According to the Global Health Assessment released
by the World Health Organization in 2020, it is estimated that in
the next 20 years, the number of global cancer cases may increase
by 60% and the tumor burden is also increasing (1). The traditional methods for treating
tumors, such as chemotherapy or radiotherapy, cause significant
harm to patients and the curative effect is not satisfactory. A
novel treatment method different from traditional therapy has been
invented, namely chimeric antigen receptor T-cell (CAR-T) therapy.
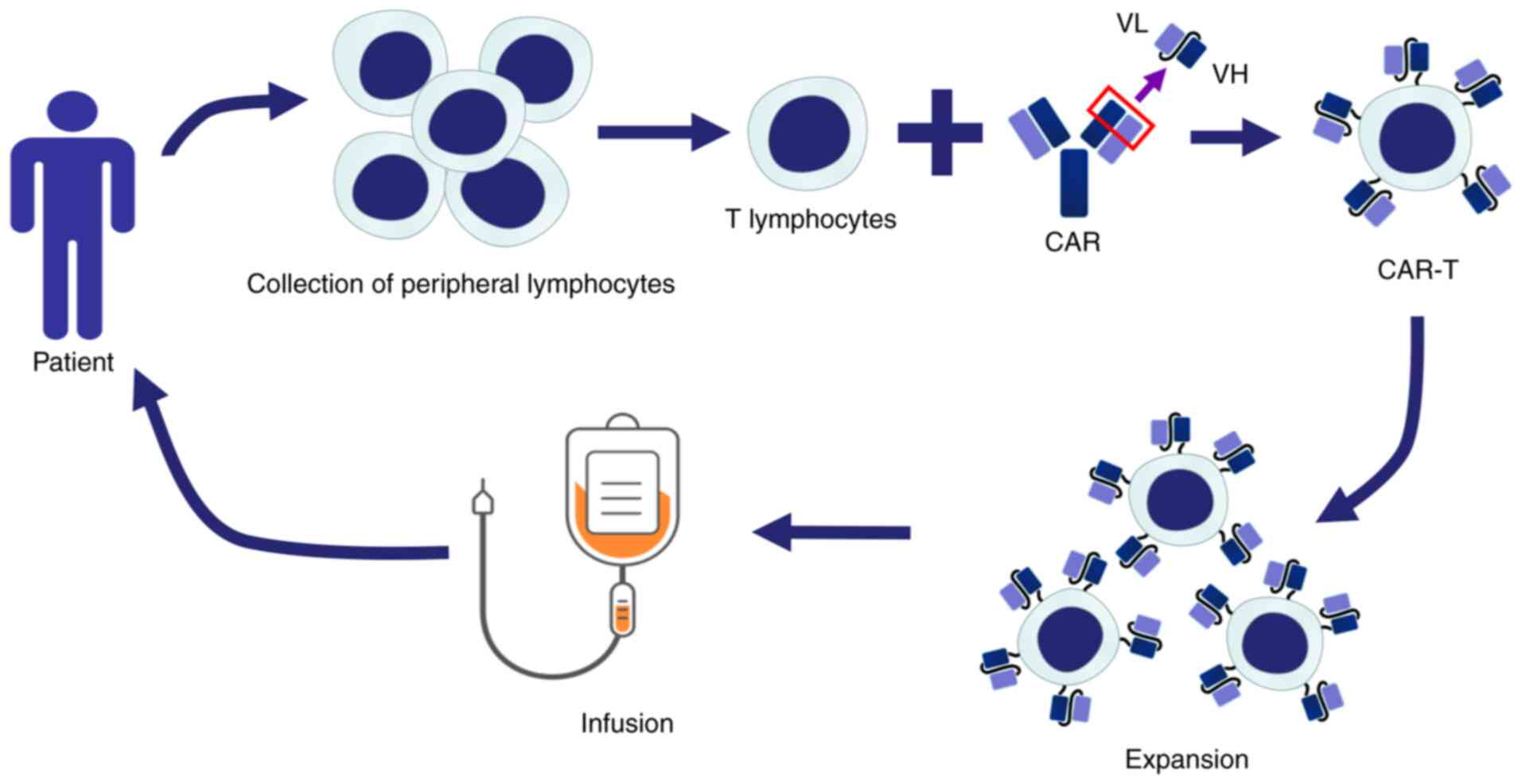
CAR-T cell therapy is a method of adoptive cell therapy that
modifies a patient's peripheral T cells in vitro by genetic
engineering, which are then infused into the patient to recognize
and kill tumor cells for treatment, as illustrated in Fig. 1. CAR-T cells have achieved
satisfactory results in treating hematopoietic malignancies
(2). CAR-T cell therapy (Kymriah),
approved by the Food and Drug Administration (FDA) to treat B-cell
acute lymphoblastic leukemia (ALL) in 2017, was the world's first
approved CAR-T product (3). CAR-T
products, including Kymirah, Yescarta, Tecartus, Breyanzi, Abecma,
Relma-cel and Carvykti, have been approved for treating
hematological malignancies, as summarized in Table I; the CAR-T treatment method and
commercialization process of CAR-T have been formally recognized by
the FDA.
However, CAR-T cell therapy has limitations in the
treatment of solid tumors due to solid tumor cells frequently
expressing different tumor antigens, which may reduce its targeting
effect, and patients receiving CAR-T cell therapy frequently
experience side effects, such as cytopenia, and have an increased
risk of infection in the presence of neutropenia, previous
immunosuppression, lymphatic clearance, tocilizumab or steroid
application (4,5). The antigens selected by CAR-T cell
therapy may be expressed on both tumor and normal tissue cells,
causing CAR-T cells to attack normal cells mistakenly and produce
off-target effects. In addition, CAR-T cells are prone to release
cytokines excessively after entering the patient's body, resulting
in cytokine release syndrome (CRS). Patients affected by CRS may
experience severe inflammation, hypotension, hypoxia and even
death, thus significantly reducing the efficacy and safety of CAR-T
therapy (6). Therefore,
summarizing the current research progress and adverse reactions of
CAR-T cell therapy is expected to provide references for improving
the efficacy and safety of this method.
T cells have an essential role in the process of
anti-tumor immunity. The activation of T cells requires multiple
signals, including T cell receptor (TCR) recognition of antigenic
peptide-major histocompatibility complex (MHC) on the surface of
antigen-presenting cells (APC), which may process antigen and
present it in conjunction with MHC molecules on the cell surface
and interact with appropriate T cell receptors, followed by
co-stimulation factor and cytokine signaling. Since the MHC mainly
activates TCRs, tumor cells frequently downregulate MHC expression
to avoid being recognized by the immune system. The traditional
TCRs must recognize MHC-antigen peptide complexes to activate T
cells, while CARs may recognize antigens directly and lead to T
cell activation. CARs are able to mimic the TCR's function and
overcome their limitation. Gross et al (7) constructed immunoglobulin-TCR chimeric
molecules as functional receptors with antibody-type specificity in
1989; this receptor is able to recognize pathogenic antigens and
transmit T cell activation signals. Since the single-chain antibody
fragment (scFv) formed by the variable regions of the heavy chain
and light chain of the antibody has the same function of
recognizing antigen as the Fab region of the antibody, Eshhar et al
(8) later replaced TCR with the
scFv fragment, which fused with CD3ζ chain; this fusion protein
effectively promoted the activation of T cells and killed lymphoma
cells expressing specific antigens in vitro, which is referred to
as the first generation of CAR-T cells. In the clinic, the
patient's T cells were extracted and integrated with genetically
engineered CARs. The cultured CAR-T cells were injected back into
the patient to attack tumor cells specifically and achieve the
purpose of treatment. The T cells were activated
antigen-specifically with no MHC restriction during the treatment.
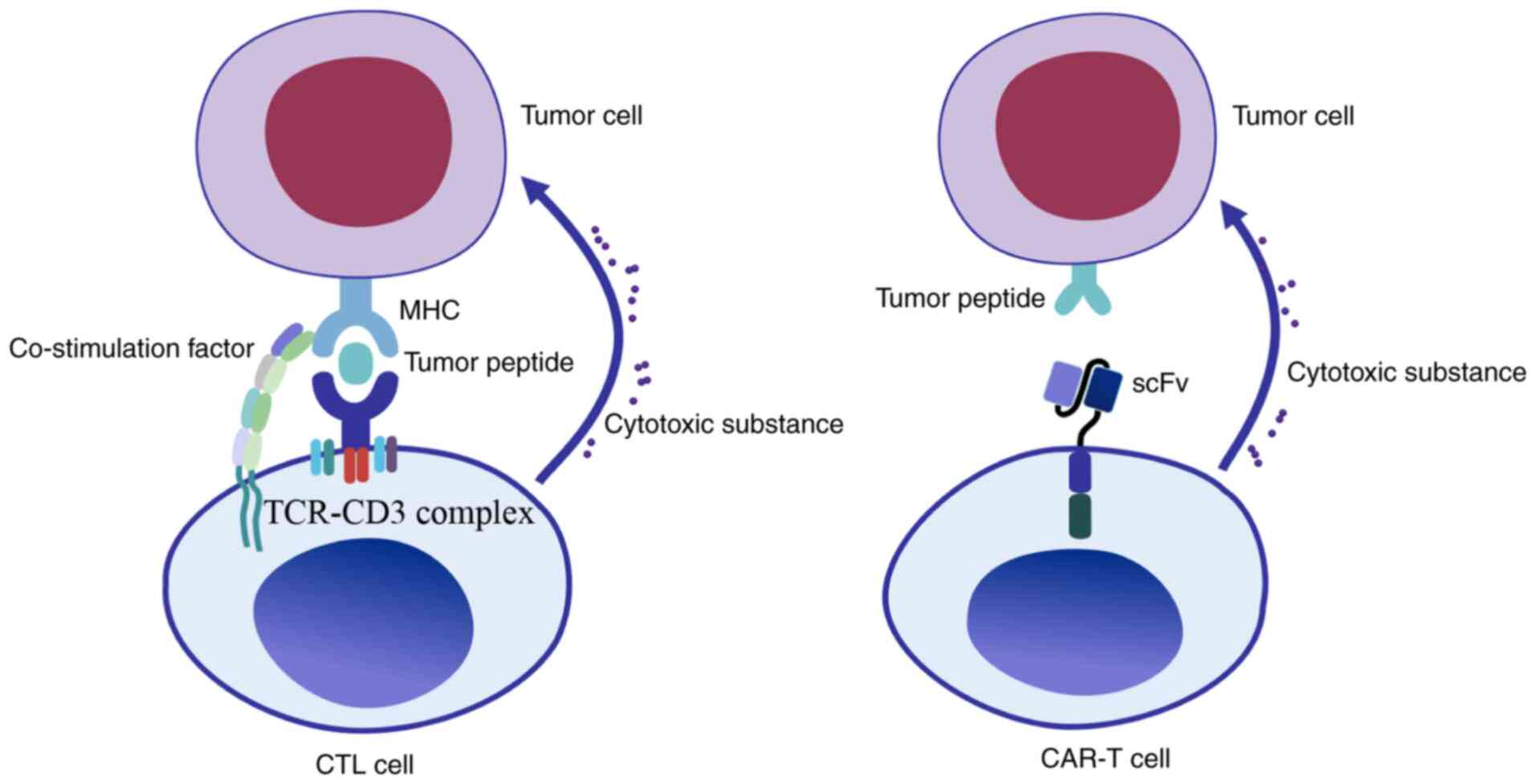
CAR-T cells are able to bind and kill tumors specifically by
releasing cytotoxic substances. Unlike cytotoxic T lymphocyte (CTL)
cells, CAR-T cells are able to recognize the antigen of tumor cells
through scFv. This recognition is not restricted by MHC molecules,
which has significant advantages over the traditional CTL cells, as
illustrated in Fig. 2.
The basic structure of CARs includes a
tumor-associated antigen (TAA) binding region, an extracellular
hinge region, a transmembrane region and an intracellular
immunoreceptor tyrosine-based activation motif (ITAM). TAA is
abundantly located on the tumor cell surface and seldom expressed
in normal tissues, and various TAAs may be used as target antigens
for CAR-T cells. The tumor target antigen is crucial for the
specificity, efficacy and safety of therapy. CAR-T cells have
developed rapidly in recent years; the extracellular regions of
these CAR-T cells are V regions of antibodies and the transmembrane
part is almost the same. The intracellular structure distinguishes
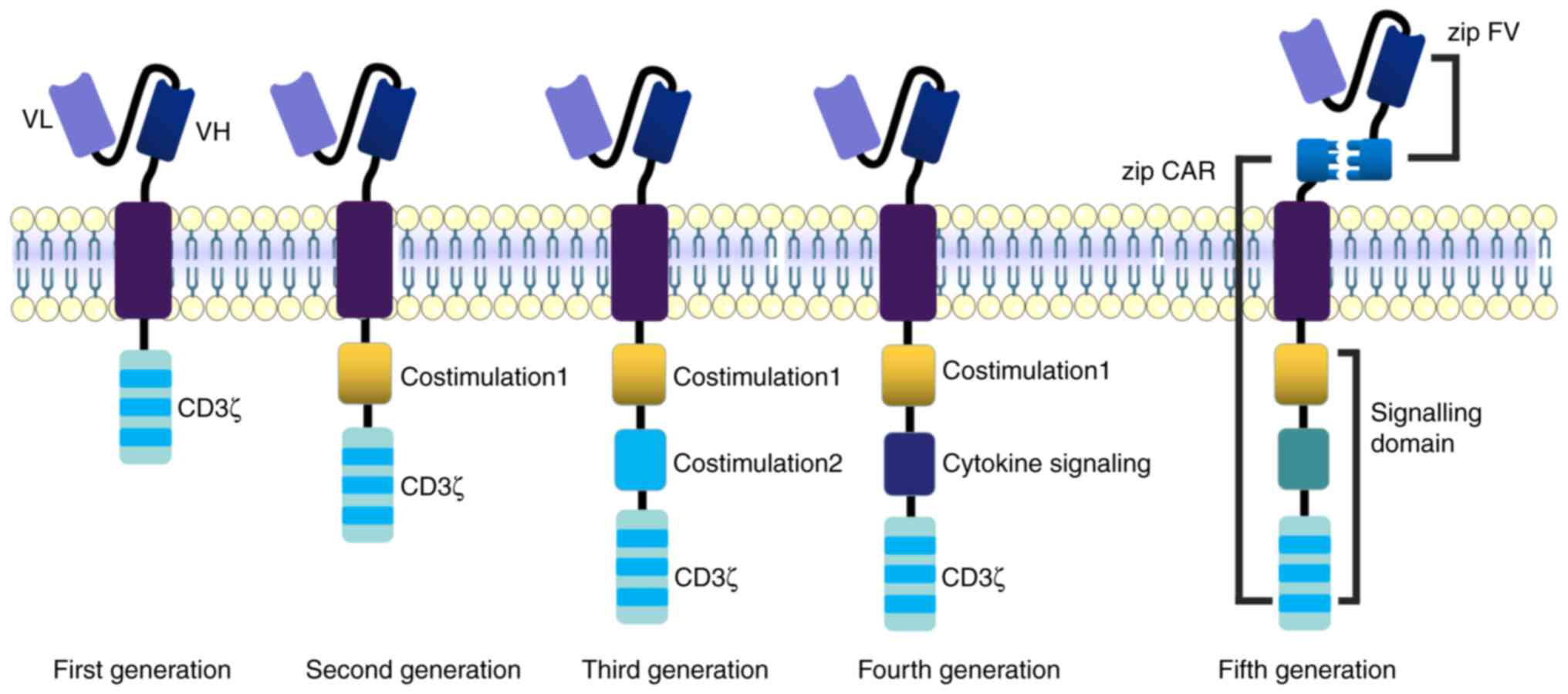
these CAR-T cells. The first-generation CAR-T cell intracellular
segment is relatively simple; it is mainly composed of the ITAM
region of the ζ chain of the CD3 molecule and there are only three
pairs of ITAMs, which lack costimulatory molecules. These
first-generation CAR-T cells may only cause transient T cell
proliferation and less cytokine secretion, cannot provide a
sustained anti-tumor effect and clinical treatment is
unsatisfactory. Due to the inefficiency of signal transmission and
short survival time of CAR-T cells in patients, various studies
have rated them as having ‘no significant effect but great
potential’ (9,10). Costimulatory molecules, such as
4-1BB (CD137), CD28, tumor necrosis factor receptor superfamily
member 4 (OX40) or inducible costimulatory molecule were added to
the intracellular region of the second-generation CAR, these
costimulatory molecules activate JNK, ERK and NF-κB signaling
pathways in T cells, having a more substantial killing effect on
tumor cells. However, most vectors used in second-generation CAR-T
cells are retroviruses that may only carry limited gene fragments,
which may restrict the function of these CAR-T cells. The
third-generation CAR-T cells use a lentivirus as a transfection
vector, which may carry larger gene segments into T cells. The
intracellular part frequently contains two or more costimulatory
signal regions to stimulate T cells to produce numerous cytokines.
Third-generation CAR-T cells have a more potent anti-tumor effect
compared with second-generation (11,12).
However, certain studies have indicated that the killing activity
of third-generation CAR-T cells has not been improved
significantly; this may be because the activation signal generated
by ITAM has reached its threshold in terms of lymphocyte activation
and only adding costimulatory molecules may not improve the
efficacy for CAR-T cells (13). In
the fourth generation of CAR-T technology, the IL-12 gene was
introduced into CAR-T cells to enhance the killing ability. When
CAR-T cells recognize tumor cells and activate, they secrete a
large amount of IL-12 at the tumor site and recruit various innate
immune cells to kill the tumor cell. Considering the side effects
of CAR-T cells, a suicide gene added to CAR may control the
survival time of CAR-T cells in vivo. CAR-T cells are mainly used
to treat hematological tumors and have not achieved satisfactory
results in treating solid tumor tissue. the main reason is that
CAR-T cells cannot enter the solid tumor. Therefore, the receptor
of certain cytokines or chemokines is added into CAR to increase
the infiltration ability of T lymphocytes in tumor tissue, thereby
enhancing the killing effects of solid tumors. This is also one of
the design ideas for the fourth generation of CAR-T cells (14–16).
The third-generation and fourth-generation CAR-T cells contain more
costimulatory signals to secrete more cytokines, which may cause a
robust immune response in the body, so the second-generation
technology is rather used to treat hematological tumors based on
safety considerations. These autologous CAR-T cell therapies have
numerous limitations, including high cost, long manufacturing cycle
and limited cell sources. Furthermore, certain patients are
unsuitable for autologous CAR-T cell therapy due to physical
reasons such as an advanced tumour and/or physical weakness,
accompanied by severe infection or poor immune activity after
repeated chemotherapy and radiotherapy. The fifth-generation CAR-T
technology called Universal CAR-T (UCAR-T) cell therapy is an
attractive breakthrough (17).
CAR-T cell therapy is expensive, e.g., Kymriah costs
$475,000 per patient in the US, which is unaffordable for an
ordinary family. The high price is not only due to the increased
investment cost of CAR-T, but CAR-T therapy also requires to be
prepared according to the individual differences of patients and it
is similar to a personalized medical service. To overcome the high
cost of CAR-T therapy, scientists proposed the concept of UCAR-T.
There are two strategies for UCAR-T: Universal T cells and UCAR.
The autologous CAR-T cells for immunotherapy frequently have
numerous limitations and it is difficult to generate sufficient
numbers of effective CAR-T cells for various unpredictable reasons.
Allogeneic CAR-T cells derived from healthy donors may have
clinical efficacy; however, these allogeneic cells may induce
graft-vs. -host disease (GVHD) after infusion, while UCAR-T cells
may overcome this obstacle. The steps include isolation of T
lymphocytes from healthy donors, transfer of CAR genes and other
genes (such as costimulatory or suicide genes) into T cells through
viral vectors and knockout of GVHD-related genes [TCR or human
leukocyte antigen (HLA)] through gene-editing technology. T cells
edited by transcription activator-like effector nuclease technology
performed well and CRISPR/Cas9 technology is also undergoing
clinical trials (18,19).
A novel UCAR-T contains a two-receptor system; the
universal receptor part is T cells with leucine adapters (Zipcar)
and the ligand part is single chain fragment variable fused to a
cognate leucine zipper with leucine adapters that is able to target
tumor antigens. This UCAR-T may perform receptor switching against
different tumor antigens or recognize multiple tumor antigens by
combining the universal receptor part without further modification
of T cells (20). UCAR T-19 cells
developed using lentivirus and CRISPR/Cas9 gene-editing systems
express mutated B2M-HLA-E and mutated B2M-HLA-G fusion proteins
that may prevent the destruction of host cells by allogeneic NK
cells (21). UCAR-T technology
currently faces numerous challenges such as GVDH reaction caused by
trace amounts of TCR-positive CAR-T cells, off-target effects and
difficult amplification in vivo due to immune system rejection.
Thus, most UCAR-T projects are in the preclinical stage and perfect
technology is expected to ensure the safety and efficacy of UCAR-T
cell therapy (22). The
development of CAR-T cell products is presented in Fig. 3.
B-ALL is a common hematological disease with a poor
prognosis for most patients. Currently, bone marrow transplantation
is the best treatment. Early CD19-targeted CAR-T cell therapy was
unsatisfactory in treating relapsed or refractory (R/R) B-ALL, as
more than half of the patients relapsed after CD19 CAR-T therapy;
however, CAR-T cell therapy combined with allogeneic stem cell
transplantation (SCT) may increase patient survival (23). In a study including 30 adult
patients with B-ALL receiving CD19+ CAR-T therapy, the
median time to progression was 5.5 months, median survival was 7.5
months and 13 patients (43%) achieved a complete response (CR). Of
note, 7 of 12 patients (58.3%) receiving blinatumomab/inotuzumab
after CAR-T treatment failure achieved a CR, and thus, new
treatment strategies are required in order to reduce the risk
associated with treatment and improve patient prognosis (24). Patients with B-ALL are prone to
relapse after receiving CD19-targeted immunotherapy and a new
alternative strategy is thus required. CD72 is a cell marker of
poor prognosis in patients with B-ALL and synthetic CD72-specific
nanobodies incorporated into CARs demonstrated robust activity
against B-cell malignancy models, which may offer new ideas for
treatment (25). CD19-targeting
CAR-T cell therapy is limited in certain conditions, while
bispecific CAR-T cells targeting both CD19 and CD22 may overcome
this limitation and all 6 patients with R/R B-ALL achieved minimal
residual disease (MRD)-negative CR after receiving this bispecific
CAR-T cell therapy (26). After
patients with B-ALL received allogenic hematopoietic SCT (HSCT),
only those who achieved a second remission without MRD were able to
obtain satisfactory long-term survival; all of those 5 patients
with B-ALL achieved rapid tumor eradication and MRD(−) complete
remission (CR) after CD19+ CAR-T cell therapy. Elevated
cytokines are directly related to tumor burden during CAR-T cell
infusion and certain patients require steroids to alleviate
cytokine-mediated toxicity (27).
Another study indicated that among 30 patients with relapsed ALL
who received CD19+CAR-T therapy, 27 patients (90%)
achieved CR, the 6-month event-free survival rate was 67% and the
overall survival rate was 78%. However, 27% of patients exhibited
severe cytokine release syndrome, which was alleviated after
treatment with an anti-IL-6 receptor antibody; this may be
associated with a high disease burden prior to infusion (28). Patients with B-ALL are prone to CRS
after CAR-T therapy and corticosteroids or IL-6 receptor inhibitors
may alleviate it effectively. In addition, serum C-reactive protein
is a reliable indicator for judging the severity of CRS (29). The relapses of patients with ALL
after single-targeted CAR-T therapy may be related to tumor antigen
escape. CD19 and CD22 dual-targeted CAR-T cells (CTA101) based on
CRISPR/Cas9 technology have advantages as a novel therapy; the CR
was 83.3% after 6 patients with R/R ALL accepted CTA101 infusion
and 5 patients achieved CR or CR with incomplete hematologic
recovery, remaining MRD negative (30).
Despite encouraging results for CAR-T in treating
B-ALL, significant challenges remain in T-ALL treatment. T-ALL is a
hematological malignancy caused by the malignant transformation and
clonal expansion of T-lineage precursor cells in the bone marrow
and thymus. It mainly occurs in children and adolescents and has a
higher recurrence rate, lower remission rate and lower long-term
survival rate than B-ALL (31).
Studies have indicated that anti-CD7 CAR-T has an important role in
treating T-ALL. In a phase I clinical trial of T-ALL, 20
participants received anti-CD7 CAR-T treatment and 90% of patients
(n=18) achieved CR. Most patients experienced adverse reactions
such as cytokine release syndrome; most adverse reactions were
reversible and only one patient died of fungal pneumonia-related
pulmonary hemorrhage (32).
Another study confirmed that 20 patients with R/R T-ALL (n=14) and
lymphoblastic lymphoma (n=6) accepted anti-CD7 CAR-T therapy, of
which 19 patients achieved MRD negative CR in the bone marrow by
day 28 (33). CD7 is a promising
therapeutic target due to its wide expression in almost all T-cell
malignancies. A study reported that a patient with T-ALL received
anti-CD7 CAR-T therapy, was injected with 5×106/kg
anti-CD7 CAR-T cells, and the patient's blood and bone marrow
achieved remission (34). Another
patient with T-ALL received autologous anti-CD7 CAR-T cells and
achieved remission on day 17; although symptoms of cytopenias
occurred from days 14 to 21, this patient accepted HSCT after
achieving CR, suggesting that anti-CD7 CAR-T may be a safe and
effective strategy for the treatment of patients with T-ALL
(35). In a study using an
anti-CD7 nanobody fragment coupled with an endoplasmic
reticulum/Golgi retention domain demonstrated that cells transduced
with CD7-CAR were able to prevent fratricide and achieve expansion;
these cells exhibited antitumor potential in CD7-positive malignant
cell lines and patient-derived xenograft models (36). Research has indicated that anti-CD7
CAR-T may cause T cells to fratricide and Png et al (37) conjugated protein expression blocker
(PEBL) to CD7 fragment; this anti-CD7 PEBL eliminates CD7
expression in T cells, effectively alleviating CAR-T-mediated
fratricide without damaging T cell proliferation or the
interferon-γ and tumor necrosis factor secretion ability. PEBL-CAR
T cells are cytotoxic to CD7+ leukemia cells in vitro,
both in leukemia cell lines and patient-derived T-ALL
xenografts.
NHL is a heterogeneous disease and most NHLs are B
cell types that account for 70–85%. Despite decades of advances in
chemotherapy regimens and new treatments, the disease remains
incurable. The CD19+ CAR-T cell therapy axicabtagene
ciloleucel (Axi-Cel) has a significant effect in patients with
refractory large B-cell lymphoma. After failure of conventional
therapy, 111 patients received Axi-Cel therapy, the objective
response rate was 82%, the CR rate was 54%, 42% of patients had
sustained remission and the overall survival rate of all patients
was 52% at 18 months of follow-up in this trial. The most common
adverse events during treatment were neutropenia (78%), anemia
(43%) and thrombocytopenia (38%). Patients who received CAR T-cell
therapy with Axi-Cel had high levels of durable response (38). A long-term study indicated that 60%
of patients with B-cell lymphoma treated with CD19+
CAR-T cells (CTL019) were still in remission after 5 years and the
clinical effect of CTL019 was not affected, although different
patients accepted different lymphodepletion regimens (39). CD28 and 4-1BB may activate
different signaling pathways and combining them may overcome the
limitations of costimulation alone. A total of 16 patients with
relapsed/refractory lymphoma accepted infusion of 2G CAR-T cells
(with CD28 only) and 3G CAR-T cells (CD28 and 4-1BB); of these, 3
patients achieved CR and 3 patients partial remission. CRS occurred
in 6 patients, but the symptoms were mild and did not require
anti-IL-6 therapy, indicating CD28 combined with 4-1BB may produce
satisfactory therapeutic effects in patients with lymphoma
(40). A CAR-T that is able to
recognize C-X-C motif chemokine receptor 5 (CXCR5) with high
affinity may target B-NHL cells effectively based on the
characteristic that CXCR5 is highly expressed on the surface of
lymphoma cells; application of CXCR5+ CAR-T cells in a
mouse lymphoma model was able to kill malignant B lymphoma cells
specifically, providing a promising strategy for the treatment of
B-NHL (41).
CAR-T cell therapy in B-NHL may cause various
adverse reactions and the prognosis of patients is not as
satisfactory as that of ALL; the lower response rate may be related
to the physical barrier of the tumor inhibitory microenvironment.
In addition, certain patients relapsed after treatment, which may
be related to limited CAR-T cell persistence and CD19 antigen
escape (42). Studies suggested
that about half of the patients with R/R diffuse large B-cell
lymphoma (DLBCL) experience severe side effects after CAR-T cell
therapy. Axi-Cel and tisagenlecleucel are new CAR-T cell therapies
that combine CAR T-cells with a fusion protein between interferon
and anti-CD20 monoclonal antibody with checkpoint inhibitors or
sensitizers that have apoptotic-regulatory effects; the remission
rate of patients may reach 83%, the complete response rate is
between 40 and 58% and the side effects of CAR-T cell therapy are
significantly reduced (43).
Although the therapeutic effect of CAR-T cells in NHL is
impressive, the single-targeted CAR-T therapy still has its
limitations. The most common reason is tumor cell immune escape due
to target antigen loss and multi-targeted CAR-T may achieve a
better therapeutic effect (44).
MM is a hematological malignancy of B cells caused
by the abnormal proliferation of plasma cells. Tumor cells express
a large amount of B cell maturation antigen (BCMA) instead of CD19,
which is different from B cell leukemia and lymphoma. Therefore,
BCMA is an important therapeutic target for MM. A variety of
BCMA-targeted therapies have achieved encouraging results in the
treatment of MM (45). The first
human trial of BCMA-CAR-T cells in MM was performed by the National
Cancer Institute in 2016, in which 12 patients received BCMA-CAR-T
cell treatment. In this trial, two patients accepted a dose level
of 9×106 CAR-T cells/kg body weight treatment, bone
marrow plasma cells became undetectable by flow cytometry after
treatment and they entered a stringent complete remission. However,
two patients experienced toxicities related to cytokine release,
including fever, hypotension and dyspnea. These results were the
first to indicate the anti-tumor activity of CAR-BCMA-T cells in
patients with MM (46). The FDA
approved idecabtagene vicleucel for the treatment of patients with
myeloma in 2021. It is the first FDA-approved cell therapy for MM,
which is expected to prolong the remission period and even cure
patients with MM (47). In a
clinical trial conducted by the Shanghai Institute of Hematology
targeting BCMA (LCAR-B38M) in 17 patients with R/R MM, the overall
response rate was 88.2% and 13 patients reached CR, 2 patients had
a very good partial response (VGPR) and 1 was a nonresponder. Of
note, 8 patients maintained CR or VGPR after 417 days of follow-up.
It was also observed that anti-CAR antibody positivity is a
high-risk factor for relapse. Bi-epitopic CAR-T cells targeting
BCMA represent a promising therapy for R/R MM and most adverse
effects were manageable (48).
Since the changes in BCMA expression levels reduce
the activity of CAR-T cells, methods to minimize tumor cell escape
are under consideration. Antigens other than BCMA on the surface of
plasma cells may be new targets for CAR-T cell recognition. CS1
(also known as CD319, CRACC or SLAMF7) is a newly discovered target
and BCMA-CS1 bispecific CAR-T cells demonstrated a superior tumor
cell clearance ability as compared with BCMA and CS1
single-targeted CAR-T therapy. Combining this treatment with
anti-programmed death 1 (PD-1) antibodies may accelerate tumor cell
clearance in vivo (49). Targeting
CD38 antigen in patients with MM using CAR-T cells in which the
intracellular region is composed of CD28 and 4-1BB may stimulate T
cell proliferation and improve the anti-tumor function
significantly, which may provide a new target for MM therapy
(50).
The latest global cancer data in 2021 pointed out
that most cancers are solid tumors, which are therefore the major
focus in the fight against cancer (51). However, CAR-T cell therapy has not
received a substantial breakthrough in treating solid tumors and
most clinical trials for solid tumors are at an early stage. One of
the important reasons is that solid tumor cells frequently express
multiple tumor antigens, which are less targeted than hematological
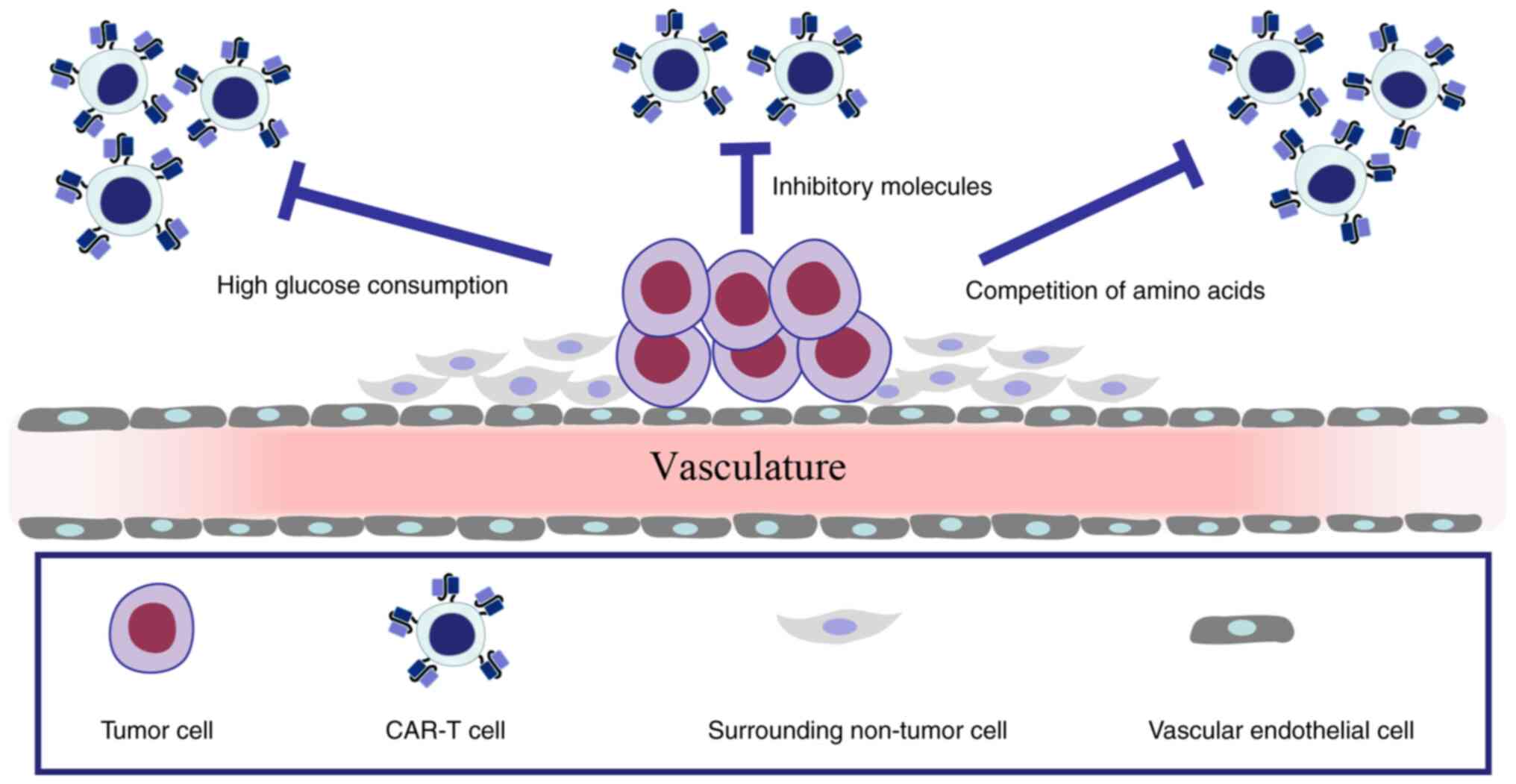
tumors. Tumor cells and surrounding non-tumor cells form a tumor
microenvironment (TME) with hypoxia and insufficient nutrients.
Furthermore, inhibitory factors of tumor cells, high glucose
consumption and competition of amino acids in the TME deprive T
cells of adequate energy support and lead to cell unresponsiveness
or exhaustion (52–55). The TME is thus unfavorable for the
survival of CAR-T cells, as outlined in Fig. 4. Currently, the improvement of
CAR-T cells in the treatment of solid tumors mainly focuses on
finding suitable targets to enhance safety and efficacy.
Solid tumors that are responsible for >85% of
cancer-associated deaths require angiogenesis for tumor cell growth
and metastasis. Targeting vascular endothelial growth factor
receptor 2, which is overexpressed in the tumor vasculature, is a
promising strategy for anti-angiogenic tumor treatment; the solid
tumor may be inhibited by targeting blood vessels in clinical
treatment (56,57). The heterogeneity of specific tumor
antigens is an obstacle to CAR-T cell therapy. Tumor antigen escape
may be overcome by multispecific targeting and CAR-T cells with
three specific antigen recognition domains may recognize the tumor
antigens specifically. Epidermal growth factor receptor (EGFR),
epithelial cell adhesion molecule and human epidermal growth factor
receptor 2 (HER2) have been linked together in a single CAR via
ankyrin repeat proteins and these CAR-T cells had a strong killing
ability of tumor cells such as K562, MDA-MB-231 and NCI-H1975
(58). The immunosuppressive TME
is an environment for tumor cells to survive and tumor-associated
macrophages (TAM) are an important part of the TME. TAMs expressing
folate receptor beta (FRβ) may promote the growth of tumor cells.
mFRβ CAR-T cells may selectively reduce the enrichment of
pro-inflammatory monocytes, delay tumor progression and prolong
survival in an ovarian cancer mouse model (59). OX40 is a potent CAR-T enhancer that
may reduce CAR-T cell apoptosis by upregulating Bcl-2 family
members and enhancing CAR-T cell cytotoxicity through NF-κB, MAPK
and PI3K-AKT signaling. CAR-T cells with OX40 exhibited enhanced
amplification and antitumor activity in mouse tumor models
(60). CAR-T cells targeting
prostate-specific membrane antigen (PSMA) are able to mediate tumor
vascular destruction, inhibit tumor cell growth and accelerate the
regression of prostate cancer. These CAR-T cells may also ablate
PSMA+ blood vessels, leading to the depletion of tumor
cells and reducing the tumor burden in the ovarian cancer mouse
model (61). Reports have
confirmed that CAR-T cells targeting tumor endothelial marker TEM-8
have a satisfactory effect in triple-negative breast cancer
treatment, as the patients' tumor volume was significantly reduced
after CAR-T cell therapy and the blood vessels in the cancer had
decreased significantly after 2 months (62).
Glypican-3 (GPC3) is a carcinoembryonic glycoprotein
highly expressed in hepatocellular carcinoma (HCC). GPC3 CAR-T
cells exhibited potent cytotoxicity against HCC cell lines, reduced
the tumor burden in the Huh-7 ×enograft mouse model and secreted
high levels of IFN-γ to inhibit tumor cell growth (63,64).
Natural killer group 2 member D (NKG2D) ligand (NKG2DL) is low
expressed on the normal cell surface but overexpressed on malignant
cells, providing an ideal target for CAR-T therapy. A CAR-T cell
based on NKG2D, 4-1BB and CD3ζ was able to kill the HCC cell lines
SMMC-7721 and MHCC97H with high expression levels of NKG2DL
effectively, which may provide new treatment ideas for
NKG2DL+ HCC (65).
Tumor-associated-mucin 1 (tMUC1) is a mucosal mucin molecule
abnormally expressed in pancreatic ductal adenocarcinoma (PDA).
tMUC1-CAR T cells are able to bind to tMUC1 on PDA cells
specifically and control the growth of pancreatic tumors in vivo
effectively (66). Carbonic
anhydrase IX (CAIX) is a membrane protein that is highly expressed
in glioblastoma. CAR-T cells targeting CAIX inhibited the growth of
glioblastoma cell lines LN229, T98G, A172 and U251 in vitro, and
intratumoral injection of CAIX CAR-T cells significantly inhibited
tumor growth in a glioblastoma mouse model, indicating the
feasibility of the use of CAIX CAR-T cells in the treatment of
glioblastoma (67). MAGE family
member A1 (MAGE-A1) is a member of the cancer-testis antigen
family, which is highly expressed in lung adenocarcinoma (LUAD).
MAGE-A1 CAR-T cells can inhibit the growth of LUAD cell lines PC9,
H1299, GLC82 and A549, which may be a strategy for the treatment of
MAGE-A1+ LUAD (68).
Carcinoembryonic antigen (CEA) is a tissue non-specific tumor
antigen expressed in numerous tumor types, such as colon cancer,
esophageal cancer and non-small cell lung cancer. In a study, 10
patients with CEA+ colorectal cancer received CEA CAR-T
cell therapy, of which 7 patients reached stable disease and 2
patients remained stable for >30 weeks; the serum CEA level
decreased significantly (69).
Malignant glioma is a common brain tumor type and
the traditional treatment, such as surgical resection, radiotherapy
or chemotherapy, is not satisfactory. A novel CAR-T cell (GCT02)
that may recognize epidermal growth factor receptor variant III
(EGFRvIII) on the tumor cells' surface may mediate tumor cell
elimination, indicating a promising treatment for glioblastoma
(70). In a study, 10 patients
with recurrent glioblastoma accepted EGFRvIII-CAR-T cell infusion;
the EGFRv expression level decreased in 5 patients, and 1 patient
had no disease progression at the 18-month follow-up (71). However, certain glioblastoma tumor
cells do not express any mutated forms of EGFR but express normal
EGFR abundantly due to the heterogeneity of tumor cells. Only
relying on CAR-T cells that recognize EGFRvIII alone may not
eliminate tumors. A novel type of CAR-T cells carry a bispecific
T-cell engager (BiTE) that may target both mutant EGFR and
wild-type EGFR, enhancing the binding ability between effector T
cells and tumor cells and significantly attenuating the effect of
EGFRvIII antigen loss on CAR-T cells effectively. Model mice with
glioblastoma received BiTE CAR-T cell infusion and tumor cells were
no longer visible in 80% of the mice. Furthermore, these BiTE CAR-T
cells had no toxicity to human skin grafts in vivo (72). A new type of synNotch CAR-T cells
is able to selectively kill neuroblastoma cells; the synNotch
protein on the surface of T cells is modified to recognize tumor
antigen GD2 and then instructs T cells to recognize the tumor
antigen B7H3, leading to the tumor cells' regression. These CAR-T
cells had high specificity in mouse models and did not exhaust
quickly after killing tumor cells, demonstrating a long-lasting
anti-tumor effect, indicating that this new therapy may help
patients achieve long-term survival (73). The popular targets of CAR-T cells
in solid tumors are summarized in Table II.
Although CAR-T cell therapy has made significant
progress in clinical trials, adverse reactions during the treatment
also affected the efficacy of this treatment. CRS, on-target
off-tumor effects, allergic reactions and neurological toxicity
syndrome are common adverse reactions during the therapy. This may
be due to the insufficient structure of CAR-T cells, differences in
patient constitution and the dose of CAR-T cells used. The side
effects during CAR-T treatment may lead to different responses in
different systems and most of the toxic and side effects are
reversible if patients receive a timely and correct
intervention.
CRS is the most common side effect, including
neutropenia, anemia and thrombocytopenia. CRS may be divided into
four grades according to its severity. The symptoms of first-grade
CRS are mild and do not threaten the patient's life; the patient
will get relief after receiving antipyretics, antiemetics and other
treatments. Symptoms of grade 2 CRS include fever, neutropenia and
elevated creatinine and patients require hospitalization. Symptoms
of grade 3 CRS include hypotension, coagulation disorders and
hypoxia, and affected patients require supportive treatments such
as vasopressors, supplemental oxygen and fibrinogen infusion. Grade
4 CRS may cause hypotension or severe organ toxicity and is
frequently life-threatening; patients require mechanical
ventilation and high-dose vasopressor therapy (74). Certain patients experience
cytopenias after receiving CAR-T cell therapy; neutropenia or
thrombocytopenia is the most common cytopenia and is called
prolonged hematologic toxicity (PHT) if persisting for >30 days.
In a clinical trial of CD19 CAR-T cell treatment of patients with
R/R DLBCL, PHT was reported in 18 of 31 (58%) patients and the
1-year overall survival was significantly reduced in patients with
PHT compared to patients without PHT (75). Red blood cell and platelet
transfusions are recommended for patients with prolonged severe
anemia and thrombocytopenia (76).
There are 2 strategies for CRS, a preventive strategy to reduce the
risk and a remedial strategy in the event of lethal toxicity.
Cytoreductive chemotherapy to reduce the disease burden of patients
prior to CAR-T cell infusion is recommended for the prevention
strategies. The dosage of CAR-T cells should be strictly controlled
in the treatment of patients with a high disease burden, predictive
biomarkers such as IFN-γ, IL-2, IL-2Rα, IL-6, TNF-α, C-reactive
protein should be closely monitored and in high-risk patients,
intervention should be performed early (77). Tocilizumab, an IL-6 receptor
antagonist, is recommended for patients with severe CRS as a
remedial strategy. In a study, the symptoms were rapidly alleviated
in all patients after receiving tocilizumab infusion; however, this
may increase the possibility of infection in certain patients and
corticosteroids may be considered if patients have no response to
tocilizumab (78). In addition,
Koristka et al (79) designed a
technology to incorporate an elimination tag (E-tag) into CAR-T
cells; if there are extra CAR-T cells in the patient, a
single-chain antibody corresponding to E-tag may be infused to kill
E-tagged CAR-T cells while sparing cells lacking the E-tag,
reducing the number of CAR-T cells in patients in a short time. In
most cases, CRS is alleviated after administration of steroids or
tocilizumab treatment. CAR-T cells may also be eliminated
selectively by introducing suicide genes, which induce apoptosis of
CAR-T cells to reduce their toxicity if adverse reactions occur.
The suicide gene encodes herpes simplex virus thymidine kinase,
which may act on DNA polymerase, prevent DNA synthesis and lead to
cell apoptosis, preventing the overactivation of CAR-T cells
(80). The common cell suicide
gene Caspase 9 may also be introduced into CAR-T cells and external
chemicals such as FK506-binding protein may induce Caspase 9
dimerization and initiate the mitochondrial apoptosis pathway,
promoting CAR-T cell apoptosis and improving the safety of CAR-T
cell therapy (81). The occurrence
of CRS is closely related to the infusion dose and the patient's
physical condition. Due to the difference in CAR-T cell culture
methods and patient inclusion criteria in each clinical trial, it
is difficult to set a uniform dose and clinicians should make
corresponding adjustments based on clinical test results, combined
with the tumor type and the patient's tumor burden.
Antigens on tumor cell surfaces may be divided into
tumor-specific antigens and TAAs. TAAs are recognized by CAR-T
cells and these antigens are not unique to tumor cells; when CAR-T
cells get in contact with non-tumor tissue, target antigens cause
‘on-target off-tumor effects’. This adverse reaction affects the
gastrointestinal tract, blood, respiratory and other systems. The
main method to manage the impact of adverse reactions on patients
is to control the amount of CAR-T cells and attenuate the function
of CAR-T cells. However, this also weakens the anti-tumor effect of
CAR-T cells simultaneously. One of the promising approaches is to
find intracellular targets to overcome the off-target effects,
which may overcome the limitations of tumor cell surface targets.
Recognizing intracellular target peptides presented by MHC
molecules on the tumor cell surface by CAR-T cells may be a
promising therapeutic direction (82). Only targeting a single tumor
antigen may increase the risk of targeting normal tissues; a case
report indicated that a patient with colon cancer died after
receiving HER2 CAR-T cell therapy, possibly due to CAR-T cells
immediately localizing to the lung and triggering the release of
cytokines by recognizing ERBB2 on lung epithelial cells (83). Fedorov et al (84) designed antigen-specific inhibitory
CARs derived from PD-1 or CTLA-4 molecules, which may competitively
inhibit the function of CAR-T cells. This molecule is able to
effectively prevent CAR-T cells' toxicity while maintaining their
anti-tumor activity. Designing a CAR-T cell that targets multiple
tumor antigens simultaneously appears to be a feasible way to avoid
the off-target effects. Combining antibodies that may recognize
different tumor antigens in the same CAR-T cell may significantly
enhance the specificity. A co-transduced CAR-T T cell targeting two
prostate tumor antigens, PSMA and prostate stem cell antigen, is
able to destroy tumors that express both antigens but does not
affect tumors expressing either antigen alone; this ‘tumor-sensing’
strategy may help avoid certain side effects of single-targeted
therapy (85). The novel
BCMA-OR-CD38 Tan CAR-T cell that exhibits cytotoxicity against BCMA
or CD38-positive tumor cells exhibited superior cytotoxicity and
proliferation ability in vitro. Importantly, these CAR-T cells
achieved complete tumor clearance in myeloma-bearing mice and no
recurrence was observed (86).
CAR-T cells may cause tumor lysis syndrome (TLS)
once entering the patient's body. Intracellular substances and
metabolites are released into the blood if tumor cells are killed
by CAR-T cells. Kidneys are not able to filter these substances,
ultimately resulting in severe metabolic disorders, the main
clinical manifestation of which include hyperkalemia,
hyperphosphatemia, hypocalcemia, acute renal failure and severe
cardiac arrhythmias that may occur in extreme cases. The incidence
of TLS in hematological malignancies is significantly higher than
that in patients with solid tumors, and metabolically active
cancers, such as B-cell lymphoma, have the highest risk of TLS.
Early identification of patients with a risk of TLS and targeted
prevention may reduce patient mortality (87). Rasburicase is a recombinant uric
acid oxidase that converts uric acid to soluble allantoin; it is
the first-choice therapeutic drug for patients with TLS.
Allopurinol may prevent uric acid formation by inhibiting xanthine
and hypoxanthine oxidase and may also be used in TLS (88). Neurotoxic syndrome is frequently
associated with CAR-T cell therapy, including somnolence, aphasia,
ataxia, disorientation and seizures; the occurrence of seizures may
be related to corticosteroids or IL-1/IL-6 signaling antagonists
applied during treatment (89).
The lentiviral vectors are mainly used to mediate the expression of
chimeric antigen receptors. However, lentiviruses are not safe in
certain cases and lentivirus may cause T lymphoma in mice,
considering the characteristics of lentivirus integration into the
host's chromosomes (90). There is
a specific error probability in the production of CAR-T cells using
the lentiviral packaging system; it has been reported that the TET2
gene is integrated randomly into CAR and this integration endows
CAR-T cells with enhanced proliferation and anti-tumor ability,
indicating that the TET2 gene may help to improve CAR-T cell
immunotherapy (91). A report
described a patient with B-ALL who accepted CAR-T cell therapy, and
during T cell manufacturing, the CAR gene was unintentionally
introduced into a single leukemic B cell and its product was bound
in cis mode to the CD19 epitope on the surface of leukemic cells,
masking it from recognition by CAR-T cells. The patient ultimately
died of complications related to progressive leukemia. Therefore,
finding a safer delivery system for producing CAR-T cells is
necessary for the future (92).
DNA-carrying biodegradable nanoparticles may introduce
leukemia-targeting CAR genes into the nucleus of T cells
effectively compared with the traditional process; these
nanoparticles are effective for transfection of CAR genes, and they
are safer and less expensive (93). Electro-transfection is a
theoretically safer method for CAR-T cell production that does not
promote the transgene integration into the host genome; CAR-T cells
may be generated within 6 days and exhibit potent cytotoxicity
against target cells (94). With
the development of modern medical technology, CAR-T cell therapy
has undergone rigorous clinical trials prior to application and its
safety has been guaranteed. A small number of patients may
experience severe side effects; although most of the side effects
are relatively mild, patients require to receive timely and
effective interventions to alleviate the toxic effects once adverse
effects occur.
Clinical trials have proved that CAR-T cell therapy
is encouraging in the treatment of hematological malignancies.
However, it is still difficult in solid tumors. The problems in the
rapid development of CAR-T cell therapy are both obstacles and
opportunities. Target antigen escape, a tumor-suppressive
microenvironment and adverse reactions during the treatment may
influence the efficacy and safety. Improving the anti-tumor effect
and reducing the incidence of adverse reactions is necessary for
future applications, and a more detailed understanding of CAR-T and
tumor cells is essential. The next generation of CAR-T cells should
solve the problems of expensive treatment, side effects during the
treatment and long preparation time. The application prospect of
CAR-T therapy is broad and it will bring a breakthrough in treating
human tumors and other diseases.
Not applicable.
This research was supported by the Shandong Province Health
Department (grant nos. 2019WS589 and 2017WS407) and the Shandong
Province Traditional Chinese Medicine Science and Technology
Development Plan (grant no. 2017-216).
Not applicable.
DLD conceived and designed the review. LC wrote the
first draft. LC, TX and BW participated in writing the manuscript.
All authors contributed to the article and read and approved the
final version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Cao W, Chen HD, Yu YW, Li N and Chen WQ:
Changing profiles of cancer burden worldwide and in China: A
secondary analysis of the global cancer statistics 2020. Chin Med J
(Engl). 134:783–791. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rafei H, Kantarjian HM and Jabbour EJ:
Recent advances in the treatment of acute lymphoblastic leukemia.
Leuk Lymphoma. 60:2606–2621. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kansagra AJ, Frey NV, Bar M, Laetsch TW,
Carpenter PA, Savani BN, Heslop HE, Bollard CM, Komanduri KV,
Gastineau DA, et al: Clinical utilization of chimeric antigen
receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia
(ALL)-an expert opinion from the European society for blood and
marrow transplantation (EBMT) and the American society for blood
and marrow transplantation (ASBMT). Bone Marrow Transplant.
54:1868–1880. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Larson RC and Maus MV: Recent advances and
discoveries in the mechanisms and functions of CAR T cells. Nat Rev
Cancer. 21:145–161. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bupha-Intr O, Haeusler G, Chee L, Thursky
K, Slavin M and Teh B: CAR-T cell therapy and infection: A review.
Expert Rev Anti Infect Ther. 19:749–758. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giavridis T, van der Stegen SJC, Eyquem J,
Hamieh M, Piersigilli A and Sadelain M: CAR T cell-induced cytokine
release syndrome is mediated by macrophages and abated by IL-1
blockade. Nat Med. 24:731–738. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gross G, Waks T and Eshhar Z: Expression
of immunoglobulin-T-cell receptor chimeric molecules as functional
receptors with antibody-type specificity. Proc Natl Acad Sci USA.
86:10024–10028. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eshhar Z, Waks T, Gross G and Schindler
DG: Specific activation and targeting of cytotoxic lymphocytes
through chimeric single chains consisting of antibody-binding
domains and the gamma or zeta subunits of the immunoglobulin and
T-cell receptors. Proc Natl Acad Sci USA. 90:720–724. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siegler EL and Wang P: Preclinical models
in chimeric antigen receptor-engineered T-cell therapy. Hum Gene
Ther. 29:534–546. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grigor EJM, Fergusson D, Kekre N, Montroy
J, Atkins H, Seftel MD, Daugaard M, Presseau J, Thavorn K, Hutton
B, et al: Risks and benefits of chimeric antigen receptor T-cell
(CAR-T) therapy in cancer: A systematic review and meta-analysis.
Transfus Med Rev. 33:98–110. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roselli E, Frieling JS, Thorner K, Ramello
MC, Lynch CC and Abate-Daga D: CAR-T engineering: Optimizing signal
transduction and effector mechanisms. BioDrugs. 33:647–659. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu W, Huang X, Huang X, Chen W, Hao L and
Chen Z: Chimeric antigen receptor modified T cell (CAR-T)
co-expressed with ICOSL-41BB promote CAR-T proliferation and tumor
rejection. Biomed Pharmacother. 118:1093332019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramello MC, Benzaïd I, Kuenzi BM,
Lienlaf-Moreno M, Kandell WM, Santiago DN, Pabón-Saldaña M,
Darville L, Fang B, Rix U, et al: An immunoproteomic approach to
characterize the CAR interactome and signalosome. Sci Signal.
12:eaap97772019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abramson JS: Anti-CD19 CAR T-cell therapy
for B-cell non-hodgkin lymphoma. Transfus Med Rev. 34:29–33. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chmielewski M and Abken H: TRUCKs: The
fourth generation of CARs. Expert Opin Biol Ther. 15:1145–1154.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kagoya Y, Tanaka S, Guo T, Anczurowski M,
Wang CH, Saso K, Butler MO, Minden MD and Hirano N: A novel
chimeric antigen receptor containing a JAK-STAT signaling domain
mediates superior antitumor effects. Nat Med. 24:352–359. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin H, Cheng J, Mu W, Zhou J and Zhu L:
Advances in universal CAR-T cell therapy. Front Immunol.
12:7448232021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao J, Lin Q, Song Y and Liu D: Universal
CARs, universal T cells, and universal CAR T cells. J Hematol
Oncol. 11:1322018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qasim W, Zhan H, Samarasinghe S, Adams S,
Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, et
al: Molecular remission of infant B-ALL after infusion of universal
TALEN gene-edited CAR T cells. Sci Transl Med. 9:eaaj20132017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cho JH, Collins JJ and Wong WW: Universal
chimeric antigen receptors for multiplexed and logical control of T
cell responses. Cell. 173:1426–1438.e11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo Y, Xu B, Wu Z, Bo J, Tong C, Chen D,
Wang J, Wang H, Wang Y and Han W: Mutant B2M-HLA-E and B2M-HLA-G
fusion proteins protects universal chimeric antigen
receptor-modified T cells from allogeneic NK cell-mediated lysis.
Eur J Immunol. 51:2513–2521. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Depil S, Duchateau P, Grupp SA, Mufti G
and Poirot L: ‘Off-the-shelf’ allogeneic CAR T cells: Development
and challenges. Nat Rev Drug Discov. 19:185–199. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martino M, Alati C, Canale FA, Musuraca G,
Martinelli G and Cerchione C: A review of clinical outcomes of CAR
T-cell therapies for B-acute lymphoblastic leukemia. Int J Mol Sci.
22:21502021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wudhikarn K, Flynn JR, Rivière I, Gönen M,
Wang X, Senechal B, Curran KJ, Roshal M, Maslak PG, Geyer MB, et
al: Interventions and outcomes of adult patients with B-ALL
progressing after CD19 chimeric antigen receptor T-cell therapy.
Blood. 138:531–543. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nix MA, Mandal K, Geng H, Paranjape N, Lin
YT, Rivera JM, Marcoulis M, White KL, Whitman JD, Bapat SP, et al:
Surface proteomics reveals CD72 as a target for in vitro-evolved
nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALL. Cancer
Discov. 11:2032–2049. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D,
Han X, Liu Y, Zhang W, Wang C, et al: Bispecific CAR-T cells
targeting both CD19 and CD22 for therapy of adults with relapsed or
refractory B cell acute lymphoblastic leukemia. J Hematol Oncol.
13:302020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brentjens RJ, Davila ML, Riviere I, Park
J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska
M, et al: CD19-targeted T cells rapidly induce molecular remissions
in adults with chemotherapy-refractory acute lymphoblastic
leukemia. Sci Transl Med. 5:177ra382013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maude SL, Frey N, Shaw PA, Aplenc R,
Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et
al: Chimeric antigen receptor T cells for sustained remissions in
leukemia. N Engl J Med. 371:1507–1517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davila ML, Riviere I, Wang X, Bartido S,
Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska
M, et al: Efficacy and toxicity management of 19-28z CAR T cell
therapy in B cell acute lymphoblastic leukemia. Sci Transl Med.
6:224ra252014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L,
Wei G, Han L, Wang H, Yu S, et al: CRISPR/Cas9-engineered universal
CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory
B-cell acute lymphoblastic leukemia. Clin Cancer Res. 27:2764–2772.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Raetz EA and Teachey DT: T-cell acute
lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program.
2016:580–588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan J, Tan Y, Wang G, Deng B, Ling Z, Song
W, Seery S, Zhang Y, Peng S, Xu J, et al: Donor-derived CD7
chimeric antigen receptor T cells for T-cell acute lymphoblastic
leukemia: First-in-human, phase I trial. J Clin Oncol.
39:3340–3351. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu P, Liu Y, Yang J, Zhang X, Yang X, Wang
H, Wang L, Wang Q, Jin D, Li J and Huang X: Naturally selected CD7
CAR-T therapy without genetic manipulations for T-ALL/LBL:
First-in-human phase 1 clinical trial. Blood. 140:321–334.
2022.PubMed/NCBI
|
|
34
|
Dai HP, Cui W, Cui QY, Zhu WJ, Meng HM,
Zhu MQ, Zhu XM, Yang L, Wu DP and Tang XW: Haploidentical CD7 CAR
T-cells induced remission in a patient with TP53 mutated relapsed
and refractory early T-cell precursor lymphoblastic
leukemia/lymphoma. Biomark Res. 10:62022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xie L, Ma L, Liu S, Chang L and Wen F:
Chimeric antigen receptor T cells targeting CD7 in a child with
high-risk T-cell acute lymphoblastic leukemia. Int Immunopharmacol.
96:1077312021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen D, You F, Xiang S, Wang Y, Li Y, Meng
H, An G, Zhang T, Li Z, Jiang L, et al: Chimeric antigen receptor T
cells derived from CD7 nanobody exhibit robust antitumor potential
against CD7-positive malignancies. Am J Cancer Res. 11:5263–5281.
2021.PubMed/NCBI
|
|
37
|
Png YT, Vinanica N, Kamiya T, Shimasaki N,
Coustan-Smith E and Campana D: Blockade of CD7 expression in T
cells for effective chimeric antigen receptor targeting of T-cell
malignancies. Blood Adv. 1:2348–2360. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Neelapu SS, Locke FL, Bartlett NL, Lekakis
LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T,
Lin Y, et al: Axicabtagene ciloleucel CAR T-cell therapy in
refractory large B-cell lymphoma. N Engl J Med. 377:2531–2544.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chong EA, Ruella M and Schuster SJ;
Lymphoma Program Investigators at the University of Pennsylvania, :
Five-year outcomes for refractory B-cell lymphomas with CAR T-cell
therapy. N Engl J Med. 384:673–674. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ramos CA, Rouce R, Robertson CS, Reyna A,
Narala N, Vyas G, Mehta B, Zhang H, Dakhova O, Carrum G, et al: In
vivo fate and activity of second-versus third-generation
CD19-specific CAR-T cells in B cell non-Hodgkin's lymphomas. Mol
Ther. 26:2727–2737. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bunse M, Pfeilschifter J, Bluhm J,
Zschummel M, Joedicke JJ, Wirges A, Stark H, Kretschmer V,
Chmielewski M, Uckert W, et al: CXCR5 CAR-T cells simultaneously
target B cell non-Hodgkin's lymphoma and tumor-supportive
follicular T helper cells. Nat Commun. 12:2402021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yin Z, Zhang Y and Wang X: Advances in
chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin
lymphoma. Biomark Res. 9:582021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mihăilă RG: Chimeric antigen
receptor-engineered T-cells-a new way and era for lymphoma
treatment. Recent Pat Anticancer Drug Discov. 14:312–323. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shah NN, Maatman T, Hari P and Johnson B:
Multi targeted CAR-T cell therapies for B-cell malignancies. Front
Oncol. 9:1462019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yu B, Jiang T and Liu D: BCMA-targeted
immunotherapy for multiple myeloma. J Hematol Oncol. 13:1252020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ali SA, Shi V, Maric I, Wang M, Stroncek
DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG,
et al: T cells expressing an anti-B-cell maturation antigen
chimeric antigen receptor cause remissions of multiple myeloma.
Blood. 128:1688–1700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ferment B and Arnulf B: CAR-T cells
immunotherapy in multiple myeloma: Present and future. Bull Cancer.
108 (10 Suppl):S65–S72. 2021.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu
YF, Xu J, Zhuang Y, Zhang W, Weng XQ, et al: Exploratory trial of a
biepitopic CAR T-targeting B cell maturation antigen in
relapsed/refractory multiple myeloma. Proc Natl Acad Sci USA.
116:9543–9551. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zah E, Nam E, Bhuvan V, Tran U, Ji BY,
Gosliner SB, Wang X, Brown CE and Chen YY: Systematically optimized
BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous
multiple myeloma. Nat Commun. 11:22832020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Drent E, Poels R, Ruiter R, van de Donk
NWCJ, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM,
Groen RWJ, et al: Combined CD28 and 4-1BB costimulation potentiates
affinity-tuned chimeric antigen receptor-engineered T cells. Clin
Cancer Res. 25:4014–4025. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Springuel L, Lonez C, Alexandre B, Van
Cutsem E, Machiels JH, Van Den Eynde M, Prenen H, Hendlisz A, Shaza
L, Carrasco J, et al: Chimeric antigen receptor-T cells for
targeting solid tumors: Current challenges and existing strategies.
Biodrugs. 33:515–537. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jiang X, Xu J, Liu M, Xing H, Wang Z,
Huang L, Mellor AL, Wang W and Wu S: Adoptive CD8+ T
cell therapy against cancer: Challenges and opportunities. Cancer
Lett. 462:23–32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Irving M, Vuillefroy de Silly R, Scholten
K, Dilek N and Coukos G: Engineering chimeric antigen receptor
T-cells for racing in solid tumors: Don't forget the fuel. Front
Immunol. 8:2672017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Junttila MR and de Sauvage FJ: Influence
of tumour micro-environment heterogeneity on therapeutic response.
Nature. 501:346–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hajari Taheri F, Hassani M, Sharifzadeh Z,
Behdani M, Arashkia A and Abolhassani M: T cell engineered with a
novel nanobody-based chimeric antigen receptor against VEGFR2 as a
candidate for tumor immunotherapy. IUBMB Life. 71:1259–1267. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Akbari P, Huijbers EJM, Themeli M,
Griffioen AW and van Beijnum JR: The tumor vasculature an
attractive CAR T cell target in solid tumors. Angiogenesis.
22:473–475. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Balakrishnan A, Rajan A, Salter AI,
Kosasih PL, Wu Q, Voutsinas J, Jensen MC, Plückthun A and Riddell
SR: Multispecific targeting with synthetic ankyrin repeat motif
chimeric antigen receptors. Clin Cancer Res. 25:7506–7516. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rodriguez-Garcia A, Lynn RC, Poussin M,
Eiva MA, Shaw LC, O'Connor RS, Minutolo NG, Casado-Medrano V, Lopez
G, Matsuyama T and Powell DJ: J: CAR-T cell-mediated depletion of
immunosuppressive tumor-associated macrophages promotes endogenous
antitumor immunity and augments adoptive immunotherapy. Nat Commun.
12:8772021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang H, Li F, Cao J, Wang X, Cheng H, Qi
K, Wang G, Xu K, Zheng J, Fu YX and Yang X: A chimeric antigen
receptor with antigen-independent OX40 signaling mediates potent
antitumor activity. Sci Transl Med. 13:eaba73082021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Santoro SP, Kim S, Motz GT, Alatzoglou D,
Li C, Irving M, Powell DJ Jr and Coukos G: T cells bearing a
chimeric antigen receptor against prostate-specific membrane
antigen mediate vascular disruption and result in tumor regression.
Cancer Immunol Res. 3:68–84. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Byrd TT, Fousek K, Pignata A, Szot C,
Samaha H, Seaman S, Dobrolecki L, Salsman VS, Oo HZ, Bielamowicz K,
et al: TEM8/ANTXR1-specific CAR T cells as a targeted therapy for
triple-negative breast cancer. Cancer Res. 78:489–500. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nishida T and Kataoka H: Glypican
3-targeted therapy in hepatocellular carcinoma. Cancers (Basel).
11:13392019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang P, Qin W, Liu T, Jiang D, Cui L, Liu
X, Fang Y, Tang X, Jin H and Qian Q: PiggyBac-engineered T cells
expressing a glypican-3-specific chimeric antigen receptor show
potent activities against hepatocellular carcinoma. Immunobiology.
225:1518502020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sun B, Yang D, Dai H, Liu X, Jia R, Cui X,
Li W, Cai C, Xu J and Zhao X: Eradication of hepatocellular
carcinoma by NKG2D-based CAR-T cells. Cancer Immunol Res.
7:1813–1823. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yazdanifar M, Zhou R, Grover P, Williams
C, Bose M, Moore LJ, Wu ST, Maher J, Dreau D and Mukherjee AP:
Overcoming immunological resistance enhances the efficacy of a
novel anti-tMUC1-CAR T cell treatment against pancreatic ductal
adenocarcinoma. Cells. 8:10702019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Cui J, Zhang Q, Song Q, Wang H, Dmitriev
P, Sun MY, Cao X, Wang Y, Guo L, Indig IH, et al: Targeting hypoxia
downstream signaling protein, CAIX, for CAR T-cell therapy against
glioblastoma. Neuro Oncol. 21:1436–1446. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mao Y, Fan W, Hu H, Zhang L, Michel J, Wu
Y, Wang J, Jia L, Tang X, Xu L, et al: MAGE-A1 in lung
adenocarcinoma as a promising target of chimeric antigen receptor T
cells. J Hematol Oncol. 12:1062019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang C, Wang Z, Yang Z, Wang M, Li S, Li
Y, Zhang R, Xiong Z, Wei Z, Shen J, et al: Phase I escalating-dose
trial of CAR-T therapy targeting CEA+ metastatic
colorectal cancers. Mol Ther. 25:1248–1258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Abbott RC, Verdon DJ, Gracey FM,
Hughes-Parry HE, Iliopoulos M, Watson KA, Mulazzani M, Luong K,
D'Arcy C, Sullivan LC, et al: Novel high-affinity EGFRvIII-specific
chimeric antigen receptor T cells effectively eliminate human
glioblastoma. Clin Transl Immunology. 10:e12832021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
O'Rourke DM, Nasrallah MP, Desai A,
Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem
S, Maloney E, Shen A, et al: A single dose of peripherally infused
EGFRvIII-directed CAR T cells mediates antigen loss and induces
adaptive resistance in patients with recurrent glioblastoma. Sci
Transl Med. 9:eaaa09842017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Choi BD, Yu X, Castano AP, Bouffard AA,
Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick
MB, et al: CAR-T cells secreting BiTEs circumvent antigen escape
without detectable toxicity. Nat Biotechnol. 37:1049–1058. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Moghimi B, Muthugounder S, Jambon S,
Tibbetts R, Hung L, Bassiri H, Hogarty MD, Barrett DM, Shimada H
and Asgharzadeh S: Preclinical assessment of the efficacy and
specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma.
Nat Commun. 12:5112021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Porter D, Frey N, Wood PA, Weng Y and
Grupp SA: Grading of cytokine release syndrome associated with the
CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 11:352018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nagle SJ, Murphree C, Raess PW, Schachter
L, Chen A, Hayes-Lattin B, Nemecek E and Maziarz RT: Prolonged
hematologic toxicity following treatment with chimeric antigen
receptor T cells in patients with hematologic malignancies. Am J
Hematol. 96:455–461. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Schubert ML, Schmitt M, Wang L, Ramos CA,
Jordan K, Müller-Tidow C and Dreger P: Side-effect management of
chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol.
32:34–48. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang Z and Han W: Biomarkers of cytokine
release syndrome and neurotoxicity related to CAR-T cell therapy.
Biomark Res. 6:42018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Szenes V and Curran KJ: Utilization of CAR
T cell therapy in pediatric patients. Semin Oncol Nurs.
35:1509292019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Koristka S, Ziller-Walter P, Bergmann R,
Arndt C, Feldmann A, Kegler A, Cartellieri M, Ehninger A, Ehninger
G, Bornhäuser M and Bachmann MP: Anti-CAR-engineered T cells for
epitope-based elimination of autologous CAR T cells. Cancer Immunol
Immunother. 68:1401–1415. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bonini C, Ferrari G, Verzeletti S, Servida
P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F,
Traversari C and Bordignon C: HSV-TK gene transfer into donor
lymphocytes for control of allogeneic graft-versus-leukemia.
Science. 276:1719–1724. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Straathof KC, Pulè MA, Yotnda P, Dotti G,
Vanin EF, Brenner MK, Heslop HE, Spencer DM and Rooney CM: An
inducible caspase 9 safety switch for T-cell therapy. Blood.
105:4247–4254. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gerber HP, Sibener LV, Lee LJ and Gee M:
Intracellular targets as source for cleaner targets for the
treatment of solid tumors. Biochem Pharmacol. 168:275–284. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Morgan RA, Yang JC, Kitano M, Dudley ME,
Laurencot CM and Rosenberg SA: Case report of a serious adverse
event following the administration of T cells transduced with a
chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Fedorov VD, Themeli M and Sadelain M:
PD-1- and CTLA-4-based inhibitory chimeric antigen receptors
(iCARs) divert off-target immunotherapy responses. Sci Transl Med.
5:215ra1722013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kloss CC, Condomines M, Cartellieri M,
Bachmann M and Sadelain M: Combinatorial antigen recognition with
balanced signaling promotes selective tumor eradication by
engineered T cells. Nat Biotechnol. 31:71–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Feng Y, Liu X, Li X, Zhou Y, Song Z, Zhang
J, Shi B and Wang J: Novel BCMA-OR-CD38 tandem-dual chimeric
antigen receptor T cells robustly control multiple myeloma.
Oncoimmunology. 10:19591022021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wilson FP and Berns JS: Tumor lysis
syndrome: New challenges and recent advances. Adv Chronic Kidney
Dis. 21:18–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Miao L, Zhang Z, Ren Z and Li Y: Reactions
related to CAR-T cell therapy. Front Immunol. 12:6632012021.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gust J, Ponce R, Liles WC, Garden GA and
Turtle CJ: Cytokines in CAR T cell-associated neurotoxicity. Front
Immunol. 11:5770272020. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Newrzela S, Cornils K, Li Z, Baum C,
Brugman MH, Hartmann M, Meyer J, Hartmann S, Hansmann ML, Fehse B
and von Laer D: Resistance of mature T cells to oncogene
transformation. Blood. 112:2278–2286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fraietta JA, Nobles CL, Sammons MA, Lundh
S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, Denizio JE,
Reddy S, et al: Disruption of TET2 promotes the therapeutic
efficacy of CD19-targeted T cells. Nature. 558:307–312. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ruella M, Xu J, Barrett DM, Fraietta JA,
Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR,
Kulikovskaya I, et al: Induction of resistance to chimeric antigen
receptor T cell therapy by transduction of a single leukemic B
cell. Nat Med. 24:1499–1503. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Smith TT, Stephan SB, Moffett HF, McKnight
LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS and
Stephan MT: In situ programming of leukaemia-specific T cells using
synthetic DNA nanocarriers. Nat Nanotechnol. 12:813–820. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang Z, Qiu S, Zhang X and Chen W:
Optimized DNA electroporation for primary human T cell engineering.
Bmc Biotechnol. 18:42018. View Article : Google Scholar : PubMed/NCBI
|