Introduction
Paragangliomas (PGLs) are rare vascular,
neuroendocrine tumors of paraganglia cell clusters originating from
the neural crest tissue of the autonomic nervous system (1). According to the World Health
Organization (WHO) 2017 classification, tumors arising from the
adrenals are considered pheochromocytomas, while those associated
with extra-adrenal sympathetic tissue are considered as
paragangliomas (1). PGLs are
accompanied by catecholamine oversecretion-related symptoms such as
hypertension, palpitations and headache. In addition, several
patients with PGLs suffer from abdominal discomfort (2,3).
Arising from paraganglionic tissue that runs along
the axis of the body that is distributed in areas from the base of
the skull to the pelvis, the most common site of PGLs is the
retroperitoneal space (55.2%) and head and neck regions (25.6%).
Occasionally, PGLs develop in the bladder and mediastinum (2). The incidence of PGLs is 2–10 per
million people. Surgical resection of the tumor is the main
therapy. However, primary hepatic PGL, and gastrointestinal
disease-related primary hepatic PGL, is very rare (2,4–12). To
the best of our knowledge, the present case report is the first to
describe a female patient with primary hepatic PGL accompanied by
megacolon in terms of clinical, radiological and histopathological
features.
Case report
A 21-year-old female patient was referred to Beijing
Tiantan Hospital (Beijing, China) with hypoferric anemia during
September 2021. The patient was referred to another local hospital
due to bilateral lower limb swelling. The patient did not complain
of dizziness and weakness, but described symptoms of abdomen
distension. After the diagnosis of anemia, the patient was treated
with iron supplements. The patient exhibited a complicated medical
history that included constipation, with an average defecation of
once every 5 days to 1 month, weight loss of 5 kg over a period of
3 months and intestinal obstruction that improved following 3 years
of conservation treatment.
Physical examination revealed microcytic hypochromic
anemia, blood pressure of 75–120 mmHg and an abdomen circumference
of up to 100 cm. Laboratory tests showed mild anemia with
hemoglobin levels of 96 g/l. The values for carcinoembryonic
antigen, neuron-specific enolase (NSE) and CYFRA 21-1 were 8.92,
27.59, and 5.79 ng/ml, respectively. The levels of urinary
vanillylmandelic acid were not determined since the patient had no
symptoms of a functional tumor.
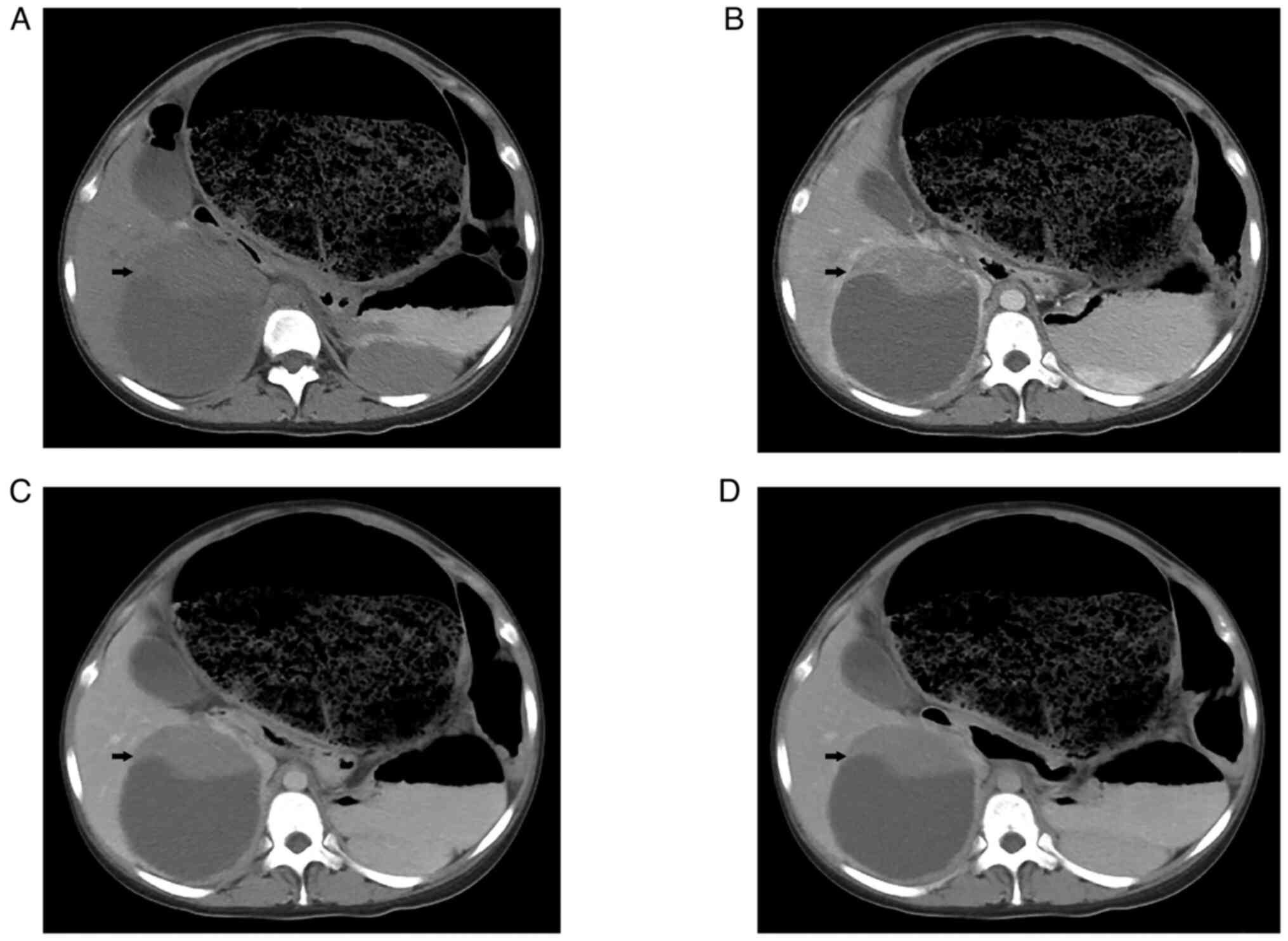
Following admission, ultrasound examination showed a
solid-cystic mass with a size of 11.4×10.2 cm in the right lobe of
the liver, which was considered benign disease. A CT scan of the
entire abdomen showed a large, round, hypodense mass with a solid
and well-marginated periphery. A contrast-enhanced CT scan revealed
that the solid portion of the abdominal circumference displayed
strong enhancement in the arterial phase. In addition, several
tortuous blood vessels were observed (Fig. 1). The sigmoid colon and rectum were
obviously distended and expanded, filled with gas and intestinal
contents. The maximum diameter of the colon was 20 cm. The patient
did not undergo MRI examination. In addition, gastrointestinal
angiography and colonoscopy were not performed due to intestinal
contents. Finally, the patient was diagnosed with iron-deficiency
anemia, liver damage and megacolon.
The patient was transferred to the General Surgery
Department of Beijing Tiantan Hospital and was routinely
administered an enema while fasting. Following symptomatic
treatment for 15 days, the patient underwent partial hepatectomy,
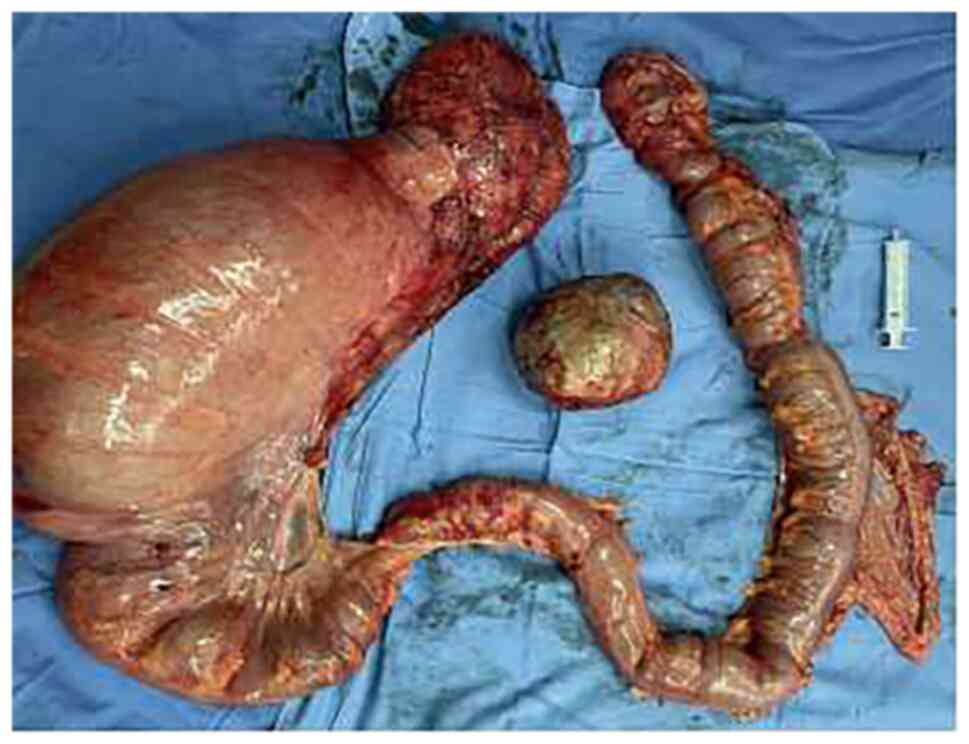
total colectomy and enterostomy. Intraoperative examination
revealed a huge sigmoid colon with a diameter of 15 cm. The rest of
the colon was dilated with a mean diameter of 6 cm. A hard mass
with a diameter of 10 cm and a smooth surface was found in the
right posterior lobe of the liver (Fig.
2). The surgical procedure was completed without any
complications based on detailed preoperative planning.
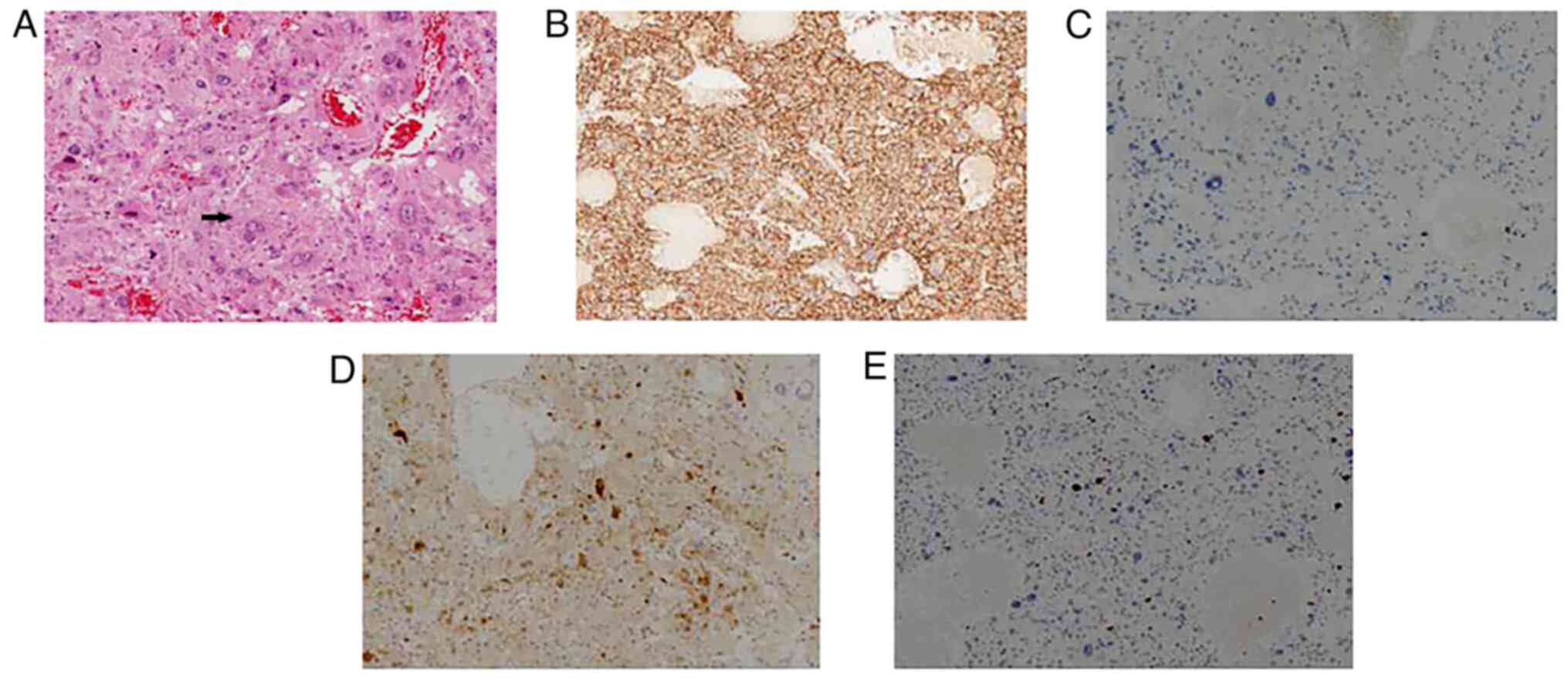
Pathological examination following surgery showed
that the mass had a nested and trabecular architecture with a
prominent vascular network. In addition, tumor cells with
pleomorphic morphology were present (Fig. 3). Giant tumor cells were also
observed, while mitotic cells were rare. For light microscope, 4 um
sections were routinely mounted. The tissues were use
formalin-fixed, paraffin-embedded (FFPE) and use 10% neutral buffer
formalin fixed for 12–24 h. We used DAB for color development (DAB
staining kit, ZLI-9019, Beijing Zhongshan Jinqiao Biotechnology
Co., Ltd.). Immunohistochemical staining of the resected liver
tissues revealed that liver cells were positive for CD56,
chromogranin A, vimentin, S-100, melan-A and NSE. However, liver
cells were negative for hepatocyte paraffin-1, α fetoprotein,
arginase-1, cytokeratin, HMB-45, CD68, epithelial membrane antigen,
desmin and smooth muscle actin. The Ki-67 labeling index was <5%
(Fig. 3). The patient was
postoperatively followed-up for 6 months and no evidence of
recurrence or metastasis was recorded.
Discussion
PGLs are rare neuroendocrine tumors arising from the
autonomic nervous system and are classified into functional and
non-functional PGLs based on secretion levels of catecholamine.
Catecholamine levels in patients with functional PGL are commonly
4× higher than normal level (10).
These patients commonly present characteristic symptoms, such as
hypertension, palpitations and headache (10). A total of 10–15% of patients with
PGLs are asymptomatic and the painless mass is identified οn
abdominal imaging (13).
In the present case report, the patient did not
display typical symptoms associated with catecholamine secretion
and only experienced hypoferric anemia. This indicated that the
accurate diagnosis of primary hepatic PGL is difficult when relying
only on age, medical history and symptoms.
PGLs are accompanied by several clinical symptoms
associated with the secretion of catecholamines. Catecholamines act
on α, β and dopaminergic receptors to modulate processes including
heart rate, blood pressure and myocardial contractility (14). In addition to effects on the
cardiovascular system, catecholamines affect the gastrointestinal
system via stimulating smooth muscle α-1, α-2 and β-2 adrenergic
receptors (3). Sustained secretion
of high catecholamine levels may result in intestinal smooth muscle
relaxation (15). Furthermore,
prolonged vasoconstriction of the gastrointestinal tract leads to
intestinal wall ischemia, which further contributes to decreased
gastrointestinal motility (3).
Clinically, patients with PGLs may initially experience
intermittent constipation followed by intestinal obstruction and
megacolon for a long period (16).
Megacolon, a rare complication of PGL, consists of
three types, namely congenital Hirschsprung's disease, idiopathic
Hirschsprung's megacolon and acquired megacolon (AMC).
Hirschsprung's disease is caused by intestinal ganglion cell
dysplasia, while AMC is associated with secretion of catecholamines
that affect the gastrointestinal system via stimulating smooth
muscle receptors (16).
In the present case report, the initial symptoms of
the patient were associated with the gastrointestinal system,
including anemia and abdomen distension. The patient's condition
was not obviously relieved following symptomatic treatment. In
addition, the patient had a medical history of intestinal
obstruction 3 years before the final hospital admission. However,
the patient had not undergone imaging examination during the
aforementioned period.
Imaging examination serves a critical role in the
management of PGLs and commonly guides clinical treatment. PGL is
commonly present as a homogenous mass with a soft tissue density
>10 Hounsfield units and uniform contrast enhancement οn CT
images (17). However, larger PGLs
undergo hemorrhage and necrosis, thus resulting in areas of lower
density. On an MRI scan, this tumor type commonly appears as a
free-fat mass on chemical shift with high signal on T2 sequences.
In previous studies, the tumor might display high signal intensity
on T2-weighted images, while increased necrosis within the contrast
enhanced tumor could be observed (14,17,18).
Anatomical imaging, such as CT and MRI scanning, show excellent
sensitivity, are helpful in delineating anatomic significance, but
less helpful in metastatic disease. Therefore, positron emission
tomography may be more useful in evaluating PGLs (17).
Distinguishing PGLs from other liver tumors via
imaging examination is difficult. Previous studies have suggested
that primary hepatic PGLs could initially be misdiagnosed as
hepatocellular carcinoma (HCC) (5,6,8,12),
fibrolamellar HCC (FHC) (4,7) or cavernous hemangioma of the liver
(CHL) (9). However, HCC is
typically characterized by arterial enhancement followed by portal
venous phase washout. Additionally, FHC commonly presents with
enhanced central scar on delayed-phase images. CHL is commonly
characterized by contrast enhancement at the periphery of the mass
in the early phase. In the present case study, the mass presented
with delayed persistent enhancement and a massive cystic change.
Given the rarity of primary hepatic PGLs and their non-specific
imaging features, it is difficult to make an accurate diagnosis of
this disease without pathological examination and laboratory
tests.
Surgical resection is considered the optimal
treatment approach for primary hepatic PGL. However, surgery may
lead to massive release of catecholamines and is therefore a major
mortality risk factor (19).
Furthermore, previous studies have reported extreme fluctuations in
blood pressure following surgery-mediated tumor induction (5,12).
Therefore, it is important for the preoperative diagnosis of PGLs
to be accurate to avoid perioperative complications and to improve
surgical safety.
Primary hepatic PGL is commonly considered a benign
disease. To the best of our knowledge, however, currently, there is
no available reliable pathological or imaging method to clarify
whether a tumor is benign or malignant (20). A previous case report showed hepatic
PGL metastasis in the spleen and liver after surgical treatment
with a 3-year follow-up (12). In
the present case report, recurrence or metastasis was not observed.
However, a long-term follow-up period is required.
PGLs usually occur sporadically and ~10% of cases
are hereditary (13). Multiple
genes are associated with PGL and up to 40% of patients exhibit a
disease-causing germ-line mutation (21). Classic tumor syndromes such as
neurofibromatosis type 1 (NF1), multiple endocrine neoplasia type 2
and Von-Hippel Lindau syndrome (VHL) are associated with a
germ-line mutation in the tumor suppressor gene NF1, proto-oncogene
RET and tumor suppressor gene VHL, respectively (22). Hereditary PGLs usually develop at a
younger age and more frequently grow bilaterally (19).
PGLs have a genetic predisposition. Once diagnosis
is established, genetic testing is recommended for all patients.
The specific gene may guide the imaging observation and subsequent
treatment (23). The first-degree
relatives of a mutation carrier should be offered predictive
testing to make a diagnosis in the early stages and to provide a
targeted preventive therapy (23).
In the present case report, the patient had no family history of
similar disease. In addition, the monitoring and treatment of
helicobacter pylori and physical examinations using gastroscopy and
colonoscopy are important methods for early detection of digestive
cancer. Prophylactic probiotics may help digestion of food and
absorption of nutrients to alleviate the burden of the intestine
and colon (24).
In summary, primary hepatic PGL is rare type of
tumor. It less commonly causes intestinal obstruction and megacolon
compared with common catecholamine secretion-associated symptoms,
such as hypertension, palpitations and headaches. The present case
report described the complex course of pathological changes
involved in the development of megacolon. Overall, the results
suggested that comprehensive imaging evaluation may be of
significance for the diagnosis and treatment of non-hypertensive
hepatic PGLs. Medical and surgical teams may be able to treat the
perioperative condition and decrease complication rates of this
rare neoplasm.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JZ contributed to the conception and design of this
study. JPB discovered this case in clinical work and summarized the
case. JPB collected clinical information and performed the
literature review. JPB and JZ wrote and revised the manuscript. MXS
performed the pathological examination. NZ collected imaging data
and revised the content of imaging examination. JZ, JPB, MXS and NZ
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from the patient.
Patient consent for publication
Informed consent was obtained from the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lloyd RV, Osamura YR, Kloppel G and Rosa
J: WHO classification of tumours of endocrine organs. WHO
Classification of Tumours. 10. 4th edition. World Health
Organization; Geneva: 2017
|
|
2
|
Liao W, Ding ZY, Zhang B, Chen L, Li GX,
Wu JJ, Zhang B, Chen XP and Zhu P: Primary functioning hepatic
paraganglioma mimicking hepatocellular carcinoma: A case report and
literature review. Medicine (Baltimore). 97:e02932018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thosani S, Ayala-Ramirez M, Román-González
A, Zhou S, Thosani N, Bisanz A and Jimenez C: Constipation: an
overlooked, unmanaged symptom of patients with pheochromocytoma and
sympathetic paraganglioma. Eur J Endocrinol. 173:377–387. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corti B, D'Errico A, Pierangeli F,
Fiorentino M, Altimari A and Grigioni WF: Primary paraganglioma
strictly confined to the liver and mimicking hepatocellular
carcinoma: An immunohistochemical and in situ hybridization study.
Am J Surg Pathol. 26:945–949. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan MR, Raza R, Jabbar A and Ahmed A:
Primary non-functioning paraganglioma of liver: A rare tumour at an
unusual location. J Pak Med Assoc. 6:814–816. 2011.PubMed/NCBI
|
|
6
|
Lin CS and Hsu YH: A primary paraganglioma
of the liver mimicking hepatocellular carcinoma. Ci Ji Yi Xue Za
Zhi. 31:286–288. 2019.PubMed/NCBI
|
|
7
|
Vella I, De Carlis R, Lauterio A and De
Carlis L: Extremely rare presentation of primary nonfunctioning
hepatic paraganglioma. Dig Liver Dis. 54:838–839. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bachmeier CAE, Haque M, Barrett HL and
Morton A: Hepatic paraganglioma hiding as a slowly growing lesion
for 24 years: A diagnostic conundrum. BMJ Case Rep. 12:e2289472019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koh PS, Koong JK, Westerhout CJ and Yoong
BK: Education and imaging. Hepatobiliary and pancreatic: A huge
liver paraganglioma. J Gastroenterol Hepatol. 28:10752013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miller ME, Vietor NO, Park EJ, Sweeney SP,
Katz M and Vietor RC: Paraganglioma masquerading as a primary liver
lesion: A rare entity discovered during surgery. Clin Case Rep.
10:e053102022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang H, Xu L and Mu Q: Primary
functioning hepatic paraganglioma: A case report. Adv Ther.
23:817–820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
You Z, Deng Y, Shrestha A, Li F and Cheng
N: Primary malignant hepatic paraganglioma mimicking liver tumor: A
case report. Oncol Lett. 10:1176–1178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Corssmit EP and Romijn JA: Clinical
management of paragangliomas. Eur J Endocrinol. 171:R231–R243.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gunawardane PTK and Grossman A:
Phaeochromocytoma and paraganglioma. Adv Exp Med Biol. 956:239–259.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Szakacs JE and Cannon A: L-Norepinephrine
myocarditis. Am J Clin Pathol. 30:425–434. 1958. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sweeney AT, Malabanan AO, Blake MA, de las
Morenas A, Cachecho R and Melby JC: Megacolon as the presenting
feature in pheochromocytoma. J Clin Endocrinol Metab. 85:3968–3972.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carrasquillo JA, Chen CC, Jha A, Ling A,
Lin FI, Pryma DA and Pacak K: Imaging of pheochromocytoma and
paraganglioma. J Nucl Med. 62:1033–1042. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baez JC, Jagannathan JP, Krajewski K,
O'Regan K, Zukotynski K and Ramaiya NH: Pheochromocytoma and
paraganglioma: Imaging characteristics. Cancer Imaging. 12:153–162.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jain A, Baracco R and Kapur G:
Pheochromocytoma and paraganglioma-an update on diagnosis,
evaluation, and management. Pediatr Nephrol. 35:581–594. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka S, Ito T, Tomoda J, Higashi T,
Yamada G and Tsuji T: Malignant pheochromocytoma with hepatic
metastasis diagnosed 20 years after resection of the primary
adrenal lesion. Intern Med. 32:789–794. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Crona J, Taïeb D and Pacak K: New
perspectives on pheochromocytoma and paraganglioma: Toward a
molecular classification. Endocr Rev. 38:489–515. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fishbein L: Pheochromocytoma and
paraganglioma: Genetics, diagnosis, and treatment. Hematol Oncol
Clin North Am. 30:135–150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neumann HPH, Young WF Jr and Eng C:
Pheochromocytoma and paraganglioma. N Engl J Med. 381:552–565.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karimi P, Islami F, Anandasabapathy S,
Freedman ND and Kamangar F: Gastric cancer: Descriptive
epidemiology, risk factors, screening, and prevention. Cancer
Epidemiol Biomarkers Prev. 23:700–713. 2014. View Article : Google Scholar : PubMed/NCBI
|