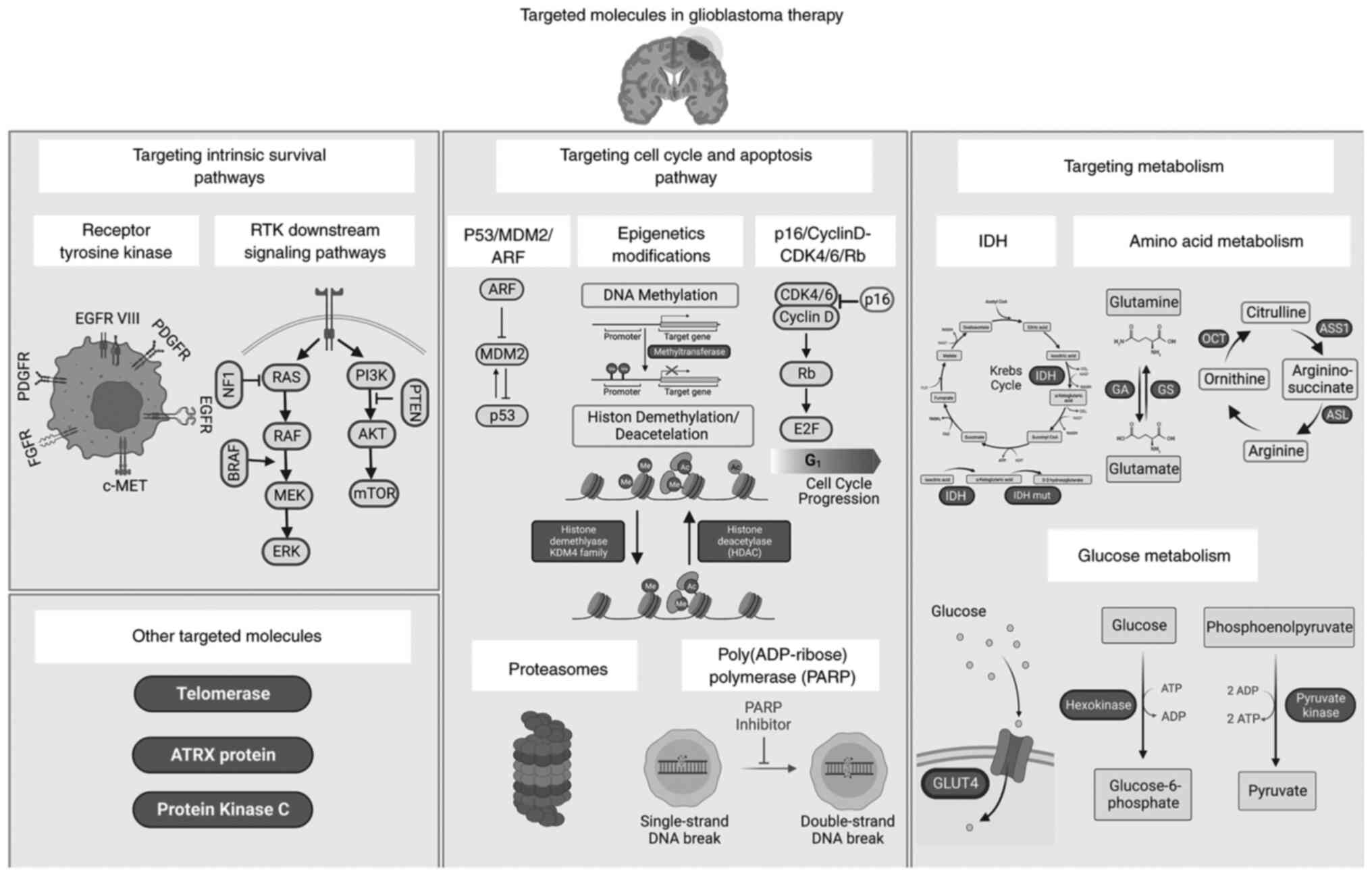
Glioblastoma (GBM) is the most aggressive and deadly
form of malignant brain cancers. It accounts for ~80% of all
primary brain gliomas and ~60% of all adult brain tumors (1). Currently, surgical resection of the
tumor, followed by radiotherapy and temozolomide, is the typical
treatment used for GBM (2). GBM is
associated with poor survival rate, despite the enhancements in
diagnosis tools, surgical techniques and therapeutic approaches;
the median survival rate is ~14–20 months and fewer than 5% of the
patients survive 5 years post-treatment (3). Moreover, the survival rates for
patients with GBMs have not shown statistically significant
improvements in the last three decades (4). Thus, there is a need of new
therapeutic approaches that inhibit GBM growth, impair its
migration and invasion ability and sensitize it to therapy.
The use of high throughput technology in the last
decade allowed researchers to classify GBM according to their
genomic signature. Based on The Cancer Genome Atlas analysis, four
different GBM sub-classes are defined: i) The neural subtype that
represents 16% of GBM and are characterized by the expression of
neuron markers such as NEFL, GABRA1, SYT1, and SLC1A5, ii) the
pro-neural subtype that shows an alteration of PGFRA, a point
mutation in IDH1and TP3 and an overexpression in development genes
such as NKX2-2, OLIG2, iii) the mesenchymal subtype that is
distinguished with alterations in neurofibromatosis type 1 (NF1),
phosphatase and tensin homolog (PTEN) point mutation and expression
of mesenchymal genes such as MET, CD44, iv) the classical subtype
that displays EGFR amplification, CDKN2 A deletion, and p53
mutations (5). Subsequently,
researchers have detected several molecular and genomic alterations
in core pathways that regulate GBM cell viability, growth,
metastasis and invasion. It is thus important to target these
altered molecules in order to inhibit GBM proliferation and
progression. The present review highlights current understanding of
the molecular alterations commonly involved in GBM (Fig. 1). Thorough understanding of
molecular alterations facilitates the development of current
therapeutic targeted therapy, and opens the way to novel
therapeutic approaches. The present review also highlight molecular
elements that are currently targeted in GBM clinical trials and
others that represent promising potential new targets.
RTKs are transmembrane proteins that consist of an
extracellular ligand-binding domain, a single transmembrane helix,
and an intracellular catalytic domain (6). RTK superfamily includes epidermal
growth factor receptor (EGFR), platelet-derived growth factors
(PDGPR), hepatocyte growth factor receptor (c-MET), and fibroblast
growth factor receptor (FGFR) (7).
Under normal physiological conditions, RTKs are involved in
maintaining cellular homeostasis by regulating cell-cell
communication, cell survival, migration, proliferation,
differentiation, metabolism, and cell cycle. Hence, dysregulation
of the RTK pathway is thought to play an important role in GBM
initiation, development, and progression (8,9).
Genomic analysis revealed that 57% of GBM cells
harbor EGFR genetic alterations (10). EGFR amplification and
overexpression were identified in 40 and 60% of primary
glioblastoma respectively. Other types of genetic alterations were
also detected; these include EGFR rearrangement, point mutations,
and deletions such as the deletion of exons 2–7 which leads to the
truncated mutant variant III (EGFRvIII) (11). EGFR amplification and
overexpression lead to constitutive activation of the receptor and
enhance GBM cell proliferation, survival, invasion and resistance
to treatments (12–15). Talasila et al (16) demonstrated that cells from patients
with GBM and with EGFR gene amplification are able to invade the
tumor microenvironment (TME) in an angiogenesis independent manner.
Additionally, EGFRvIII mutations lacking an extra cellular domain,
tend to maintain the EGFR signaling pathway constitutively active
in a ligand independent manner. These mutations are detected in 25%
of GBM cases and promote survival, tumor growth, migration,
invasion and angiogenesis (17–21).
Despite the lack of an extracellular domain, EGFRvIII mutation
maintains the EGFR signaling It is important to note that 50–60% of
GBMs overexpressing wild-type EGFR also express EGFRvIII (22,23).
GFRvIII is therefore a potential therapeutic target for GBM.
The receptor tyrosine kinase PDGFR is the second
therapeutic target in GBM proneural subtype. PDGFR gene
amplification is found in 15% of GBM cases (10). Overexpression of PDGFR and its
ligands (PDGF-AA/-AB/-BB/-CC/-DD) is observed in gliomas of all
grades and associated with poor prognosis (10,29).
Once activated, PDGFR triggers intracellular signaling cascades
that regulate cancer cell survival, growth and progression
(30,31). Therefore, dysregulation of PDGF
signaling stimulates malignant transformation of normal neural stem
cells into glioblastoma and enhances GBM cell growth and motility
through autocrine signaling (32–34).
In clinical trials, PDGFR is either targeted by
multikinase inhibitors or specific anti-PDGFR antibodies (Table I). To date, multikinase inhibitors
such as Sunitinib, Imatinib and Dasatinib have not shown promising
clinical benefits (24). In
addition, two Phase II clinical trials assessed the tolerance and
efficacy of Ramucirumab (IMC-3G3) and MEDI-575 anti-PDGFR
antibodies (NCT00895180 and NCT01268566 respectively) in patients
with recurrent glioblastoma multiforme. However, these
monotherapies did not show improved survival.
MET is a transmembrane receptor with a tyrosine
kinase activity. Different types of MET genetic alterations are
detected in GBM cells. MET gene amplification and overexpression
are detected in 5–13% of GBM cases (35), and MET fusion genes are found in
pediatric GBM (36).
Overexpression of MET and its ligand HGF promotes tumor growth,
migration, invasion and drug resistance (37–40).
Increased activation of MET/HGF pathway is strongly and selectively
associated with highly anaplastic GBM cells. The c-MET pathway is
either targeted directly by c-MET antibodies or inhibitors or
through antibodies targeting its ligand, HGF (Table I). Treatment with MET inhibitors
can potentially be viable from the standpoint of drug selectivity,
thus avoiding toxicity to normal cells (41). However, and in order to avoid drug
resistance, combination of both PI3K inhibitors along with MET
inhibitors can be favorably considered in the upcoming trials to
efficiently target patients with GBMs (42). On the other hand, one promising
result for c-MET antibodies was shown by a randomized,
double-blind, placebo-controlled, multicenter Phase II study
(NCT01632228) assessing Onartuzumab (MetMAb, an anti-cMET antibody)
with or without Bevacizumab. Survival benefits were reported in
patients with high HGF expression (43).
Ras/Raf/MEK/Erk (also known as the Ras/MAPK pathway)
is a chain of effectors downstream of RTKs that regulate cell
survival and proliferation (46).
Alterations in various components of this pathway are detected in
GBM.
BRAF is a serine/threonine kinase that belongs to
the RAF family. Multiple BRAF gene alterations are associated with
GBM; however, the BRAF V600E mutation is the most relevant. This
missense mutation leads to constitutive activation of
Ras/Raf/MEK/Erk pathway, promoting tumor cell proliferation,
survival and inhibit apoptosis (47).
The NF1 gene encodes neurofibromin, a GTPase
activating protein that controls cell growth and survival. It
regulates the conversion of active GTP-bound Ras to its inactive
GDP-bound form, thus inhibiting Ras/Raf/MEK/Erk signaling pathway
(48). NF1 mutation or deletion is
found in 10% of glioblastoma cases, especially in the mesenchymal
GBM subtype (10). Despite the
fact that genomic alterations of this tumor suppressor gene enhance
the epithelial-mesenchymal transition in neurofibromatosis and can
induce malignant transformation (49), its role in glioblastoma is not
fully understood.
In glioblastoma, MEK inhibitors are common drugs
used to target Ras/Raf/MEK/Erk signaling pathway (Table II). For instance, Atorvastatin, a
Ras/MAPK inhibitor, showed encouraging results when evaluated in
combination with radiotherapy and TMZ in patients with GBMs (Phase
II NCT02029573) (50).
Additionally, BRAF inhibitors such as Dabrafenib and Encorafenib
are evaluated in clinical trials in combination with MEK inhibitors
trametinib (Phase II NCT03919071) and Binimetinib, respectively
(Phase II NCT03973918).
Phosphatidylinositol-3-kinase (PI3K) is a family of
lipid kinases that regulates cell survival, growth, motility and
metabolism (51). It consists of
three subclasses depending on their structure and substrate
specificities (51). Activated
PI3Ks phosphorylate the lipid phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate
phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (51). PI3K signaling pathway dysregulation
is predominant in glioblastoma and is mainly caused by
gain-of-function mutations in PIK3CA gene, PIK3R1
gene and loss of PTEN gene.
PTEN mediates the conversion of PIP3 to PIP2 which
regulates cell proliferation, apoptosis, metabolism, motility and
angiogenesis (52). PTEN deletion
or mutation, found in 5–40% of GBM (53), inhibit AKT (protein kinase B)
phosphorylation and induce the hyperactivation of PI3K signaling
pathway, which in turn accelerates tumor growth, progression, and
metastasis (54).
Heterodimeric Class IA PI3Ks consist of a catalytic
subunit (p110α, p110β, or p110) and a p85-type regulatory subunit
(55). PIK3CA and PIK3R genes
encode for p110α and p85α respectively (55,56).
Several studies show that mutations in PIK3R1, identified in 8–10%
of GBM cases, are found to be mutually exclusive with mutations in
PIK3CA in primary GBM cases (57,58).
Constitutively active PIK3CA and PIK3R1 upregulate the PI3K/AKT
signaling pathway and promote tumorigenesis (59,60).
Somatic mutations in PIK3CA occurs in 6–17% of GBM (57,58,61)
and PIK3CA activating mutations are correlated with poor prognosis,
aggressive and more disseminated phenotype and with shorter
survival rate (62). Alterations
in PIK3CD (p110δ) and PIK3CB (p110β) are also detected in GBM
(56).
The PI3K pathway is generally targeted by several
PI3K pan-inhibitors in clinical trials (Table III). One example is Pictilisib, a
PI3K isoform inhibitor shown to sensitize tumors to radio- and
chemotherapy. Effectiveness of Pictilisib is being compared with
immunotherapeutic Pembrolizumab (MK-3475) in Phase II clinical
trial in patients with glioblastoma (NCT02430363). Paxalisib
(GDC-0084) is another small molecule inhibitor of PI3K currently
evaluated as an adjuvant therapy after surgical resection and
concomitant chemoradiation therapy with TMZ in patients with GBM
and unmethylated MGMT promoter status (Phase II NCT03522298).
Preliminary results show enhanced overall survival (OS; 17.7 months
for treated group compared with 12.7 months for patients treated
with TMZ) (24). The
PI3K/AKT/mammalian target of rapamycin (mTOR) pathway is also
targeted by mTOR inhibitors. While several mTOR inhibitors do not
show clinical benefits, AZD2014 is proposed as a promising drug for
radiosensitizing glioblastoma stem cells (GSCs) in vitro and
in vivo, and is currently being evaluated in clinical trials
(Phase I NCT02619864). Moreover, another clinical trial is
evaluating the efficiency of ABI-009 (Nab-Rapamycin), a novel
albumin-bound mTOR inhibitor, in recurrent and newly diagnosed
glioblastoma (Phase II NCT03463265).
The p53/MDM2/ARF pathway is one of the major core
pathways deregulated in 84% of patients with GBMs. p53, the key
protein of this pathway and also known as ‘guardian of the genome’,
protects cells from external or internal stress signals by
regulating cell processes such as cell cycle, DNA repair,
angiogenesis, metabolism, cell death (apoptosis and autophagy) and
senescence (63).
p53 genetic alterations were detected in 30% of
primary GBM and 65% of secondary GBM (64). Mainly two different types of
genomic alteration are detected in GBM cells; point mutation
associated with overexpression of the mutant version of p53 and
deletion that provokes the loss of function of p53. These
alterations result in either gain or loss of p53 function (65). The oncogenic potential of p53 is
mediated by the accumulation of p53 mutants in cells. Indeed,
mutations in the MDM2 binding domain of p53 affect its regulation
by MDM2, leading to the accumulation of p53 inside the cell and the
subsequent gain of function phenotype. This enhances tumorigeneses
in GBM by promoting inflammation, genomic instability, tumor
growth, invasion, metastasis and neo-angiogenesis (66,67).
Pedrote et al (68)
revealed that accumulation of amyloid-like p53 with p53 gain of
function phenotype leads to increased chemo-resistant GBM cells.
Thus, p53 gain of function mutations are associated with great
oncogenic potential and aggressive GBM phenotype.
Loss of p53 tumor suppression function is not only
mediated by p53 mutations. Previous studies demonstrated that
different components in the p53/MDM2/ARF pathway, such as MDM2/MDM4
protein, negatively regulate the activity of p53 (69–72).
Under normal physiological conditions, MDM2 protein, which is an E3
ubiquitin ligase, binds p53 transcriptional domain and controls its
activity by preventing its transcriptional function and promoting
its degradation. However, the activity of p53 is positively
regulated by the alternative reading frame tumor suppressor (ARF),
an upstream molecule that suppresses the MDM2 activity (73). p53 mutations or deletions, MDM2
amplification or overexpression and ARF homozygous deletion
inactivate p53 and trigger the p53 loss of function phenotype in
glioblastoma, thus contributing to tumor growth, progression and
therapy resistance (65,74).
RG7388 (Idasanutlin) is a small molecule antagonist
of MDM2 used to target p53/MDM2 pathway. Recent Phase I/II trials
are recruiting to test for molecularly matched targeted therapies
(APG101, Alectinib, Idasanutlin, Atezolizumab, Vismodegib,
Temsirolimus and Palbociclib) in combination with radiotherapy in
patients with newly diagnosed glioblastoma without MGMT promoter
methylation (NCT03158389). A more recent Phase I clinical trial is
studying the uptake and tolerance of BI 907828 (an MDM2 inhibitor)
in combination with radiotherapy in newly diagnosed glioblastoma
patients (NCT05376800). Markedly, several different strategies are
currently employed for targeting p53 using gene therapy in
different cancer types. In GBM, only one clinical trial reported
using SGT-53, a human wild type p53 DNA sequence encapsulated in
nanodelivery liposome. However, this study was terminated due to
the very small number of participants (Phase II NCT02340156).
In addition to genetic alterations, epigenetic
modulators affect expression by interacting with drivers of GBM
cell proliferation, without causing any changes in the DNA sequence
(75). In GBM, DNA methylation is
strongly correlated with responses to TMZ treatment that methylates
adenine in position N3 and guanine in position O6 and N7 (76). Guanine methylation at O6 position
leads to strand breaks, activating p53-mediated apoptosis through
Fas/CD95/Apo-1 receptor or by the mitochondrial pathway (77). To decrease the effects of
O6-methylguanine DNA methyltransferase (MGMT)
methylation, synthetic inhibitors of MGMT entered human trials
(78). Nevertheless, several
studies revealed that inhibitors such as O6-benzylguanine and
PaTrim-2 (Lomeguatrib) did not show survival improvement in
response to TMZ (79–81).
Histone demethylases (KDM)4C is often overexpressed
in GBM and is associated with epigenetic regulation of both tumor
suppressor genes and oncogenes (82). Lee et al (82) showed that KDM4C binds to the
promoter of c-Myc oncogene inducing its expression/activation and
suppressing the functions of p53 by demethylating p53K372me1, thus
inducing apoptosis. KDM4C knockdown significantly suppresses both
the proliferation and tumorigenesis of glioblastoma cells in
vitro and in vivo, thus suggesting KDM4C inhibition as a
promising tool in targeting glioblastoma.
Histone deacetylases (HDAC) also serve an important
role in regulating cell growth and survival of cancer cells
(83). Inhibition of HDACs leads
to cell cycle arrest as well as apoptosis, and, in GBM, causes the
rebalance of histones acetylation (84,85).
In clinical trials, testing Romidepsin (FR901228, an HDAC
inhibitor) exhibited failure in treating patients with GBMs
(NCT00085540) (24). Other HDAC
inhibitors, such as Vorinostat. did not show improvement in the
median OS or PFS both alone or in combination with Bortezomib
(NCT00641706) or Bevacuzimab (NCT01738646) (24).
Proteasomes are protein complexes that are central
for degradation of unneeded or damaged proteins (90). They regulate cell cycle and
homeostasis in normal and cancer cells by regulating p53 and
endoplasmic reticulum stress. Proteasomes can thus influence drug
resistance in tumor cells (91).
Due to the complexity of GBM physiology, there is a need for
therapeutic options that could target broad systems contributing to
the tumorgenicity in GBM. Proteasome inhibition is thus identified
as a promising strategy for GBM treatments. Currently, four
proteasome inhibitors are tested in clinical trials to target
proteasomes in GBM: Bortezomib, Ixazomib, Marizomib and Disulfiram
(Table IV). Bortezomib is the
first-generation proteasome inhibitor and showed promising results
when studied in combination with TMZ and radiotherapy (NCT00998010)
(92). Marizomib is a
second-generation proteasome inhibitor that has the ability to
cross the BBB (93). It is
currently being evaluated in ongoing clinical trials combined with
Bevacizumab, TMZ, or ABI-009 (Nab-rapamycin, nanoparticle
albumin-bound rapamycin). Encouraging observations are reported in
Phase III study (NCT03345095) for Marizomib administered in
combination with radiotherapy and TMZ to treat patients with newly
diagnosed glioblastoma (94).
Ixazomib, on the other hand, was evaluated in early Phase I
clinical trial (NCT02630030) for its ability to reach brain tumors
due to its distinctive permeability to tumor tissues (95). Disulfiram is another interesting
proteasome inhibitor that has an improved BBB penetration to employ
its anti-tumor action (96).
Disulfiram was well tolerated in Phase II clinical trial in
combination with TMZ but showed limited activity for unselected
population (97).
Elements of DNA damage repair mechanisms are
considered promising radio-sensitizing agents for cancer therapy
(98). PARP is an important
protein in DNA repair pathways and high PARP-1 mRNA expression is
associated with poor survival in classic GBMs (99). A few PARP-1 inhibitors have been
evaluated in clinical studies (Table
IV). For instance, a Phase I/II clinical trial (NCT00687765)
recently completed a study to evaluate the safety and efficiency of
Iniparib (BSI-201), a PARP1 inhibitor (results as yet unpublished).
Olaparib is another inhibitor of PARP that shows an effective
radio-sensitizing result in GBM cell lines and preclinical glioma
models (98,100). A Phase I trial OPARATIC
(NCT01390571) reported that Olaparib penetrates core and margin
regions of GBM at radio-sensitizing concentrations. It was also
reported to be safe when used with continuous low-dose of TMZ
(101). PARP inhibitor Veliparib
was also evaluated in several clinical trials. However, results
showed that Veliparib did not show improved clinical survival when
combined with TMZ (Phase I/II NCT01026493) (102), nor when added to radiation
followed by TMZ (Phase I/II NCT01514201) (103).
Three different isoforms of IDH exist in human
cells: IDH1, detected in the cytoplasm and in peroxisomes, whereas
IDH2 and IDH3 are both present in mitochondria (108). IDH mutations were found in 6% of
primary GBM and in 54% of secondary GBM (109). Missense mutations, caused by the
substitution of arginine residue in codons 132 and 172 in the
enzymatic site of the IDH1 and IDH2, are frequently detected in
glioblastoma cells (58,110,111). IDH mutants catalyze the
production of 2-hydroxyglutaric acid (2-HG) from isocitrate. 2-HG
affects cellular metabolism, enhances the oxidation of NADPH in
NADP+ which, in turn, disrupts cellular homeostasis and increases
the level of ROS (112). It has
been suggested that the accumulation of this oncometabolite
inhibits the activity of α-KG dependent dehydrogenase, a family of
enzymes involved in methylation of histones and DNA, which includes
KDMs and 10–11 translocation (TET, a family of DNA
hydroxylases that leads to a hypermethylation state and genetic
instability) (113). Moreover,
modification of the epigenetic profile is associated with the
inhibition of glioma stem cell differentiation (114). Furthermore, overexpression of
hypoxia-inducible factor 1-α (HIF-1α), VEGF and platelet-derived
growth factor subunit A (PDGF-A) are detected in GBM cells holding
IDH mutation (115–117). Hence, these alterations induced
by the high levels 2-HG are associated with an aggressive and
invasive carcinogenic phenotype.
Patients with GBMs and IDH mutations are often
treated with inhibitors such as Olaparib targeting PARP (Phase II
NCT03212274). More clinical trials are investigating other PARP
inhibitors such as Nivolumab (Phase II NCT03718767, recruiting),
Talazoparib (Phase II NCT04740190, recruiting) and BGB-290 (Phase
II NCT03914742, active, not recruiting/Phase I NCT03749187) to
treat patients with advanced gliomas including glioblastoma.
Notably, a recently completed study investigated the safety and
clinical activity of Enasidenib (AG-221), a small molecule
inhibitor of IDH2 in patients with advanced solid tumors, including
glioma (Phase I/II NCT02273739). Patients with similar cancer types
were treated in a recently completed study (Phase I/II NCT03684811)
with Olutasidenib (FT-2102), another agent that specifically
inhibits mutant IDH1 at arginine R132. Currently, a number of other
clinical trials are evaluating IDH inhibitors, therefore, more time
and research are needed to conclude about their efficiency to treat
gliomas (Table V).
Normal cells rely on oxidative phosphorylation as
the main energy source. However, cancer cells shift to aerobic
glycolysis even in the presence of oxygen. This phenomenon, known
as Warburg effect, protects cancer cells from apoptosis, stimulates
the production of new precursors and increases invasion capacity
(118–121). Therefore, targeting glucose
transporters, such as glucose transporter (GLUT), and metabolic
enzymes, such as Hexokinase 2 and Pyruvate kinase muscle, may
provide a novel avenue in glioblastoma treatment.
GLUT is a transmembrane protein that belongs to the
major facilitator superfamily (122), Currently, 14 different isoforms
are identified in human tissues and divided into three classes
(123). Class I, which consists
of GLUT 1/2/3/4, is mainly expressed in brain cells. In GBM, GLUT-1
and GLUT-3 are the predominantly expressed isoforms and associated
with poor survival rates. These transporters, characterized by
their high affinity for glucose, promote glycolysis metabolism by
increasing glucose uptake to maintain the survival, proliferation
and growth of cancer cells. GLUT-3, also known as neural glucose
transporter, is highly expressed by brain tumor initiating cells
(BTICs) and has higher affinity for glucose compared with GLUT-1
(124).
Overexpression of GLUT-3 enhances chemoresistance
and survival of BTICs in glucose deficient microenvironment
(124). Recently, Libby et
al (125) proposed that
overexpression of GLUT-3 is positively correlated with the invasion
phenotype of GBM. They found that the C-terminal tail of GLUT-3
reduces invasion (125).
Accordingly, the design of drugs targeting GLUT-3 could be improved
to inhibit molecular functions outside its role in metabolism as a
means to limit potential brain toxicity. This can be achieved by
potentially targeting the C-terminal tail or the protein
interactions driving GLUT-3 mediated invasion. While glucose
transporters provide promising results in research laboratories,
they are not yet targeted in clinical trials.
Hexokinase 2 (HK2), the driver of the aerobic
glycolysis, regulates the first step of glucose metabolism by
converting glucose to glucose 6-phosphate. In GBM, HK2 promotes
tumor progression by enhancing cancer cell growth, lactate
production and chemoresistance (126,127). It also binds to the mitochondrial
membrane and controls the release of cytochrome c, protecting
cancer cells from apoptosis (128). Notably, depletion of HK2 induces
the shift to oxidative glucose metabolism, impairs cancer cell
proliferation, reduces angiogenesis and sensitizes GBM cells to
chemoradiotherapy (129).
Microarray analysis reveals that HK2 expression is negligible in
normal cells but predominant in GBM (126). Therefore, targeting HK2 or its
activity may be a novel therapeutic strategy to selectively kill
cancer cells without damaging normal cells. Ketoconazole and
Posaconazole are antifungals known to inhibit tumor metabolism.
These drugs also selectively target HK2 in glioblastoma cells
(129). Two recent recruiting
early Phase I clinical trials are studying the delivery and
activity of Ketoconazole and Posaconazole in brain tumors
(NCT04869449 and NCT04825275, respectively).
Three different forms of PKM2 exist in mammalian
cells: A tetrameric form, known as the metabolic/glycolytic form
and is involved in aerobic glycolysis, and less active monomeric
and dimeric forms. The latter two forms promote GBM tumor growth
(134,135) and protect it from apoptosis
(136,137). This is achieved either by
diverting the glycolysis pathway to an anabolic pentose phosphate
pathway, or by activating the transcription of a number of
oncogenes such as c-Myc and cyclin D (138). The ratio of monomeric/dimeric and
tetrameric forms of PKM2 decides whether cells will undergo
glycolysis or phosphate pathway (139). In addition, a study conducted by
Sizemore et al (140)
reveals that the ATM-PKM2-CtIP axis enhances DNA double-stand break
repair efficiency and resistance to genotoxic damage caused by
radiation. Overexpression of PKM2 is hence associated with a
radio-resistant phenotype. Therefore, targeting PKM2 might be a
promising therapeutic strategy for glioblastoma treatment.
Amino acid metabolism is another intriguing
metabolism route important for GBM cells. Data suggest that GBM
cells produce an increased pool of free amino acids associated with
poor disease prognosis (141).
Research provided evidence that the hypoxic stress in glioma and
GBM cells increases protein catabolism (142,143). Under hypoxic conditions, and due
to elevated levels of redox stress, hypoxic tumor cells maintain
redox homeostasis that is dependent on increased glutathione
synthesis. Thus, GBM cells want to maintain high levels of
glutathione, consequently favoring conditions that are resistant to
chemoradiotherapy (142).
Arginine, asparagine, glutamine and lysine are among the relevant
amino acids studied, however, the present review will discuss
metabolic pathways of both glutamine and arginine that are targeted
to therapeutically control cancer cell proliferation and
spreading.
In GBM, glutamine is an important source for the TCA
cycle and nucleotide and fatty acid synthesis, thereby sustaining
tumor growth and stimulating glutathione synthesis (148). Concentrations of glutamine and
its related metabolites are proportional to decreased cell survival
(149). Due to developments in
noninvasive image collection and analysis, in vivo glutamine
measurement is now clinically feasible. Previous research
demonstrated that glutamine imaging may have prognostic
significance (150). In the line
of the role of glutamine in cancer cell growth and promoting
chemo-and radiotherapy resistance, targeting glutamine metabolism
constitutes an attractive research area for the treatment of brain
tumors.
Research strategies to target glutamine metabolism
included targeting enzymes involved in their synthesis (GA and GS),
or targeting transporters for glutamine/glutamate uptake (147,151–153). Modulating the GA enzyme was
performed using the RNA interference approach or the use of
allosteric inhibitors. GLS, a gene encoding GA, was targeted
in GBM cell lines, patient derived GBM cells and mouse xenografts
(147,154). Several research groups report a
decreased cancer cell viability by silencing GLS, whereas
GLS2 isoform shows an opposite effect as its overexpression
reverses the aggressive GBM phenotype (155–157). These data suggest approached for
a combinatorial therapeutic approach of both GLS inhibition
and GLS2 overexpression thereby enhancing the antitumor effect. On
the other hand, GS enzyme is also targeted, and its overexpression
resulted in decreased proliferation and migration in rat glioma
cell lines (158). Along with
similar supporting data, GS is suggested to have an anti-glioma
effect.
To date, targeting glutamine uptake by neural cells
through silencing SNAT transporters has not shown significant
effects on the proliferation of GBM cells (147). Conversely, targeting glutamate
transporters such as GLT-1 and GLAST showed more promising results
in GBM cell lines and xenografts [reviewed in details in (147)]. Another promising target is
system xc- (SXC), a cysteine-glutamate
exchanger that is shown to mediate >50% of glutamate transport
in GBM cell lines (159).
Notably, SXC is targeted and inhibited by an FDA approved
sulfasalazine (SAS) for the treatment of bowl disease (159). While SAS resulted in increased
cell death of GBM cells in vitro and in xenografts (160), Phase I/II clinical trials were
terminated due to incidents of severe side effects (161). Research studies using SAS in
gliomas, in combination with radiotherapy, are still in progress
(162) and more studies are
needed to reveal the effectiveness of the combined treatment.
Arginine is a semi-essential amino acid synthesized
from citrulline via urea cycle enzymes, argininosuccinate
synthetase-1 (ASS1) and argininosuccinate lyase (ASL) (164). Cancer cells, which are
characterized with a high proliferation rate, depend on exogenous
arginine in their microenvironment (165). Therefore, reduced expression of
urea cycle enzymes ASS1, ASL and OCT makes cancer cells auxotrophic
and completely reliant on extracellular arginine sources (166,167). Arginine deprivation has been
shown to promote cell death and impairs cell motility and invasion.
Hence, arginine deprivation is a promising potential therapy target
for selective destruction of tumor cells.
Arginine deprivation serves a role in enhancing
endoplasmic reticulum stress and inducing the expression of genes
involved in unfolded protein responses, thus resulting in cancer
cell death (168). Furthermore,
arginine deprivation reduces the extent of β-actin arginylation
which disturbs cell cytoskeletal organization, alters cell adhesion
and impairs cell migration and invasion (169). HuArgI (Co)-PEG5000 is human
arginase I, characterized by the addition of two cobalt ions and
polyethylene glycol that results in improved enzymatic catalytic
activity, increased stability, decreased immunogenicity and lower
dissociation constant at neutral pH (170,171). Promising effects were shown when
using HuArgI (Co)-PEG5000 to target arginine auxotrophy in
different tumor types (166,172–175). Our laboratory research found
evidence that HuArgI (Co)-PEG5000 induces autophagy cell death in
hepatocellular carcinoma (176),
pancreatic carcinoma (173,176), acute lymphoblastic leukemia
(174), renal cell carcinoma
(177), prostate cancer (178), colorectal cancer (CRC) (172), ovarian carcinoma (175) and breast cancer (179). In GBM, we observed that HuArgI
(Co)-PEG5000 induces autophagy-dependent cancer cell death. The
addition of exogenous citrulline failed to rescue any of the GBM
cell lines from arginine depletion-induced cytotoxicity, indicating
a high level of arginine dependence (166). A recent recruiting Phase I
clinical trial (NCT04587830) is currently assessing the safety and
tolerability of ADI-PEG 20, an arginine deprivation agent, in
combination with radiotherapy and TMZ in patients newly diagnosed
with GBM (180). Taken together,
targeting GBM cells using arginine depletion is a potent and
selective potential treatment for GBM.
Other molecules such as human telomerase reverse
transcriptase (hTERT), ATRX and protein kinase C, have also gained
attention as potential targets for GBM cancer therapy. This is due
to the finding that alterations in these molecules enhance GBM
cells survival and progression.
Telomerase is a ribonucleoprotein that consists of
two subunits: i) the protein catalytic hTERT that mediates reverse
transcriptase activity and ii) the human telomerase RNA template.
Telomerase is mainly active in embryonic cells, stem cells and
precursor cells. Notably, studies show that hTERT expression is
upregulated in glioblastoma (181,182). In addition, ~80% of primary
glioblastoma cells harbor a mutation or methylation in the hTERT
promoter (183). Genomic
sequencing revealed a high incidence of mutually exclusive point
mutations C228T and C250T in hTERT promotor of glioblastoma cells
(183). Arita et al
(181) show that hTERT expression
is 6.1 times higher in tumors carrying these mutations than in
wild-type tumors and therefore leads to the upregulation of TERT.
Upregulation of hTERT enhances telomerase activity and promotes an
immortal phenotype (183). In
addition, hTERT mutations are associated with poor prognosis and a
multifocal invasive phenotype in GBM (184,185). Thus, hTERT promotor mutation can
be recognized as a novel biomarker and therapeutic target molecule
for GBMs (184,186,187). Currently, two immunization
strategies are currently targeting hTERT in clinical trials. A
Recruiting Phase II/III study (NCT03548571) is investigating the
efficiency of immunization with IMP dendritic cells that are
transfected with mRNA hTERT, as well as autologous tumor stem cells
and survivin, compared with standard adjuvant chemoradiotherapy.
Additionally, an active, not recruiting Phase I/II study,
(NCT03491683) is evaluating the safety of INO-5401 (a combination
of three separate DNA plasmids targeting hTERT, Wilms tumor gene-1
antigen, and prostate-specific membrane antigen) in patients with a
glioblastoma tumor with an unmethylated MGMT promote.
PKC is a serine threonine kinase that serves a
crucial role in GBM growth and progression. PKC consists of 11
isoforms and is divided into three subfamilies: Classical, novel
and atypical classes (191).
Classical PKCs consist of two members, PKCα and PKCβ (192). Leirdal et al (193) show that PKC enhances GBM survival
via MAK-ERK12 pathway. PKCα induces GBM progression via
PKC-progesterone pathway (194).
Once stimulated with 12-O tetradecanoylphorbol-13-acetate or
lysophosphatidic acid, PKCa translocates to the nucleus, enhances
progesterone receptor phosphorylation and promotes the migration
and invasion capacity of GBM (194,195). PKCb serves an essential role in
enhancing angiogenesis, thus facilitating GBM progression (196). Moreover, PKCβ leads to GBM cell
death. Conversely, Liu et al (197) show that in an orthotopic mouse
xenograft model, PKCβ II expression suppresses GBM tumor growth and
extends mouse survival by inhibiting YAP/TAZ. Thus, PKCβ has two
opposite roles in GBM.
The novel PKC subfamily contains four members:
PKCδ, PKCɛ, PKCη and PKCθ. Inhibition of PKCδ suppresses tumor
spheroid formation in vitro and tumor development in
vivo (198). In glioblastoma,
PKCδ is involved in tumor initiation (199), cancer cell migration and invasion
(194,200) and radio-chemoresistance (199). Activation of PKCδ phosphorylates
glycerol-3-phosphate dehydrogenase enzyme and serves an essential
role in glucose metabolism and phospholipid synthesis (201). PKCɛ, on the other hand, is
overexpressed in glioblastoma cell (202) and controls tumor survival
(203) cancer cell adhesion and
motility via activation of the ERK pathway (204,205). PKCη promotes GBM proliferation
(206,207) and reduces cell sensitivity to
radiotherapy (208).
Enzastaurin is a kinase inhibitor that particularly
inhibits protein kinase C, thus decreasing tumor growth and cell
proliferation (212). Results
from recent clinical studies (Phase II NCT00586508 and Phase III
NCT03776071) did not show improved PFS compared with bevacizumab
and other therapeutic agents (212,213).
Targeting intrinsically altered molecules that
trigger GBM malignant phenotype is an innovative therapeutic
strategy that aims to destroy tumor cells without damaging normal
cells. However, despite the promising in vitro results, this
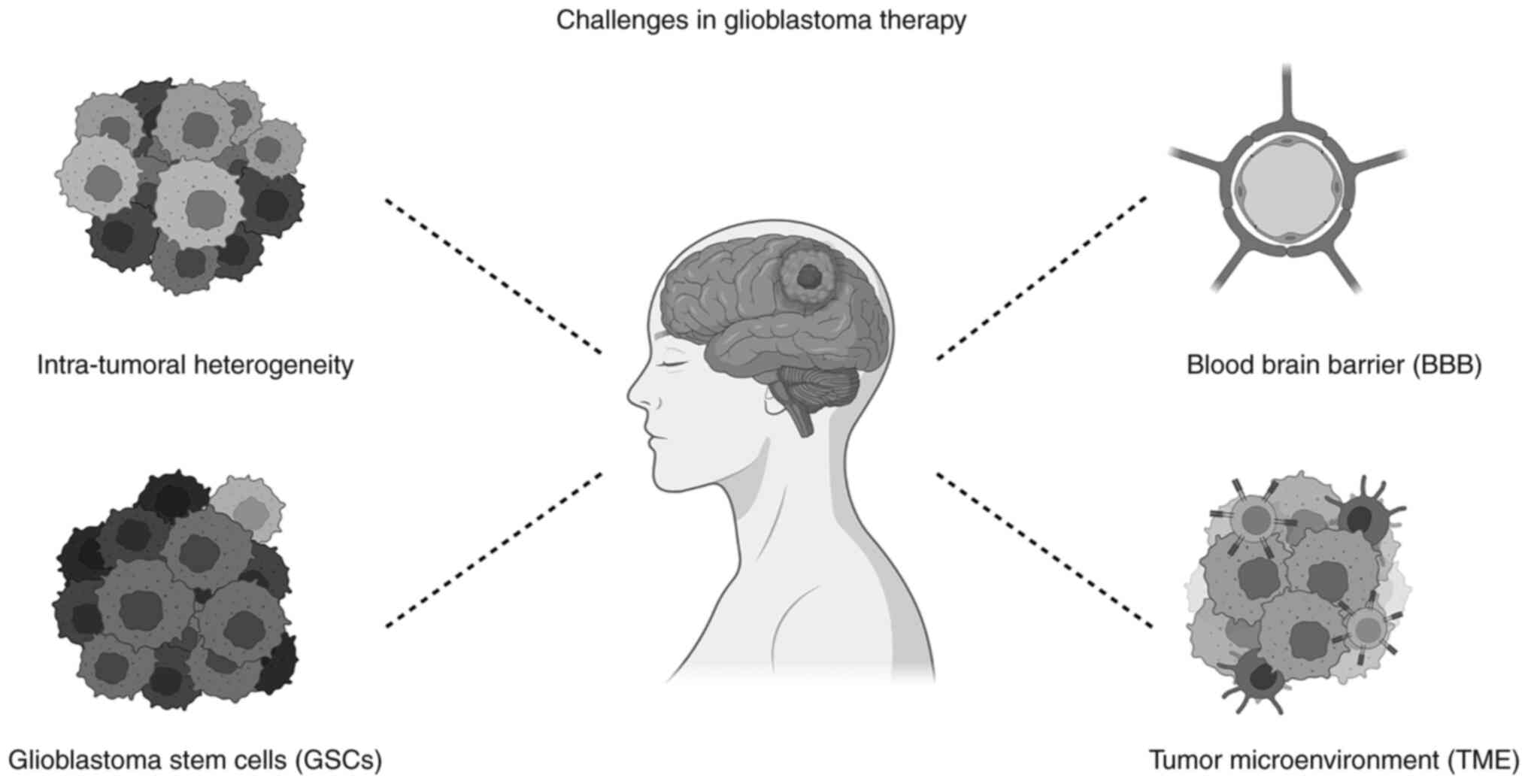
novel therapy faces various barriers in vivo (Fig. 2).
One reason behind the complexity of GBM management
is that the genetic profiles of recurrent glioma cells and the
initial cancer derived from the same patient are different
(214). Thus, the plasticity and
heterogeneity of GBM cell populations can explain the failure of
clinical trials based on monotherapy in vivo. Clones of the
genes that are identified by different number of chromosomal sets,
exhibit a high level of heterogeneity in the expression of cell
membrane markers. This leads to phenotypic heterogeneity at the
level of the population providing an advantage for tumor survival
(215). Neftel et al
(216) found that gliomas rarely
have identical proportions of single-cell transcriptomic states and
this proportion is skewed by genetic associations that are specific
to each patient leading to abundant heterogeneity. Therefore, it is
recommended to carefully weigh the genetic, epigenetic, and
molecular profile of each patient. Single-cell RNA-sequencing
reveals the presence of various molecular and genetic cell subtypes
within the same patient biopsy (215,217). These subtypes can change
progressively and become more resistant to chemoradiotherapy
(214). Neftel et al
(216) describe multiple cellular
states and determine a correlation between the cellular states,
plasticity and genetic signature of tumor cells. They define four
cellular states of malignant GBM tumor cells:
Neural-progenitor-like, oligodendrocyte-progenitor-like,
astrocyte-like and mesenchymal-like states. These states are
characterized by the overexpression of CDK4, PDGPRα, epidermal
growth factor (EGF) and NF1 alterations, respectively. Neftel et
al (216) also showed the
co-existence of multiple cellular states and their transition
potential within the same patient sample and evidenced the ability
of a single cell to generate all cellular states. Amplification of
the EGFR and PDGFRA genes that specifically encode for RTKs has
been recognized in GBM tumors (218). Snuderl et al (219) established that the amplification
of different RTKs was infrequently found in the same region of the
tumor; yet, different RTKs (MET, EGFR and PDGFRA) were amplified in
diverse subpopulations of the GBM tumoral cells.
Another advancing technology that expands the
understanding of the heterogeneity of GBM is the whole genome
amplification (WGA) methods. WGA helps to identify different
imbalances on the level of the chromosomes (218). In one study, Nobusawa et
al (220) applied the WGA
method on GBM samples (14 different primary samples) from two to
five locations within each tumor. They recognized not only common
modifications between all studied locations, but also changes that
were specific to a certain region among the studied GBM tumor
samples. A later study targeted 33 cancer genes with single
molecular inversion probes and reported regional mutational
heterogeneity among different patients with GBMs, as well as among
the same patient (221).
Efficient treatment options necessitate building a
model the mimics a patient's GBM biology. Jacob et al
(224) generated a new organoid
model from patient-derived primary cancer cells that conserved the
original tumor's histological, genetic and molecular signatures in
addition to its cellular heterogeneity. In order to generate
patient-derived glioblastoma organoids (GBOs), Jacob et al
(224) obtained tissues along the
tumor margin. These tissues were refined from necrosis and
surrounding brain tissues, dissected and cultured in optimized
serum-free GBO medium. When allowed to grow larger, GBO created
gradients of hypoxia reflecting a hallmark of GBM.
Immunohistological analyses reported markers for glia, immature
neurons, neural progenitors and glioma stem cells, resembling the
cellular composition of parental tumors. Also, single-cell
transcriptome analysis confirmed cell-type heterogeneity and
suggested that specific elements of the TME were preserved. Upon
transplantation into adult rodent brains, GBOs showed reliable
engraftment and aggressive infiltration (224). This suggested that a biobank of
patient-derived GBO can be used as a prescreening drug model to
investigate drug response and to evaluate its effectiveness on
patient cells.
‘Cancer stem cell likes’, also referred to as
‘cancer stem cells’ (CSCs) are self-autonomous units that play a
significant role in tumor initiation and growth as well as
therapeutic resistance. GSCs are derived from the malignant
transformation of normal neural stem cells or the dedifferentiation
of tumor cells after radiotherapy or chemotherapy (225,226). GSCs exhibit prolonged
proliferation and are able to metastasize and suppress
anti-inflammatory responses and confer resistance to therapeutic
treatments (227). Thus, GSCs are
important factors that limit clinical options and treatments of GBM
(228,229). Numerous studies that are
currently being undertaken to target GSC were made possible by
improved understanding of the biology of GCS (24). GSCs represent a hot topic for
improved GBM treatment.
Some studies aim to target and kill GSCs directly
by targeting stem cell biomarkers. For instance, GSCs that are
CD133 positive are important for sustaining and spreading GBM
(230). Patients with GBM have a
higher survival rate when CD133 positive stem cells are eliminated
(231). CD133 stem cells may be
eliminated by BMI1 gene suppression, an oncogene involved in the
control of stem cell self-renewal and differentiation (232). Inducing apoptosis in GSCs can be
also achieved by targeting the signaling pathways that play a role
in their self-renewal. For instance, the Wnt signaling pathway has
been targeted since 2014 as it is involved in neural stem cell
development (233). Targeting
glycogen synthase kinase-3β (GSK-3β) and β-catenin is suggested to
inhibit the Wnt pathway in vitro (234). AR-A01441, LiCl and SEN461 are
examples of GSK-β inhibitors that lead to increased apoptosis of
GBMs cells (24). Notably,
Celecoxib is a β-catenin inhibitor that is already studied in
clinical trials, however, no promising results have been reported
yet (Table VI).
The Notch pathway is another targeted pathway for
GBM treatment as it is involved in resistance to immunotherapies
(235). γ-secretase is an enzyme
that mediates Notch's cleavage and translocation to the nucleus.
Therefore, γ-secretase is suggested as a plausible target for Notch
inhibition (236). In fact,
several clinical trials are testing the effect of RO4929097, a
γ-secretase inhibitor, as a monotherapy for GBM treatment or in
combination with other treatments. In addition to the
aforementioned pathways, Hedgehog (SHH) pathway is also targeted to
inhibit the renewal of GSCs, as it confers resistance to
conventional GBM treatment. SHH pathway is targeted by inhibiting
smoothened (SMO), its downstream effector (24). Vismodegib is one of the SMO
inhibitors used in several clinical trials and it showed enhanced
PFS when used before surgical resection (NCT00980343). Finally, the
STAT3 pathway is also targeted to control neural stem development
(237). WP1066 and Napabucasin
(BBI608) are STAT3 inhibitors currently in Phase I (NCT01904123)
and II (NCT02315534) clinical trials, respectively (no results are
reported yet).
Notably, GSCs can be targeted by targeting the
mitochondria or via specific antibiotics. For instance, oxidative
phosphorylation (OXPHOS) is a major source of ATP in a number of
types of cancer and plays a significant role in carcinogenesis and
tumor growth (238). GSCs are
OXPHOS-dependent GBM cells that require mitochondrial translation
(239). The bacterial antibiotic
quinupristin/dalfopristin (Q/D) blocks mitochondrial translation,
which not only inhibits the formation of GSCs but also disrupts the
cell cycle and increases apoptosis (239). These results suggest that Q/D may
be taken into consideration for the treatment of GBM and that
reducing mitochondrial translation may be examined to inhibit GSC
growth. On the other hand, Salinomycin, is a K+
ionophore antibiotic that causes lysosomal iron sequestration, and
leads to the production of ROS and lysosome membrane
permeabilization (240).
Salinomycin is shown to favorably kill GSCs and other types of CSCs
(240,241). Although clinical trials examining
the possible efficiency of Salinomycin in the treatment of
glioblastoma have yet to be published, Salinomycin and its
derivatives are now being researched intensively as anti-CSCs
treatments for a variety of malignancies (241). Collectively, these results imply
that focusing on GSCs is a promising approach for treating GBM more
effectively.
The BBB forms an interface between the brain and
the systematic blood circulation. It consists of endothelial cells
(ECs) that line blood vessels, are surrounded by astrocytic
perivascular pseudopodium and pericytes, and interconnected by
neuronal ending and microglia (242,243). Under normal physiological
conditions, ECs, connected by tight junction proteins, create a
compact barrier. In addition, efflux transporters, carried by BBB
cells, regulate molecular and cellular transport across the BBB and
protect the brain from toxins, xenobiotics, and pathogens (243). The development and progression of
GBM impair the integrity and function of the BBB, resulting in a
heterogenous resistant barrier known as Blood Brain Tumor Barrier
(BBTB). In glioblastoma, the BBTB is characterized by the
downregulation of tight junction proteins such as claudin and
occludin and overexpression of efflux transporters (244–247). These tight junction proteins
increase the permeability of the BBTB, enhance paracellular
transport and thus facilitate the penetration of cells and small
molecules to the interstitial space of the brain. Accumulation of
such molecules might have neuro-toxic effects. In addition, the
heterogeneous permeability of the BBTB caused by the ‘leaking’
structure of the new blood vessels as against the compact structure
of the local blood vessels leads to unequal distribution of the
drug (248–250). Several FDA-approved drugs,
including chemotherapy drugs, have affinity for efflux
transporters, including P-gp and BCRP (251–256). Hence, overexpression of efflux
transporters in glioblastoma restricts drug delivery to the
brain.
In the line of BBB heterogeneity and overexpression
of efflux transporters, cellular, chemical and physical approaches
are being investigated to enhance drug delivery through the BBTB.
One of these advanced approaches is the nanomaterial system which
is based on different types of nanocarriers including,
polymer-based, viral, drug-conjugated, lipid-based and inorganic
nanoparticles (NPs) (257). NPs
are usually loaded with drugs and are known to have a small size
within a range between 1–100 nm (258). Budama-Kilinc et al
(259) used a nanoparticle system
based on a poly (ε-caprolactone) to deliver a tripeptide,
glycyl-L-histidyl-L-lysine (GHK), to the GBM cells. GHK is known as
a natural growth modulating tripeptide in vitro; thus, it
decreases the viability of GBM cells to ~65%. Another study was
based on a nanobubble system that consists of NPs made up of iron
and platinum. NPs were either surface-functionalized with
transferrin to target the glioma cells or loaded with doxorubicin
(DOX) in a mouse model. These NPs resulted in reduced growth of the
tumor by ~70% (260). On the
other hand, Ambruosi et al (261) used a rat glioma model to test
poly (n-butyl cyanoacrylate) (PBCA) as a potential therapy for GBM.
PBCA NPs were loaded with DOX chemotherapeutic drug and coated with
polysorbate 80 (surfactant). NPs successfully delivered the drug
across the BBB and increased the median survival rate by 35% among
tested rats living for the whole time of the study period.
On the molecular level, transferrin receptor (TfR)
is a protein commonly targeted to enhance the delivery of
therapeutical drugs through the BBB (262). Poly (lactic-co-glycolic acid) NPs
loaded with TMZ and coated with TfR-monoclonal antibodies showed
higher internalization in GBM cells compared with control cells
(263). Kuang et al
(264) examined the use of
polymer-based NPs coupled to a peptide having the ability to target
TfR on both the BBB and GBM cells to transport doxorubicin and RNA.
Results showed higher efficiency in in vitro tumor targeting
and cellular uptake, with a decline in the tumor growth leading to
improved median survival time.
Focused ultrasound therapy (FUS) is another method
used to facilitate transport through the BBB. It is a non-invasive
and image-guided method that enhances the efficacy of drug delivery
to GBM cells (265). Wei et
al (266) showed an
enhancement in the local delivery of TMZ to tumor cells through the
FUS-mediated disruption of the BBB. Results reported an increase in
the OS of rats with experimentally induced gliomas. Notably,
MRI-guided FUS (MRgFUS) was evaluated to attain enhanced tissue
delivery of TMZ in mice (267),
liposome-encapsulated doxorubicin in rats (266) and cisplatin-conjugated gold NPs
in mice (268). This method is
advantageous as it reduces the systematic toxic effects of the
drugs used (low doses of therapeutic drugs can be used) (269) and leads to temporary opening of
the BBB rather than long-term opening associated with BBB
disruptions (270). These studies
show that the use of different NPs (nanomaterial system) or FUS has
the potential to enhance the efficiency of therapeutical drug
delivery in GBM management.
The TME comprises fibroblasts, endothelial cells,
immune cells, ECM and soluble factors surrounding the tumor. It
provides biophysical and biochemical support for the tumor and
contributes significantly to tumor initiation, growth, progression,
and resistance to therapy (271).
GBM is classified as a hypoxic solid tumor where
hypoxia is considered one of the non-cellular TME components. The
hypoxic microenvironment is a major inducer of angiogenesis; a
process in which ECs branch from preexisting small vessels to form
sprouts of capillaries. Angiogenesis thus promotes tumor growth by
supplying cancer cells with O2 and nutrients and
facilitates their metastasis (272,273). Moreover, hypoxia induces the
activation of a number of cellular processes that promote the
migration, invasion and radio-chemoresistance of GBM cells
(274–280). This is due to the overexpression
of VEGF/HIF-1α protein. Therefore, and in order to control the
hypoxic environment, researchers developed Bevacizumab, an FDA
approved anti-VEGF antibody, for GBM treatment. Bevacizumab showed
encouraging results as monotherapy or when combined with
traditional treatment, as reviewed in the study by Cruz Da Silva
et al (24).
GBM secretes a wide range of chemo-attractants and
cytokines that recruit immune cells to the TME. For instance, GBM
stem cells recruit macrophages and promote their transition from
the antitumor (M1) to the pro-tumor (M2) phenotype (281). Tumor associated macrophages (TAM)
are the most abundant cells inflating tumor lesion and accounting
for ≤40% of the tumor mass. Indeed, the disrupted structure of BBTB
facilitates the infiltration of TAM. In addition, GBM cells inhibit
the proliferation of cytotoxic T cells and activate regulatory T
cells (282). Occurrence of
immune cells in the TME leads to the formation of an extensively
immune-suppressive microenvironment and enhances GBM cell migration
and invasion.
At present, researchers are investigating new
therapeutic approaches that aim to reactivate the immune system
against GBM and restore the immune surveillance in GBM. These
approaches include reversing the polarization of M2 macrophages to
M1, targeting immune checkpoints using checkpoint inhibitors to
rebut T cell responses, and implementing cytokine therapy. Yang
et al (283), discovered
that the dual-targeting of IL-6, which stimulates different
macrophages activation in GBM, and CD40 may help in reversing tumor
immunosuppression mediated by macrophages thus informing the
immunotherapy based on T-cells against GBM. In their study, it was
shown that CD40 stimulation and IL-6 inhibition reverse tumor
immunosuppression mediated by macrophages and increase the survival
rates of the animals in GBM models (283). Similar findings supported the use
of macrophages-based therapies as effective therapeutic approaches
when targeting GBM.
Cytokine therapy represents a hot topic for
immunotherapeutic approaches since cytokines are molecular
messengers of innate and adaptive immunity (284). This approach promotes human
natural killer (NK) and T cell activity, survival and proliferation
thus coordinating immune responses against malignancies (285). TGF-β, IFN-α, IFN-γ and IL-12 and
other cytokines have anti-tumor effect in GBM, thus reducing tumor
size and inhibiting carcinogenesis at early stages (286). Tumoral injection of Ad-RTS-hIL-12
in combination with veledimex (VDX, an hIL-12 activator) and an FDA
approved antibody cemiplimab was tolerated well and exhibited
increased circulating cytotoxic T cells in patients with recurrent
or progressive GBM (Phase II NCT04006119). IFN-β immunotherapy in
combination with TMZ was also evaluated in clinical trials
(287). Han et al
(288), used cytokine-induced
killer therapy with TMZ on patients with GBMs and reported higher
PFS rates in treated vs. control untreated group. The therapeutic
efficacy of various cytokines in the treatment of gliomas is still
being investigated. The cytokines' safety and tolerance are being
defined, and some are thought to be a promising supplement to
conventional treatments (289).
Glioma cells frequently employ a variety of
strategies to avoid immune surveillance. Therefore, several
therapeutic approaches have been developed to activate immune
responses in the TME. Cytotoxic T lymphocyte-associated protein 4
(CTLA-4), programmed cell death protein 1 (PD-1), and PD ligand 1
(PD-L1) are immune checkpoint proteins targeted for monoclonal
antibody inhibition (290,291).
Monoclonal antibodies act by freeing T lymphocytes from their
negative regulation thus suppressing cancer evasion of the immune
system (292,293). Several immune checkpoint
inhibitor antibodies have been approved by the FDA for the
treatment of several types of cancer (294). Neoadjuvant therapy with
pembrolizumab, an anti-PD-1 monoclonal antibody, resulted in
improved OS and PFS in patients with recurrent surgically
respectable GBM (Phase II NCT02337686) (43). Wang et al (295) demonstrated that genetically
engineered multifunctional NK cells (CD73.mCARpNKs) can lead to
efficient anti-GBM activity mainly by bypassing the heterogeneity
and the immunosuppressive structures of GBM tumors. Immune
responses in the TME can be also stimulated by adoptive cell
therapy (ACT). This method involves isolating autologous T
lymphocytes with anti-tumor activity from cancer patients,
expanding them ex vivo, and transferring amplified
engineered T cells to the patients (296,297). Among the different ACT methods
developed, chimeric antigen receptor (CAR)-T cell gene therapy has
attracted the most attention for killing cancer cells. CAR-modified
T cells may identify different types of antigens regardless of how
they are presented on MHC molecules (298). To date, there are currently 24
clinical trials examining the efficacy of CAR-T cell treatment for
GBM according to Clinicaltrials.gov (accessed on 10 August 2022;
Table VII). Several CARs have
been generated to specifically target GBM such as EGFRvIII, HER2,
IL13R2, and CD70 (290).
Collectively, studies using CAR-T cell immunotherapy exhibit
encouraging results. However, obstacles such as tumor
heterogeneity, the heterogenous expression of antigens, and the
role of T cells at the tumor's sites make it challenging to
efficiently eradicate the tumor (290).
Glioblastoma is characterized with high cell
proliferation and neovascularization leading to tumor infiltration
reaching crucial structures in the brain. This increases the
possibility of tumor recurrence, scoring low survival rates
(299,300). Therefore, more effective
treatment procedures are required to extend the lives of patients.
Conventional treatments such as radiotherapy and chemotherapy are
regarded as a two-edged sword. This is because these treatments are
frequently accompanied with normal tissue damage or genetic
changes, nonspecific drug distribution and multidrug resistance.
However, chemoradiotherapy promises improved survival and quality
of life that may outweigh their disadvantages; thus, these
modalities remain the primary cancer weapons. Substantial research
has been done, and continues to be done, to develop novel
therapeutic approaches that are less toxic for normal tissues and
endow increased survival and improved quality of the lives of
patients. Researchers have discovered genetic and subsequent
molecular changes influencing critical pathways that cause
aggressive behavior of tumors (24,214). Therefore, targeting these altered
molecules and pathways is seen as a promising new approach to GBM
treatment.
Over the previous few decades, biomarker-driven
methods, such as small molecule inhibitors and monoclonal
antibodies, have been developed. These methods aim to target key
altered players that are directly correlated with tumor progression
and invasion. EGFRs constitute one of the mostly targeted molecules
in clinical trials to treat patients with GBMs (Table I). However, targeted therapy has
shown little to no promising results due the intra-tumoral
heterogeneity of GBM and drug resistance (216,218). Tumor heterogeneity could be a
plausible explanation for GBM resistance to EGFR-targeted
treatments. Moreover, resistance to EGFR therapy can also be caused
by upregulation of redundant RTKs and downregulation of EGFR
downstream molecules (218).
Consequently, co-targeting of EGFR and other RTKs such as c-MET
could offer a solution as seen in vivo (301). Multi-targeted therapeutic
approaches offer an advantage where drugs can target numerous
important nodes for GBM growth and progression, compensating for
the inefficiency and quick acquisition of resistance seen with
monotherapies (302).
Multi-targeted therapeutic approaches also include the
administration of multi-kinase inhibitors (303,304). Moreover, more complicated
pathways have been identified and targeted in vitro.
In-depth-investigation of these pathways helps to identify novel
drugs.
Glioblastomas are intrinsically immunologically
‘cold’ tumors due to presence of few T cells but predominant
pro-tumor immune cells in the TME, constituting a common reason for
drug resistance (305).
Immunotherapies thus aim to use a patient's immune system to
re-direct immune cells against a tumor. Researchers have
established several other immunotherapeutic strategies to target
patients with GBMs with immunostimulatory conditions. These include
immune check-point inhibition, autologous stimulated lymphocytes,
cytokine therapy and dendritic cell therapy (306). Except for certain cases,
immunotherapy in glioblastoma clinical trials has been
disappointing overall. CAR-T cell therapy, for instance, faces a
significant hurdle since glioblastoma tumors can rapidly adapt via
antigen escape (307). CAR-NK
cell therapy, on the other hand, offers an advantage as it can be
administered to an HLA-mismatched patient, and Phase I clinical
trial is showing promising preliminary results (NCT03383978)
(308). Yet, NK cell expansion
and manufacturing are still time and money consuming (309). Akin to targeted therapy, tumor
heterogeneity is also a limiting factor for immunotherapy.
Moreover, combing immunotherapy with chemotherapy may also lead to
drug resistance (310).
Anti-inflammatory corticosteroids, for example, may counteract the
therapeutic effects of immunotherapy (311). Additionally, glioblastoma cells
can adapt to immune checkpoint inhibition by increasing the
expression of alternative checkpoints (312). The immunosuppressive TME is
another limitation for immunotherapy, thus, combinatorial
immunotherapy approaches may overcome this issue (313,314). In fact, preclinical research on
combination immunotherapy yielded promising outcomes [reviewed in
(314)]. The most successful
approaches are those that influence the cancer-immunity cycle at
different times and/or sites, leading to the activation of immune
response and the inhibition of immunosuppressive elements (314).
Cancer vaccines are one of the very promising
immunotherapies and are being thoroughly researched as a method to
reduce tumor development and eliminate tumor cells in cancer
patients. Peptide-based, dendritic cell (DC)-based, and
personalized vaccines are being explored as potential vaccine
therapies in GBM (305,315) (Table VII). Cancer vaccine design
depends on selecting an ideal antigen specifically expressed and
present on all cancer cells, highly immunogenic and essential for
cancer cell survival (316).
These antigens are therefore processed by DCs aiding in recruiting
and activating CD8+ and CD4+ T cells that
will promote tumor eradication (317). DCVax®-L and ICT-107
are DC-based vaccines tested in clinical trials for safety,
tolerance and efficacy in patients with GBMs (NCT00045968 and
NCT01280552 respectively). Personalized vaccination is now made
possible using multi- epitope-based patient-specific vaccine to
target neoantigens only (Phase I NCT02287428) (318) or both neoantigens and unmutated
tumor-specific antigens (Phase I NCT02149225) (319). Corresponding results reported
neoantigen-specific CD4+ and CD8+ T cell
responses migrating into the tumor with increased number of
infiltrating T cells (318,319). AV-GBM-1 is another personalized
cancer vaccine based on autologous dendritic cells full of
autologous tumor neoantigens. Phase II clinical study (NCT03400917)
reported an enhanced PFS (320)
demonstrating a promising positive effect of vaccination in
patients with GBM.
Most of the current therapeutic strategies need to
overcome the obstacles of drug delivery due to BBB, tumor and TME
heterogeneity and abundant GSC niches. Burkhardt et al
(321) propose promising results
for an intra-arterial brain administration of Bevacizumab after
temporary destruction of the BBB by mannitol. Followed by
intravenous administration, their technique is able to overcome the
poor intracerebral availability of the drug and enhanced PFS in
patients with recurrent GBMs. Several studies suggest that
molecular subgroups exist within histologically similar GBM tumors
(322,323). Research into the dynamics of
various glioblastoma subtypes would help understand the survival
features of the TME. This highlights that future treatment options
should no longer be based exclusively on morphological assessments,
but must now take molecular and cellular heterogeneity into
account. Furthermore, the plasticity of GBM cells caused by the
bidirectional communication between GSCs and their differentiated
counterparts complicates heterogeneity (227). These two populations respond
differently to radio-, chemo- and targeted therapies. Thus,
targeting both components should be well considered during the
course of treatment.
Due to genetic heterogeneity and tumor growth,
single-target therapy generates recurrence and consequent
resistance to initial treatment. Therefore, therapeutic strategies
are implementing targeted therapy along with immunotherapy.
Notably, immunostimulatory and immunosuppressive effects are also
mediated by targeted therapy (324). Inhibition of several molecular
targets, such as anti-angiogenic agents, has already been proved to
improve immunotherapy clinical results in a variety of types of
cancer (325). It is worth noting
here that, when it comes to combinatorial therapeutic approaches,
greater laboratory and clinical efforts are required to validate
their efficiency.
The increased understanding of GBM biology outlined
above was made possible in part by the growth of murine preclinical
GBM models. These models were key for translatable research from
preclinical to clinical studies. Glioblastoma cell-line xenografts,
patient-derived xenografts, and genetically engineered mouse models
are currently available. However, these models fail to recapitulate
tumoral and TME heterogeneity of GBM. Thus, the effects of advanced
therapies in animal models rarely replicate clinical data, which is
a persistent issue in glioblastoma research. The success of such
therapies may rely on the development of reliable GBM models.
Several reproducible glioma models in pigs are evolving as
preclinical large-animal models (326–329). Therefore, xenograft and
genetically engineered porcine models are suggested as reliable
prospective models to allow for a transitional step between murine
models and human clinical trials (330).
Effective drug delivery systems that can cross the
BBB are also attracting attention. This includes non-viral vectors
such as nanocarriers and viral vectors such as oncolytic viruses.
Due to advanced research and promising outcomes, chemoradiotherapy
nanomedicine is expected to be progressively applied in the clinic
in the coming years (331).
Targeted gene therapy employing nano-based carriers is being
explored and its applicability for a number of forms of cancer are
currently under Phase I–III development (332). It is thus expected that combing
monoclonal antibodies and nano-carriers could also improve the
efficiency of immunotherapy. On the other hand, oncolytic viruses
are vectors of targeted gene therapy that can cross the BBB and are
considered a promising method for treating GBM (333–336). While drug and gene delivery via
oncolytic viruses are currently in clinical trials, more research
and large randomized controlled Phase II/III trials are needed to
assess their beneficial clinical outcome.
An efficient GBM treatment starts by understanding
the molecular alterations in cells derived from the patient and
that are driving the malignant phenotype. This is followed by
studying the effect of a selected treatment in vitro using
the ‘ideal’ model that can conserve the molecular signature of
patient cells. Prior to incorporating the patient in any clinical
trial, it is also recommended that researchers or physicians
analyze the structure of the BBB to evaluate the ability of a
selected drug to pass across the BBB. Finally, the use of a
combined therapy strategy that targets tumor cells and cells of the
surrounding microenvironment may increase the efficiency of the GBM
treatment.
Not applicable.
Funding: No funding was received.
Not applicable.
OEA and RN equally designed the review and wrote
most of the article. MA researched references and contributed to
the writing of the manuscript. RAH and MES (PIs) wrote the final
draft and edited the manuscript. Data authentication is not
applicable. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Hanif F, Muzaffar K, Perveen K, Malhi SM
and Simjee SHU: Glioblastoma multiforme: A review of its
epidemiology and pathogenesis through clinical presentation and
treatment. Asian Pac J Cancer Prev. 18:3–9. 2017.PubMed/NCBI
|
|
2
|
Karachi A, Dastmalchi F, Mitchell DA and
Rahman M: Temozolomide for immunomodulation in the treatment of
glioblastoma. Neuro Oncol. 20:1566–1572. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delgado-López PD and Corrales-García EM:
Survival in glioblastoma: A review on the impact of treatment
modalities. Clin Transl Oncol. 18:1062–1071. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tamimi AF and Juweid M: Epidemiology and
outcome of glioblastoma. Glioblastoma. De Vleeschouwer S: Codon
Publications; Brisbane, AU: 2017, Available from:. http://www.ncbi.nlm.nih.gov/books/NBK470003/
View Article : Google Scholar
|
|
5
|
Verhaak RGW, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hubbard SR: Structural analysis of
receptor tyrosine kinases. Prog Biophys Mol Biol. 71:343–358. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Montor WR, Salas AROSE and Melo FHM:
Receptor tyrosine kinases and downstream pathways as druggable
targets for cancer treatment: The current arsenal of inhibitors.
Mol Cancer. 17:552018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hunter T: Tyrosine phosphorylation: Thirty
years and counting. Curr Opin Cell Biol. 21:140–146. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brennan CW, Verhaak RGW, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chakravarti A, Chakladar A, Delaney MA,
Latham DE and Loeffler JS: The epidermal growth factor receptor
pathway mediates resistance to sequential administration of
radiation and chemotherapy in primary human glioblastoma cells in a
RAS-dependent manner. Cancer Res. 62:4307–4315. 2002.PubMed/NCBI
|
|
13
|
Mazzoleni S, Politi LS, Pala M, Cominelli
M, Franzin A, Sergi Sergi L, Falini A, De Palma M, Bulfone A,
Poliani PL and Galli R: Epidermal growth factor receptor expression
identifies functionally and molecularly distinct tumor-initiating
cells in human glioblastoma multiforme and is required for
gliomagenesis. Cancer Res. 70:7500–7513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Dutra A, Pak E, Labrie JE III,
Gerstein RM, Pandolfi PP, Recht LD and Ross AH: EGFRvIII expression
and PTEN loss synergistically induce chromosomal instability and
glial tumors. Neuro Oncol. 11:9–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hatanpaa KJ, Burma S, Zhao D and Habib AA:
Epidermal growth factor receptor in glioma: Signal transduction,
neuropathology, imaging, and radioresistance. Neoplasia.
12:675–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Talasila KM, Soentgerath A, Euskirchen P,
Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO,
Sakariassen PØ, Eskilsson E, et al: EGFR wild-type amplification
and activation promote invasion and development of glioblastoma
independent of angiogenesis. Acta Neuropathol. 125:683–698. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bonavia R, Inda MM, Vandenberg S, Cheng
SY, Nagane M, Hadwiger P, Tan P, Sah DW, Cavenee WK and Furnari FB:
EGFRvIII promotes glioma angiogenesis and growth through the NF-κB,
interleukin-8 pathway. Oncogene. 31:4054–4066. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Katanasaka Y, Kodera Y, Kitamura Y,
Morimoto T, Tamura T and Koizumi F: Epidermal growth factor
receptor variant type III markedly accelerates angiogenesis and
tumor growth via inducing c-myc mediated angiopoietin-like 4
expression in malignant glioma. Mol Cancer. 12:312013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng H, Hu B, Vuori K, Sarkaria JN,
Furnari FB, Cavenee WK and Cheng SY: EGFRvIII stimulates glioma
growth and invasion through PKA-dependent serine phosphorylation of
Dock180. Oncogene. 33:2504–2512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eskilsson E, Rosland GV, Talasila KM,
Knappskog S, Keunen O, Sottoriva A, Foerster S, Solecki G, Taxt T,
Jirik R, et al: EGFRvIII mutations can emerge as late and
heterogenous events in glioblastoma development and promote
angiogenesis through Src activation. Neuro Oncol. 18:1644–1655.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roos A, Dhruv HD, Peng S, Inge LJ, Tuncali
S, Pineda M, Millard N, Mayo Z, Eschbacher JM, Loftus JC, et al:
EGFRvIII-Stat5 signaling enhances glioblastoma cell migration and
survival. Mol Cancer Res. 16:1185–1195. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sugawa N, Ekstrand AJ, James CD and
Collins VP: Identical splicing of aberrant epidermal growth factor
receptor transcripts from amplified rearranged genes in human
glioblastomas. Proc Natl Acad Sci USA. 87:8602–8606. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Frederick L, Wang XY, Eley G and James CD:
Diversity and frequency of epidermal growth factor receptor
mutations in human glioblastomas. Cancer Res. 60:1383–1387.
2000.PubMed/NCBI
|
|
24
|
Cruz Da Silva E, Mercier MC,
Etienne-Selloum N, Dontenwill M and Choulier L: A systematic review
of glioblastoma-targeted therapies in phases II, III, IV clinical
trials. Cancers (Basel). 13:17952021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Van Den Bent M, Eoli M, Sepulveda JM,
Smits M, Walenkamp A, Frenel JS, Franceschi E, Clement PM, Chinot
O, De Vos F, et al: INTELLANCE 2/EORTC 1410 randomized Phase II
study of Depatux-M alone and with temozolomide vs temozolomide or
lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol.
22:684–693. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Solomon MT, Miranda N, Jorrín E, Chon I,
Marinello JJ, Alert J, Lorenzo-Luaces P and Crombet T: Nimotuzumab
in combination with radiotherapy in high grade glioma patients: A
single institution experience. Cancer Biol Ther. 15:504–509. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reardon DA, Nabors LB, Mason WP, Perry JR,
Shapiro W, Kavan P, Mathieu D, Phuphanich S, Cseh A, Fu Y, et al:
Phase I/randomized Phase II study of afatinib, an irreversible ErbB
family blocker, with or without protracted temozolomide in adults
with recurrent glioblastoma. Neuro Oncol. 17:430–439.
2015.PubMed/NCBI
|
|
28
|
Sampson JH, Aldape KD, Archer GE, Coan A,
Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE,
McLendon RE, et al: Greater chemotherapy-induced lymphopenia
enhances tumor-specific immune responses that eliminate
EGFRvIII-expressing tumor cells in patients with glioblastoma.
Neuro Oncol. 13:324–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paulsson J, Lindh MB, Jarvius M, Puputti
M, Nistér M, Nupponen NN, Paulus W, Söderberg O, Dresemann G, von
Deimling A, et al: Prognostic but not predictive role of
platelet-derived growth factor receptors in patients with recurrent
glioblastoma. Int J Cancer. 128:1981–1988. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Plate KH, Breier G, Farrell CL and Risau
W: Platelet-derived growth factor receptor-beta is induced during
tumor development and upregulated during tumor progression in
endothelial cells in human gliomas. Lab Invest. 67:529–534.
1992.PubMed/NCBI
|
|
31
|
Camorani S, Esposito CL, Rienzo A,
Catuogno S, Iaboni M, Condorelli G, de Franciscis V and Cerchia L:
Inhibition of receptor signaling and of glioblastoma-derived tumor
growth by a novel PDGFRβ aptamer. Mol Ther. 22:828–841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vassbotn FS, Ostman A, Langeland N,
Holmsen H, Westermark B, Heldin CH and Nistér M: Activated
platelet-derived growth factor autocrine pathway drives the
transformed phenotype of a human glioblastoma cell line. J Cell
Physiol. 158:381–389. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lokker NA, Sullivan CM, Hollenbach SJ,
Israel MA and Giese NA: Platelet-derived growth factor (PDGF)
autocrine signaling regulates survival and mitogenic pathways in
glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D
ligands may play a role in the development of brain tumors. Cancer
Res. 62:3729–3735. 2002.PubMed/NCBI
|
|
34
|
Cattaneo MG, Gentilini D and Vicentini LM:
Deregulated human glioma cell motility: Inhibitory effect of
somatostatin. Mol Cell Endocrinol. 256:34–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kwak Y, Kim SI, Park CK, Paek SH, Lee ST
and Park SH: C-MET overexpression and amplification in gliomas. Int
J Clin Exp Pathol. 8:14932–14938. 2015.PubMed/NCBI
|
|
36
|
Bender S, Gronych J, Warnatz HJ, Hutter B,
Gröbner S, Ryzhova M, Pfaff E, Hovestadt V, Weinberg F, Halbach S,
et al: Recurrent MET fusion genes represent a drug target in
pediatric glioblastoma. Nat Med. 22:1314–1320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wen PY, Schiff D, Cloughesy TF, Raizer JJ,
Laterra J, Smitt M, Wolf M, Oliner KS, Anderson A, Zhu M, et al: A
Phase II study evaluating the efficacy and safety of AMG 102
(rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol.
13:437–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Li A, Glas M, Lal B, Ying M, Sang Y,
Xia S, Trageser D, Guerrero-Cázares H, Eberhart CG, et al: c-Met
signaling induces a reprogramming network and supports the
glioblastoma stem-like phenotype. Proc Natl Acad Sci USA.
108:9951–9956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jahangiri A, De Lay M, Miller LM,
Carbonell WS, Hu YL, Lu K, Tom MW, Paquette J, Tokuyasu TA, Tsao S,
et al: Gene expression profile identifies tyrosine kinase c-Met as
a targetable mediator of antiangiogenic therapy resistance. Clin
Cancer Res. 19:1773–1783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Johnson J, Ascierto ML, Mittal S, Newsome
D, Kang L, Briggs M, Tanner K, Marincola FM, Berens ME, Vande Woude
GF and Xie Q: Genomic profiling of a hepatocyte growth
factor-dependent signature for MET-targeted therapy in
glioblastoma. J Transl Med. 13:3062015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cruickshanks N, Zhang Y, Yuan F, Pahuski
M, Gibert M and Abounader R: Role and therapeutic targeting of the
HGF/MET pathway in glioblastoma. Cancers (Basel). 9:872017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang
Z, Dai Z, Zhang X, Zhang L, Peng Y, et al: Glioma targeted therapy:
Insight into future of molecular approaches. Mol Cancer. 21:392022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cloughesy TF, Mochizuki AY, Orpilla JR,
Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA,
Sanders CM, et al: Neoadjuvant anti-PD-1 immunotherapy promotes a
survival benefit with intratumoral and systemic immune responses in
recurrent glioblastoma. Nat Med. 25:477–486. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Torre M, Vasudevaraja V, Serrano J,
DeLorenzo M, Malinowski S, Blandin AF, Pages M, Ligon AH, Dong F,
Meredith DM, et al: Molecular and clinicopathologic features of
gliomas harboring NTRK fusions. Acta Neuropathol Commun. 8:1072020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jimenez-Pascual A, Mitchell K,
Siebzehnrubl FA and Lathia JD: FGF2: A novel druggable target for
glioblastoma? Expert Opin Ther Targets. 24:311–318. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Feldkamp MM, Lau N and Guha A: The
farnesyltransferase inhibitor L-744,832 inhibits the growth of
astrocytomas through a combination of antiproliferative,
antiangiogenic, and proapoptotic activities. Ann N Y Acad Sci.
886:257–260. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kowalewski A, Durślewicz J, Zdrenka M,
Grzanka D and Szylberg Ł: Clinical relevance of BRAF V600E mutation
status in brain tumors with a focus on a novel management
algorithm. Target Oncol. 15:531–540. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dischinger PS, Tovar EA, Essenburg CJ,
Madaj ZB, Gardner EE, Callaghan ME, Turner AN, Challa AK, Kempston
T, Eagleson B, et al: NF1 deficiency correlates with estrogen
receptor signaling and diminished survival in breast cancer. NPJ
Breast Cancer. 4:292018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Arima Y, Hayashi H, Kamata K, Goto TM,
Sasaki M, Kuramochi A and Saya H: Decreased expression of
neurofibromin contributes to epithelial-mesenchymal transition in
neurofibromatosis type 1. Exp Dermatol. 19:e136–e141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Altwairgi AK, Alghareeb WA, AlNajjar FH,
Alhussain H, Alsaeed E, Balbaid AAO, Aldanan S, Orz Y and Alsharm
AA: Atorvastatin in combination with radiotherapy and temozolomide
for glioblastoma: A prospective Phase II study. Invest New Drugs.
39:226–231. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–519. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mondin VE, Ben El Kadhi K, Cauvin C,
Jackson-Crawford A, Bélanger E, Decelle B, Salomon R, Lowe M,
Echard A and Carréno S: PTEN reduces endosomal
PtdIns(4,5)P2 in a phosphatase-independent manner via a
PLC pathway. J Cell Biol. 218:2198–2214. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Han F, Hu R, Yang H, Liu J, Sui J, Xiang
X, Wang F, Chu L and Song S: PTEN gene mutations correlate to poor
prognosis in glioma patients: A meta-analysis. Onco Targets Ther.
9:3485–3492. 2016.PubMed/NCBI
|
|
54
|
Chen C, Zhu S, Zhang X, Zhou T, Gu J, Xu
Y, Wan Q, Qi X, Chai Y, Liu X, et al: Targeting the synthetic
vulnerability of PTEN-deficient glioblastoma cells with MCL1
inhibitors. Mol Cancer Ther. 19:2001–2011. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF
and Roberts TM: The oncogenic properties of mutant p110alpha and
p110beta phosphatidylinositol 3-kinases in human mammary epithelial
cells. Proc Natl Acad Sci USA. 102:18443–18448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
McLendon R, Friedman A, Bigner D, Van Meir
EG, Brat DJ, Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N,
Aldape K, et al: Comprehensive genomic characterization defines
human glioblastoma genes and core pathways. Nature. 455:1061–1068.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Parsons DW, Jones S, Zhang X, Lin JCH,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Weber GL, Parat MO, Binder ZA, Gallia GL
and Riggins GJ: Abrogation of PIK3CA or PIK3R1 reduces
proliferation, migration, and invasion in glioblastoma multiforme
cells. Oncotarget. 2:833–849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Quayle SN, Lee JY, Cheung LWT, Ding L,
Wiedemeyer R, Dewan RW, Huang-Hobbs E, Zhuang L, Wilson RK, Ligon
KL, et al: Somatic mutations of PIK3R1 promote gliomagenesis. PLoS
One. 7:e494662012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gallia GL, Rand V, Siu IM, Eberhart CG,
James CD, Marie SKN, Oba-Shinjo SM, Carlotti CG, Caballero OL,
Simpson AJ, et al: PIK3CA gene mutations in pediatric and adult
glioblastoma multiforme. Mol Cancer Res. 4:709–714. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tanaka S, Batchelor TT, Iafrate AJ,
Dias-Santagata D, Borger DR, Ellisen LW, Yang D, Louis DN, Cahill
DP and Chi AS: PIK3CA activating mutations are associated with more
disseminated disease at presentation and earlier recurrence in
glioblastoma. Acta Neuropathol Commun. 7:662019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
England B, Huang T and Karsy M: Current
understanding of the role and targeting of tumor suppressor p53 in
glioblastoma multiforme. Tumour Biol. 34:2063–2074. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ohgaki H: Genetic pathways to
glioblastomas. Neuropathology. 25:1–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Olafson LR, Gunawardena M, Nixdorf S,
McDonald KL and Rapkin RW: The role of TP53 gain-of-function
mutation in multifocal glioblastoma. J Neurooncol. 147:37–47. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fontemaggi G, Dell'Orso S, Trisciuoglio D,
Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany
E, et al: The execution of the transcriptional axis mutant p53,
E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol.
16:1086–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK,
Shin YJ, Lee Y, Kim SH, Lee SY, Seo S, et al: TP53 gain-of-function
mutation promotes inflammation in glioblastoma. Cell Death Differ.
26:409–425. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pedrote MM, Motta MF, Ferretti GDS,
Norberto DR, Spohr TCLS, Lima FRS, Gratton E, Silva JL and de
Oliveira GAP: Oncogenic gain of function in glioblastoma is linked
to mutant p53 amyloid oligomers. iScience. 23:1008202020.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tao W and Levine AJ: P19(ARF) stabilizes
p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl
Acad Sci USA. 96:6937–6941. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Nakamura M, Watanabe T, Klangby U, Asker
C, Wiman K, Yonekawa Y, Kleihues P and Ohgaki H: p14ARF deletion
and methylation in genetic pathways to glioblastomas. Brain Pathol.
11:159–168. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Reifenberger G, Liu L, Ichimura K, Schmidt
EE and Collins VP: Amplification and overexpression of the MDM2
gene in a subset of human malignant gliomas without p53 mutations.
Cancer Res. 53:2736–2739. 1993.PubMed/NCBI
|
|
72
|
Riemenschneider MJ, Büschges R, Wolter M,
Reifenberger J, Boström J, Kraus JA, Schlegel U and Reifenberger G:
Amplification and overexpression of the MDM4 (MDMX) gene from 1q32
in a subset of malignant gliomas without TP53 mutation or MDM2
amplification1. Cancer Res. 59:6091–6096. 1999.PubMed/NCBI
|
|
73
|
Xiong Y, Zhang Y, Xiong S and
Williams-Villalobo AE: A glance of p53 functions in brain
development, neural stem cells, and brain cancer. Biology (Basel).
9:2852020.PubMed/NCBI
|
|
74
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Allen BK, Stathias V, Maloof ME, Vidovic
D, Winterbottom EF, Capobianco AJ, Clarke J, Schurer S, Robbins DJ
and Ayad NG: Epigenetic pathways and glioblastoma treatment:
Insights from signaling cascades. J Cell Biochem. 116:351–363.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Romani M, Pistillo MP and Banelli B:
Epigenetic targeting of glioblastoma. Front Oncol. 8:4482018.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Roos WP, Batista LFZ, Naumann SC, Wick W,
Weller M, Menck CFM and Kaina B: Apoptosis in malignant glioma
cells triggered by the temozolomide-induced DNA lesion
O6-methylguanine. Oncogene. 26:186–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yu W, Zhang L, Wei Q and Shao A:
O6-methylguanine-DNA methyltransferase (MGMT): Challenges and new
opportunities in glioma chemotherapy. Front Oncol. 9:15472020.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang J, Chen L, Han L, Shi Z, Zhang J, Pu
P and Kang C: EZH2 is a negative prognostic factor and exhibits
pro-oncogenic activity in glioblastoma. Cancer Lett. 356:929–936.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sharma V, Malgulwar PB, Purkait S, Patil
V, Pathak P, Agrawal R, Kulshreshtha R, Mallick S, Julka PK, Suri
A, et al: Genome-wide ChIP-seq analysis of EZH2-mediated H3K27me3
target gene profile highlights differences between low- and
high-grade astrocytic tumors. Carcinogenesis. 38:152–161.
2017.PubMed/NCBI
|
|
81
|
Mohammad F, Weissmann S, Leblanc B, Pandey
DP, Højfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT,
et al: EZH2 is a potential therapeutic target for H3K27M-mutant
pediatric gliomas. Nat Med. 23:483–492. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lee DH, Kim GW, Yoo J, Lee SW, Jeon YH,
Kim SY, Kang HG, Kim DH, Chun KH, Choi J and Kwon SH: Histone
demethylase KDM4C controls tumorigenesis of glioblastoma by
epigenetically regulating p53 and c-Myc. Cell Death Dis. 12:892021.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ceccacci E and Minucci S: Inhibition of
histone deacetylases in cancer therapy: Lessons from leukaemia. Br
J Cancer. 114:605–611. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lee P, Murphy B, Miller R, Menon V, Banik
NL, Giglio P, Lindhorst SM, Varma AK, Vandergrift WA III, Patel SJ
and Das A: Mechanisms and clinical significance of histone
deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer
Res. 35:615–625. 2015.PubMed/NCBI
|
|
85
|
Chen R, Zhang M, Zhou Y, Guo W, Yi M,
Zhang Z, Ding Y and Wang Y: The application of histone deacetylases
inhibitors in glioblastoma. J Exp Clin Cancer Res. 39:1382020.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Harbour JW, Luo RX, Santi AD, Postigo AA
and Dean DC: Cdk phosphorylation triggers sequential intramolecular
interactions that progressively block Rb functions as cells move
through G1. Cell. 98:859–869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Matsushime H, Ewen ME, Strom DK, Kato JY,
Hanks SK, Roussel MF and Sherr CJ: Identification and properties of
an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type
G1 cyclins. Cell. 71:323–334. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cooper S and Shayman JA: Revisiting
retinoblastoma protein phosphorylation during the mammalian cell
cycle. Cell Mol Life Sci. 58:580–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Taylor JW, Parikh M, Phillips JJ, James
CD, Molinaro AM, Butowski NA, Clarke JL, Oberheim-Bush NA, Chang
SM, Berger MS and Prados M: Phase-2 trial of palbociclib in adult
patients with recurrent RB1-positive glioblastoma. J Neurooncol.
140:477–483. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Goldberg AL: Protein degradation and
protection against misfolded or damaged proteins. Nature.
426:895–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Narayanan S, Cai CY, Assaraf YG, Guo HQ,
Cui Q, Wei L, Huang JJ, Ashby CR Jr and Chen ZS: Targeting the
ubiquitin-proteasome pathway to overcome anti-cancer drug
resistance. Drug Resist Updat. 48:1006632020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kong XT, Nguyen NT, Choi YJ, Zhang G,
Nguyen HN, Filka E, Green S, Yong WH, Liau LM, Green RM, et al:
Phase 2 study of bortezomib combined with temozolomide and regional
radiation therapy for upfront treatment of patients with newly
diagnosed glioblastoma multiforme: safety and efficacy assessment.
Int J Radiat Oncol Biol Phys. 100:1195–1203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Di K, Lloyd GK, Abraham V, MacLaren A,
Burrows FJ, Desjardins A, Trikha M and Bota DA: Marizomib activity
as a single agent in malignant gliomas: Ability to cross the
blood-brain barrier. Neuro Oncol. 18:840–848. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Roth P, Mason WP, Richardson PG and Weller
M: Proteasome inhibition for the treatment of glioblastoma. Expert
Opin Investig Drugs. 29:1133–1141. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Quillin J, Patel R, Herzberg E, Alton D,
Bikzhanova G, Geisler L and Olson J: A phase 0 analysis of ixazomib
in patients with glioblastoma. Mol Clin Oncol. 13:432020.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Huang J, Campian JL, Gujar AD, Tsien C,
Ansstas G, Tran DD, DeWees TA, Lockhart AC and Kim AH: Final
results of a Phase I dose-escalation, dose-expansion study of
adding disulfiram with or without copper to adjuvant temozolomide
for newly diagnosed glioblastoma. J Neurooncol. 138:105–111. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Huang J, Chaudhary R, Cohen AL, Fink K,
Goldlust S, Boockvar J, Chinnaiyan P, Wan L, Marcus S and Campian
JL: A multicenter Phase II study of temozolomide plus disulfiram
and copper for recurrent temozolomide-resistant glioblastoma. J
Neurooncol. 142:537–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Carruthers R and Chalmers AJ: Improving
the therapeutic ratio of radiotherapy by targeting the DNA damage
response. Increasing the Therapeutic Ratio of Radiotherapy. Series:
Cancer Drug Discovery and Development. Tofilon PJ and Camphausen K:
Humana Press; Cham: pp. 1–34. 2017, View Article : Google Scholar
|
|
99
|
Murnyák B, Kouhsari MC, Hershkovitch R,
Kálmán B, Marko-Varga G, Klekner Á and Hortobágyi T: PARP1
expression and its correlation with survival is tumour molecular
subtype dependent in glioblastoma. Oncotarget. 8:46348–46362. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chalmers AJ: Overcoming resistance of
glioblastoma to conventional cytotoxic therapies by the addition of
PARP inhibitors. Anticancer Agents Med Chem. 10:520–533. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hanna C, Kurian KM, Williams K, Watts C,
Jackson A, Carruthers R, Strathdee K, Cruickshank G, Dunn L,
Erridge S, et al: Pharmacokinetics, safety, and tolerability of
olaparib and temozolomide for recurrent glioblastoma: Results of
the Phase I OPARATIC trial. Neuro Oncol. 22:1840–1850. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Robins HI, Zhang P, Gilbert MR,
Chakravarti A, de Groot JF, Grimm SA, Wang F, Lieberman FS, Krauze
A, Trotti AM, et al: A randomized Phase I/II study of ABT-888 in
combination with temozoflomide in recurrent temozolomide resistant
glioblastoma: An NRG oncology RTOG group study. J Neurooncol.
126:309–316. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Baxter PA, Su JM, Onar-Thomas A, Billups
CA, Li XN, Poussaint TY, Smith ER, Thompson P, Adesina A, Ansell P,
et al: A Phase I/II study of veliparib (ABT-888) with radiation and
temozolomide in newly diagnosed diffuse pontine glioma: A pediatric
brain tumor consortium study. Neuro Oncol. 22:875–885. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organi-zation
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jo SH, Son MK, Koh HJ, Lee SM, Song IH,
Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, et al: Control of
mitochondrial redox balance and cellular defense against oxidative
damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J
Biol Chem. 276:16168–16176. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lee SM, Koh HJ, Park DC, Song BJ, Huh TL
and Park JW: Cytosolic NADP(+)-dependent isocitrate dehydrogenase
status modulates oxidative damage to cells. Free Radic Biol Med.
32:1185–1196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kim SY and Park JW: Cellular defense
against singlet oxygen-induced oxidative damage by cytosolic
NADP+-dependent isocitrate dehydrogenase. Free Radic Res.
37:309–316. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Krell D, Assoku M, Galloway M, Mulholland
P, Tomlinson I and Bardella C: Screen for IDH1, IDH2, IDH3, D2HGDH
and L2HGDH mutations in glioblastoma. PLoS One. 6:e198682011.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Arita H, Narita Y, Yoshida A, Hashimoto N,
Yoshimine T and Ichimura K: IDH1/2 mutation detection in gliomas.
Brain Tumor Pathol. 32:79–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Capper D, Weissert S, Balss J, Habel A,
Meyer J, Jäger D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H,
et al: Characterization of R132H mutation-specific IDH1 antibody
binding in brain tumors. Brain Pathol. 20:245–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan
S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et
al: IDH mutation impairs histone demethylation and results in a
block to cell differentiation. Nature. 483:474–478. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yalaza C, Ak H, Cagli MS, Ozgiray E, Atay
S and Aydin HH: R132H mutation in IDH1 gene is associated with
increased tumor HIF1-alpha and serum VEGF levels in primary
glioblastoma multiforme. Ann Clin Lab Sci. 47:362–364.
2017.PubMed/NCBI
|
|
116
|
Flavahan WA, Drier Y, Liau BB, Gillespie
SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML and Bernstein BE:
Insulator dysfunction and oncogene activation in IDH mutant
gliomas. Nature. 529:110–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fu Y, Zheng S, Zheng Y, Huang R, An N,
Liang A and Hu C: Glioma derived isocitrate dehydrogenase-2
mutations induced up-regulation of HIF-1α and β-catenin signaling:
Possible impact on glioma cell metastasis and chemo-resistance. Int
J Biochem Cell Biol. 44:770–775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Stern R, Shuster S, Neudecker BA and
Formby B: Lactate stimulates fibroblast expression of hyaluronan
and CD44: The Warburg effect revisited. Exp Cell Res. 276:24–31.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer's Achilles' heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Saier MH Jr: Families of transmembrane
sugar transport proteins. Mol Microbiol. 35:699–710. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Scheepers A, Joost H and Schürmann A: The
glucose transporter families SGLT and GLUT: Molecular basis of
normal and aberrant function. JPEN J Parenter Enteral Nutr.
28:364–371. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim
Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, et al:
Brain tumor initiating cells adapt to restricted nutrition through
preferential glucose uptake. Nat Neurosci. 16:1373–1382. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Libby CJ, Gc S, Benavides GA, Fisher JL,
Williford SE, Zhang S, Tran AN, Gordon ER, Jones AB, Tuy K, et al:
A role for GLUT3 in glioblastoma cell invasion that is not
recapitulated by GLUT1. Cell Adh Migr. 15:101–115. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wolf A, Agnihotri S, Micallef J, Mukherjee
J, Sabha N, Cairns R, Hawkins C and Guha A: Hexokinase 2 is a key
mediator of aerobic glycolysis and promotes tumor growth in human
glioblastoma multiforme. J Exp Med. 208:313–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Vartanian A, Agnihotri S, Wilson MR,
Burrell KE, Tonge PD, Alamsahebpour A, Jalali S, Taccone MS,
Mansouri S, Golbourn B, et al: Targeting hexokinase 2 enhances
response to radio-chemotherapy in glioblastoma. Oncotarget.
7:69518–69535. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Pastorino JG, Shulga N and Hoek JB:
Mitochondrial binding of hexokinase II inhibits Bax-induced
cytochrome c release and apoptosis. J Biol Chem. 277:7610–7618.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Agnihotri S, Mansouri S, Burrell K, Li M,
Mamatjan Y, Liu J, Nejad R, Kumar S, Jalali S, Singh SK, et al:
Ketoconazole and posaconazole selectively target HK2-expressing
glioblastoma cells. Clin Cancer Res. 25:844–855. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Harada K, Saheki S, Wada K and Tanaka T:
Purification of four pyruvate kinase isozymes of rats by affinity
elution chromatography. Biochim Biophys Acta. 524:327–339. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Verma H, Cholia RP, Kaur S, Dhiman M and
Mantha AK: A short review on cross-link between pyruvate kinase
(PKM2) and glioblastoma multiforme. Metab Brain Dis. 36:751–765.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Uyeda K: Pyruvate kinase. Encyclopedia of
Biological Chemistry. (2nd edition). Lennarz WJ and Lane MD:
Academic Press; Waltham: pp. 719–721. 2013, Available from:.
https://www.sciencedirect.com/science/article/pii/B9780123786302000530
View Article : Google Scholar
|
|
133
|
Mukherjee J, Phillips JJ, Zheng S, Wiencke
J, Ronen SM and Pieper RO: Pyruvate kinase M2 expression, but not
pyruvate kinase activity, is up-regulated in a grade-specific
manner in human glioma. PLoS One. 8:e576102013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Alfred Yung WK and Lu Z: PKM2
phosphorylates histone H3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Jiang Y, Li X, Yang W, Hawke DH, Zheng Y,
Xia Y, Aldape K, Wei C, Guo F, Chen Y and Lu Z: PKM2 regulates
chromosome segregation and mitosis progression of tumor cells. Mol
Cell. 53:75–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Cholia RP, Dhiman M, Kumar R and Mantha
AK: Oxidative stress stimulates invasive potential in rat C6 and
human U-87 MG glioblastoma cells via activation and cross-talk
between PKM2, ENPP2 and APE1 enzymes. Metab Brain Dis.
33:1307–1326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Liang J, Cao R, Wang X, Zhang Y, Wang P,
Gao H, Li C, Yang F, Zeng R, Wei P, et al: Mitochondrial PKM2
regulates oxidative stress-induced apoptosis by stabilizing Bcl2.
Cell Res. 27:329–351. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Chaneton B and Gottlieb E: Rocking cell
metabolism: Revised functions of the key glycolytic regulator PKM2
in cancer. Trends Biochem Sci. 37:309–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sizemore ST, Zhang M, Cho JH, Sizemore GM,
Hurwitz B, Kaur B, Lehman NL, Ostrowski MC, Robe PA, Miao W, et al:
Pyruvate kinase M2 regulates homologous recombination-mediated DNA
double-strand break repair. Cell Res. 28:1090–1102. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Rieger J, Bähr O, Maurer GD, Hattingen E,
Franz K, Brucker D, Walenta S, Kämmerer U, Coy JF, Weller M and
Steinbach JP: ERGO: A pilot study of ketogenic diet in recurrent
glioblastoma. Int J Oncol. 44:1843–1852. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Kucharzewska P, Christianson HC and
Belting M: Global profiling of metabolic adaptation to hypoxic
stress in human glioblastoma cells. PLoS One. 10:e01167402015.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ogunrinu TA and Sontheimer H: Hypoxia
increases the dependence of glioma cells on glutathione. J Biol
Chem. 285:37716–37724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Robertson DS: The physical chemistry of
brain and neural cell membranes: An overview. Neurochem Res.
35:681–687. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Newsholme P, Lima MMR, Procopio J,
Pithon-Curi TC, Doi SQ, Bazotte RB and Curi R: Glutamine and
glutamate as vital metabolites. Braz J Med Biol Res. 36:153–163.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Bak LK, Schousboe A and Waagepetersen HS:
The glutamate/GABA-glutamine cycle: Aspects of transport,
neurotransmitter homeostasis and ammonia transfer. J Neurochem.
98:641–653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Obara-Michlewska M and Szeliga M:
Targeting glutamine addiction in gliomas. Cancers (Basel).
12:3102020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Michalak KP, Maćkowska-Kędziora A,
Sobolewski B and Woźniak P: Key roles of glutamine pathways in
reprogramming the cancer metabolism. Oxid Med Cell Longev.
2015:9643212015. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Rubin H: Deprivation of glutamine in cell
culture reveals its potential for treating cancer. Proc Natl Acad
Sci USA. 116:6964–6968. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ekici S, Nye JA, Neill SG, Allen JW, Shu
HK and Fleischer CC: Glutamine imaging: A new avenue for glioma
management. AJNR Am J Neuroradiol. 43:11–18. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
de Groot JF, Liu TJ, Fuller G and Yung
WKA: The excitatory amino acid transporter-2 induces apoptosis and
decreases glioma growth in vitro and in vivo. Cancer Res.
65:1934–1940. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lyons SA, Chung WJ, Weaver AK, Ogunrinu T
and Sontheimer H: Autocrine glutamate signaling promotes glioma
cell invasion. Cancer Res. 67:9463–9471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Dranoff G, Elion GB, Friedman HS and
Bigner DD: Combination chemotherapy in vitro exploiting glutamine
metabolism of human glioma and medulloblastoma. Cancer Res.
45:4082–4086. 1985.PubMed/NCBI
|
|
154
|
Seltzer MJ, Bennett BD, Joshi AD, Gao P,
Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS,
Rabinowitz JD, et al: Inhibition of glutaminase preferentially
slows growth of glioma cells with mutant IDH1. Cancer Res.
70:8981–8987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Majewska E, Márquez J, Albrecht J and
Szeliga M: Transfection with GLS2 glutaminase (GAB) sensitizes
human glioblastoma cell lines to oxidative stress by a common
mechanism involving suppression of the PI3K/AKT pathway. Cancers
(Basel). 11:1152019. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Szeliga M, Obara-Michlewska M, Matyja E,
Łazarczyk M, Lobo C, Hilgier W, Alonso FJ, Márquez J and Albrecht
J: Transfection with liver-type glutaminase cDNA alters gene
expression and reduces survival, migration and proliferation of
T98G glioma cells. Glia. 57:1014–1023. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Szeliga M, Zgrzywa A, Obara-Michlewska M
and Albrecht J: Transfection of a human glioblastoma cell line with
liver-type glutaminase (LGA) down-regulates the expression of
DNA-repair gene MGMT and sensitizes the cells to alkylating agents.
J Neurochem. 123:428–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Yin Y, Sun W, Xiang J, Deng L, Zhang B,
Xie P, Qiao W, Zou J and Liu C: Glutamine synthetase functions as a
negative growth regulator in glioma. J Neurooncol. 114:59–69. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ye ZC, Rothstein JD and Sontheimer H:
Compromised glutamate transport in human glioma cells:
Reduction-mislocalization of sodium-dependent glutamate
transporters and enhanced activity of cystine-glutamate exchange. J
Neurosci. 19:10767–10777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chung WJ, Lyons SA, Nelson GM, Hamza H,
Gladson CL, Gillespie GY and Sontheimer H: Inhibition of cystine
uptake disrupts the growth of primary brain tumors. J Neurosci.
25:7101–7110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Robe PA, Martin DH, Nguyen-Khac MT, Artesi
M, Deprez M, Albert A, Vanbelle S, Califice S, Bredel M and Bours
V: Early termination of ISRCTN45828668, a phase 1/2 prospective,
randomized study of sulfasalazine for the treatment of progressing
malignant gliomas in adults. BMC Cancer. 9:3722009. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Sleire L, Skeie BS, Netland IA, Førde HE,
Dodoo E, Selheim F, Leiss L, Heggdal JI, Pedersen PH, Wang J and
Enger PØ: Drug repurposing: Sulfasalazine sensitizes gliomas to
gamma knife radiosurgery by blocking cystine uptake through system
Xc-, leading to glutathione depletion. Oncogene. 34:5951–5959.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Choi YK and Park KG: Targeting glutamine
metabolism for cancer treatment. Biomol Ther (Seoul). 26:19–28.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Morris SM Jr: Enzymes of arginine
metabolism. J Nutr. 134 (Suppl 10):2743S–2747S. 2765S–2767S. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Kuo MT, Savaraj N and Feun LG: Targeted
cellular metabolism for cancer chemotherapy with recombinant
arginine-degrading enzymes. Oncotarget. 1:246–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Khoury O, Ghazale N, Stone E, El-Sibai M,
Frankel AE and Abi-Habib RJ: Human recombinant arginase I
(Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is
selectively cytotoxic to human glioblastoma cells. J Neurooncol.
122:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Choy CT, Wong CH and Loong HHF: Low
expressions of ASS1 and OTC in glioblastoma suggest the potential
clinical use of recombinant human arginase (rhArg). J Neurooncol.
129:579–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Bobak Y, Kurlishchuk Y,
Vynnytska-Myronovska B, Grydzuk O, Shuvayeva G, Redowicz MJ,
Kunz-Schughart LA and Stasyk O: Arginine deprivation induces
endoplasmic reticulum stress in human solid cancer cells. Int J
Biochem Cell Biol. 70:29–38. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Pavlyk I, Rzhepetskyy Y, Jagielski AK,
Drozak J, Wasik A, Pereverzieva G, Olchowik M, Kunz-Schugart LA,
Stasyk O and Redowicz MJ: Arginine deprivation affects glioblastoma
cell adhesion, invasiveness and actin cytoskeleton organization by
impairment of β-actin arginylation. Amino Acids. 47:199–212. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Stasyk OV, Boretsky YR, Gonchar MV and
Sibirny AA: Recombinant arginine-degrading enzymes in metabolic
anticancer therapy and bioanalytics. Cell Biol Int. 39:246–252.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Stone EM, Chantranupong L and Georgiou G:
The second-shell metal ligands of human arginase affect
coordination of the nucleophile and substrate. Biochemistry.
49:10582–10588. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Al-Koussa H, Al-Haddad M, Abi-Habib R and
El-Sibai M: Human recombinant arginase I [HuArgI
(Co)-PEG5000]-induced arginine depletion inhibits colorectal cancer
cell migration and invasion. Int J Mol Sci. 20:60182019. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Khalil N and Abi-Habib RJ: [HuArgI
(co)-PEG5000]-induced arginine deprivation leads to autophagy
dependent cell death in pancreatic cancer cells. Invest New Drugs.
38:1236–1246. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Tanios R, Bekdash A, Kassab E, Stone E,
Georgiou G, Frankel AE and Abi-Habib RJ: Human recombinant arginase
I(Co)-PEG5000 [HuArgI(Co)-PEG5000]-induced arginine depletion is
selectively cytotoxic to human acute myeloid leukemia cells. Leuk
Res. 37:1565–1571. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Nasreddine G, El-Sibai M and Abi-Habib RJ:
Cytotoxicity of [HuArgI (co)-PEG5000]-induced arginine deprivation
to ovarian cancer cells is autophagy dependent. Invest New Drugs.
38:10–19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Glazer ES, Stone EM, Zhu C, Massey KL,
Hamir AN and Curley SA: Bioengineered human arginase I with
enhanced activity and stability controls hepatocellular and
pancreatic carcinoma xenografts. Transl Oncol. 4:138–146. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Yoon CY, Sung DJ, Lee JH, Kim AR, Oh CW,
Je JH, Weon BM, Seol SK, Pyun A, Hwu Y, et al: Imaging of renal and
prostate carcinoma with refractive index radiology. Int J Urol.
14:96–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Hsueh EC, Knebel SM, Lo WH, Leung YC,
Cheng PNM and Hsueh CT: Deprivation of arginine by recombinant
human arginase in prostate cancer cells. J Hematol Oncol. 5:172012.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang Z, Shi X, Li Y, Fan J, Zeng X, Xian
Z, Wang Z, Sun Y, Wang S, Song P, et al: Blocking autophagy
enhanced cytotoxicity induced by recombinant human arginase in
triple-negative breast cancer cells. Cell Death Dis. 5:e15632014.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Hou X, Chen S, Zhang P, Guo D and Wang B:
Targeted arginine metabolism therapy: A dilemma in glioma
treatment. Front Oncol. 12:9388472022. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Arita H, Narita Y, Fukushima S, Tateishi
K, Matsushita Y, Yoshida A, Miyakita Y, Ohno M, Collins VP,
Kawahara N, et al: Upregulating mutations in the TERT promoter
commonly occur in adult malignant gliomas and are strongly
associated with total 1p19q loss. Acta Neuropathol. 126:267–276.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Nonoguchi N, Ohta T, Oh JE, Kim YH,
Kleihues P and Ohgaki H: TERT promoter mutations in primary and
secondary glioblastomas. Acta Neuropathol. 126:931–937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda
C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL,
Giovanella BC, et al: TERT promoter mutations occur frequently in
gliomas and a subset of tumors derived from cells with low rates of
self-renewal. Proc Natl Acad Sci USA. 110:6021–6066. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Arita H, Yamasaki K, Matsushita Y,
Nakamura T, Shimokawa A, Takami H, Tanaka S, Mukasa A, Shirahata M,
Shimizu S, et al: A combination of TERT promoter mutation and MGMT
methylation status predicts clinically relevant subgroups of newly
diagnosed glioblastomas. Acta Neuropathol Commun. 4:792016.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Kikuchi Z, Shibahara I, Yamaki T, Yoshioka
E, Shofuda T, Ohe R, Matsuda KI, Saito R, Kanamori M, Kanemura Y,
et al: TERT promoter mutation associated with multifocal phenotype
and poor prognosis in patients with IDH wild-type glioblastoma.
Neurooncol Adv. 2:vdaa1142020.PubMed/NCBI
|
|
186
|
Vuong HG, Nguyen TQ, Ngo TNM, Nguyen HC,
Fung KM and Dunn IF: The interaction between TERT promoter mutation
and MGMT promoter methylation on overall survival of glioma
patients: A meta-analysis. BMC Cancer. 20:8972020. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Patel B, Taiwo R, Kim AH and Dunn GP:
TERT, a promoter of CNS malignancies. Neurooncol Adv.
2:vdaa0252020.PubMed/NCBI
|
|
188
|
Picketts DJ, Higgs DR, Bachoo S, Blake DJ,
Quarrell OW and Gibbons RJ: ATRX encodes a novel member of the SNF2
family of proteins: Mutations point to a common mechanism
underlying the ATR-X syndrome. Hum Mol Genet. 5:1899–1907. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Jiao Y, Killela PJ, Reitman ZJ, Rasheed
BA, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo
SM, Nagahashi Marie SK, et al: Frequent ATRX, CIC, FUBP1 and IDH1
mutations refine the classification of malignant gliomas.
Oncotarget. 3:709–722. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Koschmann C, Calinescu AA, Nunez FJ,
Mackay A, Fazal-Salom J, Thomas D, Mendez F, Kamran N, Dzaman M,
Mulpuri L, et al: ATRX loss promotes tumor growth and impairs
nonhomologous end joining DNA repair in glioma. Sci Transl Med.
8:328ra282016. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
do Carmo A, Balça-Silva J, Matias D and
Lopes MC: PKC signaling in glioblastoma. Cancer Biol Ther.
14:287–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Steinberg SF: Structural basis of protein
kinase C isoform function. Physiol Rev. 88:1341–1378. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Leirdal M and Sioud M: Protein kinase
Calpha isoform regulates the activation of the MAP kinase ERK1/2 in
human glioma cells: Involvement in cell survival and gene
expression. Mol Cell Biol Res Commun. 4:106–110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Marquina-Sánchez B, González-Jorge J,
Hansberg-Pastor V, Wegman-Ostrosky T, Baranda-Ávila N, Mejía-Pérez
S, Camacho-Arroyo I and González-Arenas A: The interplay between
intracellular progesterone receptor and PKC plays a key role in
migration and invasion of human glioblastoma cells. J Steroid
Biochem Mol Biol. 172:198–206. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Valdés-Rives SA, Arcos-Montoya D, de la
Fuente-Granada M, Zamora-Sánchez CJ, Arias-Romero LE, Villamar-Cruz
O, Camacho-Arroyo I, Pérez-Tapia SM and González-Arenas A: LPA1
receptor promotes progesterone receptor phosphorylation through
PKCα in human glioblastoma cells. Cells. 10:8072021. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Yoshiji H, Kuriyama S, Ways DK, Yoshii J,
Miyamoto Y, Kawata M, Ikenaka Y, Tsujinoue H, Nakatani T, Shibuya M
and Fukui H: Protein kinase C lies on the signaling pathway for
vascular endothelial growth factor-mediated tumor development and
angiogenesis. Cancer Res. 59:4413–4418. 1999.PubMed/NCBI
|
|
197
|
Liu Z, Wei Y, Zhang L, Yee PP, Johnson M,
Zhang X, Gulley M, Atkinson JM, Trebak M, Wang HG and Li W:
Induction of store-operated calcium entry (SOCE) suppresses
glioblastoma growth by inhibiting the Hippo pathway transcriptional
coactivators YAP/TAZ. Oncogene. 38:120–139. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Chen Z, Forman LW, Williams RM and Faller
DV: Protein kinase C-δ inactivation inhibits the proliferation and
survival of cancer stem cells in culture and in vivo. BMC Cancer.
14:902014. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Kim MJ, Kim RK, Yoon CH, An S, Hwang SG,
Suh Y, Park MJ, Chung HY, Kim IG and Lee SJ: Importance of PKCδ
signaling in fractionated-radiation-induced expansion of
glioma-initiating cells and resistance to cancer treatment. J Cell
Sci. 124:3084–3094. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Sarkar S and Yong VW: Reduction of protein
kinase C delta attenuates tenascin-C stimulated glioma invasion in
three-dimensional matrix. Carcinogenesis. 31:311–317. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Lu J, Xu Z, Duan H, Ji H, Zhen Z, Li B,
Wang H, Tang H, Zhou J, Guo T, et al: Tumor-associated macrophage
interleukin-β promotes glycerol-3-phosphate dehydrogenase
activation, glycolysis and tumorigenesis in glioma cells. Cancer
Sci. 111:1979–1990. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Sharif TR and Sharif M: Overexpression of
protein kinase C epsilon in astroglial brain tumor derived cell
lines and primary tumor samples. Int J Oncol. 15:237–243.
1999.PubMed/NCBI
|
|
203
|
Okhrimenko H, Lu W, Xiang C, Hamburger N,
Kazimirsky G and Brodie C: Protein kinase C-epsilon regulates the
apoptosis and survival of glioma cells. Cancer Res. 65:7301–7309.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Besson A, Davy A, Robbins SM and Yong VW:
Differential activation of ERKs to focal adhesions by PKC epsilon
is required for PMA-induced adhesion and migration of human glioma
cells. Oncogene. 20:7398–7407. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Besson A, Wilson TL and Yong VW: The
anchoring protein RACK1 links protein kinase cepsilon to integrin
beta chains. Requirements for adhesion and motility. J Biol Chem.
277:22073–22084. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Aeder SE, Martin PM, Soh JW and Hussaini
IM: PKC-eta mediates glioblastoma cell proliferation through the
Akt and mTOR signaling pathways. Oncogene. 23:9062–9069. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Uht RM, Amos S, Martin PM, Riggan AE and
Hussaini IM: The protein kinase C-eta isoform induces proliferation
in glioblastoma cell lines through an ERK/Elk-1 pathway. Oncogene.
26:2885–2893. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Hussaini IM, Carpenter JE, Redpath GT,
Sando JJ, Shaffrey ME and Vandenberg SR: Protein kinase C-eta
regulates resistance to UV- and gamma-irradiation-induced apoptosis
in glioblastoma cells by preventing caspase-9 activation. Neuro
Oncol. 4:9–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Phillips E, Lang V, Bohlen J, Bethke F,
Puccio L, Tichy D, Herold-Mende C, Hielscher T, Lichter P and
Goidts V: Targeting atypical protein kinase C iota reduces
viability in glioblastoma stem-like cells via a notch signaling
mechanism. Int J Cancer. 139:1776–1787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Guo H, Gu F, Li W, Zhang B, Niu R, Fu L,
Zhang N and Ma Y: Reduction of protein kinase C zeta inhibits
migration and invasion of human glioblastoma cells. J Neurochem.
109:203–213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Baldwin RM, Parolin DA and Lorimer IA:
Regulation of glioblastoma cell invasion by PKC iota and RhoB.
Oncogene. 27:3587–3595. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Odia Y, Iwamoto FM, Moustakas A, Fraum TJ,
Salgado CA, Li A, Kreisl TN, Sul J, Butman JA and Fine HA: A Phase
II trial of enzastaurin (LY317615) in combination with bevacizumab
in adults with recurrent malignant gliomas. J Neurooncol.
127:127–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sevastre AS, Costachi A, Tataranu LG,
Brandusa C, Artene SA, Stovicek O, Alexandru O, Danoiu S, Sfredel V
and Dricu A: Glioblastoma pharmacotherapy: A multifaceted
perspective of conventional and emerging treatments (review). Exp
Ther Med. 22:14082021. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Johnson BE, Mazor T, Hong C, Barnes M,
Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et
al: Mutational analysis reveals the origin and therapy-driven
evolution of recurrent glioma. Science. 343:189–193. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Stieber D, Golebiewska A, Evers L,
Lenkiewicz E, Brons NHC, Nicot N, Oudin A, Bougnaud S, Hertel F,
Bjerkvig R, et al: Glioblastomas are composed of genetically
divergent clones with distinct tumourigenic potential and variable
stem cell-associated phenotypes. Acta Neuropathol. 127:203–219.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Neftel C, Laffy J, Filbin MG, Hara T,
Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM,
et al: An integrative model of cellular states, plasticity, and
genetics for glioblastoma. Cell. 178:835–849.e21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Trapnell C, Williams BA, Pertea G,
Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter
L: Transcript assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation. Nat Biotechnol. 28:511–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Lauko A, Lo A, Ahluwalia MS and Lathia JD:
Cancer cell heterogeneity and plasticity in glioblastoma and brain
tumors. Semin Cancer Biol. 82:162–175. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Snuderl M, Fazlollahi L, Le LP, Nitta M,
Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD,
Betensky RA, et al: Mosaic amplification of multiple receptor
tyrosine kinase genes in glioblastoma. Cancer Cell. 20:810–817.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Nobusawa S, Lachuer J, Wierinckx A, Kim
YH, Huang J, Legras C, Kleihues P and Ohgaki H: Intratumoral
patterns of genomic imbalance in glioblastomas. Brain Pathol.
20:936–944. 2010.PubMed/NCBI
|
|
221
|
Kumar A, Boyle EA, Tokita M, Mikheev AM,
Sanger MC, Girard E, Silber JR, Gonzalez-Cuyar LF, Hiatt JB, Adey
A, et al: Deep sequencing of multiple regions of glial tumors
reveals spatial heterogeneity for mutations in clinically relevant
genes. Genome Biol. 15:5302014. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Bhaduri A, Di Lullo E, Jung D, Müller S,
Crouch EE, Espinosa CS, Ozawa T, Alvarado B, Spatazza J, Cadwell
CR, et al: Outer radial glia-like cancer stem cells contribute to
heterogeneity of glioblastoma. Cell Stem Cell. 26:48–63.e6. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Pang B, Xu J, Hu J, Guo F, Wan L, Cheng M
and Pang L: Single-cell RNA-seq reveals the invasive trajectory and
molecular cascades underlying glioblastoma progression. Mol Oncol.
13:2588–2603. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Jacob F, Salinas RD, Zhang DY, Nguyen PTT,
Schnoll JG, Wong SZH, Thokala R, Sheikh S, Saxena D, Prokop S, et
al: A patient-derived glioblastoma organoid model and biobank
recapitulates inter- and intra-tumoral heterogeneity. Cell.
180:188–204.e22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Iwadate Y: Plasticity in glioma stem cell
phenotype and its therapeutic implication. Neurol Med Chir (Tokyo).
58:61–70. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Koso H, Takeda H, Yew CCK, Ward JM, Nariai
N, Ueno K, Nagasaki M, Watanabe S, Rust AG, Adams DJ, et al:
Transposon mutagenesis identifies genes that transform neural stem
cells into glioma-initiating cells. Proc Natl Acad Sci USA.
109:E2998–E3007. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Wang X, Prager BC, Wu Q, Kim LJY, Gimple
RC, Shi Y, Yang K, Morton AR, Zhou W, Zhu Z, et al: Reciprocal
signaling between glioblastoma stem cells and differentiated tumor
cells promotes malignant progression. Cell Stem Cell.
22:514–528.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Hsu JF, Chu SM, Liao CC, Wang CJ, Wang YS,
Lai MY, Wang HC, Huang HR and Tsai MH: Nanotechnology and
nanocarrier-based drug delivery as the potential therapeutic
strategy for glioblastoma multiforme: An update. Cancers (Basel).
13:1952021. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Lathia JD, Heddleston JM, Venere M and
Rich JN: Deadly teamwork: Neural cancer stem cells and the tumor
microenvironment. Cell Stem Cell. 8:482–485. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Yuan X, Curtin J, Xiong Y, Liu G,
Waschsmann-Hogiu S, Farkas DL, Black KL and Yu JS: Isolation of
cancer stem cells from adult glioblastoma multiforme. Oncogene.
23:9392–9400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Brescia P, Ortensi B, Fornasari L, Levi D,
Broggi G and Pelicci G: CD133 is essential for glioblastoma stem
cell maintenance. Stem Cells. 31:857–869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Carlsson SK, Brothers SP and Wahlestedt C:
Emerging treatment strategies for glioblastoma multiforme. EMBO Mol
Med. 6:1359–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Nusse R: Wnt signaling and stem cell
control. Cell Res. 18:523–527. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Sareddy GR, Kesanakurti D, Kirti PB and
Babu PP: Nonsteroidal anti-inflammatory drugs diclofenac and
celecoxib attenuates Wnt/β-catenin/Tcf signaling pathway in human
glioblastoma cells. Neurochem Res. 38:2313–2322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Yu JB, Jiang H and Zhan RY: Aberrant Notch
signaling in glioblastoma stem cells contributes to tumor
recurrence and invasion. Mol Med Rep. 14:1263–1268. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Shih IM and Wang TL: Notch signaling,
gamma-secretase inhibitors, and cancer therapy. Cancer Res.
67:1879–1882. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
de la Iglesia N, Puram SV and Bonni A:
STAT3 regulation of glioblastoma pathogenesis. Curr Mol Med.
9:580–590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Porporato PE, Filigheddu N, Pedro JMBS,
Kroemer G and Galluzzi L: Mitochondrial metabolism and cancer. Cell
Res. 28:265–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Sighel D, Notarangelo M, Aibara S, Re A,
Ricci G, Guida M, Soldano A, Adami V, Ambrosini C, Broso F, et al:
Inhibition of mitochondrial translation suppresses glioblastoma
stem cell growth. Cell Rep. 35:1090242021. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Mai TT, Hamaï A, Hienzsch A, Cañeque T,
Müller S, Wicinski J, Cabaud O, Leroy C, David A, Acevedo V, et al:
Salinomycin kills cancer stem cells by sequestering iron in
lysosomes. Nat Chem. 9:1025–1033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Sharifzad F, Ghavami S, Verdi J, Mardpour
S, Mollapour Sisakht M, Azizi Z, Taghikhani A, Łos MJ, Fakharian E,
Ebrahimi M and Hamidieh AA: Glioblastoma cancer stem cell biology:
Potential theranostic targets. Drug Resist Updat. 42:35–45. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Saunders NR, Dreifuss JJ, Dziegielewska
KM, Johansson PA, Habgood MD, Møllgård K and Bauer HC: The rights
and wrongs of blood-brain barrier permeability studies: A walk
through 100 years of history. Front Neurosci. 8:4042014. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Daneman R and Prat A: The blood-brain
barrier. Cold Spring Harb Perspect Biol. 7:a0204122015. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Liebner S, Fischmann A, Rascher G, Duffner
F, Grote EH, Kalbacher H and Wolburg H: Claudin-1 and claudin-5
expression and tight junction morphology are altered in blood
vessels of human glioblastoma multiforme. Acta Neuropathol.
100:323–331. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Wolburg H, Wolburg-Buchholz K, Kraus J,
Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W
and Engelhardt B: Localization of claudin-3 in tight junctions of
the blood-brain barrier is selectively lost during experimental
autoimmune encephalomyelitis and human glioblastoma multiforme.
Acta Neuropathol. 105:586–592. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Ishihara H, Kubota H, Lindberg RLP,
Leppert D, Gloor SM, Errede M, Virgintino D, Fontana A, Yonekawa Y
and Frei K: Endothelial cell barrier impairment induced by
glioblastomas and transforming growth factor beta2 involves matrix
metalloproteinases and tight junction proteins. J Neuropathol Exp
Neurol. 67:435–448. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Rama AR, Alvarez PJ, Madeddu R and Aranega
A: ABC transporters as differentiation markers in glioblastoma
cells. Mol Biol Rep. 41:4847–4851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Lockman PR, Mittapalli RK, Taskar KS,
Rudraraju V, Gril B, Bohn KA, Adkins CE, Roberts A, Thorsheim HR,
Gaasch JA, et al: Heterogeneous blood-tumor barrier permeability
determines drug efficacy in experimental brain metastases of breast
cancer. Clin Cancer Res. 16:5664–5678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Taskar KS, Rudraraju V, Mittapalli RK,
Samala R, Thorsheim HR, Lockman J, Gril B, Hua E, Palmieri D, Polli
JW, et al: Lapatinib distribution in HER2 overexpressing
experimental brain metastases of breast cancer. Pharm Res.
29:770–781. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Tiwary S, Morales JE, Kwiatkowski SC, Lang
FF, Rao G and McCarty JH: Metastatic brain tumors disrupt the
blood-brain barrier and alter lipid metabolism by inhibiting
expression of the endothelial cell fatty acid transporter Mfsd2a.
Sci Rep. 8:82672018. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Elmeliegy MA, Carcaboso AM, Tagen M, Bai F
and Stewart CF: Role of ATP-binding cassette and solute carrier
transporters in erlotinib CNS penetration and intracellular
accumulation. Clin Cancer Res. 17:89–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
de Vries NA, Buckle T, Zhao J, Beijnen JH,
Schellens JHM and van Tellingen O: Restricted brain penetration of
the tyrosine kinase inhibitor erlotinib due to the drug
transporters P-gp and BCRP. Invest New Drugs. 30:443–449. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Oberoi RK, Mittapalli RK and Elmquist WF:
Pharmacokinetic assessment of efflux transport in sunitinib
distribution to the brain. J Pharmacol Exp Ther. 347:755–764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Lin F, de Gooijer MC, Roig EM, Buil LCM,
Christner SM, Beumer JH, Würdinger T, Beijnen JH and van Tellingen
O: ABCB1, ABCG2, and PTEN determine the response of glioblastoma to
temozolomide and ABT-888 therapy. Clin Cancer Res. 20:2703–2713.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
de Gooijer MC, de Vries NA, Buckle T, Buil
LCM, Beijnen JH, Boogerd W and van Tellingen O: Improved brain
penetration and antitumor efficacy of temozolomide by inhibition of
ABCB1 and ABCG2. Neoplasia. 20:710–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Choi YH and Yu AM: ABC transporters in
multidrug resistance and pharmacokinetics, and strategies for drug
development. Curr Pharm Des. 20:793–807. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Tran S, DeGiovanni PJ, Piel B and Rai P:
Cancer nanomedicine: A review of recent success in drug delivery.
Clin Transl Med. 6:442017. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Sun T, Zhang YS, Pang B, Hyun DC, Yang M
and Xia Y: Engineered nanoparticles for drug delivery in cancer
therapy. Angew Chem Int Ed Engl. 53:12320–12364. 2014.PubMed/NCBI
|
|
259
|
Budama-Kilinc Y, Kecel-Gunduz S, Cakir-Koc
R, Aslan B, Bicak B, Kokcu Y, Ozel AE and Akyuz S: Structural
characterization and drug delivery system of natural
growth-modulating peptide against glioblastoma cancer. Int J Pept
Res Ther. 27:2015–2028. 2021. View Article : Google Scholar
|
|
260
|
Pasut G: Grand challenges in nano-based
drug delivery. Front Med Technol. 1:12019. View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Ambruosi A, Gelperina S, Khalansky A,
Tanski S, Theisen A and Kreuter J: Influence of surfactants,
polymer and doxorubicin loading on the anti-tumour effect of
poly(butyl cyanoacrylate) nanoparticles in a rat glioma model. J
Microencapsul. 23:582–592. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Caraway CA, Gaitsch H, Wicks EE, Kalluri
A, Kunadi N and Tyler BM: Polymeric nanoparticles in brain cancer
therapy: A review of current approaches. Polymers (Basel).
14:29632022. View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Ramalho MJ, Sevin E, Gosselet F, Lima J,
Coelho MAN, Loureiro JA and Pereira MC: Receptor-mediated PLGA
nanoparticles for glioblastoma multiforme treatment. Int J Pharm.
545:84–92. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Kuang Y, Jiang X, Zhang Y, Lu Y, Ma H, Guo
Y, Zhang Y, An S, Li J, Liu L, et al: Dual functional
peptide-driven nanoparticles for highly efficient glioma-targeting
and drug codelivery. Mol Pharm. 13:1599–1607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
265
|
Mathew EN, Berry BC, Yang HW, Carroll RS
and Johnson MD: Delivering therapeutics to glioblastoma: Overcoming
biological constraints. Int J Mol Sci. 23:17112022. View Article : Google Scholar : PubMed/NCBI
|
|
266
|
Wei KC, Chu PC, Wang HYJ, Huang CY, Chen
PY, Tsai HC, Lu YJ, Lee PY, Tseng IC, Feng LY, et al: Focused
ultrasound-induced blood-brain barrier opening to enhance
temozolomide delivery for glioblastoma treatment: A preclinical
study. PLoS One. 8:e589952013. View Article : Google Scholar : PubMed/NCBI
|
|
267
|
Treat LH, McDannold N, Vykhodtseva N,
Zhang Y, Tam K and Hynynen K: Targeted delivery of doxorubicin to
the rat brain at therapeutic levels using MRI-guided focused
ultrasound. Int J Cancer. 121:901–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Coluccia D, Figueiredo CA, Wu MY,
Riemenschneider AN, Diaz R, Luck A, Smith C, Das S, Ackerley C,
O'Reilly M, et al: Enhancing glioblastoma treatment using
cisplatin-gold-nanoparticle conjugates and targeted delivery with
magnetic resonance-guided focused ultrasound. Nanomedicine.
14:1137–1148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
269
|
Timbie KF, Mead BP and Price RJ: Drug and
gene delivery across the blood-brain barrier with focused
ultrasound. J Control Release. 219:61–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
270
|
Burgess A, Shah K, Hough O and Hynynen K:
Focused ultrasound-mediated drug delivery through the blood-brain
barrier. Expert Rev Neurother. 15:477–491. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
271
|
Dutta D, Heo I and Clevers H: Disease
modeling in stem cell-derived 3D organoid systems. Trends Mol Med.
23:393–410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
272
|
Nishida N, Yano H, Nishida T, Kamura T and
Kojiro M: Angiogenesis in cancer. Vasc Health Risk Manag.
2:213–219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
273
|
Hillen F and Griffioen AW: Tumour
vascularization: Sprouting angiogenesis and beyond. Cancer
Metastasis Rev. 26:489–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Joseph JV, Conroy S, Pavlov K, Sontakke P,
Tomar T, Eggens-Meijer E, Balasubramaniyan V, Wagemakers M, den
Dunnen WF and Kruyt FA: Hypoxia enhances migration and invasion in
glioblastoma by promoting a mesenchymal shift mediated by the
HIF1α-ZEB1 axis. Cancer Lett. 359:107–116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
275
|
Huang W, Ding X, Ye H, Wang J, Shao J and
Huang T: Hypoxia enhances the migration and invasion of human
glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway.
Neuroreport. 29:1578–1585. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
276
|
Chen JWE, Lumibao J, Blazek A, Gaskins HR
and Harley B: Hypoxia activates enhanced invasive potential and
endogenous hyaluronic acid production by glioblastoma cells.
Biomater Sci. 6:854–862. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Rosa P, Catacuzzeno L, Sforna L, Mangino
G, Carlomagno S, Mincione G, Petrozza V, Ragona G, Franciolini F
and Calogero A: BK channels blockage inhibits hypoxia-induced
migration and chemoresistance to cisplatin in human glioblastoma
cells. J Cell Physiol. 233:6866–6877. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
278
|
Velásquez C, Mansouri S, Gutiérrez O,
Mamatjan Y, Mollinedo P, Karimi S, Singh O, Terán N, Martino J,
Zadeh G and Fernández-Luna JL: Hypoxia can induce migration of
glioblastoma cells through a methylation-dependent control of ODZ1
gene expression. Front Oncol. 9:10362019. View Article : Google Scholar : PubMed/NCBI
|
|
279
|
Huang S, Michalek JE, Reardon DA, Wen PY,
Floyd JR, Fox PT, Clarke GD, Jerabek PA, Schmainda KM, Muzi M, et
al: Assessment of tumor hypoxia and perfusion in recurrent
glioblastoma following bevacizumab failure using MRI and 18F-FMISO
PET. Sci Rep. 11:76322021. View Article : Google Scholar : PubMed/NCBI
|
|
280
|
Chédeville AL and Madureira PA: The role
of hypoxia in glioblastoma radiotherapy resistance. Cancers
(Basel). 13:5422021. View Article : Google Scholar : PubMed/NCBI
|
|
281
|
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang
X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al: Periostin
secreted by glioblastoma stem cells recruits M2 tumour-associated
macrophages and promotes malignant growth. Nat Cell Biol.
17:170–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Wei J, Barr J, Kong LY, Wang Y, Wu A,
Sharma AK, Gumin J, Henry V, Colman H, Priebe W, et al:
Glioblastoma cancer-initiating cells inhibit T-cell proliferation
and effector responses by the signal transducers and activators of
transcription 3 pathway. Mol Cancer Ther. 9:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
283
|
Yang F, He Z, Duan H, Zhang D, Li J, Yang
H, Dorsey JF, Zou W, Nabavizadeh SA, Bagley SJ, et al: Synergistic
immunotherapy of glioblastoma by dual targeting of IL-6 and CD40.
Nat Commun. 12:34242021. View Article : Google Scholar : PubMed/NCBI
|
|
284
|
Banyer JL, Hamilton NH, Ramshaw IA and
Ramsay AJ: Cytokines in innate and adaptive immunity. Rev
Immunogenet. 2:359–373. 2000.PubMed/NCBI
|
|
285
|
Conlon KC, Miljkovic MD and Waldmann TA:
Cytokines in the treatment of cancer. J Interferon Cytokine Res.
39:6–21. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
286
|
Zhu VF, Yang J, Lebrun DG and Li M:
Understanding the role of cytokines in glioblastoma multiforme
pathogenesis. Cancer Lett. 316:139–150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
287
|
Wakabayashi T, Kayama T, Nishikawa R,
Takahashi H, Hashimoto N, Takahashi J, Aoki T, Sugiyama K, Ogura M,
Natsume A and Yoshida J: A multicenter Phase I trial of combination
therapy with interferon-β and temozolomide for high-grade gliomas
(INTEGRA study): The final report. J Neurooncol. 104:573–577. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
288
|
Han MH, Kim JM, Cheong JH, Ryu JI, Won YD,
Nam GH and Kim CH: Efficacy of cytokine-induced killer cell
immunotherapy for patients with pathologically pure glioblastoma.
Front Oncol. 12:8516282022. View Article : Google Scholar : PubMed/NCBI
|
|
289
|
Iwami K, Natsume A and Wakabayashi T:
Cytokine therapy of gliomas. Prog Neurol Surg. 32:79–89. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
290
|
Xu S, Tang L, Li X, Fan F and Liu Z:
Immunotherapy for glioma: Current management and future
application. Cancer Lett. 476:1–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
291
|
Zhang Y and Zhang Z: The history and
advances in cancer immunotherapy: Understanding the characteristics
of tumor-infiltrating immune cells and their therapeutic
implications. Cell Mol Immunol. 17:807–821. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
292
|
de Mello RA, Veloso AF, Esrom Catarina P,
Nadine S and Antoniou G: Potential role of immunotherapy in
advanced non-small-cell lung cancer. Onco Targets Ther. 10:21–30.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
293
|
Dine J, Gordon R, Shames Y, Kasler MK and
Barton-Burke M: Immune checkpoint inhibitors: An innovation in
immunotherapy for the treatment and management of patients with
cancer. Asia Pac J Oncol Nurs. 4:127–135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
294
|
Mpakali A and Stratikos E: The role of
antigen processing and presentation in cancer and the efficacy of
immune checkpoint inhibitor immunotherapy. Cancers (Basel).
13:1342021. View Article : Google Scholar : PubMed/NCBI
|
|
295
|
Wang J, Toregrosa-Allen S, Elzey BD,
Utturkar S, Lanman NA, Bernal-Crespo V, Behymer MM, Knipp GT, Yun
Y, Veronesi MC, et al: Multispecific targeting of glioblastoma with
tumor microenvironment-responsive multifunctional engineered NK
cells. Proc Natl Acad Sci USA. 118:e21075071182021. View Article : Google Scholar : PubMed/NCBI
|
|
296
|
Rosenberg SA, Restifo NP, Yang JC, Morgan
RA and Dudley ME: Adoptive cell transfer: A clinical path to
effective cancer immunotherapy. Nat Rev Cancer. 8:299–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
297
|
Redeker A and Arens R: Improving adoptive
T cell therapy: The particular role of T cell costimulation,
cytokines, and post-transfer vaccination. Front Immunol. 7:3452016.
View Article : Google Scholar : PubMed/NCBI
|
|
298
|
Chruściel E, Urban-Wójciuk Z, Arcimowicz
Ł, Kurkowiak M, Kowalski J, Gliwiński M, Marjański T, Rzyman W,
Biernat W, Dziadziuszko R, et al: Adoptive cell therapy-harnessing
antigen-specific T cells to target solid tumours. Cancers (Basel).
12:6832020. View Article : Google Scholar : PubMed/NCBI
|
|
299
|
Staquicini FI, Smith TL, Tang FHF,
Gelovani JG, Giordano RJ, Libutti SK, Sidman RL, Cavenee WK, Arap W
and Pasqualini R: Targeted AAVP-based therapy in a mouse model of
human glioblastoma: A comparison of cytotoxic versus suicide gene
delivery strategies. Cancer Gene Ther. 27:301–310. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
300
|
Vigneswaran K, Neill S and Hadjipanayis
CG: Beyond the World Health Organization grading of infiltrating
gliomas: Advances in the molecular genetics of glioma
classification. Ann Transl Med. 3:952015.PubMed/NCBI
|
|
301
|
Goodwin CR, Rath P, Oyinlade O, Lopez H,
Mughal S, Xia S, Xia S, Li Y, Kaur H, Zhou X, et al: Crizotinib and
erlotinib inhibits growth of c-Met+/EGFRvIII+
primary human glioblastoma xenografts. Clin Neurol Neurosurg.
171:26–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
302
|
Sestito S, Runfola M, Tonelli M, Chiellini
G and Rapposelli S: New multitarget approaches in the War against
glioblastoma: A mini-perspective. Front Pharmacol. 9:8742018.
View Article : Google Scholar : PubMed/NCBI
|
|
303
|
Lang F, Liu Y, Chou FJ and Yang C:
Genotoxic therapy and resistance mechanism in gliomas. Pharmacol
Ther. 228:1079222021. View Article : Google Scholar : PubMed/NCBI
|
|
304
|
Lassman AB, Pugh SL, Gilbert MR, Aldape
KD, Geinoz S, Beumer JH, Christner SM, Komaki R, DeAngelis LM, Gaur
R, et al: Phase 2 trial of dasatinib in target-selected patients
with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 17:992–998.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
305
|
Baratta MG: Glioblastoma is ‘hot’ for
personalized vaccines. Nat Rev Cancer. 19:1292019. View Article : Google Scholar : PubMed/NCBI
|
|
306
|
Wiwatchaitawee K, Quarterman JC, Geary SM
and Salem AK: Enhancement of therapies for glioblastoma (GBM) using
nanoparticle-based delivery systems. AAPS PharmSciTech. 22:712021.
View Article : Google Scholar : PubMed/NCBI
|
|
307
|
Pearson JRD, Cuzzubbo S, McArthur S,
Durrant LG, Adhikaree J, Tinsley CJ, Pockley AG and McArdle SEB:
Immune escape in glioblastoma multiforme and the adaptation of
immunotherapies for treatment. Front Immunol. 11:5821062020.
View Article : Google Scholar : PubMed/NCBI
|
|
308
|
Burger MC, Zhang C, Harter PN, Romanski A,
Strassheimer F, Senft C, Tonn T, Steinbach JP and Wels WS:
CAR-engineered NK cells for the treatment of glioblastoma: Turning
innate effectors into precision tools for cancer immunotherapy.
Front Immunol. 10:26832019. View Article : Google Scholar : PubMed/NCBI
|
|
309
|
Granzin M, Wagner J, Köhl U, Cerwenka A,
Huppert V and Ullrich E: Shaping of natural killer cell antitumor
activity by ex vivo cultivation. Front Immunol. 8:4582017.
View Article : Google Scholar : PubMed/NCBI
|
|
310
|
Yu MW and Quail DF: Immunotherapy for
glioblastoma: Current progress and challenges. Front Immunol.
12:6763012021. View Article : Google Scholar : PubMed/NCBI
|
|
311
|
Dietrich J, Rao K, Pastorino S and Kesari
S: Corticosteroids in brain cancer patients: Benefits and pitfalls.
Expert Rev Clin Pharmacol. 4:233–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
312
|
Koyama S, Akbay EA, Li YY, Herter-Sprie
GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ,
Asahina H, et al: Adaptive resistance to therapeutic PD-1 blockade
is associated with upregulation of alternative immune checkpoints.
Nat Commun. 7:105012016. View Article : Google Scholar : PubMed/NCBI
|
|
313
|
Chan HY, Choi J, Jackson C and Lim M:
Combination immunotherapy strategies for glioblastoma. J
Neurooncol. 151:375–391. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
314
|
Bausart M, Préat V and Malfanti A:
Immunotherapy for glioblastoma: The promise of combination
strategies. J Exp Clin Cancer Res. 41:352022. View Article : Google Scholar : PubMed/NCBI
|
|
315
|
Oh T, Sayegh ET, Fakurnejad S, Oyon D,
Lamano JB, DiDomenico JD, Bloch O and Parsa AT: Vaccine therapies
in malignant glioma. Curr Neurol Neurosci Rep. 15:5082015.
View Article : Google Scholar : PubMed/NCBI
|
|
316
|
Hollingsworth RE and Jansen K: Turning the
corner on therapeutic cancer vaccines. NPJ Vaccines. 4:72019.
View Article : Google Scholar : PubMed/NCBI
|
|
317
|
Liu J, Fu M, Wang M, Wan D, Wei Y and Wei
X: Cancer vaccines as promising immuno-therapeutics: Platforms and
current progress. J Hematol Oncol. 15:282022. View Article : Google Scholar : PubMed/NCBI
|
|
318
|
Keskin DB, Anandappa AJ, Sun J, Tirosh I,
Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E,
et al: Neoantigen vaccine generates intratumoral T cell responses
in Phase Ib glioblastoma trial. Nature. 565:234–239. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
319
|
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur
V, Stevanović S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V,
van der Burg SH, et al: Actively personalized vaccination trial for
newly diagnosed glioblastoma. Nature. 565:240–245. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
320
|
Inc. AB, . AIVITA biomedical's phase 2
glioblastoma trial shows improved progression free survival. [cited
2022 Aug 5]. Available from:. https://www.prnewswire.com/news-releases/aivita-biomedicals-phase-2-glioblastoma-trial-shows-improved-progression-free-survival-301307757.html
|
|
321
|
Burkhardt JK, Riina H, Shin BJ, Christos
P, Kesavabhotla K, Hofstetter CP, Tsiouris AJ and Boockvar JA:
Intra-arterial delivery of bevacizumab after blood-brain barrier
disruption for the treatment of recurrent glioblastoma:
Progression-free survival and overall survival. World Neurosurg.
77:130–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
322
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
323
|
Wang Q, Hu B, Hu X, Kim H, Squatrito M,
Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al: Tumor
evolution of glioma-intrinsic gene expression subtypes associates
with immunological changes in the microenvironment. Cancer Cell.
32:42–56.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
324
|
Petroni G, Buqué A, Zitvogel L, Kroemer G
and Galluzzi L: Immunomodulation by targeted anticancer agents.
Cancer Cell. 39:310–345. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
325
|
Song Y, Fu Y, Xie Q, Zhu B, Wang J and
Zhang B: Anti-angiogenic agents in combination with immune
checkpoint inhibitors: A promising strategy for cancer treatment.
Front Immunol. 11:19562020. View Article : Google Scholar : PubMed/NCBI
|
|
326
|
Selek L, Seigneuret E, Nugue G, Wion D,
Nissou MF, Salon C, Seurin MJ, Carozzo C, Ponce F, Roger T and
Berger F: Imaging and histological characterization of a human
brain xenograft in pig: The first induced glioma model in a large
animal. J Neurosci Methods. 221:159–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
327
|
Khoshnevis M, Carozzo C, Bonnefont-Rebeix
C, Belluco S, Leveneur O, Chuzel T, Pillet-Michelland E, Dreyfus M,
Roger T, Berger F and Ponce F: Development of induced glioblastoma
by implantation of a human xenograft in Yucatan minipig as a large
animal model. J Neurosci Methods. 282:61–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
328
|
Tora MS, Texakalidis P, Neill S, Wetzel J,
Rindler RS, Hardcastle N, Nagarajan PP, Krasnopeyev A, Roach C,
James R, et al: Lentiviral vector induced modeling of high-grade
spinal cord glioma in minipigs. Sci Rep. 10:52912020. View Article : Google Scholar : PubMed/NCBI
|
|
329
|
Khoshnevis M, Carozzo C, Brown R, Bardiès
M, Bonnefont-Rebeix C, Belluco S, Nennig C, Marcon L, Tillement O,
Gehan H, et al: Feasibility of intratumoral 165Holmium siloxane
delivery to induced U87 glioblastoma in a large animal model, the
Yucatan minipig. PLoS One. 15:e02347722020. View Article : Google Scholar : PubMed/NCBI
|
|
330
|
Hicks WH, Bird CE, Pernik MN, Haider AS,
Dobariya A, Abdullah KG, Aoun SG, Bentley RT, Cohen-Gadol AA,
Bachoo RM, et al: Large animal models of glioma: Current status and
future prospects. Anticancer Res. 41:5343–5353. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
331
|
Zhao CY, Cheng R, Yang Z and Tian ZM:
Nanotechnology for cancer therapy based on chemotherapy. Molecules.
23:8262018. View Article : Google Scholar : PubMed/NCBI
|
|
332
|
Zaimy MA, Saffarzadeh N, Mohammadi A,
Pourghadamyari H, Izadi P, Sarli A, Moghaddam LK, Paschepari SR,
Azizi H, Torkamandi S and Tavakkoly-Bazzaz J: New methods in the
diagnosis of cancer and gene therapy of cancer based on
nanoparticles. Cancer Gene Ther. 24:233–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
333
|
Geletneky K, Hajda J, Angelova AL, Leuchs
B, Capper D, Bartsch AJ, Neumann JO, Schöning T, Hüsing J, Beelte
B, et al: Oncolytic H-1 parvovirus shows safety and signs of
immunogenic activity in a first Phase I/IIa glioblastoma trial. Mol
Ther. 25:2620–2634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
334
|
Angelova AL, Barf M, Geletneky K,
Unterberg A and Rommelaere J: Immunotherapeutic potential of
oncolytic H-1 parvovirus: Hints of glioblastoma microenvironment
conversion towards immunogenicity. Viruses. 9:3822017. View Article : Google Scholar : PubMed/NCBI
|
|
335
|
Suryawanshi YR and Schulze AJ: Oncolytic
viruses for malignant glioma: On the verge of success? Viruses.
13:12942021. View Article : Google Scholar : PubMed/NCBI
|
|
336
|
Li J, Wang W, Wang J, Cao Y, Wang S and
Zhao J: Viral gene therapy for glioblastoma multiforme: A promising
hope for the current dilemma. Front Oncol. 11:6782262021.
View Article : Google Scholar : PubMed/NCBI
|
|
337
|
Clement PMJ, Dirven L, Eoli M,
Sepulveda-Sanchez JM, Walenkamp AME, Frenel JS, Franceschi E,
Weller M, Chinot O, De Vos FYFL, et al: Impact of depatuxizumab
mafodotin on health-related quality of life and neurological
functioning in the Phase II EORTC 1410/INTELLANCE 2 trial for
EGFR-amplified recurrent glioblastoma. Eur J Cancer. 147:1–12.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
338
|
Sathornsumetee S, Desjardins A,
Vredenburgh JJ, McLendon RE, Marcello J, Herndon JE, Mathe A,
Hamilton M, Rich JN, Norfleet JA, et al: Phase II trial of
bevacizumab and erlotinib in patients with recurrent malignant
glioma. Neuro Oncol. 12:1300–1310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
339
|
Mellinghoff IK, Wang MY, Vivanco I,
Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute
DJ, et al: Molecular determinants of the response of glioblastomas
to EGFR kinase inhibitors. N Engl J Med. 353:2012–2024. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
340
|
Raizer JJ, Abrey LE, Lassman AB, Chang SM,
Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KD, Wen PY, et al:
A Phase I trial of erlotinib in patients with nonprogressive
glioblastoma multiforme postradiation therapy, and recurrent
malignant gliomas and meningiomas. Neuro Oncol. 12:87–94. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
341
|
Lucas JT Jr, Knapp BJ, Uh J, Hua CH,
Merchant TE, Hwang SN, Patay Z and Broniscer A: Posttreatment
DSC-MRI is predictive of early treatment failure in children with
supratentorial high-grade glioma treated with erlotinib. Clin
Neuroradiol. 28:393–400. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
342
|
Prados MD, Chang SM, Butowski N, DeBoer R,
Parvataneni R, Carliner H, Kabuubi P, Ayers-Ringler J, Rabbitt J,
Page M, et al: Phase II study of erlotinib plus temozolomide during
and after radiation therapy in patients with newly diagnosed
glioblastoma multiforme or gliosarcoma. J Clin Oncol. 27:579–54.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
343
|
Hegi ME, Diserens AC, Bady P, Kamoshima Y,
Kouwenhoven MCM, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch
A, et al: Pathway analysis of glioblastoma tissue after
preoperative treatment with the EGFR tyrosine kinase inhibitor
gefitinib-a Phase II trial. Mol Cancer Ther. 10:1102–1112. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
344
|
Wang Y, Liang D, Chen J, Chen H, Fan R,
Gao Y, Gao Y, Tao R and Zhang H: Targeted therapy with anlotinib
for a patient with an oncogenic FGFR3-TACC3 fusion and recurrent
glioblastoma. Oncologist. 26:173–177. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
345
|
Hainsworth JD, Ervin T, Friedman E, Priego
V, Murphy PB, Clark BL and Lamar RE: Concurrent radiotherapy and
temozolomide followed by temozolomide and sorafenib in the
first-line treatment of patients with glioblastoma multiforme.
Cancer. 116:3663–3669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
346
|
Shahid S, Kushner BH, Modak S, Basu EM,
Rubin EM, Gundem G, Papaemmanuil E and Roberts SS: Association of
BRAF V600E mutations with vasoactive intestinal peptide syndrome in
MYCN-amplified neuroblastoma. Pediatr Blood Cancer. 68:e292652021.
View Article : Google Scholar : PubMed/NCBI
|
|
347
|
Rahman MA, Brekke J, Arnesen V, Hannisdal
MH, Navarro AG, Waha A, Herfindal L, Rygh CB, Bratland E, Brandal
P, et al: Sequential bortezomib and temozolomide treatment promotes
immunological responses in glioblastoma patients with positive
clinical outcomes: A phase 1B study. Immun Inflamm Dis. 8:342–359.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
348
|
Morin A, Soane C, Pierce A, Sanford B,
Jones KL, Crespo M, Zahedi S, Vibhakar R and Mulcahy Levy JM:
Proteasome inhibition as a therapeutic approach in atypical
teratoid/rhabdoid tumors. Neurooncol Adv. 2:vdaa0512020.PubMed/NCBI
|
|
349
|
Gupta SK, Kizilbash SH, Carlson BL, Mladek
AC, Boakye-Agyeman F, Bakken KK, Pokorny JL, Schroeder MA, Decker
PA, Cen L, et al: Delineation of MGMT hypermethylation as a
biomarker for veliparib-mediated temozolomide-sensitizing therapy
of glioblastoma. J Natl Cancer Inst. 108:djv3692016. View Article : Google Scholar : PubMed/NCBI
|
|
350
|
Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu
Y, Wang X, Gao Y, Yu F, Su D, et al: Pamiparib is a potent and
selective PARP inhibitor with unique potential for the treatment of
brain tumor. Neoplasia. 22:431–440. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
351
|
Lowery MA, Burris HA III, Janku F, Shroff
RT, Cleary JM, Azad NS, Goyal L, Maher EA, Gore L, Hollebecque A,
et al: Safety and activity of ivosidenib in patients with
IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet
Gastroenterol Hepatol. 4:711–720. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
352
|
DiNardo CD, Hochhaus A, Frattini MG, Yee
K, Zander T, Krämer A, Chen X, Ji Y, Parikh N, Choi J and Wei AH: A
phase 1 study of IDH305 in patients with IDH1R132-mutant acute
myeloid leukemia or myelodysplastic syndrome. J Cancer Res Clin
Oncol. Mar 30–2022.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
353
|
Penas-Prado M, Hess KR, Fisch MJ, Lagrone
LW, Groves MD, Levin VA, De Groot JF, Puduvalli VK, Colman H,
Volas-Redd G, et al: Randomized Phase II adjuvant factorial study
of dose-dense temozolomide alone and in combination with
isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro
Oncol. 17:266–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
354
|
Kesari S, Schiff D, Henson JW, Muzikansky
A, Gigas DC, Doherty L, Batchelor TT, Longtine JA, Ligon KL, Weaver
S, et al: Phase II study of temozolomide, thalidomide, and
celecoxib for newly diagnosed glioblastoma in adults. Neuro Oncol.
10:300–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
355
|
Halatsch ME, Dwucet A, Schmidt CJ,
Mühlnickel J, Heiland T, Zeiler K, Siegelin MD, Kast RE and
Karpel-Massler G: In vitro and clinical compassionate use
experiences with the drug-repurposing approach CUSP9v3 in
glioblastoma. Pharmaceuticals (Basel). 14:12412021. View Article : Google Scholar : PubMed/NCBI
|
|
356
|
Morgan RA, Johnson LA, Davis JL, Zheng Z,
Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, et
al: Recognition of glioma stem cells by genetically modified T
cells targeting EGFRvIII and development of adoptive cell therapy
for glioma. Hum Gene Ther. 23:1043–1053. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
357
|
Liau LM, Ashkan K, Tran DD, Campian JL,
Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D'Andre SD, et
al: First results on survival from a large Phase 3 clinical trial
of an autologous dendritic cell vaccine in newly diagnosed
glioblastoma. J Transl Med. 16:1422018. View Article : Google Scholar : PubMed/NCBI
|
|
358
|
Weller M, Butowski N, Tran DD, Recht LD,
Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al:
Rindopepimut with temozolomide for patients with newly diagnosed,
EGFRvIII-expressing glioblastoma (ACT IV): A randomised,
double-blind, international phase 3 trial. Lancet Oncol.
18:1373–1385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
359
|
Reardon DA, Desjardins A, Vredenburgh JJ,
O'Rourke DM, Tran DD, Fink KL, Nabors LB, Li G, Bota DA, Lukas RV,
et al: Rindopepimut with bevacizumab for patients with relapsed
EGFRvIII-expressing glioblastoma (ReACT): Results of a double-blind
randomized Phase II trial. Clin Cancer Res. 26:1586–1594. 2020.
View Article : Google Scholar : PubMed/NCBI
|