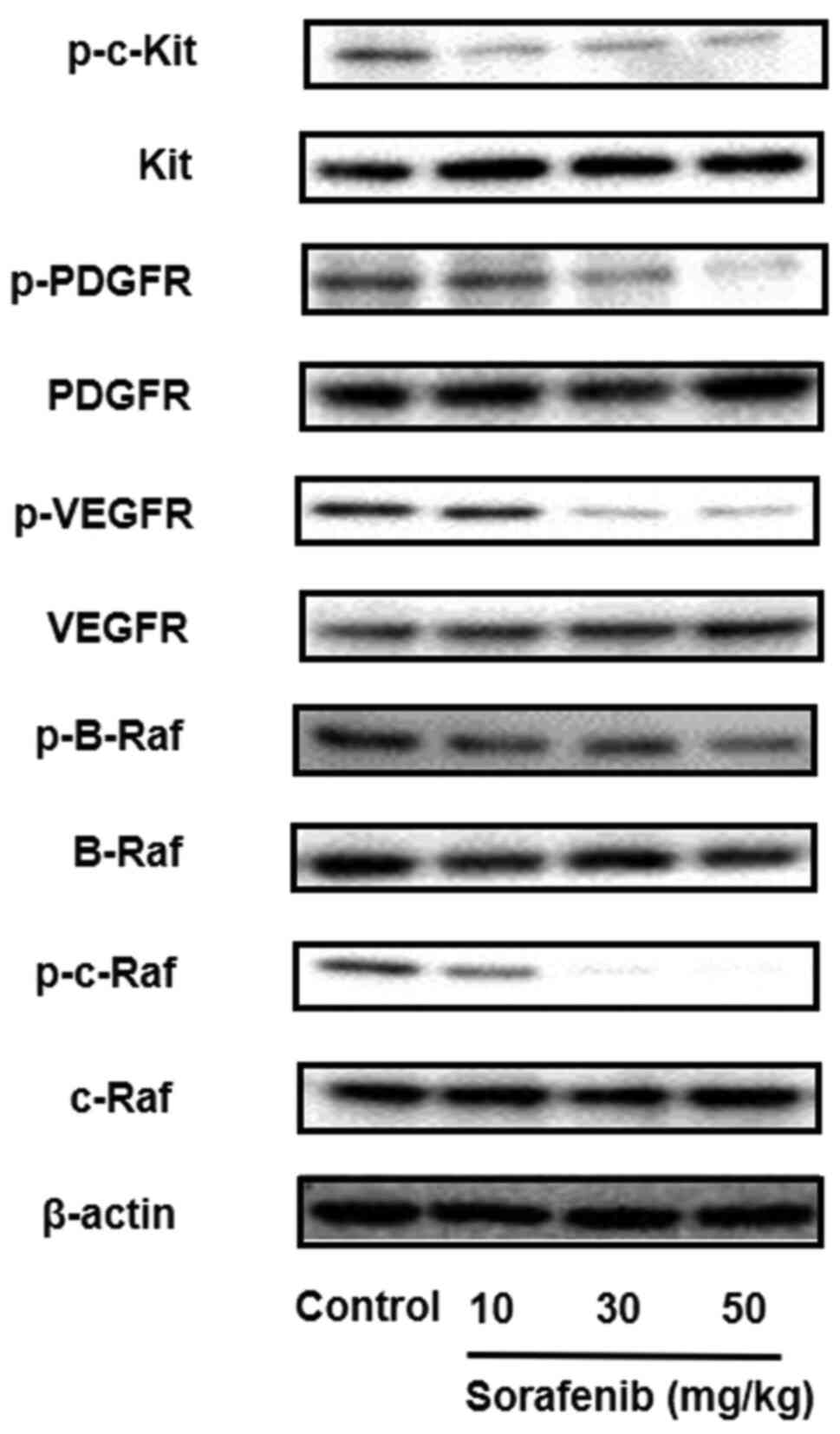 |
Figure 6.
Sorafenib inhibits the expression of p-c-Kit, p-PDGFR, p-VEGFR, p-B-Raf and p-c-Raf in vivo. B16BL6 cells were inoculated into the right hind footpad of mice and treated with sorafenib (10, 30 and 50 mg/kg). After 21 days, the mice were sacrificed, and the right footpads were harvested. The expression levels of p- and total c-Kit, PDGFR, VEGFR, B-Raf and c-Raf were analyzed via western blotting. β-actin was used as internal control. All determinations were made in western blotting from three different experiments. p-, phosphorylated; PDGFR, platelet derived growth factor receptor.
|
Sorafenib decreases the mRNA expression levels of MMPs and VLAs in vivo
The mRNA expression levels of MMPs (MMP-1, MMP-2, MMP-9 and MMP-14) and VLAs (VLA-1, VLA-2, VLA-4, VLA-5 and VLA-6) in tumor lysates were detected via RT-qPCR. Sorafenib significantly inhibited the mRNA expression levels of MMP-9 and MMP-14 (Fig. 7). Sorafenib treatment also led to a notable decrease in the mRNA expression levels of MMP-1 and MMP-2, but this was not significant. In addition, sorafenib significantly decreased the mRNA expression levels of VLA-1, VLA-2, VLA-4 and VLA-5 (Fig. 8). The mRNA expression level of VLA-6 showed a small reduction. These results suggest that sorafenib decreases metastasis by suppressing the expression levels of MMPs and VLAs.
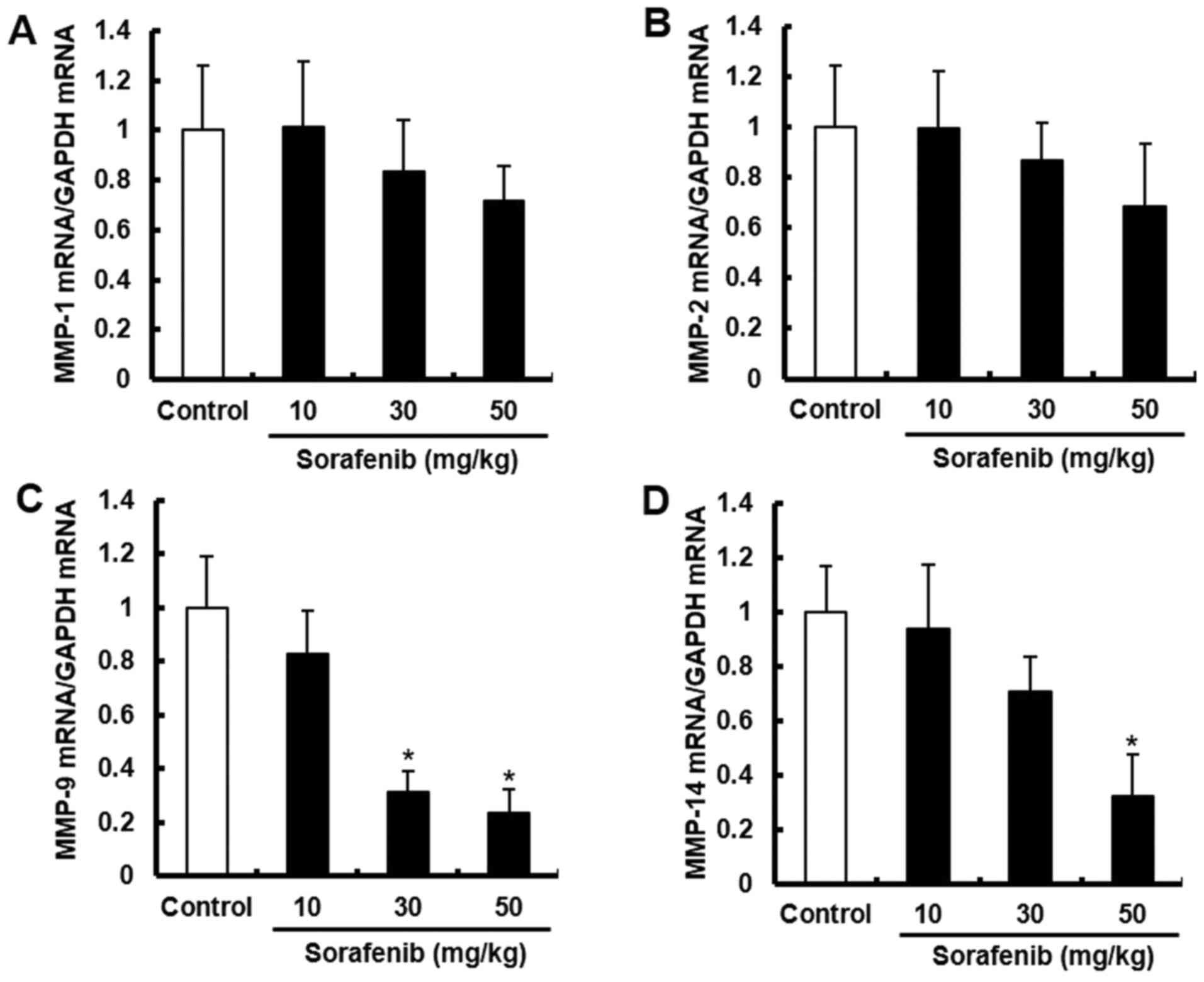 |
Figure 7.
Sorafenib decreases the mRNA expression of MMPs in vivo. B16BL6 cells were inoculated into the right hind footpad of mice, and treated with sorafenib (10, 30 and 50 mg/kg). After 21 days, the mice were sacrificed, and the right footpads were harvested. The mRNA expression levels of (A) MMP-1, (B) MMP-2, (C) MMP-9 and (D) MMP-14 were measured using reverse transcription-quantitative PCR. Data are presented as the mean ± SD of three independent experiments. *P<0.05 vs. control.
|
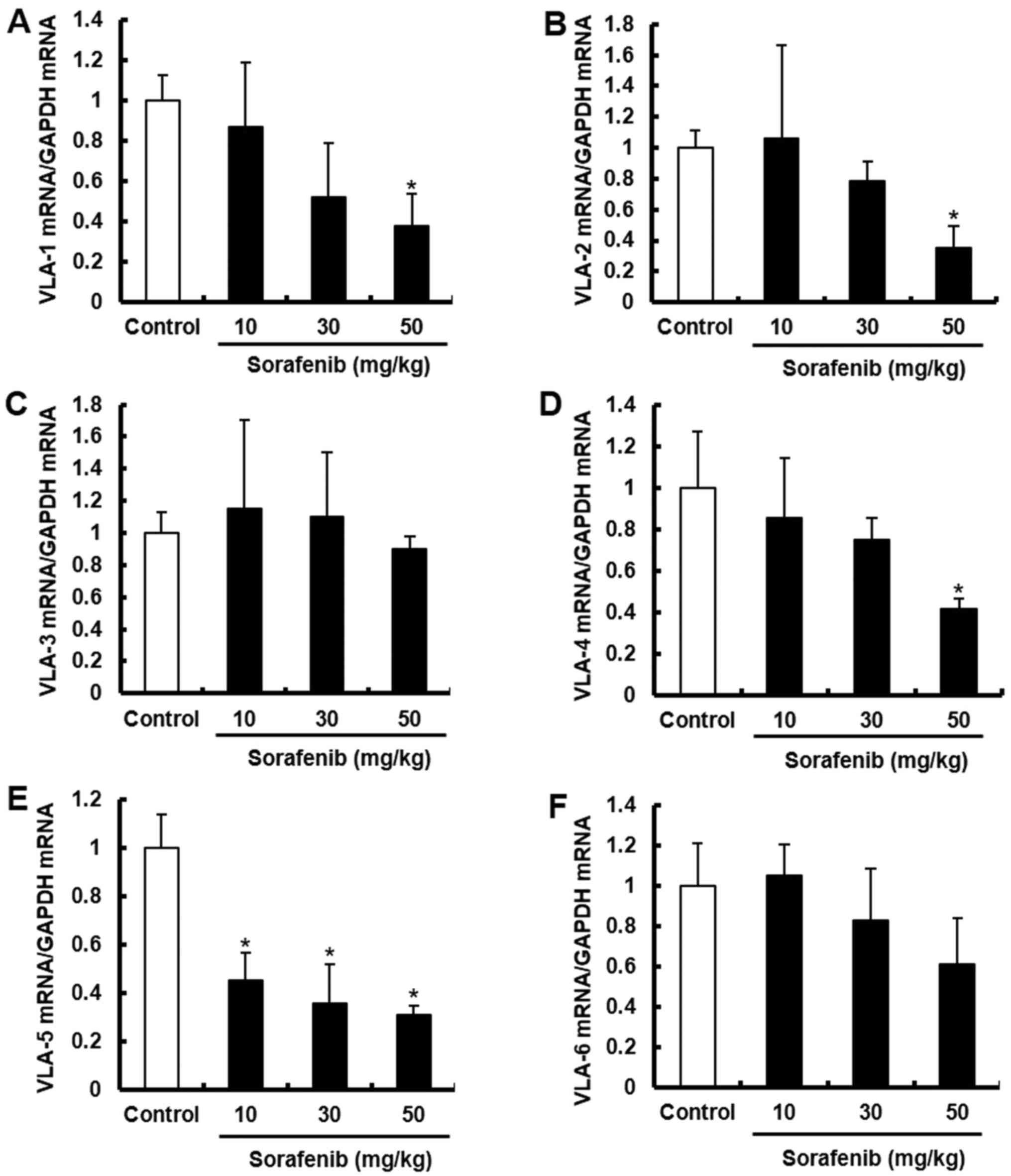 |
Figure 8.
Sorafenib decreases the mRNA expression of VLAs in vivo. B16BL6 cells were inoculated into the right hind footpad of mice, and treated with sorafenib (10, 30 and 50 mg/kg). After 21 days, the mice were sacrificed, and the right footpads were harvested. The mRNA expression levels of (A) VLA-1, (B) VLA-2, (C) VLA-3, (D) VLA-4, (E) VLA-5 and (F) VLA-6 were measured using reverse transcription-quantitative PCR. Data are presented as the mean ± SD of three independent experiments. *P<0.05 vs. control. VLA, very late antigen.
|
Discussion
Mutations or amplification of the c-Kit gene has been identified in 5–10% of melanoma, in particular mucosal melanomas, acral melanomas and melanomas as a result of chronic sun damage (CSD) (18). Previous data shows that c-Kit aberration in melanoma cells leads to increased proliferation, survival, migration and oncogenic potential (3,19). Although, inhibition of c-Kit is a potential approach for the treatment of melanoma, c-Kit inhibitors for melanomas with c-Kit mutation or amplification are not currently available. Therefore, the present study aimed to investigate the effect of sorafenib on melanoma cells that have a c-Kit aberration.
The present study demonstrated that sorafenib inhibited cell viability, migration and invasion in vitro. Furthermore, it was found that sorafenib inhibited metastasis and tumor growth in vivo. It was also shown that sorafenib inhibited c-Kit, PDGFR, VEGFR, B-Raf and c-Raf signaling pathways both in vitro and in vivo. Previous studies have reported that sorafenib prevents the tumor growth of hepatocellular, breast and pancreatic cancer types via the inhibition of c-Kit, VEGFR and B-Raf (12,20,21). Moreover, the inhibition of c-Kit suppresses tumor growth and metastasis in melanoma (22–24). The current results support the notion that sorafenib inhibits tumor growth and metastasis by suppressing c-Kit, PDGFR, VEGFR, B-Raf and c-Raf signaling pathways.
Melanomas are highly malignant tumors that readily metastasize and have poor prognosis. Previous studies have revealed that MMPs and VLAs are involved in migration, extracellular matrix degradation, adhesion and invasion of cancer cells such as melanoma (25–27). In addition, c-Kit, VEGFR and B-Raf pathways regulate the expression of MMPs and VLAs (28–30). The present study identified that sorafenib inhibited the mRNA expression levels of MMP-9 and MMP-14, whereas sorafenib did not significantly decrease the mRNA expression of MMP-1 and MMP-2. In addition, sorafenib decreased the mRNA expression levels of VLA-1, VLA-2, VLA-4 and VLA-5. These results suggest that sorafenib suppresses metastasis by decreasing the expression of MMPs and VLAs via suppressing c-Kit, PDGFR, VEGFR, B-Raf and c-Raf pathways.
In the present study, there was a significant effect of sorafenib treatment on OS. Sorafenib is currently the only Food and Drug Administration (FDA)-approved first-line therapy for patients with hepatocellular carcinoma (31). According to several clinical studies, the mean plasma concentration of sorafenib was between 5–16 µM, the FDA recommended daily dose (400 mg, orally, twice daily), and the most commonly used dose in patients with cancer (32–35). The dose of 30 mg/kg sorafenib for in vivo administration could reach plasma concentrations of ~10 µM (36,37). Therefore, doses of 30 mg/kg sorafenib in mice correspond to the peak plasma levels in humans. This data suggests that sorafenib is therapeutically useful for the treatment of melanoma.
Melanomas can be divided into specific clinical subtypes of mucosal, acral, occurring on skin with CSD and occurring on skin without CSD (38). Recent research into the molecular genetics of melanomas has revealed that these subtypes are characterized by distinct genetic mutations. In particular, mutations in B-Raf were most frequently found in lesions from non-CSD (39). The B-Raf inhibitor vemurafenib has shown an objective response rate of 48% and a survival benefit in patients with melanoma harboring a B-Raf mutation (40). However, the incidence of BRAF mutations is relatively low in mucosal, acral and CSD melanomas (41). Interestingly, KIT mutations and/or increased copy number were found in 39% of mucosal melanoma, 36% of acral melanomas and 28% of CSD melanoma, but were less frequent mutations in BRAF and NRAS (42). The present study used B16BL6 and MEWO cells. Other studies have reported that B16 cell lines express wild-type B-Raf, and offer a model system for studying alternative mechanisms that give rise to malignant melanoma (43–46). MeWo cells were also reported to express c-Kit (47). The current study demonstrated that sorafenib inhibited the viability, migration and invasion of B16BL6 and MeWo cells. Therefore, these results suggest that sorafenib may be a therapeutic strategy in patients with CSD, acral and mucosal melanoma with c-Kit aberration.
Future studies should investigate the effect of sorafenib on clinical samples that have a c-Kit aberration. In addition, the effect of sorafenib in melanoma with c-Kit mutation such as L576P, V559A and K642E (7) should be studied in vitro and in vivo.
In conclusion, the present study identified that sorafenib prolonged survival time in mice by inhibiting metastasis and tumor growth. The decrease in tumor growth and metastasis could be attributed to inhibition of c-Kit, PDGFR, B-Raf and c-Raf signaling pathways. In addition, sorafenib inhibited the expression levels of MMP-9, MMP-14, VLA-1, VLA-2, VLA-4 and VLA-5 (Fig. 9). Thus, sorafenib may have potential clinical applications for the treatment of metastatic melanoma with c-Kit aberration.
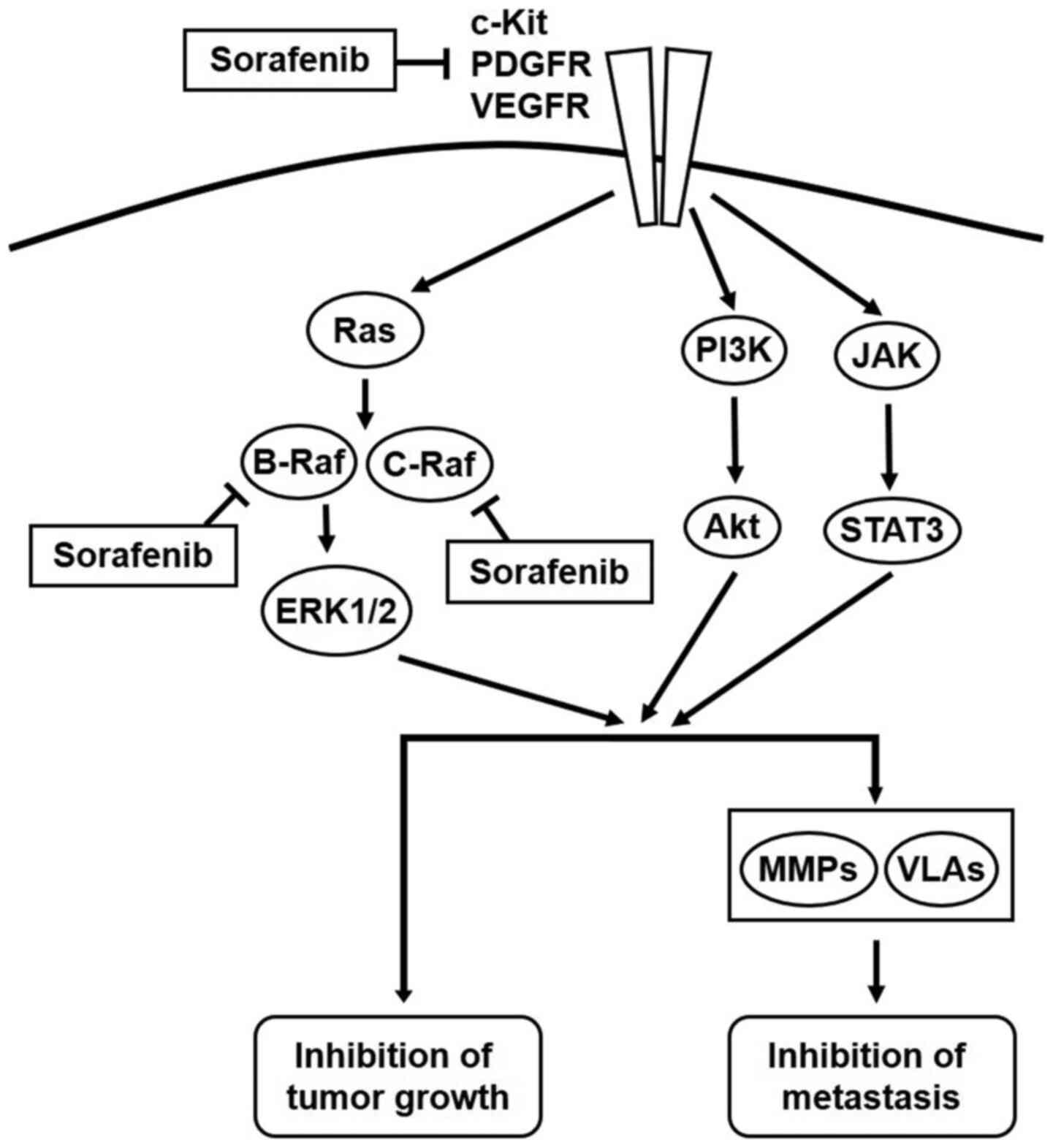 |
Figure 9.
Schematic diagram of the effects of sorafenib on melanoma cells. Sorafenib inhibited c-Kit, PDGFR, VEGFR, B-Raf and c-Raf activation melanoma cells with c-Kit aberration. This subsequently suppresses tumor growth and metastasis. Therefore, sorafenib may have potential clinical applications for the treatment of metastatic melanoma with c-Kit aberration. VLA, very late antigen; PDGFR, platelet derived growth factor receptor.
|
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported in part by Grant-in-Aid for Young Scientists from the Japan Society for the Promotion of Science (grant no. 20K16343).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
TT carried out analysis of trypan blue exclusion assay, western blotting, total RNA extraction and RT-qPCR and in vivo experiments and drafted the manuscript. MT, NK and SG carried out the migration and invasion assays, western blotting and collagenase activity assays. MT, EI and AE performed the in vivo experiments. MI and TS contributed to statistical analyses. SN designed the experiments and revised the manuscript. TT, MT and SN confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal experiments were approved by the Animal Care and Use Committee of Kindai University (project identification code KAPS-27-021; 1 April 2015).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Mahalingam D, Malik L, Beeram M, Rodon J, Sankhala K, Mita A, Benjamin D, Ketchum N, Michalek J, Tolcher A, et al: Phase II study evaluating the efficacy, safety, and pharmacodynamic correlative study of dual antiangiogenic inhibition using bevacizumab in combination with sorafenib in patients with advanced malignant melanoma. Cancer Chemother Pharmacol. 74:77–84. 2014. View Article : Google Scholar
|
|
2
|
Strickland LR, Pal HC, Elmets CA and Afaq F: Targeting drivers of melanoma with synthetic small molecules and phytochemicals. Cancer Lett. 359:20–35. 2015. View Article : Google Scholar
|
|
3
|
Luo C, Shen J, Ying J, Fang X, Wang X, Fu Z and Liu P: Case report of a KIT-mutated melanoma patient with an excellent response to apatinib and temozolomide combination therapy. OncoTargets Ther. 10:4553–4557. 2017. View Article : Google Scholar
|
|
4
|
Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, et al: KIT as a therapeutic target in metastatic melanoma. JAMA. 305:2327–2334. 2011. View Article : Google Scholar
|
|
5
|
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al: Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 363:809–819. 2010. View Article : Google Scholar
|
|
6
|
Goldinger SM, Murer C, Stieger P and Dummer R: Targeted therapy in melanoma - the role of BRAF, RAS and KIT mutations. EJC Suppl. 11:92–96. 2013. View Article : Google Scholar
|
|
7
|
Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F III, Azar S, et al: KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 14:6821–6828. 2008. View Article : Google Scholar
|
|
8
|
Carvajal RD: Another option in our KIT of effective therapies for advanced melanoma. J Clin Oncol. 31:3173–3175. 2013. View Article : Google Scholar
|
|
9
|
Huang S, Luca M, Gutman M, McConkey DJ, Langley KE, Lyman SD and Bar-Eli M: Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene. 13:2339–2347. 1996.
|
|
10
|
Montone KT, van Belle P, Elenitsas R and Elder DE: Proto-oncogene c-kit expression in malignant melanoma: Protein loss with tumor progression. Mod Pathol. 10:939–944. 1997.
|
|
11
|
Kuczynski EA, Lee CR, Man S, Chen E and Kerbel RS: Effects of sorafenib dose on acquired reversible resistance and toxicity in hepatocellular carcinoma. Cancer Res. 75:2510–2519. 2015. View Article : Google Scholar
|
|
12
|
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al: BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64:7099–7109. 2004. View Article : Google Scholar
|
|
13
|
Ishiguro T, Nakajima M, Naito M, Muto T and Tsuruo T: Identification of genes differentially expressed in B16 murine melanoma sublines with different metastatic potentials. Cancer Res. 56:875–879. 1996.
|
|
14
|
Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, et al Committee of the National Cancer Research Institute, : Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 102:1555–1577. 2010. View Article : Google Scholar
|
|
15
|
Takeda T, Tsubaki M, Sakamoto K, Ichimura E, Enomoto A, Suzuki Y, Itoh T, Imano M, Tanabe G, Muraoka O, et al: Mangiferin, a novel nuclear factor kappa B-inducing kinase inhibitor, suppresses metastasis and tumor growth in a mouse metastatic melanoma model. Toxicol Appl Pharmacol. 306:105–112. 2016. View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
17
|
Tsubaki M, Takeda T, Kino T, Obata N, Itoh T, Imano M, Mashimo K, Fujiwara D, Sakaguchi K, Satou T, et al: Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am J Cancer Res. 5:3186–3197. 2015.
|
|
18
|
Curtin JA, Busam K, Pinkel D and Bastian BC: Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 24:4340–4346. 2006. View Article : Google Scholar
|
|
19
|
Li G, Satyamoorthy K and Herlyn M: N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 61:3819–3825. 2001.
|
|
20
|
Newell P, Toffanin S, Villanueva A, Chiang DY, Minguez B, Cabellos L, Savic R, Hoshida Y, Lim KH, Melgar-Lesmes P, et al: Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J Hepatol. 51:725–733. 2009. View Article : Google Scholar
|
|
21
|
Merz M, Komljenovic D, Zwick S, Semmler W and Bäuerle T: Sorafenib tosylate and paclitaxel induce anti-angiogenic, anti-tumour and anti-resorptive effects in experimental breast cancer bone metastases. Eur J Cancer. 47:277–286. 2011. View Article : Google Scholar
|
|
22
|
Zhan Y, Guo J, Yang W, Goncalves C, Rzymski T, Dreas A, Żyłkiewicz E, Mikulski M, Brzózka K, Golas A, et al: MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J Clin Invest. 127:4179–4192. 2017. View Article : Google Scholar
|
|
23
|
Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA and Zigler AJ: Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 96:925–932. 2000. View Article : Google Scholar
|
|
24
|
Todd JR, Scurr LL, Becker TM, Kefford RF and Rizos H: The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma. Oncogene. 33:236–245. 2014. View Article : Google Scholar
|
|
25
|
Tsubaki M, Matsuoka H, Yamamoto C, Kato C, Ogaki M, Satou T, Itoh T, Kusunoki T, Tanimori Y and Nishida S: The protein kinase C inhibitor, H7, inhibits tumor cell invasion and metastasis in mouse melanoma via suppression of ERK1/2. Clin Exp Metastasis. 24:431–438. 2007. View Article : Google Scholar
|
|
26
|
Tsubaki M, Satou T, Itoh T, Imano M, Ogaki M, Yanae M and Nishida S: Reduction of metastasis, cell invasion, and adhesion in mouse osteosarcoma by YM529/ONO-5920-induced blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathway. Toxicol Appl Pharmacol. 259:402–410. 2012. View Article : Google Scholar
|
|
27
|
Qian F, Vaux DL and Weissman IL: Expression of the integrin α4β1 on melanoma cells can inhibit the invasive stage of metastasis formation. Cell. 77:335–347. 1994. View Article : Google Scholar
|
|
28
|
Braeuer RR, Zigler M, Villares GJ, Dobroff AS and Bar-Eli M: Transcriptional control of melanoma metastasis: The importance of the tumor microenvironment. Semin Cancer Biol. 21:83–88. 2011. View Article : Google Scholar
|
|
29
|
Stahtea XN, Roussidis AE, Kanakis I, Tzanakakis GN, Chalkiadakis G, Mavroudis D, Kletsas D and Karamanos NK: Imatinib inhibits colorectal cancer cell growth and suppresses stromal-induced growth stimulation, MT1-MMP expression and pro-MMP2 activation. Int J Cancer. 121:2808–2814. 2007. View Article : Google Scholar
|
|
30
|
Chiang IT, Liu YC, Wang WH, Hsu FT, Chen HW, Lin WJ, Chang WY and Hwang JJ: Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-κB pathway in hepatocellular carcinoma cells. In Vivo. 26:671–681. 2012.
|
|
31
|
Reiss KA, Yu S, Mamtani R, Mehta R, D'Addeo K, Wileyto EP, Taddei TH and Kaplan DE: Starting dose of sorafenib for the treatment of hepatocellular carcinoma: A retrospective, multi-institutional study. J Clin Oncol. 35:3575–3581. 2017. View Article : Google Scholar
|
|
32
|
Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, et al: Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol. 23:965–972. 2005. View Article : Google Scholar
|
|
33
|
Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ and Schwartz B: Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: A review of four phase I trials in patients with advanced refractory solid tumors. Oncologist. 12:426–437. 2007. View Article : Google Scholar
|
|
34
|
Chen ML, Yan BS, Lu WC, Chen MH, Yu SL, Yang PC and Cheng AL: Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int J Cancer. 134:319–331. 2014. View Article : Google Scholar
|
|
35
|
Fabian MA, Biggs WH III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al: A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 23:329–336. 2005. View Article : Google Scholar
|
|
36
|
Hu S, Niu H, Inaba H, Orwick S, Rose C, Panetta JC, Yang S, Pounds S, Fan Y, Calabrese C, et al: Activity of the multikinase inhibitor sorafenib in combination with cytarabine in acute myeloid leukemia. J Natl Cancer Inst. 103:893–905. 2011. View Article : Google Scholar
|
|
37
|
Zebary A, Omholt K, Vassilaki I, Höiom V, Lindén D, Viberg L, Kanter-Lewensohn L, Johansson CH and Hansson J: KIT, NRAS, BRAF and PTEN mutations in a sample of Swedish patients with acral lentiginous melanoma. J Dermatol Sci. 72:284–289. 2013. View Article : Google Scholar
|
|
38
|
Handolias D, Salemi R, Murray W, Tan A, Liu W, Viros A, Dobrovic A, Kelly J and McArthur GA: Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 23:210–215. 2010. View Article : Google Scholar
|
|
39
|
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, et al: Distinct sets of genetic alterations in melanoma. N Engl J Med. 353:2135–2147. 2005. View Article : Google Scholar
|
|
40
|
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al BRIM-3 Study Group, : Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar
|
|
41
|
Itoh M, Goto A, Wakasugi H, Yoshida Y, Matsunaga Y, Fujii K, Suzuki K, Yonezawa K, Abe T, Arimura Y, et al: Anorectal melanoma with a KIT-activating mutation, which is a target for tyrosine kinase inhibitor. Int J Clin Oncol. 16:428–434. 2011. View Article : Google Scholar
|
|
42
|
Lee SJ, Kim TM, Kim YJ, Jang KT, Lee HJ, Lee SN, Ahn MS, Hwang IG, Lee S, Lee MH, et al: Phase II Trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: A multicenter trial of Korean Cancer Study Group (UN10-06). Oncologist. 20:1312–1319. 2015. View Article : Google Scholar
|
|
43
|
Morris VL, Toseef T, Nazumudeen FB, Rivoira C, Spatafora C, Tringali C and Rotenberg SA: Anti-tumor properties of cis-resveratrol methylated analogs in metastatic mouse melanoma cells. Mol Cell Biochem. 402:83–91. 2015. View Article : Google Scholar
|
|
44
|
Akil H, Rouanet J, Viallard C, Besse S, Auzeloux P, Chezal JM, Miot-Noirault E, Quintana M and Degoul F: Targeted radionuclide therapy decreases melanoma lung invasion by modifying epithelial-mesenchymal transition-like mechanisms. Transl Oncol. 12:1442–1452. 2019. View Article : Google Scholar
|
|
45
|
Melnikova VO, Bolshakov SV, Walker C and Ananthaswamy HN: Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene. 23:2347–2356. 2004. View Article : Google Scholar
|
|
46
|
Sabbah M, Najem A, Krayem M, Awada A, Journe F and Ghanem GE: RTK inhibitors in melanoma: From bench to bedside. Cancers (Basel). 13:16852021. View Article : Google Scholar
|
|
47
|
Grossman D: Imatinib mesylate for melanoma: Will a new target be revealed? J Invest Dermatol. 123:xi–xiii. 2004. View Article : Google Scholar
|














