Introduction
Urothelial carcinoma (UC) is the most common
malignancy of the urinary system, with ~90% cases developing in the
urinary bladder and ~10% in the renal pelvis and ureter (upper
tract) (1). There were ~573,000
new cases and 213,000 deaths from bladder cancer worldwide in 2020
(2), with most cases being
superficial bladder cancer at initial diagnosis, and ~70%
representing non-muscle-invasive bladder cancer (NMIBC) (3). Although the survival rate in patients
with NMIBC is favorable, 30% will experience disease recurrence,
progressing to muscle-invasive bladder cancer (MIBC) (4,5).
Although radical cystectomy combined with pelvic lymph node
dissection increases the survival rate in patients with MIBC, ~50%
ultimately experience disease recurrence (6,7).
Cisplatin-based systemic therapy plays a key role in reducing
recurrence rates in patients with MIBC after local surgery
(8). Although immune checkpoint
inhibitors have been developed as second-line therapy for
programmed cell death protein-1-positive patients, cisplatin
therapy remains the standard therapy for metastatic bladder cancer.
However, due to the unfavorable toxicity of cisplatin-based
therapy, and only a fraction of patients achieving a disease-free
survival response and chemoresistance (9), there is an urgent need to develop new
drugs for the treatment of patients with UC.
Reactive oxygen species (ROS) formation and
signaling play an important role in regulating physiological
responses, such as metabolism, biosynthesis and cell survival
(10,11). Elevated ROS levels and an
alteration of the redox balance and associated signaling pathways
are common in cancer cells (12,13),
which may result from their compromised ROS-scavenging ability
(14). Moreover, increased ROS
level render cancer cells more vulnerable to ROS induction by
exhausting the antioxidant system capacity in cancer cells thereby
causing cell death (15). Drugs
that increase ROS levels have been analyzed for their anticancer
activity in biliary (16), colon
(17), prostate (18), pancreatic (19) and brain cancer (20) cells. Therefore, searching for drugs
that increase ROS levels in tumor cells without causing damage to
normal cells may be a promising strategy for cancer therapy
(21,22).
Auranofin (AF) was developed over 30 years ago for
the treatment of rheumatoid arthritis (23). Recently, it has also been described
as a potential anticancer agent that increases ROS levels in
hepatocellular carcinoma (24),
gastric cancer (25) and
colorectal cancer cells (26),
suggesting that cancer cells with elevated ROS levels may be
vulnerable to AF treatment. ROS-producing enzymes, such as NADPH
oxidase, are upregulated in UC (27,28).
Therefore, AF may be a good cytotoxic drug candidate in UC cells.
In the present study, two UC cell lines, HT 1376 (bladder UC) and
BFTC 909 (upper-tract UC) cells, were used to examine the
cytotoxicity of AF. ROS production, cell cycle profile progression
and apoptosis were analyzed following AF treatment in both cell
lines. The possible synergistic effect of AF and cisplatin, the
gold standard chemotherapeutic agent for advanced UC, was also
assessed in AF and cisplatin-treated cells. The findings of this
study may provide insight into the cytotoxicity of AF in UC
cells.
Materials and methods
Cell culture
The human UC cell lines, HT 1376 (bladder) and BFTC
909 (upper tract UC), were purchased from The Bioresource
Collection and Research Center (Hsinchu, Taiwan, China). HT 1376
cells were cultured in minimum essential medium containing
L-glutamine, non-essential amino acids and sodium pyruvate
supplemented with 10% FBS (all from Gibco; Thermo Fisher
Scientific, Inc.). BFTC 909 cells were cultured in 10% Dulbecco's
modified Eagle's medium with L-glutamine (all Gibco; Thermo Fisher
Scientific, Inc.). The two cell lines were incubated in a
humidified atmosphere containing 5% CO2 at 37°C.
Reagents and antibodies
AF and cisplatin were purchased from Sigma-Aldrich
(Merck KGaA). Mammalian Protein Extraction Reagent buffer (Thermo
Fisher Scientific, Inc.) was used for protein extraction. Protease
inhibitor was pur-chased from MilliporeSigma. Primary antibodies
against apoptosis-associated proteins, including poly (ADP-ribose)
polymerase (PARP; cat. no. 9542S, 1:1,000) and caspase 3 (cat. no.
9662S, 1:1,500), caspase 8 (9746S, 1:1,000), caspase 9 (9502S,
1:1,000) were purchased from Cell Signaling Technology, Inc.
Antibodies specific for cell cycle-associated proteins, p21 (2947S,
1:1,000 p21), p27 (cat. no. 2552S, 1:1,000), CDK2 (2546S, 1:1,000),
cyclin E2 (cat. no. 4132S, 1:1,000), CDC25A (3652S; 1:1,000),
cyclin D1 (2978S, 1:1,000) were purchased from Cell Signaling
Technology, Inc. Antibodies for cell-cycle-associated proteins CDK4
(GTX102993, 1:1,000), cyclin A2 (GTX103042, 1:1,000), cyclin E1
(GTX103045, 1:1,000), were purchased from GeneTex. Horseradish
peroxidase-conjugated secondary goat anti-rabbit IgG (AB_2307391,
1:3,000, polyclonal) and horse anti-mouse IgG (AB_10015289,
1:3,000, polyclonal), were purchased fromJackson ImmunoReseach,
Inc. The antibody against β-actin (loading control; cat. no.
sc-47778, 1:7,500) was purchased from Santa Cruz Biotechnology,
Inc. N-acetyl-L-cysteine (NAC; cat. no. A7250) and
H2DCFDA (D6883) were purchased from Sigma-Aldrich (Merck
KGaA).
Cell viability
Cell viability was assessed using the Cell Counting
Kit-8 Proliferation Assay (CCK-8; Sigma-Aldrich; Merck KGaA).
Briefly, 1×104 UC cells were seeded in 96-well plates,
then treated with AF and incubated for 24 or 48 h at 37°C in a
humidified incubator. Viability was determined according to the
manufacturer's instructions, and absorbance values were read at 490
nm 2 h after the addition of CCK-8 reagent. Values were normalized
to DMSO-treated control cells. The experiments were conducted
independently at least three times.
ROS analysis
To determine ROS levels, 1×106 UC cells
were seeded in 6-well plates at the indicated AF concentration (3
µM for HT 1376 cells and 4 µM for BFTC 909 cells) in the presence
or absence of the ROS inhibitor, 3 mM NAC. The cells were treated
with 10 µM H2DCFDA (Sigma-Aldrich; Merck KGaA) at 37°C
in the dark for 30 min, washed, then analyzed using a BD FACSDiva
flow cytometer (BD Biosciences). Cells stained with 10 µM
H2DCFDA only were used as a vehicle control group. The
data were analyzed using FlowJo software (version 10.6.1; FlowJo
LLC). The mean fluorescent intensity was calculated.
Cell cycle analysis
To determine the cell cycle distribution following
AF treatment, 1×106 UC cells were seeded in 6-well
plates. Serum-starved cells were treated with AF, then harvested at
24 and 48 h and fixed using 100% methanol overnight at 4°C. The
fixed cells were incubated with 0.05 mg/ml propidium iodide
solution containing RNase at room temperature in the dark for 30
min. The DNA content was analyzed using a BD FACSDiva flow
cytometer (BD Biosciences). The percentage of cells in each phase
of the cell cycle was analyzed using Modfit LT 3.3 cell cycle
analysis software (Verity Software House).
Apoptosis
An annexin V-FITC Apoptosis Detection Kit
(BioVision) was used to determine apoptosis in UC cells treated
with AF for 24 h. HT 1376 cells were treated with 1.5 or 3 µM AF,
BFTC 909 cells were treated with 2 or 4 µM AF at 37°C for 24 h. The
cells were then harvested and subjected to Annexin V staining (5 µl
Annexin V-FITC and 5 µl propidium iodide in 500 µl binding buffer)
at 25°C for 5 min in the dark. The apoptotic cells were analyzed
using a BD FACSDiva flow cytometer (BD Biosciences) and analyzed
using BDFACSDiva software (v6.1.3; BD Biosciences). The apoptotic
ratio was calculated based on the percentage early (annexin
V+ PI−) and late (annexin V+
PI+) apoptotic cells.
Protein expression analysis
The protein expression in cells was analyzed after
AF treatment by western blotting. The cells were lysed in Mammalian
Protein Extraction Reagent (Thermo Fisher Scientific, Inc.)
containing 0.1% protease inhibitor cocktail (Cell Signaling
Technology, Inc.), and the protein concentration was quantified
using Bio-Rad Protein Assay reagent (Bio-Rad Laboratories, Inc.). A
total of 40 µg/lane protein samples were subjected to 12% SDS-PAGE
and electrotransferred to a PVDF membrane. The membrane was blocked
in 5% BSA (Gibco; Thermo Fisher Scientific, Inc.) and 1X TBS with
0.1% Tween-20 (Sigma-Aldrich; Merck KGaA at 25°C for 1 h). The
membrane was incubated with a primary antibody at 4°C for 16 h. The
membrane was washed with three times for 5 min each with 0.1% TBST,
then incubated with a HRP-conjugated secondary antibody (Jackson
ImmunoResearch Laboratories, Inc.) for 2 h at 25°C. The membrane
was washed with three times for 5 min each with 0.1% TBST. Protein
expression was detected using an enhanced chemiluminescence HRP
substrate detection kit (cat. No. WBKLS0500, MilliporeSigma) and
visualized using the UVP BioSpectrum 800 image system (Analytik
Jena AG).
Drug combination index analysis
The Calcusyn software (Biosoft, ver. 2.0.0.0) was
used to evaluate the combined effect of AF and cisplatin by
calculating the combination index (CI) based on cell viability
rates. The CI equation of this software is based on the multiple
drug-effect equation of the Chou-Talalay method derived from
enzymatic models (29). The
non-constant ratio combination was used to determine the CI.
Briefly, CI was determined using the equation:
(D)1/(Dx)1 +
(D)2/(Dx)2 +
(D)1(D)2/(Dx)1(Dx)2.
(Dx)1. (Dx)2 represent the doses for x%
inhibition by drug 1 and drug 2, respectively. (D)1 and
(D)2 are the combinatory doses that inhibit cell growth
by x%. Fraction affect analysis was used to generate a graphic
representation of the CI. CI values <0.1 indicate very strong
synergism, 0.1-0.3 strong synergism, 0.3-0.7 synergism, 0.7-0.9
moderate to slight synergism, 1 nearly additive, 1.1-1.45 slight to
moderate antagonism, 1.45-3.3 antagonism and >3.3 strong to very
strong antagonism (30,31).
Statistical analysis
Statistical analysis was carried out using GraphPad
Prism 7.0 software (GraphPad Software, Inc.). Data are presented at
the mean ± SD of ≥3 experimental repeats. The data were analyzed
using one-way ANOVA followed by Bonferroni correction for multiple
comparisons. The in vitro experiments were performed in
triplicate. P≤0.05 was considered to indicate a statistically
significant difference.
Results
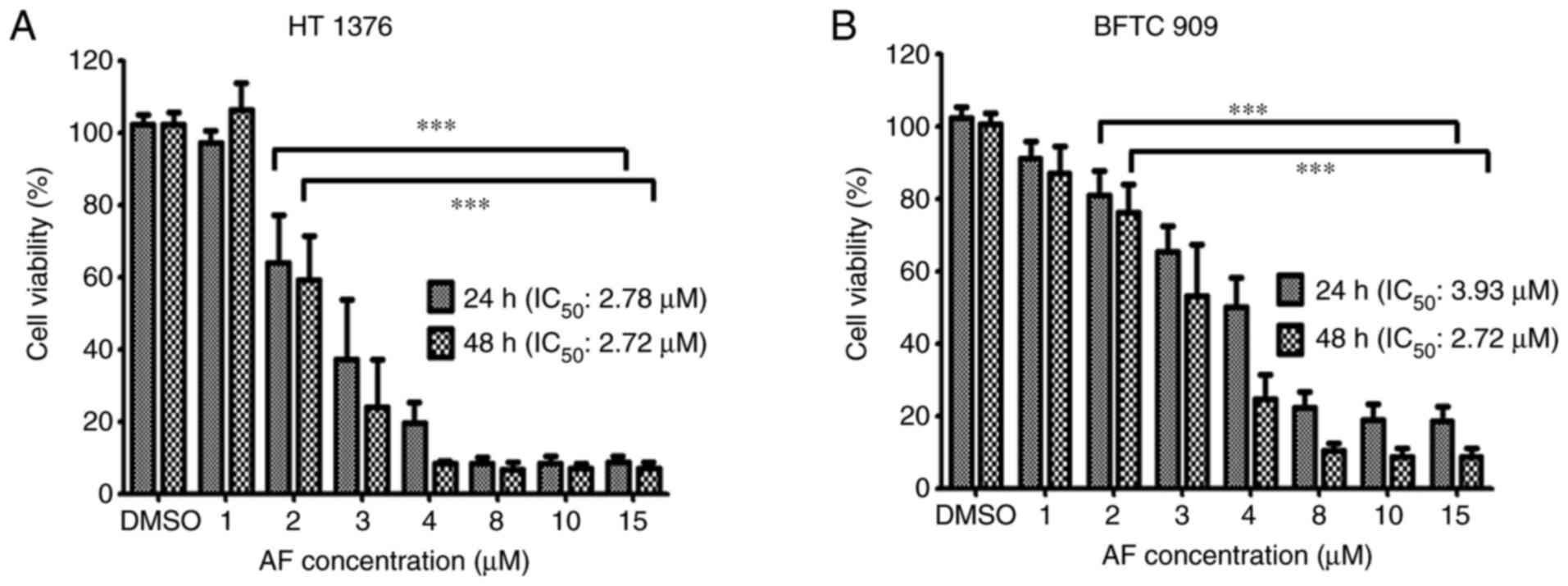
Effects of AF on the viability of UC
cells
To evaluate the effect of AF on the viability of UC
cells, 1, 2, 3, 4, 8, 10, and 15 µM AF were used to treat to HT
1376 and BFTC 909 cells for 24 and 48 h, and cell viability was
determined using a CCK-8 assay. AF exhibited cytotoxic effects on
both cell lines, with a lower IC50 in HT 1376 cells
following 24-h treatment. The IC50 values for HT 1376 at
24 and 48 h were 2.78 and 2.72 µM following AF treatment,
respectively (Fig. 1A). The
IC50 for BFTC 909 was 3.93 and 2.72 µM after AF
treatment at 24 and 48 h, respectively (Fig. 1B).
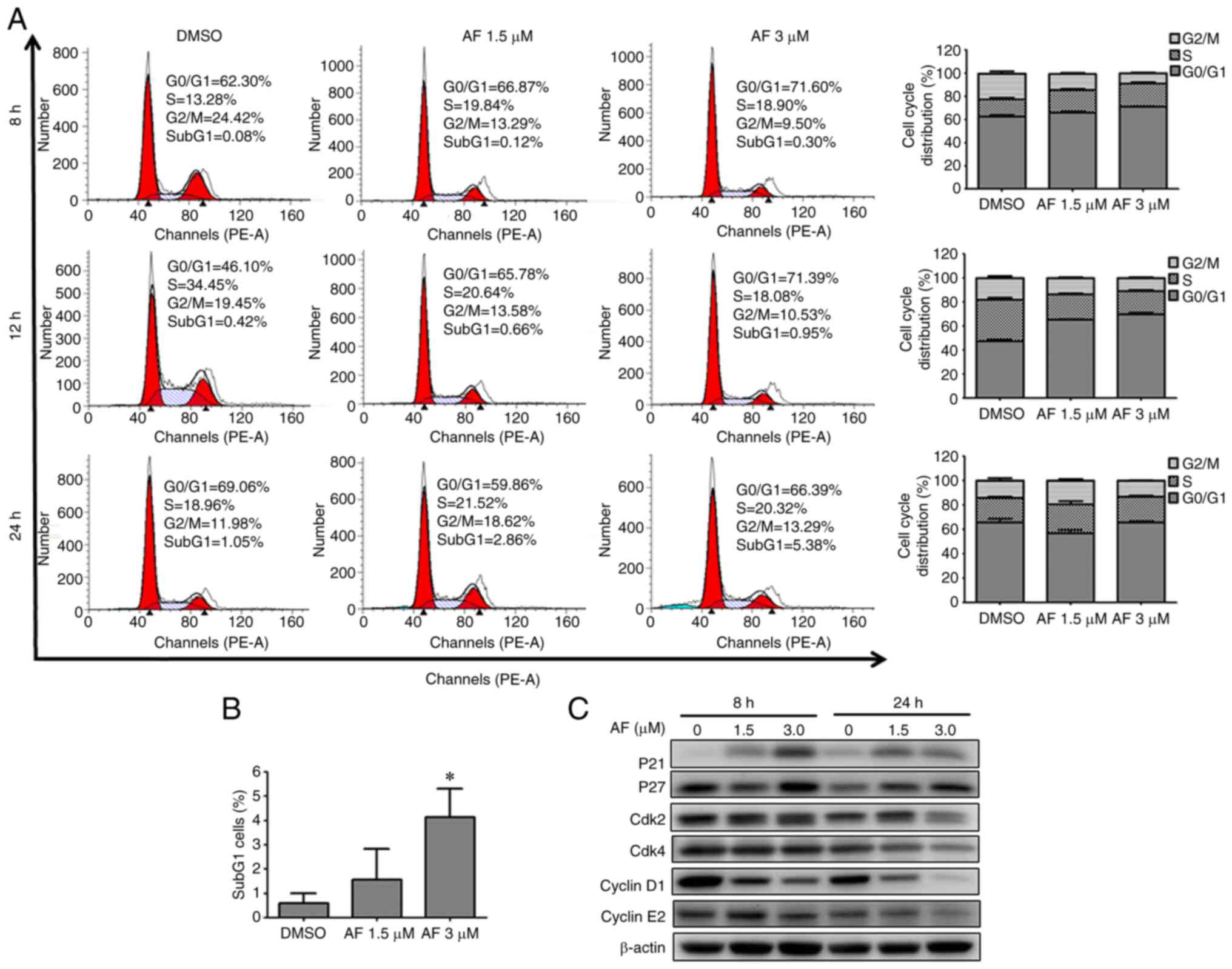
AF interferes with cell cycle
progression in HT 1376 and BFTC 909 cells
To determine whether cell cycle progression was
altered after AF treatment, HT 1376 cells were treated with 1.5 and
3 µM of AF and BFTC 909 cells were treated with 2 and 4 µM of AF,
and analyzed by flow cytometry. HT 1376 cells were blocked at the
G0/G1 phase following AF treatment, as
evidenced by a concentration-dependent increase in the proportion
of cells in the G0/G1 phase (Fig. 2A), After 1.5 and 3 µM AF treatment,
G0/G1 phase increased from 66.06 to 71.05 at
8 h; from 65.47 to 69.74% at 12 h and from 57.00 to 65.67% at 24 h.
Moreover, there was a small increase from 0.57 to 4.12% in the
frequency of subG1 cells following treatment with 3 µM
AF, but not significant in 1.5 µM AF treated group (Fig. 2B). Cell cycle progression is
regulated by the coordination of several CDKs/cyclin complexes
(32,33). Having confirmed that AF caused
G0/G1 cell cycle arrest in HT 1376 cells, the
next experiments were carried out to determine the expression
patterns of cell cycle-associated proteins. The CDK inhibitors, p21
and p27, inhibit the activity of cyclin D1, cyclin E1/E2 and CDK4,
thereby negatively regulating cell cycle progression and causing
cell arrest at the G0/G1 phase (34,35).
In HT 1376 cells, AF treatment upregulated the expression of p21
and p27 in a concentration-dependent manner compared with the
control (0 µM). In addition, decreased expression of CDK2, CDK4,
cyclin D1 and cyclin E2 was observed in AF-treated HT 1376 cells
(Fig. 2C). These data suggested AF
treatment caused HT 1376 cells arrest at the
G0/G1 phase.
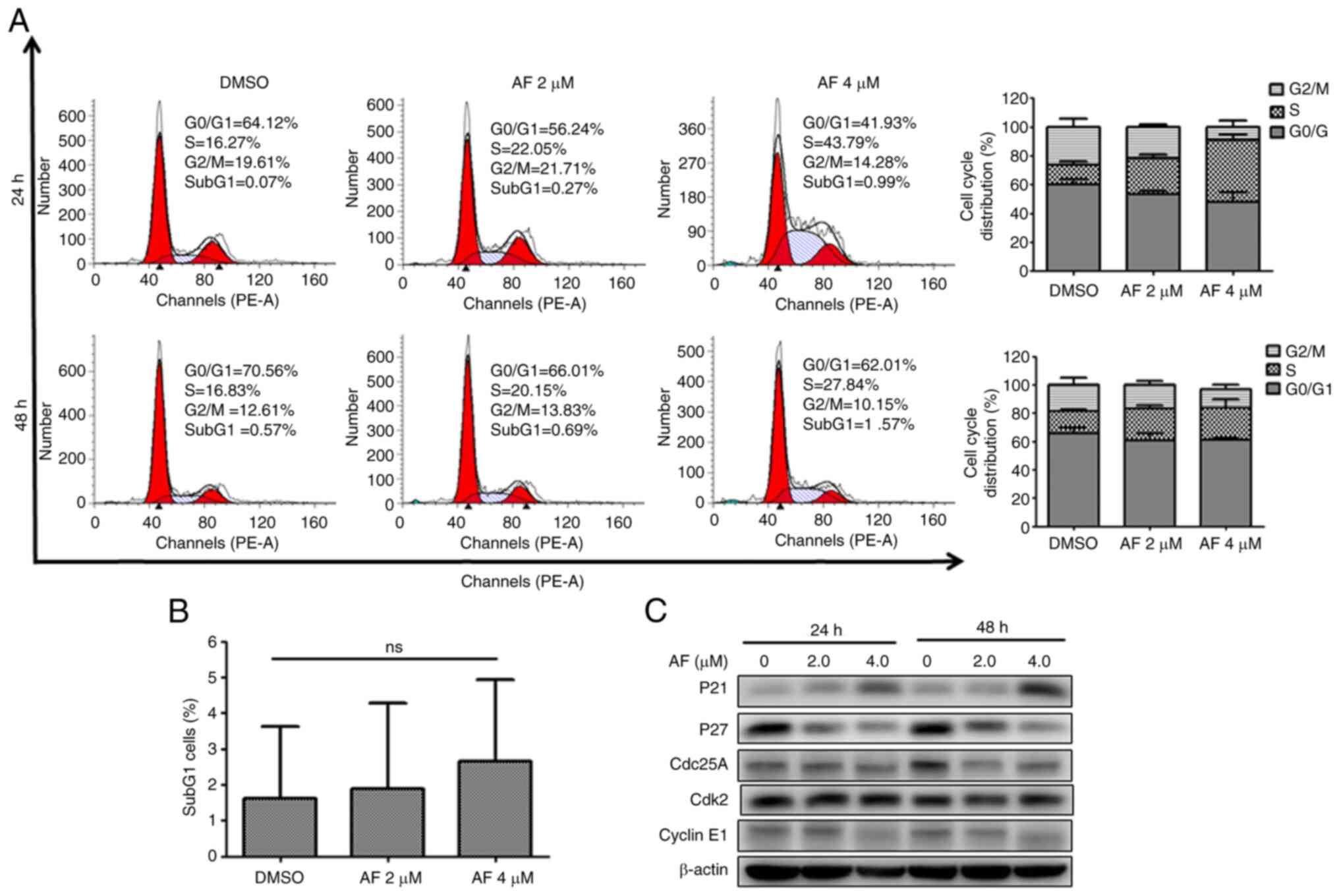
Similarly, AF treatment affected cell cycle
progression in BFTC 909 cells. Indeed, the frequency of cells in
the S phase increased from 13.61% in the DMSO group to 24.38 and
42.51% after 2 and 4 µM AF treatment for 24 h, respectively
(Fig. 3A). However, the increase
in the S phase after AF treatment for 48 h was not as evident in
BFTC 909 cells (Fig. 3A).
Additionally, although there was a small increase in the frequency
of subG1 cells, this was not statistically significant
(Fig. 3B). CDC25A is thought to
play an essential role in G1/S progression (36,37)
and the intra-S phase checkpoint by the CDC25A-CDK2 complex
(38,39). In addition to an increase of p21
expression, decreased expression of CDC25A and CDK2 was observed in
AF-treated BFTC 909 cells (Fig.
3C). The increased expression of p21 and decrease expression of
CDC25A suggested that AF treatment caused BFTC 909 cell cycle
arrest at the S phase.
Altogether, these findings suggested that AF
treatment interfered with cell cycle progression in these UC cell
lines, albeit at different phases.
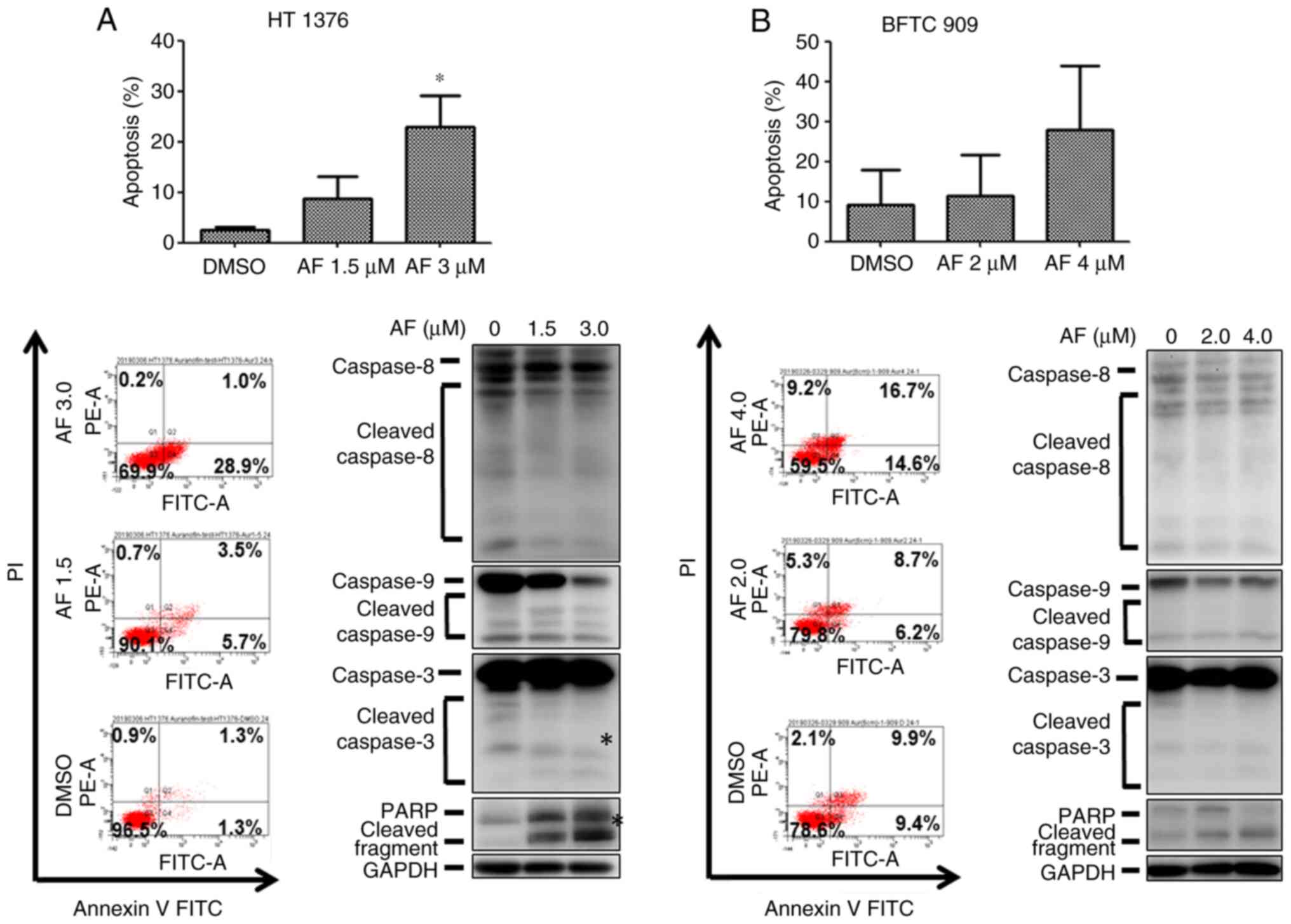
Effects of AF on apoptosis in UC
cells
The next experiments were performed to examine
whether AF could induce apoptosis in UC cells. The frequency of
annexin V+ cells increased in HT 1376 cells after
treatment with AF, from 2.4 to 8.7 and 22.8% for the vehicle, 1.5
and 3 µM AF treatment groups, respectively (Fig. 4A, upper panel). However, only the
highest concentration (3 µM) of AF treatment produced a
statistically significant increase. A small increase in caspase 3
and PARP protein expression was observed after 3 µM AF treatment
(Fig. 4A, lower panel), which was
consistent with the flow cytometry results. However, AF treatment
did not significantly increase apoptosis in BFTC 909 cells
(Fig. 4B), with only a slight
increase in annexin V+ cells (Fig. 4B, upper panel).
AF exhibits cytotoxicity in UC cells
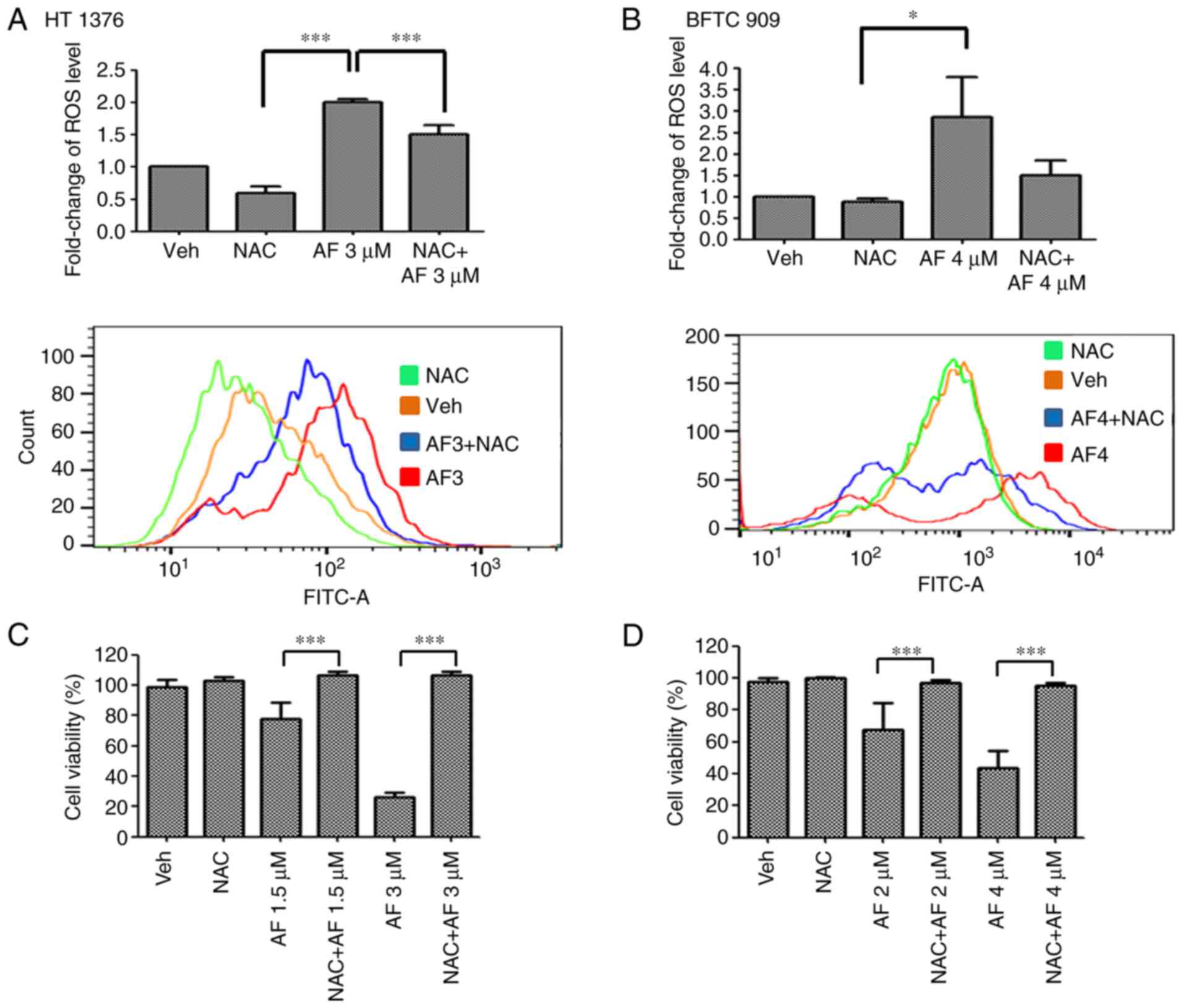
by triggering ROS production
AF has been reported to induce ROS production in
several cancer cell types (24–26).
Therefore, ROS levels were examined in UC cells after AF treatment
in the presence or absence of the ROS scavenger, NAC. AF treatment
increased ROS levels in HT 1376 and BFTC 909 cells compared with
the vehicle-treated group. However, ROS levels were reduced
following co-treatment with AF and NAC compared with AF treatment
alone (Fig. 5A). To determine if
the cell viability rate would be rescued following a reduction of
ROS levels, cell viability was determined in AF-treated HT 1376 and
BFTC 909 cells in the presence or absence of NAC. The results
showed that cell survival was significantly increased following
co-treatment with AF and NAC compared with AF treatment alone, both
in HT 1376 (Fig. 5C) and BFTC 909
(Fig. 5D) cells. Thus, these data
suggested that AF was cytotoxic to UC cells and interfered with
their redox balance.
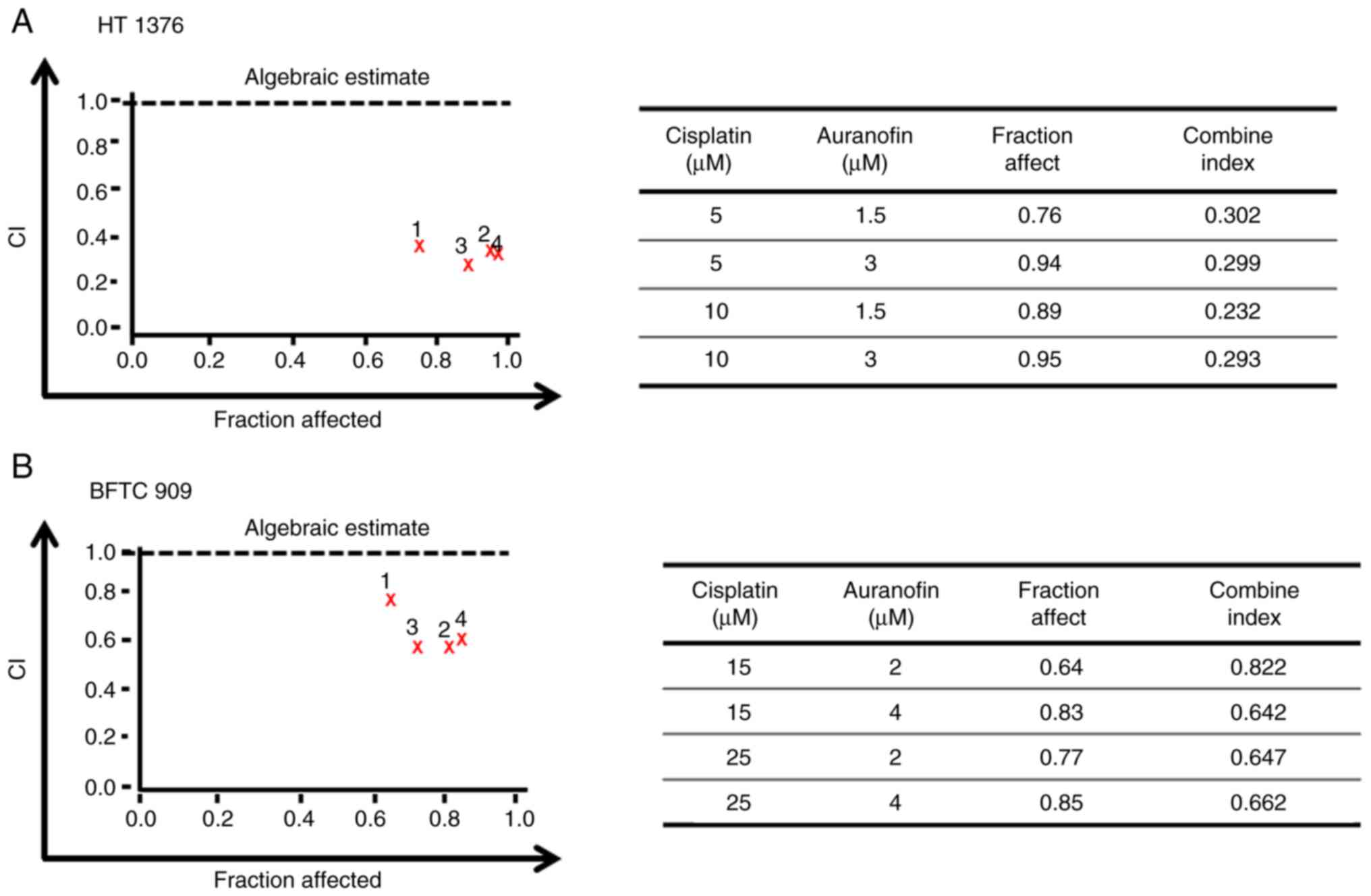
AF synergizes with cisplatin to
inhibit the viability of UC cells
Cisplatin therapy remains the standard chemotherapy
for metastatic bladder cancer; however, it is limited by
unfavorable toxicity, and only a fraction of patients achieve
disease-free survival (9). After
determining the cytotoxicity of AF (Fig. 1) and cisplatin in UC cells
(Fig. S1), we want to examine the
possible synergism of AF and cisplatin in UC cells. Different
concentrations of AF and cisplatin were combined to treat UC cells,
and cell viability was analyzed using a CCK-8 assay. The CI of AF
and cisplatin on HT 1376 cells ranged from 0.232 to 0.302 at 1.5 or
3 µM AF co-treatment with 5 or 10 µM cisplatin and from 0.642 to
0.822 for BFTC 909 cells at 2 or 4 µM AF co-treatment with 15 or 25
µM cisplatin. According to the CI thresholds set by the Calcusyn
software, this suggests there was synergism (0.3-0.7) to strong
synergism (0.1-0.3) in HT 1376 cells (Fig. 6A). For BFTC cells, there was
synergism (0.3-0.7) to moderate (0.7-0.85) (Fig. 6B). Therefore, AF, a drug approved
for the treatment of rheumatoid arthritis, may also represent a
potential candidate for UC in combination with cisplatin.
Discussion
UC is the most common malignancy in the urinary
system. Cisplatin-based combination treatment with surgery remains
the standard therapy for patients with MIBC, although its efficacy
is limited due to its adverse effects and chemoresistance (9,40).
Cancer cells may be more sensitive to ROS-accumulation due to their
elevated ROS levels. Increasing ROS generation may have selective
cytotoxicity to cancer cells by exhausting the antioxidant system
capacity without affecting normal cells (41). In the present study, AF exhibited
cytotoxicity towards HT 1376 bladder cancer cells and BFTC 909
upper tract cells by increasing ROS levels. In addition, this
compound interfered with cell cycle progression in both cell lines,
although cell cycle arrest occurred at different stages. AF induced
apoptosis in HT 1376, but not BFTC 909 cells. Furthermore,
synergistic cytotoxicity was observed in both HT 1376 and BFTC 909
cells following combined treatment with AF and cisplatin,
indicating that this drug may be used for UC treatment.
Drug repurposing (or repositioning) is an attractive
approach in the development of new medicines for cancer therapy
(42,43). AF, previously used in the treatment
of rheumatoid arthritis, has been shown to have anti-proliferation
activity in several cancer cell types (44,45).
In the present study, the effect of AF in bladder and upper tract
UC was examined in HT 1376 and BFTC 909 cells, respectively. The
results indicated that AF inhibited the viability of HT 1376 and
BFTC 909 cells with a lower IC50 in HT 1376 cells,
indicating that the HT 1376 cell line is more sensitive to AF
treatment. Cancer cells are reported to have elevated ROS levels
(12,13), rendering them more sensitive to ROS
induction (15). ROS-producing
enzymes are upregulated in UC (27,28).
Therefore, drugs that can induce ROS may represent potential
candidates for UC treatment. AF increases ROS by targeting
thioredoxin reductase (45,46).
The reasons underlying the differences observed in the bladder
cancer and upper tract urothelial carcinoma cell lines remain
unclear. Further analysis of the expression of ROS-reducing enzymes
may be needed in order to identify the factors contributing to
these difference.
The divergent responses of HT 1376 and BFTC 909
cells to AF treatment were also observed in their cell cycle
distribution. HT 1376 were blocked at the
G0/G1 phase starting at 8 h after AF
treatment, whereas BFTC 909 cells showed arrest at S phase 24 h
after AF treatment. The expression patterns of cell
cycle-associated proteins also supported these results. However,
there are limitations in the present study, since we did not check
the same panel of cell cycle regulators in both HT 1376 and BFTC
909 cells in our western blotting analysis.
A slight increase in the frequency of
subG1 cells was detected after 24-h AF treatment in HT
1376, but not in BFTC 909 cells. Apoptosis was not significantly
increased following AF treatment in HT 1376 and BFTC 909 cells. AF
treatment has been reported to increase apoptosis in several cancer
cell types (44), including lung
cancer cells (47) and acute
lymphoblastic leukemia (48);
thus, our results were inconsistent with these findings, although
these results may be due to the different cell lines used. An
increase in ROS production can cause DNA damage, which, if
continues may signal the cells to cell cycle arrest, ultimately
apoptosis will occur (49).
Therefore, cell cycle arrest may occur before apoptosis. In the
present assay conditions, AF treatment for 24 h did not trigger a
significant change in apoptosis rates in BFTC 909 cells, and only
higher concentrations of AF had an effect on HT 1376 cells. This
suggests a longer AF treatment time may have been necessary for
apoptosis to occur in BFTC 909 cells. In addition, a high apoptosis
rate was found in DMSO-treated BFTC 909 cells, possibly because the
BFTC 909 cells were sensitive to trypsin during the flow cytometric
analysis. Thus, the findings on apoptosis need to be treated with
caution, as the assay conditions require further optimization.
Another possibility is that a longer AF exposure
would be required for the UC cells to exit cell cycle arrest to
undergo apoptosis. However, ROS production was increased in both HT
1376 and BFTC 909 cells following AF treatment, and this effect was
inhibited in the presence of NAC, a ROS scavenger. In addition,
cell viability was increased in the presence of NAC. This suggests
that ROS production may cause redox imbalance, which might serve an
important role in the effect of AF on the viability of UC cells.
Further study is needed to determine the mechanism underlying
AF-mediated cytotoxicity in UC cells.
Cisplatin-based chemotherapy remains the standard
therapy for patients with advanced UC (8,40,50).
Although shifting the regimen of the combination of methotrexate,
vinblastine, doxorubicin and cisplatin to gemcitabine + cisplatin
demonstrates less toxicity (51,52),
cisplatin-based chemotherapy remains relatively toxic. Efforts are
needed to develop less toxic, cisplatin-based therapy for patients
with advanced UC. Combination therapy of two or more drugs has
several benefits, including enhancing efficacy and reducing
toxicity (53). Several molecular
mechanisms mediate the antitumor properties of cisplatin (54), such as cisplatin-induced oxidative
stress (55). AF treatment
synergizes with cisplatin cytotoxicity by ROS production, causing
mitochondrial dysfunction and DNA damage in small cell lung cancer
(56). The curcuminoid WZ35
inhibits thioredoxin reductase 1 activity, leading to ROS
production and thereby enhancing the inhibitory effects of
cisplatin on cell viability (57).
Therefore, the combination of AF and cisplatin may aggravate redox
imbalance, leading to UC cell death.
In summary, AF exhibits anticancer activity in HT
1376 and BFTC 909 cells, interfering with cell cycle progression
and redox balance. AF also shows synergism with cisplatin. To the
best of our knowledge, these findings are the first to report the
effect of AF on the viability of UC cell lines, although further
studies are needed to determine the efficacy of AF as potential
treatment for UC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The Ditmanson Medical
Foundation Chiayi Christian Hospital (grant no. R110-008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SYC designed the research and collected the data.
CNC analyzed and interpreted the data. HYH performed the
experiments. CYF designed the study and wrote the paper. HYH and
CYF confirm the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yaxley JP: Urinary tract cancers: An
overview for general practice. J Family Med Prim Care. 5:533–538.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lenis AT, Lec PM, Chamie K and Mshs MD:
Bladder cancer: A review. JAMA. 324:1980–1991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Griffiths TR; Action on Bladder Cancer, :
Current perspectives in bladder cancer management. Int J Clin
Pract. 67:435–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ritch CR, Velasquez MC, Kwon D, Becerra
MF, Soodana-Prakash N, Atluri VS, Almengo K, Alameddine M, Kineish
O, Kava BR, et al: Use and validation of the AUA/SUO risk grouping
for nonmuscle invasive bladder cancer in a contemporary cohort. J
Urol. 203:505–511. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yafi FA, Aprikian AG, Chin JL, Fradet Y,
Izawa J, Estey E, Fairey A, Rendon R, Cagiannos I, Lacombe L, et
al: Contemporary outcomes of 2287 patients with bladder cancer who
were treated with radical cystectomy: A Canadian multicentre
experience. BJU Int. 108:539–545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perera M, McGrath S, Sengupta S, Crozier
J, Bolton D and Lawrentschuk N: Pelvic lymph node dissection during
radical cystectomy for muscle-invasive bladder cancer. Nat Rev
Urol. 15:686–692. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patel VG, Oh WK and Galsky MD: Treatment
of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J
Clin. 70:404–423. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chamie K, Litwin MS, Bassett JC, Daskivich
TJ, Lai J, Hanley JM, Konety BR and Saigal CS; Urologic Diseases in
America Project, : Recurrence of high-risk bladder cancer: A
population-based analysis. Cancer. 119:3219–3227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sabharwal SS and Schumacker PT:
Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles'
heel? Nat Rev Cancer. 14:709–721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pizzino G, Irrera N, Cucinotta M, Pallio
G, Mannino F, Arcoraci V, Squadrito F, Altavilla D and Bitto A:
Oxidative stress: Harms and benefits for human health. Oxid Med
Cell Longev. 2017:84167632017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cohen Z, Maimon Y, Samuels N and Berger R:
Role of reactive oxygen species in the anticancer activity of
botanicals: Comparing sensitivity profiles. Oncol Lett.
13:2642–2648. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kumari S, Badana AK, Mohan GM, Shailender
G and Malla R: Reactive oxygen species: A key constituent in cancer
survival. Biomark Insights. 13:11772719187553912018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moloney JN and Cotter TG: ROS signalling
in the biology of cancer. Semin Cell Dev Biol. 80:50–64. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen SY, Huang HY, Lin HP and Fang CY:
Piperlongumine induces autophagy in biliary cancer cells via
reactive oxygen species-activated Erk signaling pathway. Int J Mol
Med. 44:1687–1696. 2019.PubMed/NCBI
|
|
17
|
Lin S, Li Y, Zamyatnin AA Jr, Werner J and
Bazhin AV: Reactive oxygen species and colorectal cancer. J Cell
Physiol. 233:5119–5132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang H, Xie H, Pan Y, Zheng K, Xia Y and
Chen W: Plumbagin triggers ER stress-mediated apoptosis in prostate
cancer cells via induction of ROS. Cell Physiol Biochem.
45:267–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Li J, Zong L, Chen X, Chen K,
Jiang Z, Nan L, Li X, Li W, Shan T, et al: Reactive oxygen species
and targeted therapy for pancreatic cancer. Oxid Med Cell Longev.
2016:16167812016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sharma V, Joseph C, Ghosh S, Agarwal A,
Mishra MK and Sen E: Kaempferol induces apoptosis in glioblastoma
cells through oxidative stress. Mol Cancer Ther. 6:2544–2553. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zou Z, Chang H, Li H and Wang S: Induction
of reactive oxygen species: An emerging approach for cancer
therapy. Apoptosis. 22:1321–1335. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chaffman M, Brogden RN, Heel RC, Speight
TM and Avery GS: Auranofin. A preliminary review of its
pharmacological properties and therapeutic use in rheumatoid
arthritis. Drugs. 27:378–424. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee D, Xu IM, Chiu DK, Leibold J, Tse AP,
Bao MH, Yuen VW, Chan CY, Lai RK, Chin DW, et al: Induction of
oxidative stress through inhibition of thioredoxin reductase 1 is
an effective therapeutic approach for hepatocellular carcinoma.
Hepatology. 69:1768–1786. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wen C, Wang H, Wu X, He L, Zhou Q, Wang F,
Chen S, Huang L, Chen J, Wang H, et al: ROS-mediated inactivation
of the PI3K/AKT pathway is involved in the antigastric cancer
effects of thioredoxin reductase-1 inhibitor chaetocin. Cell Death
Dis. 10:8092019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oh BM, Lee SJ, Cho HJ, Park YS, Kim JT,
Yoon SR, Lee SC, Lim JS, Kim BY, Choe YK and Lee HG: Cystatin SN
inhibits auranofin-induced cell death by autophagic induction and
ROS regulation via glutathione reductase activity in colorectal
cancer. Cell Death Dis. 8:e30532017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shimada K, Fujii T, Anai S, Fujimoto K and
Konishi N: ROS generation via NOX4 and its utility in the
cytological diagnosis of urothelial carcinoma of the urinary
bladder. BMC Urol. 11:222011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miyata Y, Matsuo T, Sagara Y, Ohba K,
Ohyama K and Sakai H: A mini-review of reactive oxygen species in
urological cancer: Correlation with NADPH oxidases, angiogenesis,
and apoptosis. Int J Mol Sci. 18:22142017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chou TC and Talaly P: A simple generalized
equation for the analysis of multiple inhibitions of
michaelis-menten kinetic systems. J Biol Chem. 252:6438–6442. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Otto T and Sicinski P: Cell cycle proteins
as promising targets in cancer therapy. Nat Rev Cancer. 17:93–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Malumbres M: Cyclin-dependent kinases.
Genome Biol. 15:1222014. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sgambato A, Cittadini A, Faraglia B and
Weinstein IB: Multiple functions of p27(Kip1) and its alterations
in tumor cells: A review. J Cell Physiol. 183:18–27. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roskoski R Jr: Cyclin-dependent protein
serine/threonine kinase inhibitors as anticancer drugs. Pharmacol
Res. 139:471–488. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen T and Huang S: The role of Cdc25A in
the regulation of cell proliferation and apoptosis. Anticancer
Agents Med Chem. 12:631–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartek J and Lukas J: Mammalian G1- and
S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol.
13:738–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Falck J, Petrini JH, Williams BR, Lukas J
and Bartek J: The DNA damage-dependent intra-S phase checkpoint is
regulated by parallel pathways. Nat Genet. 30:290–294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Falck J, Mailand N, Syljuåsen RG, Bartek J
and Lukas J: The ATM-Chk2-Cdc25A checkpoint pathway guards against
radioresistant DNA synthesis. Nature. 410:842–847. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Drayton RM and Catto JW: Molecular
mechanisms of cisplatin resistance in bladder cancer. Expert Rev
Anticancer Ther. 12:271–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim SJ, Kim HS and Seo YR: Understanding
of ROS-inducing strategy in anticancer therapy. Oxid Med Cell
Longev. 2019:53816922019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kirtonia A, Gala K, Fernandes SG, Pandya
G, Pandey AK, Sethi G, Khattar E and Garg M: Repurposing of drugs:
An attractive pharmacological strategy for cancer therapeutics.
Semin Cancer Biol. 68:258–278. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hernandez JJ, Pryszlak M, Smith L, Yanchus
C, Kurji N, Shahani VM and Molinski SV: Giving drugs a second
chance: Overcoming regulatory and financial hurdles in repurposing
approved drugs as cancer therapeutics. Front Oncol. 7:2732017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Onodera T, Momose I and Kawada M:
Potential anticancer activity of auranofin. Chem Pharm Bull
(Tokyo). 67:186–191. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang X, Selvaraju K, Saei AA, D'Arcy P,
Zubarev RA, Arnér ES and Linder S: Repurposing of auranofin:
Thioredoxin reductase remains a primary target of the drug.
Biochimie. 162:46–54. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hwang-Bo H, Jeong JW, Han MH, Park C, Hong
SH, Kim GY, Moon SK, Cheong J, Kim WJ, Yoo YH and Choi YH:
Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis
in hepatocellular carcinoma Hep3B cells by generation of reactive
oxygen species. Gen Physiol Biophys. 36:117–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cui XY, Park SH and Park WH: Auranofin
inhibits the proliferation of lung cancer cells via necrosis and
caspase-dependent apoptosis. Oncol Rep. 44:2715–2724. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Karsa M, Kosciolek A, Bongers A, Mariana
A, Failes T, Gifford AJ, Kees UR, Cheung LC, Kotecha RS, Arndt GM,
et al: Exploiting the reactive oxygen species imbalance in
high-risk paediatric acute lymphoblastic leukaemia through
auranofin. Br J Cancer. 125:55–64. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kuczler MD, Olseen AM, Pienta KJ and Amend
SR: ROS-induced cell cycle arrest as a mechanism of resistance in
polyaneuploid cancer cells (PACCs). Prog Biophys Mol Biol. 165:3–7.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nadal R and Bellmunt J: Management of
metastatic bladder cancer. Cancer Treat Rev. 76:10–21. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
von der Maase H, Sengelov L, Roberts JT,
Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Roberts JT, von der Maase H, Sengeløv L,
Conte PF, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine/cisplatin and
methotrexate/vinblastine/doxorubicin/cisplatin in patients with
locally advanced and metastatic bladder cancer. Ann Oncol. 17
(Suppl 5):v118–v122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Narayan RS, Molenaar P, Teng J,
Cornelissen FMG, Roelofs I, Menezes R, Dik R, Lagerweij T, Broersma
Y, Petersen N, et al: A cancer drug atlas enables synergistic
targeting of independent drug vulnerabilities. Nat Commun.
11:29352020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Saad SY, Najjar TA and Alashari M: Role of
non-selective adenosine receptor blockade and phosphodiesterase
inhibition in cisplatin-induced nephrogonadal toxicity in rats.
Clin Exp Pharmacol Physiol. 31:862–867. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu X, Wang W, Yin Y, Li M, Li H, Xiang H,
Xu A, Mei X, Hong B and Lin W: A high-throughput drug screen
identifies auranofin as a potential sensitizer of cisplatin in
small cell lung cancer. Invest New Drugs. 37:1166–1176. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
He W, Xia Y, Cao P, Hong L, Zhang T, Shen
X, Zheng P, Shen H, Liang G and Zou P: Curcuminoid WZ35 synergize
with cisplatin by inducing ROS production and inhibiting TrxR1
activity in gastric cancer cells. J Exp Clin Cancer Res.
38:2072019. View Article : Google Scholar : PubMed/NCBI
|