Introduction
Pancreatic cancer is associated with early
metastases and an extremely poor prognosis, and ranks as the eighth
most common cause of cancer-associated deaths globally. Less than
9% of people survive for 5 years on average (1). When diagnosed, the majority of
patients have localized, regional or distant metastases (2). Gemcitabine is currently a standard
first-line agent for the treatment of pancreatic cancer.
The PI3K/AKT/mTOR signaling pathway is established
as being crucial for tumor formation and is frequently active in
pancreatic cancer (3). AKT has been
shown to contribute to cancer development when activated by various
stimuli, including PI3K and growth factors, while downstream
effectors such as mTOR lead to signal transduction (4). PI3K is a downstream effector of
oncogenic KRAS, which is nearly ubiquitous in pancreatic cancer
(5). A study that analyzed 32
cancer types in The Cancer Genome Atlas identified that KRAS exerts
a pro-tumorigenic effect via activation of the PI3K/AKT/mTOR
pathway in pancreatic cancer (6).
There is evidence suggesting that an interaction exists between
KRAS and the PI3K/AKT/mTOR pathway. In a study of bladder tumors,
PI3K catalytic subunit α (PIK3CA) mutations were found to be
associated with mutated KRAS (7).
In another study, patient-derived xenograft models of
PIK3CA-mutated metaplastic breast cancers treated with a
combination of PI3K and MEK inhibitors were shown to undergo a
durable remission (8). A study
conducted by Candido et al (9) suggested that patients with melanoma
who are treated with agents targeting the MAPK signaling pathway
may experience tumor progression due to the development of
resistance to the targeted therapy; as mutations affecting the
PI3K-AKT pathway appeared to favor drug resistance, crosstalk may
occur between the MAPK and PI3K-AKT pathways.
The efficacy of gemcitabine in pancreatic cancer has
been linked with mTOR activation (10). The high aggressiveness of pancreatic
cancer may be partly attributed to the chemotherapy-resistant
characteristics of pancreatic cancer cells, which are associated
with the epithelial-mesenchymal transition (EMT) phenotype and
cancer stem cells (11). EMT is a
process that alters cell-cell and cell-extracellular matrix (ECM)
interactions, inducing a fibroblast-like appearance in cancer
tissue and enabling neoplastic cells to migrate and invade
(12,13). EMT is essentially the transition
between two types of completely differentiated and mature cells
(14). E-cadherin and other
epithelial cell phenotype marker proteins are expressed at reduced
levels and mesenchymal cell phenotype marker proteins including
vimentin and N-cadherin are expressed at increased levels in tumor
cells. Additionally, homocellular tight junctions and cell polarity
are lacking (15,16). Notably, some studies have suggested
that EMT alters the sensitivity of neoplastic cells to chemotherapy
agents (17,18). Therefore, therapy aimed at
inhibiting or minimizing EMT may increase the efficacy of
chemotherapy.
Several types of chemotherapeutic agents are widely
used as first-line treatments for pancreatic cancer, including
5-fluouracil (5-FU), according to National Comprehensive Cancer
Network (NCCN) guidelines (2021.V2) (19). In the NCCN Guidelines, gemcitabine
is included in first-line therapy or the preferred neoadjuvant
therapy regimens for resectable/borderline resectable disease,
adjuvant therapy, locally advanced disease and metastatic disease,
i.e., gemcitabine is a standard first-line chemotherapeutic drug
for pancreatic cancer. Other types of local therapies, including
radiofrequency ablation, irreversible electroporation,
high-intensity focused ultrasound, microwave ablation and local
anti-KRAS therapy using a small interfering RNA targeting KRA G12D
in a biodegradable polymeric matrix are under investigation
(20). Numerous researchers are
focused on the development of cell therapies, antitumor vaccines
and new biotechnological drugs, which have shown promising results
in preclinical studies (21).
According to NCCN guidelines (2021.V2) (19), FOLFIRINOX, which includes 5-FU,
irinotecan, oxaliplatin and taxanes, is also an important
first-line treatment (22,23), but it is not recommended for
patients with a low performance status (24). Moreover, the majority of patients
experience side effects when treated with FOLFIRINOX, which
restricts the use of aspirin (ASA). Adding ASA to chemotherapy may
increase the risk of bleeding, which is a serious issue that could
lead to death and commonly causes anemia; therefore, (ASA) was not
combined with FOLFIRINOX in the present study. However, ASA has
been reported to inhibit the proliferation of pancreatic cancer
cells via inhibition of the PI3K/AKT/mTOR signaling pathway and to
show therapeutic potential via the targeted inhibition of tumor
growth and angiogenesis (25).
Considering these findings, gemcitabine may be more suitable than
5-FU, irinotecan, oxaliplatin, taxanes and other chemotherapeutic
agents for investigation in combination with ASA.
ASA is a weak organic acid that shows promise as a
cancer chemoprevention agent due to its anti-inflammatory
properties (26). A pooled analysis
of 25,570 patients in eight trials reported that the daily use of
ASA reduced the deaths associated with several common cancers,
including pancreatic cancer, with the greatest benefit observed
after 5 years of the scheduled treatment (27), while in a clinic-based case-control
study, the use of ASA was associated with a lowered risk of
pancreatic cancer development (28). Furthermore, in China, a
population-based study of 761 cases and 794 control subjects showed
that regular use of ASA was associated with a reduced risk of
pancreatic cancer: Odds ratio, 0.54; 95% CI, 0.40-0.73 (29). However, the evidence that ASA lowers
the risk of pancreatic cancer is conflicting and the ability of ASA
to improve the efficacy of gemcitabine in pancreatic cancer remains
unclear, as does the molecular mechanism by which ASA may provide a
benefit. Therefore, these were investigated in the present
study.
Materials and methods
Cell lines and cell culture
Human pancreatic cancer cell lines SW1990 and BxPC-3
were obtained from the Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences and Changhai Hospital Affiliated to the
Second Military Medical University, respectively. Mycoplasma
testing was performed for both cell lines. Cells were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) containing
10% fetal bovine serum (FBS) (Biological Industries) and maintained
in a humid atmosphere at 37°C with 5% CO2. The cell
medium was changed every 2 or 3 days. Cell passage was performed
with trypsin when the confluence of monolayer cells reached
70–80%.
Main reagents
ASA was purchased from Sigma-Aldrich (Merck KGaA)
and was dissolved in dimethyl sulfoxide (DMSO; SRL Chemical) to
make a 5-M stock solution. This solution was stored at −20°C.
Gemcitabine was obtained from Eli Lilly and Company and dissolved
in a sterile saline solution to make a 1g/l stock solution. This
solution was stored at −20°C. Even though different solvents were
used for gemcitabine and ASA due to their different solubilities,
this was not anticipated to significantly influence the results
obtained. In addition, the concentration of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Merck KGaA) was set to 5 mg/ml. The Annexin V-fluorescein
isothiocyanate (FITC) apoptosis detection kit was obtained from
R&D Systems, Inc.
Cell proliferation assay
Cells were cultured in RPMI-1640 medium containing
10% FBS and maintained in a humid atmosphere at 37°C with 5%
CO2 for 72 h. The cells were then harvested and
inoculated into 96-well plates with 3×103 cells/well 24
h before treatment. After that, the cells were divided into four
groups as follows: i) RPMI-1640 containing 10% FBS and 2 mmol/l
ASA; ii) RPMI-1640 containing 10% FBS and 1 mg/l gemcitabine; iii)
RPMI-1640 medium containing 10% FBS, 2 mmol/l ASA and 1 mg/l
gemcitabine; and iv) negative control comprising RPMI-1640 with 10%
FBS. After 24 h of culture, the cells were incubated at 37°C with
5% CO2 for 0, 24, 48 and 72 h. Then, 10% MTT (5 mg/ml)
was added to each well. After 4 h of treatment with MTT, 150 µl
DMSO was added to each well and the samples were vibrated for 10
min. The optical density at 570 nm was then measured using a
microplate reader. Each cell proliferation experiment was repeated
three times and the readings were averaged for statistical
analysis.
Wound healing assay
BxPC-3 and SW1990 cells were seeded in 6-well
plates. When the cell confluence was ~100%, scratches were made on
the monolayer cell surface with 200-µl pipette tips. After washing
twice with phosphate-buffered saline (PBS), the cells were divided
into four groups as described in the cell proliferation assay. The
four groups include the combination of ASA and gemcitabine, ASA or
gemcitabine alone, and the control group, with RPMI-1640 containing
1% FBS.
Images of the scratches and the cells on each side
were captured with an inverted microscope at 0 and 24 h. Finally,
the migration was analyzed in three randomly selected fields of
view in images captured at ×100 magnification under an inverted
microscope (Olympus Corporation). The width (W) of the scratch was
measured and the percentage of the wound remaining was calculated
as follows: W24 h/W0 h ×100. All experiments
were performed in triplicate.
Transwell assay
Transwell assays were performed using 6.5-mm
diameter Transwell chambers each with an 8-µm pore polycarbonate
membrane insert (Corning, Inc.). BxPC-3 and SW1990 cells
(3×104) were plated on the upper chambers. The upper
chambers were coated at 4°C with 60 µl Matrigel (Corning, Inc.) in
serum-free medium and then left overnight at 37°C. RPMI-1640
containing 10% FBS was added to the lower chambers. After
incubation with the aforementioned treatments for 24 h at 37°C, the
cells were fixed with 4% paraformaldehyde at 25°C for 30 min before
being stained with crystal violet solution at 25°C for 30 min. Then
counted under a light microscope (×200 magnification). The assay
was repeated three times in duplicate. The numbers of cells counted
in five random fields were averaged.
Cell apoptosis
BxPC-3 and SW1990 cells were seeded in 6-well plates
and cultured with RPMI-1640 medium containing 10% FBS. When the
cells had attached to the plate for 24 h, they were divided into
four groups as described in the cell proliferation assay and
cultured for 24 h at 37°C. The cells were then collected and washed
with PBS solution twice or thrice. In order to count the number of
apoptotic cells, the cells were stained with annexin V-FITC and
propidium iodide (PI). The stained cells were analyzed using
FlowJo_V10 (FlowJo, LLC). All experiments were carried out in
triplicate.
Western blot analysis
Cells from the aforementioned four groups were
harvested after culture for 24 h, lysed with RIPA lysis buffer
(Thermo Fisher Scientific, Inc.) and quantified by bicinchoninic
acid analysis (Beyotime Institute of Biotechnology). Next, the
proteins released from the cells were separated on 10% gels using
SDS-PAGE, and then transferred to PVDF membranes for western
blotting. The membranes were blocked with 5% non-fat milk at 25°C
for 1 h. Next, the membranes were incubated with primary antibodies
against β-actin, Bax, Bcl-2, E-cadherin, vimentin, PI3K, phospho
(p)-PI3K, AKT, p-AKT, mTOR and p-mTOR at a dilution of 1:1,000 at
4°C overnight. Monoclonal antibodies against human E-cadherin (cat.
no. 14-3249-82; Thermo Fisher Scientific, Inc.), vimentin (cat. no.
14-9897-82; Thermo Fisher Scientific, Inc.), Bcl-2 (cat. no.
MA5-11757; Thermo Fisher Scientific, Inc.), Bax (cat. no.
MA5-14003; Thermo Fisher Scientific, Inc.), PI3K (cat. no. 4249;
Cell Signaling Technology, Inc.), p-PI3K (cat. no. ab32089; Abcam),
AKT (cat. no. 60203-2-Ig; ProteinTech Group, Inc.), p-AKT (cat. no.
66444-1-Ig; ProteinTech Group, Inc.), mTOR (cat. no. 66888-1-Ig;
ProteinTech Group, Inc.), p-mTOR (cat. no. 67778-1-Ig; ProteinTech
Group, Inc.) and β-actin (cat. no. AF0003; Beyotime Institute of
Biotechnology). Horseradish peroxidase (HRP)-labeled
goat-anti-rabbit and goat-anti-mouse IgG secondary antibodies were
obtained from Abcam (cat. nos. ab205719 and ab205718). The
membranes were incubated with the HRP-labeled secondary antibody at
a dilution of 1:5,000 at 37°C for 1 h. The western blots were
visualized using Immobilon Western Chemiluminescent HRP Substrate
(MilliporeSigma) followed by film exposure. β-actin was used as the
internal reference. Using ImageJ v1.8.0 (National Institutes of
Health), the gray levels were analyzed, and the results were
calculated as the gray level of the target protein divided by the
gray level of the internal reference.
Statistical analysis
All data are expressed as the mean ± standard
deviation. SPSS version 20.0 (IBM Corp.) was used to perform the
statistical analyses. One-way analysis of variance followed by
Dunnett's post hoc test was used for the analysis of multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Combination of ASA and gemcitabine
inhibits cell proliferation more than ASA or gemcitabine alone
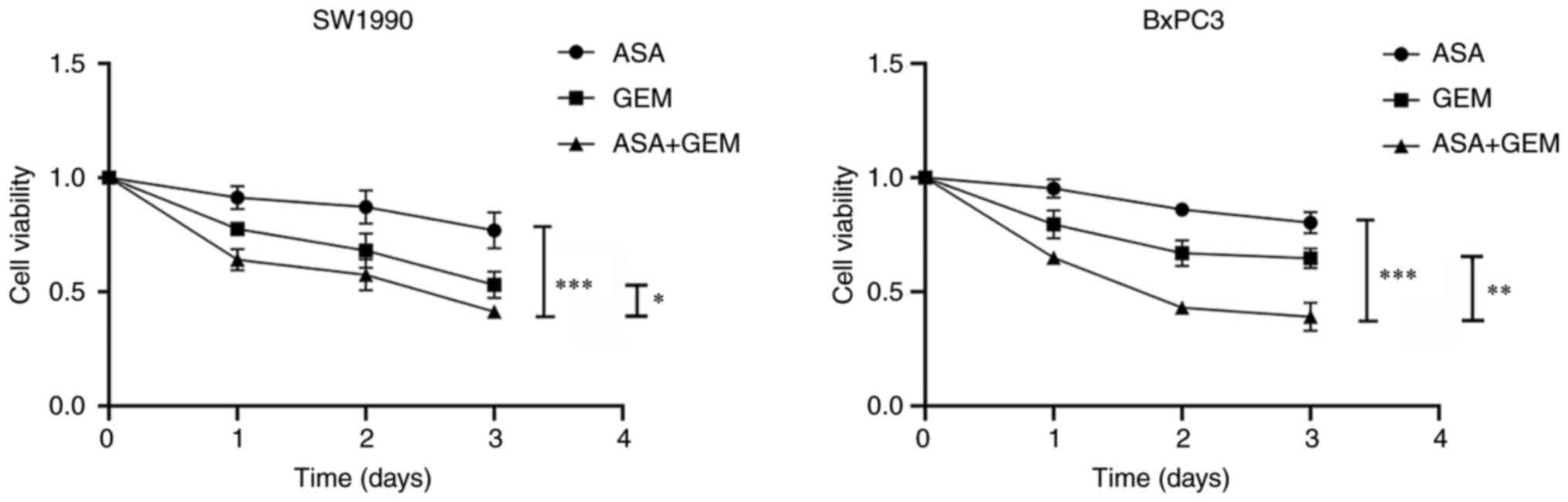
As shown in Fig. 1,
the viability of SW1990 cells was more strongly reduced after
treatment with a combination of 2 mmol/l ASA and 1 mg/l gemcitabine
than with gemcitabine or ASA alone (P<0.05). Similar results
were observed with BxPC-3 cells, in which the combination of ASA
and gemcitabine inhibited cell viability more strongly compared
with ASA or gemcitabine alone. These results indicate that ASA
increases the antiproliferative efficacy of gemcitabine in SW1990
and BxPC-3 cells.
Combination of ASA and gemcitabine
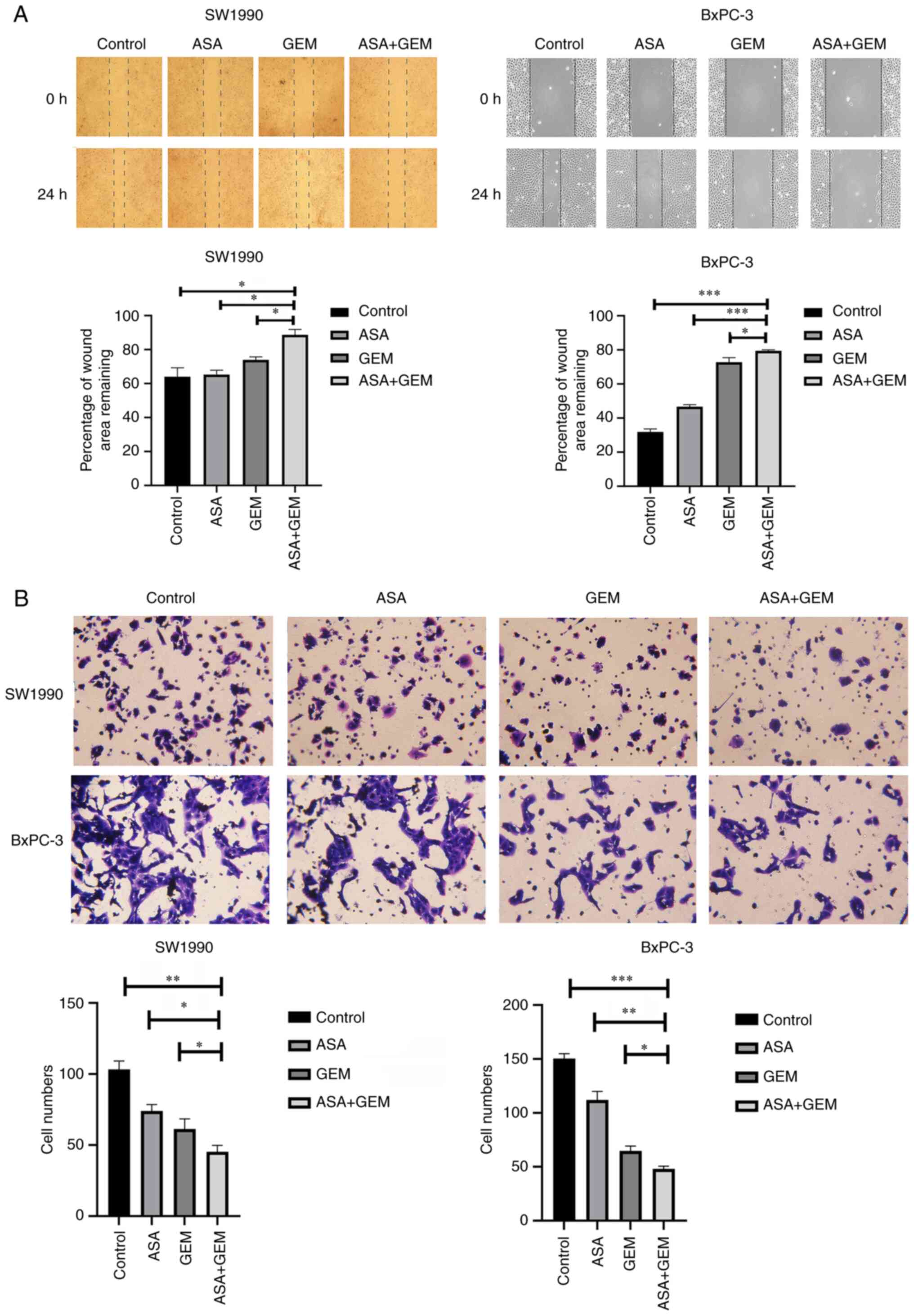
inhibits pancreatic cell migration and invasion
Wound healing and Transwell assays were performed to
examine the effect of ASA and gemcitabine on cell migration and
invasion, respectively. The migration and invasion of the cells
were significantly reduced following treatment with the combination
of ASA and gemcitabine compared with either ASA or gemcitabine
alone (P<0.05; Fig. 2). This
indicates that treatment with a combination of ASA and gemcitabine
strongly suppressed the cell migration and invasion abilities of
SW1990 and BxPC-3 pancreatic cancer cells.
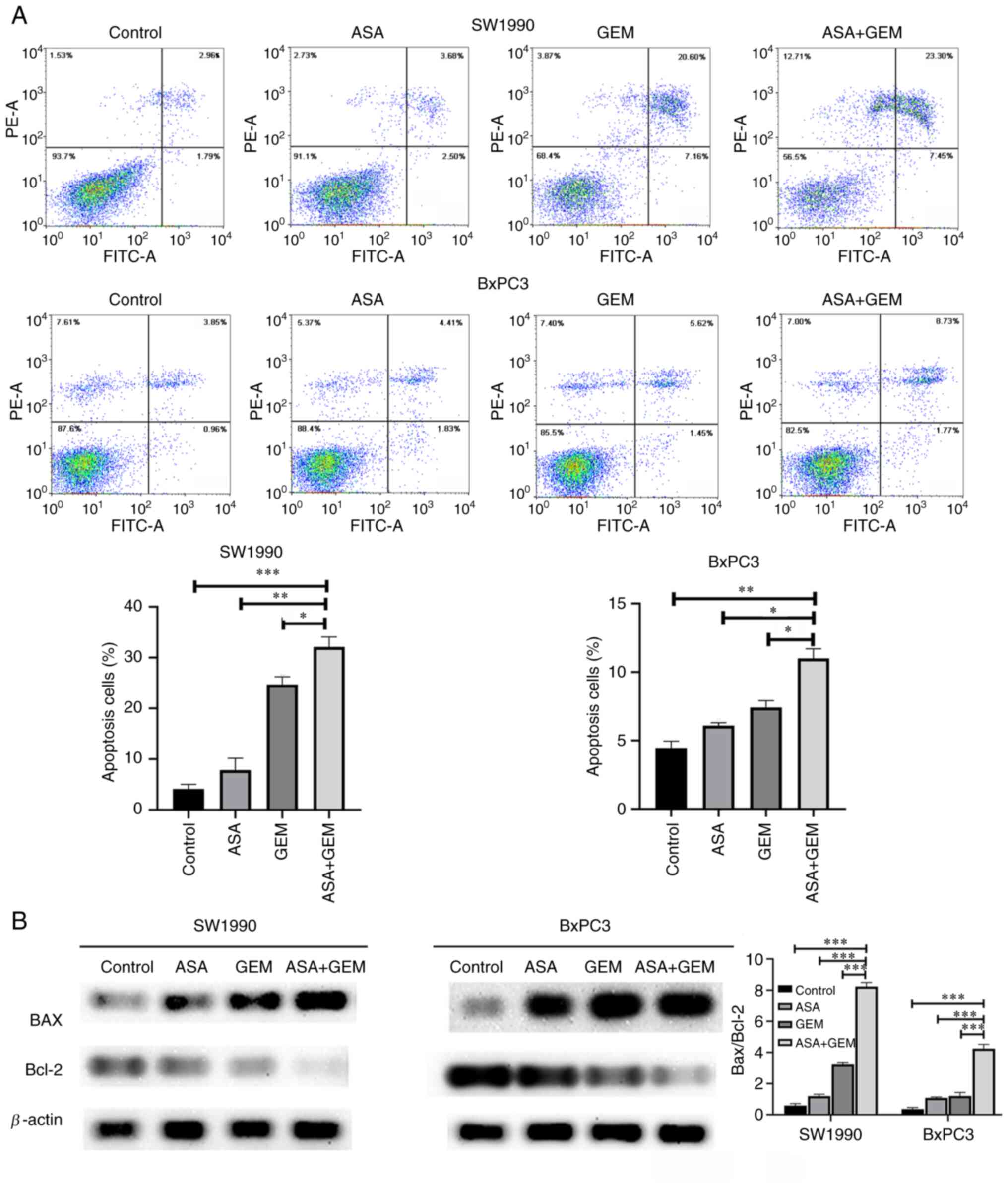
Effect of the combination of ASA and
gemcitabine on the apoptosis of pancreatic cancer cells
The proportion of apoptotic cells in the two
pancreatic cancer cell lines was significantly higher when they
were treated with the combination of ASA and gemcitabine than when
they were treated with ASA or gemcitabine alone (P<0.05;
Fig. 3A). In the control SW1990 and
BxPC3 cells, the percentages of apoptosis were 4.13±0.88 and
4.79±0.65%, respectively. In cells treated with ASA alone, these
values were 7.84±2.35 and 6.12±0.23%, respectively; in cells
treated with gemcitabine alone, they were 24.68±1.53 and
7.31±0.83%, respectively; and in cells treated with the combination
of ASA and gemcitabine, they were 32.13±1.95 and 11.32±0.98%,
respectively. Furthermore, western blotting revealed that the
protein expression level of Bcl-2 was markedly reduced and that of
Bax was increased in the cells treated with the combination of ASA
and gemcitabine, and the increase in the Bax/Bcl-2 ratio in cells
treated with the combination was significantly greater than that
obtained with either ASA or gemcitabine alone (P<0.05; Fig. 3B). These experimental results
indicate that the combination of ASA and gemcitabine induced the
apoptosis of pancreatic cancer cells to a greater extent than ASA
or gemcitabine alone. These findings suggest that ASA promoted the
antitumor effect of gemcitabine in SW1990 and BxPC-3 cells.
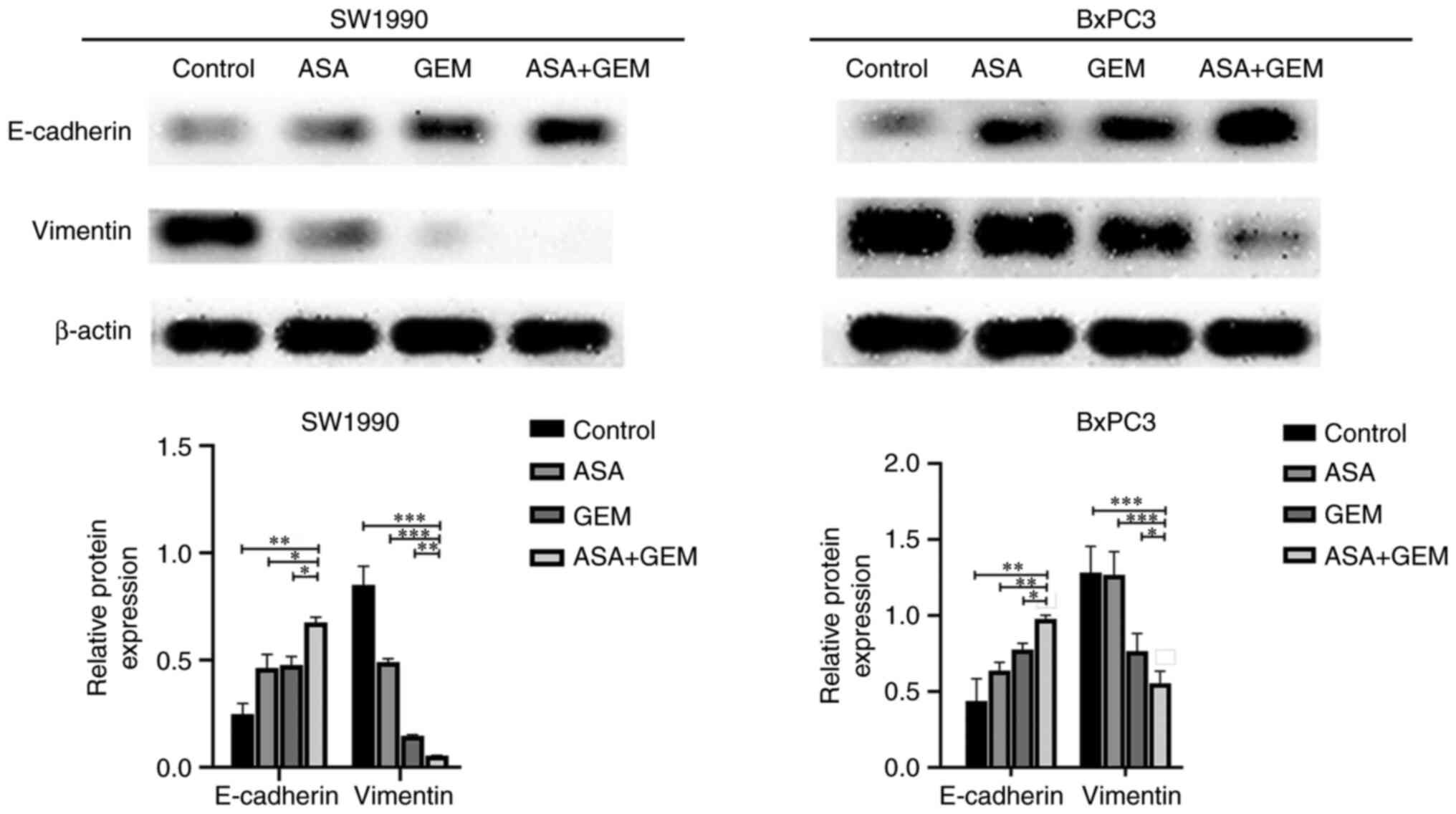
Combination of ASA and gemcitabine
significantly increases the expression of E-cadherin and decreases
the expression of vimentin
The expression of EMT biomarkers was assessed in the
pancreatic cancer cells in vitro, and the results revealed
that the expression of E-cadherin was significantly increased and
that of vimentin was significantly decreased in the ASA and
gemcitabine combination group compared with the ASA and gemcitabine
alone groups (P<0.05). These results indicate that the cell
apoptosis and growth inhibition induced by the combination
treatment may be closely associated with EMT (Fig. 4).
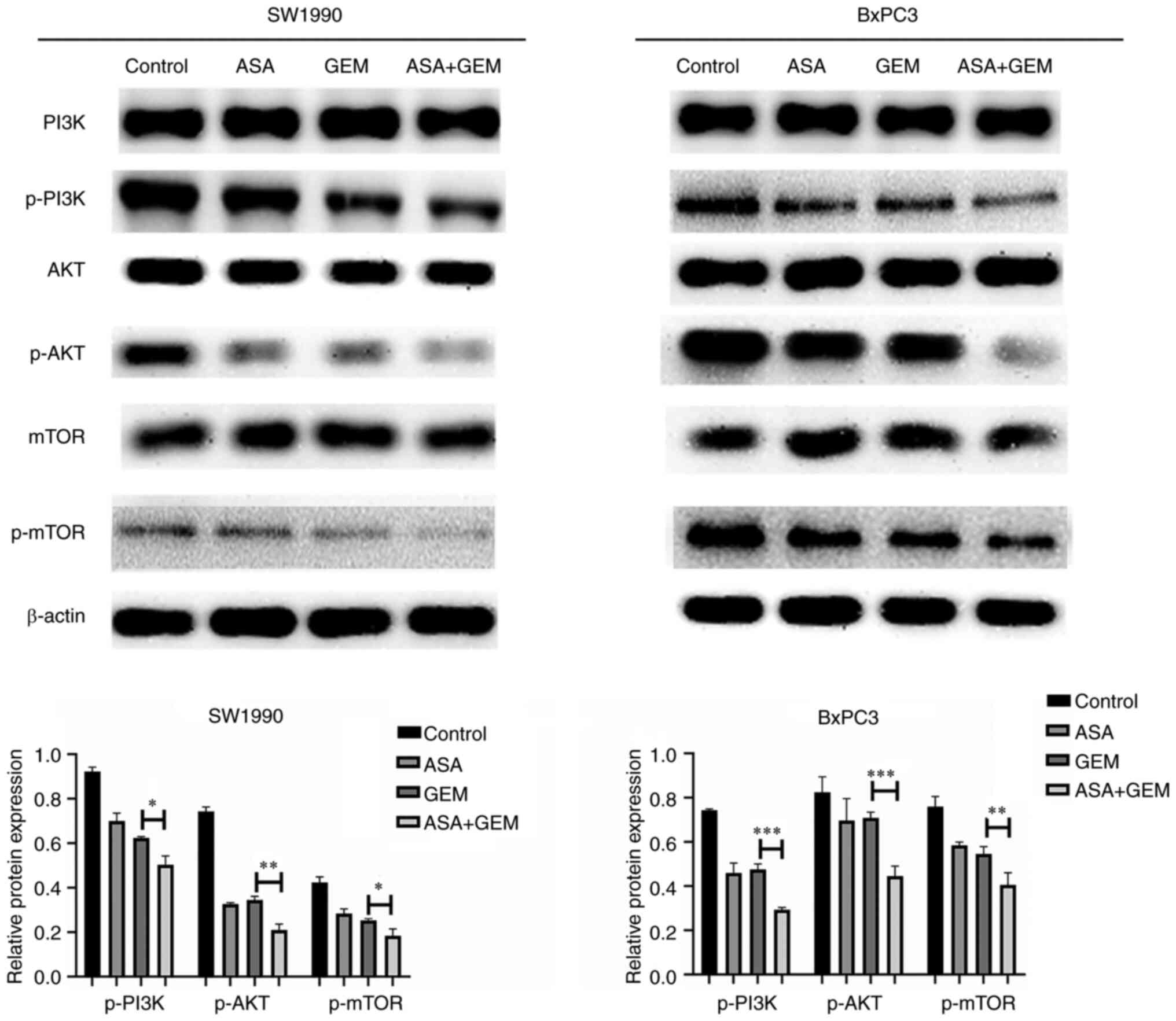
Combination of ASA and gemcitabine
downregulates PI3K/AKT/mTOR signaling
The pancreatic cancer cells presented a significant
reduction in the levels of p-PI3K and p-AKT after treatment with
the combination of ASA (2 mmol/l) and gemcitabine (1 mg/l) for 24 h
compared with those observed in untreated control cells. The p-mTOR
protein level of the combination treatment group was also
significantly reduced by the combination treatment. These findings
suggest that downregulation of PI3K/AKT/mTOR pathway signaling may
be involved in the cell apoptosis and growth inhibition induced by
the combination treatment (Fig.
5).
Discussion
The present study revealed the potential of ASA
combined with gemcitabine against pancreatic cancer in
vitro. The findings support those from a previous study, in
which the non-steroidal anti-inflammatory drug ASA was shown to
prevent the growth of pancreatic cancer by blocking the mTOR
signaling pathway (30). The
regular use of ASA has been observed to be associated with a
decreased risk of various types of cancer, including hepatocellular
carcinoma and breast cancer (31,32).
In addition, ASA has been shown to inhibit the activation of GSK-3β
in the pancreatic cancer cell lines Capan-1 and PANC-1 when
combined with gemcitabine (33).
Another study demonstrated that a combination of ASA and metformin
significantly inhibited pancreatic cancer cell growth in
vitro and in vivo via the regulation of pro- and
anti-apoptotic Bcl-2 gene family members (34). Studies have also indicated that
adenomas in the large bowel can be prevented to a certain extent by
low-dose ASA, as patients who have had colorectal cancer in the
past are significantly less likely to develop colorectal adenomas
when they take ASA every day (35,36).
Other studies have shown that ASA decreases various tumor
characteristics, such as tumor cell migration, and lowers the risk
of cancer initiation and development (37,38).
However, to the best of our knowledge, no detailed study has been
published in which the effect of ASA on gemcitabine
monotherapy-associated chemotherapy in pancreatic cancer cells has
been assessed. The upregulation of ECM quantity in pancreatic
cancer prevents drugs from penetrating the tumor (39), which may be associated with a
requirement for an increased ASA concentration to treat the
disease. However, scientists have been exploring several approaches
to address this issue. Collagenase nanoparticles have been
suggested to improve the access of medications to pancreatic cancer
tissue. The thick collagen stroma of pancreatic cancer can be
disassembled by pretreatment with a proteolytic-enzyme nanoparticle
system, which promotes the penetration of medication into the tumor
(40). Furthermore, to improve drug
penetration, a dendrimer-camptothecin conjugate was developed that
actively penetrated deep into pancreatic cancer via γ-glutamyl
transpeptidase-triggered cell endocytosis and transcytosis. This
conjugate exhibited high antitumor activity in multiple pancreatic
cancer xenograft mouse models compared with gemcitabine (41). In summary, advancements in materials
science, biology, pharmacology and other domains are necessary to
increase the concentration of medications in the pancreatic cancer
microenvironment.
The ability of ASA to inhibit the tumorigenesis and
development of pancreatic cancer and the underlying mechanism
remain unclear. The goal of the present study was to evaluate
whether ASA increases the efficacy of gemcitabine in pancreatic
cancer. In addition, the possible underlying mechanism was
investigated.
Previous research has addressed the role of EMT in
tumor cell drug efficacy (42).
Therefore, research into the mechanisms of EMT in pancreatic cancer
is necessary to facilitate the development of new therapeutic
strategies (43). The present study
showed that ASA increased the expression level of E-cadherin and
decreased that of vimentin in SW1990 and BxPC-3 cells, indicating
that ASA increased the efficacy of gemcitabine in pancreatic cancer
cells via the inhibition of EMT.
The PI3K/AKT pathway is among the most frequently
mutated in human cancer, and multiple genetic events have been
described that lead to activation of the PI3K/AKT/mTOR pathway in
cancer (44). Inhibitors of
AKT/mTOR pathways, including rapamycin and LY294002, are able to
inhibit EMT progression in pancreatic cancer (45). Previous research has shown that
reactive oxygen species (ROS) regulate EMT (46). ROS have roles in cellular integrity
and cell death, and several anticancer drugs have been shown to
induce high levels of ROS and reduce AKT/mTOR signaling (47–49).
The present study suggested that ASA increased the efficacy of
gemcitabine in pancreatic cancer via the downregulation of
PI3K/AKT/mTOR signaling, that is, by reducing PI3K, AKT and mTOR
phosphorylation, which is consistent with a previous study which
demonstrated that gemcitabine inhibited cancer growth, metastasis
and EMT in pancreatic cancer cells with involvement of the
PI3K/AKT/mTOR pathway (50). The
present study demonstrated that the combination of ASA and
gemcitabine exerted a remarkable inhibitory effect on cell
proliferation and markedly promoted apoptosis in SW1990 and BxPC-3
cell lines via a mechanism involving the PI3K/AKT/mTOR signaling
pathway.
The difference in response rates between the two
cell lines merits discussion. Previous literature supports a
difference in response rates between the BxPC-3 and SW1990 cell
lines; specifically, when treated with gemcitabine, the
half-maximal inhibitory concentration (IC50) of the
SW1990 cell line was reported to be 6.27 mmol/l while that of the
BxPC-3 cell line was 4.01 mmol/l (51). In the present study, preliminary MTT
experiments indicated that the IC50 of ASA was 1.93
mmol/l in SW1990 cells and 2.26 mmol/l in BxPC-3 cell lines.
According to a study conducted by Lin et al (52), PANC-1 cells treated with 2 mmol/l
ASA for 24 h had a growth suppression rate of ~40% compared with
untreated cells, which is similar to the findings for the cell
lines used in the present study. According to these data, it was
decided to use a 2 mmol/l concentration of ASA in the current
study. However, a 2 mmol/l concentration appears to be a little
higher than that used in certain other studies. The optimum
concentration of ASA for use in pancreatic cancer is unclear and
may differ from that in other cancers.
The present study found that low-dose ASA combined
with gemcitabine significantly reduced cell proliferation and
migration, increased apoptosis, increased the expression of
E-cadherin and suppressed that of vimentin when compared with
gemcitabine alone. When ASA plus gemcitabine combination therapy
was compared with ASA or gemcitabine alone, the results revealed a
significant downregulation of activation of the PI3K/AKT/mTOR
pathway in SW1990 and BxPC-3 cells, as indicated by effective
reductions in the phosphorylation of PI3K, AKT and mTOR. These data
suggested that the complementary effect of ASA plus gemcitabine
therapy in pancreatic cancer may occur by attenuation of EMT and
the downregulation of PI3K/AKT/mTOR signaling. These findings are
similar to those observed in a previous study, in which ASA
inhibited the EMT and migration of oncogenic KRAS-expressing
non-small cell lung carcinoma cells via downregulation of the
E-cadherin repressor Slug (53).
In summary, the results of the present demonstrate
that ASA increased the efficacy of gemcitabine in pancreatic cancer
via the inhibition of cell proliferation and migration and the
induction of apoptosis. The study also reveals that ASA increased
the ability of gemcitabine to inhibit the PI3K/AKT/mTOR signaling
pathway and reverse the EMT in pancreatic cancer cells, which is a
new insight. The results suggest that ASA combined with gemcitabine
has the potential to be a promising therapeutic option for patients
with pancreatic carcinoma.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ conceived and designed the study. XY performed
the experiments. KX analyzed the data. YS made contributions to the
interpretation of the data. All authors read and approved the final
version of the manuscript. KX and YS confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tuveson DA and Neoptolemos JP:
Understanding metastasis in pancreatic cancer: A call for new
clinical approaches. Cell. 148:21–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mann KM, Ying H, Juan J, Jenkins NA and
Copeland NG: KRAS-related proteins in pancreatic cancer. Pharmacol
Ther. 168:29–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song M, Bode AM, Dong Z and Lee MH: AKT as
a therapeutic target for cancer. Cancer Res. 79:1019–1031. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Kwok-Shing Ng P, Kucherlapati M,
Chen F, Liu Y, Tsang YH, de Velasco G, Jeong KJ, Akbani R,
Hadjipanayis A, et al: A pan-cancer proteogenomic atlas of
PI3K/AKT/mTOR pathway alterations. Cancer Cell. 31:820–832.
e8232017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Juanpere N, Agell L, Lorenzo M, de Muga S,
López-Vilaró L, Murillo R, Mojal S, Serrano S, Lorente JA, Lloreta
J and Hernández S: Mutations in FGFR3 and PIK3CA, singly or
combined with RAS and AKT1, are associated with AKT but not with
MAPK pathway activation in urothelial bladder cancer. Hum Pathol.
43:1573–1582. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coussy F, El Botty R, Lavigne M, Gu C,
Fuhrmann L, Briaux A, de Koning L, Dahmani A, Montaudon E, Morisset
L, et al: Combination of PI3K and MEK inhibitors yields durable
remission in PDX models of PIK3CA-mutated metaplastic breast
cancers. J Hematol Oncol. 13:132020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Candido S, Salemi R, Piccinin S, Falzone L
and Libra M: The PIK3CA H1047R Mutation confers resistance to BRAF
and MEK inhibitors in A375 melanoma cells through the
cross-activation of MAPK and PI3K-Akt pathways. Pharmaceutics.
14:5902022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng M, Xiong G, Cao Z, Yang G, Zheng S,
Qiu J, You L, Zheng L, Zhang T and Zhao Y: LAT2 regulates
glutamine-dependent mTOR activation to promote glycolysis and
chemoresistance in pancreatic cancer. J Exp Clin Cancer Res.
37:2742018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou P, Li B, Liu F, Zhang M, Wang Q, Liu
Y, Yao Y and Li D: The epithelial to mesenchymal transition (EMT)
and cancer stem cells: Implication for treatment resistance in
pancreatic cancer. Mol Cancer. 16:522017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marais R, Sellers W, Livingston D and
Mihich E: Twenty-fourth annual Pezcoller symposium: Molecular basis
for resistance to targeted agents. Cancer Res. 73:1046–1049. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Craene B, Gilbert B, Stove C, Bruyneel
E, van Roy F and Berx G: The transcription factor snail induces
tumor cell invasion through modulation of the epithelial cell
differentiation program. Cancer Res. 65:6237–6244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chaw SY, Majeed AA, Dalley AJ, Chan A,
Stein S and Farah CS: Epithelial to mesenchymal transition (EMT)
biomarkers-E-cadherin, beta-catenin, APC and Vimentin-in oral
squamous cell carcinogenesis and transformation. Oral Oncol.
48:997–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo T, Wang L, Wu P, Gong W, Chen W, Zhao
H and Zheng Z: Downregulated vimentin and upregulated E-cadherin in
T1 stage non-small-cell lung cancer: Does it suggest a
mesenchymal-epithelial transition? Neoplasma. 64:693–699. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang W, Wang L, Mizokami A, Shi J, Zou C,
Dai J, Keller ET, Lu Y and Zhang J: Down-regulation of E-cadherin
enhances prostate cancer chemoresistance via Notch signaling. Chin
J Cancer. 36:352017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mallini P, Lennard T, Kirby J and Meeson
A: Epithelial-to-mesenchymal transition: What is the impact on
breast cancer stem cells and drug resistance. Cancer Treat Rev.
40:341–348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tempero MA, Malafa MP, Al-Hawary M,
Behrman SW, Benson AB, Cardin DB, Chiorean EG, Chung V, Czito B,
Chiaro MD, et al: Pancreatic adenocarcinoma, version 2.2021, NCCN
clinical practice guidelines in oncology. J Natl Compr Canc Netw.
19:439–457. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Springfeld C, Jager D, Buchler MW, Strobel
O, Hackert T, Palmer DH and Neoptolemos JP: Chemotherapy for
pancreatic cancer. Presse Med. 48:e159–e174. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Falzone L, Salomone S and Libra M:
Evolution of cancer pharmacological treatments at the turn of the
third millennium. Front Pharmacol. 9:13002018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Conroy T, Hammel P, Hebbar M, Abdelghani
MB, Wei AC, Raoul JL, Choné L, Francois E, Artru P, Biagi JJ, et
al: FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic
cancer. New Engl J Med. 379:2395–2406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khorana AA, McKernin SE, Berlin J, Hong
TS, Maitra A, Moravek C, Mumber M, Schulick R, Zeh HJ and Katz MHG:
Potentially curable pancreatic adenocarcinoma: ASCO clinical
practice guideline update. J Clin Oncol. 37:2082–2088. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tempero MA: NCCN Guidelines updates:
Pancreatic cancer. J Natl Compr Cancer Netw. 17:603–605.
2019.PubMed/NCBI
|
|
25
|
Zelenay S, van der Veen AG, Bottcher JP,
Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais
R, Quezada SA, et al: Cyclooxygenase-dependent tumor growth through
evasion of immunity. Cell. 162:1257–1270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ulrich CM, Bigler J and Potter JD:
Non-steroidal anti-inflammatory drugs for cancer prevention:
Promise, perils and pharmacogenetics. Nat Rev Cancer. 6:130–140.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rothwell PM, Fowkes FG, Belch JF, Ogawa H,
Warlow CP and Meade TW: Effect of daily aspirin on long-term risk
of death due to cancer: Analysis of individual patient data from
randomised trials. Lancet. 377:31–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tan XL, Lombardo KM, Bamlet WR, Oberg AL,
Robinson DP, Anderson KE and Petersen GM: Aspirin, nonsteroidal
anti-inflammatory drugs, acetaminophen, and pancreatic cancer risk:
A clinic-based case-control study. Cancer Prev Res (Phila).
4:1835–1841. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang P, Lan TH, Zhou YM, Deng JP, Wei CZ,
Wang GH and Tian L: Risk factor analysis of perioperative
complications in patients with radical gastrectomy for gastric
cancer. Zhonghua Wei Chang Wai Ke Za Zhi. 22:736–741. 2019.(In
Chinese). PubMed/NCBI
|
|
30
|
Yue W, Yang CS, DiPaola RS and Tan XL:
Repurposing of metformin and aspirin by targeting AMPK-mTOR and
inflammation for pancreatic cancer prevention and treatment. Cancer
Prev Res (Phila). 7:388–397. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Simon TG, Ma Y, Ludvigsson JF, Chong DQ,
Giovannucci EL, Fuchs CS, Meyerhardt JA, Corey KE, Chung RT, Zhang
X and Chan AT: Association between aspirin use and risk of
hepatocellular carcinoma. JAMA Oncol. 4:1683–1690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kehm RD, Hopper JL, John EM, Phillips KA,
MacInnis RJ, Dite GS, Milne RL, Liao Y, Zeinomar N, Knight JA, et
al: Regular use of aspirin and other non-steroidal
anti-inflammatory drugs and breast cancer risk for women at
familial or genetic risk: A cohort study. Breast Cancer Res.
21:522019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ou YQ, Zhu W, Li Y, Qiu PX, Huang YJ, Xie
J, He SM, Zheng XK, Leng TD, Xu D and Yan GM: Aspirin inhibits
proliferation of gemcitabine-resistant human pancreatic cancer
cells and augments gemcitabine-induced cytotoxicity. Acta Pharm
Sin. 31:73–80. 2010. View Article : Google Scholar
|
|
34
|
Yue W, Zheng X, Lin Y, Yang CS, Xu Q,
Carpizo D, Huang H, DiPaola RS and Tan XL: Metformin combined with
aspirin significantly inhibit pancreatic cancer cell growth in
vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and
Bcl-2. Oncotarget. 6:21208–21224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baron JA, Cole BF, Sandler RS, Haile RW,
Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R,
Burke CA, et al: A randomized trial of aspirin to prevent
colorectal adenomas. N Engl J Med. 348:891–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sandler RS, Halabi S, Baron JA, Budinger
S, Paskett E, Keresztes R, Petrelli N, Pipas JM, Karp DD, Loprinzi
CL, et al: A randomized trial of aspirin to prevent colorectal
adenomas in patients with previous colorectal cancer. N Engl J Med.
348:883–890. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao Y, Nishihara R, Wu K, Wang M, Ogino S,
Willett WC, Spiegelman D, Fuchs CS, Giovannucci EL and Chan AT:
Population-wide impact of long-term use of aspirin and the risk for
cancer. JAMA Oncol. 2:762–769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fraser DM, Sullivan FM, Thompson AM and
McCowan C: Aspirin use and survival after the diagnosis of breast
cancer: A population-based cohort study. Br J Cancer. 111:623–627.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qorri B, Mokhtari RB, Harless WW and
Szewczuk MR: Next generation of cancer drug repurposing:
therapeutic combination of aspirin and oseltamivir phosphate
potentiates gemcitabine to disable key survival pathways critical
for pancreatic cancer progression. Cancers (Basel). 14:13742022.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zinger A, Koren L, Adir O, Poley M, Alyan
M, Yaari Z, Noor N, Krinsky N, Simon A, Gibori H, et al:
Collagenase nanoparticles enhance the penetration of drugs into
pancreatic tumors. ACS Nano. 13:11008–11021. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang G, Zhou Z, Zhao Z, Li Q, Wu Y, Yan S,
Shen Y and Huang P: Enzyme-triggered transcytosis of dendrimer-drug
conjugate for deep penetration into pancreatic tumors. ACS Nano.
14:4890–4904. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu F, Kai J, Chen L, Wu M, Dong J, Wang Q
and Zeng LH: Akt inhibitor perifosine prevents epileptogenesis in a
rat model of temporal lobe epilepsy. Neurosci Bull. 34:283–290.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:7–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Quan C, Sun J, Lin Z, Jin T, Dong B, Meng
Z and Piao J: Ezrin promotes pancreatic cancer cell proliferation
and invasion through activating the Akt/mTOR pathway and inducing
YAP translocation. Cancer Manag Res. 11:6553–6566. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Samuel EL, Marcano DC, Berka V, Bitner BR,
Wu G, Potter A, Fabian RH, Pautler RG, Kent TA, Tsai AL and Tour
JM: Highly efficient conversion of superoxide to oxygen using
hydrophilic carbon clusters. Proc Natl Acad Sci USA. 112:2343–2348.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Trachootham D, Lu W, Ogasawara MA, Nilsa
RD and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
He K, Wu L, Ding Q, Haider F, Yu H, Wang H
and Xiang G: Apatinib promotes apoptosis of pancreatic cancer cells
through downregulation of hypoxia-inducible factor-1alpha and
increased levels of reactive oxygen species. Oxid Med Cell Longev.
2019:51520722019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zou P, Zhang J, Xia Y, Kanchana K, Guo G,
Chen W, Huang Y, Wang Z, Yang S and Liang G: ROS generation
mediates the anti-cancer effects of WZ35 via activating JNK and ER
stress apoptotic pathways in gastric cancer. Oncotarget.
6:5860–5876. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang F, Wang Q, Zhou ZW, Yu SN, Pan ST, He
ZX, Zhang X, Wang D, Yang YX, Yang T, et al: Plumbagin induces cell
cycle arrest and autophagy and suppresses epithelial to mesenchymal
transition involving PI3K/Akt/mTOR-mediated pathway in human
pancreatic cancer cells. Drug Des Devel Ther. 9:537–560.
2015.PubMed/NCBI
|
|
51
|
Gu WJ and Liu HL: Induction of pancreatic
cancer cell apoptosis, invasion, migration, and enhancement of
chemotherapy sensitivity of gemcitabine, 5-FU, and oxaliplatin by
hnRNP A2/B1 siRNA. Anticancer Drugs. 24:566–576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lin S, Pan Y and Xu C: Effects of aspirin
on pancreatic cancer cells PANC-1 and its potential molecular
mechanism. J BUON. 25:2449–2455. 2020.PubMed/NCBI
|
|
53
|
Drew DA, Cao Y and Chan AT: Aspirin and
colorectal cancer: The promise of precision chemoprevention. Nat
Rev Cancer. 16:173–186. 2016. View Article : Google Scholar : PubMed/NCBI
|