Introduction
Lung cancer is considered as one of the most lethal
tumors, having the most increased incidence rate among tumors, with
the highest mortality rate worldwide. Lung cancer remains the
leading cause of cancer-related mortality, ranking first in
percentage due to cancer in 2020 (1). According to the pathological type,
lung cancer can be divided into small cell lung cancer and
non-small cell lung cancer (NSCLC), of which NSCLC accounts for 80%
of all, and lung adenocarcinoma (LUAD) accounts for the majority of
NSCLC. The majority of patients with NSCLC, patients with LUAD in
particular, exhibit symptoms not earlier than the middle or late
stages of the disease, since the etiology remains unclear and early
symptoms are not evident. In spite of several advancements being
made in the treatment of LUAD, the average overall survival of
patients with LUAD is limited to <5 years (2). Therefore, it is of utmost urgency to
further identify novel key molecules for the development of novel
therapeutic targets.
Several LUAD molecular markers have been identified
in previous studies (3–7); however, a single gene cannot
accurately represent the characteristics of LUAD due to its complex
pathophysiology. Unlike the differential expression analysis that
focuses on a single gene, co-expression network analysis provides
new insight into understanding the pathogenesis of diseases and
opportunities for therapeutic intervention by unsupervised
identification of co-expressed gene modules (8,9). It
has been successfully applied to the study of various biological
processes, including chronic obstructive pulmonary disease and
cancer, and has been proven to be quite effective in identifying
candidate biological markers and therapeutic targets (9,10).
Currently, several studies have identified genes
that are closely associated with LUAD development through
comprehensive analysis of single or multiple microarray datasets in
the currently available in the Gene Expression Omnibus (GEO)
database. For example, Dong et al (11) identified aurora kinase A
(AURKA) and DNA topoisomerase II alpha (TOP2A) as the
two genes with the highest lymph node stage (N), which may be
targets for the diagnosis and treatment of LUAD. Zhang et al
(12) observed mitotic
spindle-related features that may be used as independent prognostic
indicators for patients with LUAD. Wang et al (13) observed that TOP2A may be one of the
key protein-coding genes for LUAD possibly serving as a biomarker
and therapeutic target for LUAD. Li et al (14) suggested that eight genes, including
TOP2A, marker of proliferation Ki-67 (MKI67),
platelet and endothelial cell adhesion molecule 1 (PECAM1),
CDK1, secreted phosphoprotein 1 (SPP1), checkpoint
kinase 1 (CHEK1), cyclin B1 (CCNB1), and
ribonucleotide reductase regulatory subunit M2 (RRM2) may be novel
pivotal genes closely associated with the progression and prognosis
of LUAD. Wang et al (15)
revealed that CCNB1, BUB1 mitotic checkpoint
serine/threonine kinase B (BUB1B), cell division cycle 20
(CDC20), TTK protein kinase (TTK) and mitotic arrest
deficient 2 like 1 (MAD2L1) may be potential targets for the
treatment of LUAD. Chen et al (16) demonstrated that 10 gene targets
including CDK1 and CDC20 were associated with a poor
prognosis of patients with lung cancer. Fan et al (17) suggested that TOP2A, G
protein-coupled receptor kinase 5 (GRK5), sirtuin 7
(SIRT7), minichromosome maintenance complex component 7
(MCM7), EGFR and collagen type I alpha 2 chain
(COL1A2) may be used as predictors for the diagnosis of
LUAD. Guo et al (18)
proposed that TOP2A and UBE2C were independent prognostic factors
for LUAD. Regardless of the abundance of studies on this topic, the
mechanisms responsible for the development of LUAD remain unclear
and have not yet been systematically studied, with further studies
required.
In the present study, the gene expression profile
dataset, GSE140797, was acquired from the GEO database, containing
gene expression data from 14 samples, including seven normal lung
and seven LUAD tissues for analysis. Following normalized data
preprocessing, the differentially expressed genes (DEGs) between
the two sample sets were analyzed. Concurrently, weighted gene
co-expression network analysis (WGCNA) was performed to construct a
gene co-expression network of LUAD and identify co-expression
modules. Subsequently, eight cancer tissue and eight adjacent
tissue samples were collected from patients with LUAD and reverse
transcription-quantitative PCR (RT-qPCR) and western blot analysis
were performed, in order to verify the WGCNA analysis, and the
expression analysis of the three key genes, AURKA, TOP2A and
maternal embryonic leucine zipper kinase (MELK), was
evaluated.
AURKA is a cyclin whose activation is required for
the process of cell division through the regulation of mitosis. The
ectopic overexpression of the AURKA gene results in the
inactivation of the G2-phase DNA damage checkpoint and the mitotic
spindle assembly checkpoint, as well as tetraploid and centrosome
expansion, particularly in cells with defective p53-dependent DNA
damage checkpoints upstream of AURKA. At the transporter level, the
EGF-induced expression of AURKA is dependent on the interaction of
nuclear EGFR and STAT5. At the downstream end of AURKA, certain
substrates of AURKA play critical inhibitory roles, with p53 and
large tumor suppressor kinase 2 being the most important substrates
of AURKA. AURKA substrates have received widespread attention as
tumor suppressors (19).
TOP2A has been demonstrated to be related to the
progression of several cancer types, such as hepatocellular
carcinoma (20), breast cancer
(21), bladder cancer (22), ovarian cancer (23), cervical cancer (24), pancreatic cancer (25), stomach cancer (26), including NSCLC (27,28).
Increased expression of MELK has been observed in
various cancer cells and tissues, playing a crucial and critical
role in the proliferation and self-renewal of progenitor and tumor
stem cells and is overexpressed in LUAD, increasing the probability
of tumorigenesis. Among them, MELK increases the proliferation of
cervical, breast, colorectal and pancreatic cancer cell lines
(29), while it is also involved in
and affects the development of hepatocellular carcinoma (30) and bladder cancer (31).
Materials and methods
Data source and preprocessing
GSE gene expression profile data and clinical
information were obtained from the GEO database at the National
Center for Bioinformatics. Gene expression data from 14 samples in
the GSE140797 dataset were analyzed, including seven normal lung
tissue and seven LUAD tissue samples. The annotation information of
the GPL13497 (Agilent-026652 Whole Human Genome Microarray 4×44Kv2)
platform was used as a reference to convert the probe to the
corresponding gene symbol, and the Limma software (version 3.54.2)
package was used to normalize the data for further analysis.
DEG analysis
The samples were divided into the normal control and
LUAD groups, and the conditions |log2FC|>1 and P<0.05 were
set to screen for genes with significant differences in
expression.
Data filtering
Co-expression networks were constructed using the
WGCNA package in the R language. To obtain a valid co-expression
network, the expression variance of each gene in all samples was
calculated, and the genes with the same variance were considered
for the construction of the co-expression network. Cluster analysis
was performed, in order to detect and remove outliers.
Construction of gene co-expression
network
Scale-free networks were constructed by selecting an
appropriate weighting coefficient (soft threshold) to make the
connections between genes adhere to the scale-free distribution of
network connection requirements, and the correlation coefficient
between genes was used to construct hierarchical clustering tree.
Different branches of the clustering tree represented different
gene modules, and different colors represented different gene
modules. Subsequently, genes were categorized according to their
expression patterns based on their weighted correlation
coefficients. The genes that exhibited similar gene expression
patterns were then grouped into a module, and then classified by
gene expression pattern for further analysis. Lastly, by applying
this coefficient, the correlation matrix was converted into an
adjacency matrix, which was then converted into a topological
overlap matrix.
Module and clinical feature
correlation analysis
The Pearson's correlation coefficients and P-values
of the matrices composed of gene and sample and clinical
correlations per module were calculated using WGCNA, and the
Pearson's correlation coefficients were used to measure the
correlation between different modules and clinical traits, and the
module with the highest correlation coefficient was used in
subsequent analysis. The correlation between gene expressed in the
module and the phenotype [gene significance (GS)] and the
correlation between gene expressed in the module and the module
membership (MM) were analyzed, and the genes were screened
according to GS >0.8 and MM >0.8.
Functional enrichment analysis
The cross section of modules with the highest
correlation between WGCNA and DEGs were selected, and Kyoto
Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO)
analyses were performed on this part of the gene set using the R
package cluster profiler (https://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html).
Construction of protein-protein
interaction (PPI) networks
The STRING database (https://string-db.org/) was used to select
intersecting genes to construct the PPI network. PPI pairs in the
network were visualized with a combined confidence score of ≥0.4.
Hub genes in the PPI network were identified using cytohubba, a
plug-in for Cytoscape software (version 3.7.2. http://cytoscape.org/) that identifies the top 10 hub
genes.
Verification of the central gene
The Gene Expression Profiling Interactive Analysis
Database (http://gepia.cancer-pku.cn/) (32) is an online analysis tool which can
be used to validate the top 10 central genes selected through
protein-protein interaction networks, which are based on The Cancer
Genome Atlas (TCGA) of Lung Adenocarcinoma (33) and the Genotype-Tissue Expression
(GTEx) LUAD database, which provides differential expression
analysis, profiling, and survival analysis for central gene
expression analysis, receiver operating characteristic (ROC) curve
analysis, and survival analysis.
Collection and processing of clinical
tissue samples
A total of eight fresh frozen clinical samples were
obtained from lung adenocarcinoma patients in Renmin Hospital of
Wuhan University. In addition, three male and five female patients,
ranging in age from 51 to 80 years, were recruited between December
14 and December 28, 2020. The specific age, sex, and disease stage
were i) male 70 years old, 2020.12.14, IIB stage; ii) male 63 years
old, 2020.12.16, IA2 stage; iii) female 62 years old, 2020.12.16,
IA stage; iv) female 59 years old, 2020.12.16, A stage; v) male 51
years old, 2020.12.17, A stage; vi) female 80 years old,
2020.12.24, IA3 stage; vii) Female 73 years old, 2020.12.25, IA
stage; viii) female 73 years old, 2020.12.28, IA stage). The
samples were obtained with patient consent and ethical approval
(approval no.WDRY2022-K231) from Renmin Hospital of Wuhan
University (Wuhan, China).
RT-qPCR
RNA was obtained from frozen fresh samples of lung
cancer and normal paracancerous lung tissue from eight lung
adenocarcinoma patients. RNA extraction was conducted using
TRIzol® reagent (cat. no. 15596026, Invitrogen; Thermo
Fisher Scientific, Inc.) and reverse transcribed into cDNA using
the PrimeScript RT Reagent kit according to the manufacturer's
instructions (cat. no. RR037A; Takara Bio, Inc.). Candidate primers
for each gene were designed using Premier 5 design program (PREMIER
Biosoft). PCR reaction was performed with the quantitative TB
Green-based PCR kit (cat. no. RR420A; Takara Bio, Inc.) using a CFX
Connect PCR machine (CFX Connect TM; Bio-Rad Laboratories, Inc.).
The following conditions were applied: Pre-denaturation stage:
95°C, 1 min for 1 cycle; amplification stage: denaturation at 95°C,
5 sec and annealing at 58°C, 30 sec, 40 cycles; melting curve
stage: 65°C to 95°C, increment 0.5°C for 5 second. The results were
analyzed using the 2−ΔΔCq method (34), and the primer pair sequences for
each gene are listed in Table
I.
 | Table I.Oligonucleotide primers used in the
present study. |
Table I.
Oligonucleotide primers used in the
present study.
| Gene |
| Oligonucleotide
primer sequence (5′-3′) |
|---|
| GAPDH | Sense |
GGAAGCTTGTCATCAATGGAAATC |
|
| Antisense |
TGATGACCCTTTTGGCTCCC |
| CDK1 | Sense |
AAGGGTAGACACAAAACTACAGGTC |
|
| Antisense |
ATGTACTGACCAGGAGGGATAGA |
| TOP2A | Sense |
CCTTCTATGGTGGATGGTTTGA |
|
| Antisense |
ATGGGCTGCAAGAGGTTTAGAT |
| MELK | Sense |
GATGTTCCCAAGTGGCTCTCTC |
|
| Antisense |
TCCTCCATTGTTTGCCTGTTG |
| NUSAP1 | Sense |
CTGCTGCTGTTATTACCCCATTC |
|
| Antisense |
CTTTCTTCTCCTTTCGTTCTTGC |
| BUB1 | Sense |
GAAGAAATACCACAATGACCCAAG |
|
| Antisense |
TGGGTTTCAGTGAGGCGTGT |
| AURKA | Sense |
TGCCCTGTCTTACTGTCATTCG |
|
| Antisense |
AAAGGAGGCTTCCCAACTAAAA |
| CCNB1 | Sense |
GCCTATTTTGGTTGATACTGCCTC |
|
| Antisense |
CTCCATCTTCTGCATCCACATC |
| PBK | Sense |
TGACTGCTCCTGCCTTCATAAC |
|
| Antisense |
TAACACCATTCTCCTCCACAGC |
Western blot analysis
Western blot analysis of relative protein expression
levels was performed as described as follows: Lung adenocarcinoma
and parapulmonary carcinoma were lysed with RIPA (cat. no. P0013B;
Beyotime Institute of Biotechnology) buffer to extract total
proteins, and the protein concentrations were then detected using a
BCA kit (cat. no. P0012S; Beyotime Institute of Biotechnology). The
protein samples were denatured in a dry heater at 95°C and
subsequently subjected to electrophoresis; 10% SDS gel (cat. no.
P0012A; Beyotime Institute of Biotechnology) was used for
electrophoresis and 25 µg of protein was loaded in each strip
Following electrophoresis, the separated proteins were transferred
to polyvinylidene difluoride membranes (cat. no. FFP2; Beyotime
Institute of Biotechnology) by the wet transfer membrane method.
Non-specific proteins on the membrane were blocked for 1 h at room
temperature and then incubated with primary monoclonal antibodies
corresponding to the proteins overnight at 4°C. The antibodies used
are as follows: A rabbit anti-AURKA polyclonal antibody (cat. no.
A15728), a rabbit anti-BUB1 mitotic checkpoint serine/threonine
kinase (BUB1) polyclonal antibody (cat. no. A1929), a rabbit
anti-CCNB1 polyclonal antibody (cat. no. A16800), a rabbit
anti-CDK1 polyclonal antibody (cat. no. A0220), a rabbit anti-MELK
monoclonal antibody (cat. no. A3530), a rabbit anti-nucleolar and
spindle associated protein 1 (NUSAP1) polyclonal antibody (cat. no.
A16000), a rabbit anti-TOP2A polyclonal antibody (cat. no. A16440)
and a mouse monoclonal antibody for β-actin (cat. no. AC004) (all
from ABclonal Biotech Co., Ltd. and all at 1:1,000).
The following day, the membranes were incubated for
1 h at room temperature using the corresponding secondary antibody;
Goat Anti-Rabbit IgG H&L (HRP; cat. no. ab205719)and Goat
Anti-Mouse IgG H&L (HRP; cat. no. ab205719 all from Abcam and
all at 1:10,000. This was followed by a brief incubation with ECL
Western Blotting Detection Reagent (cat. no. P0018S; Beyotime
Institute of Biotechnology) and a final exposure with an iBright
imaging system (Thermo Fisher Scientific, Inc.). Density
measurement was by ImageJ (version V1.8.0.112; National Institutes
of Health).
Statistical analysis
For the statistical calculations, the R (version
3.6) and WGCNA packages were used. The calculation of the
correlation coefficient between the relevant clinical
characteristics of LUAD tissue and the ME of each co-expression
module used in this article was based on the R language platform
Rstudio (version 8.9.173593; http://support-rstudio-com.netlify.app/products/rstudio/download/).
WGCNA was used to identify genes with similar functions. For each
gene pair, WGCNA determines the likelihood of association by using
a soft threshold. A weighted network of co-expression was formed
based on this concept. The data are expressed as the mean ± SE.
Parametric data were analyzed using the Student's paired t-test and
non-parametric data were analyzed using the Mann-Whitney U test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Data filtering
A co-expression network was constructed by including
5,435 genes with 25% of the maximum variation in the present study.
No significant outliers were observed by building hierarchical
clustering trees for 5,435 genes from 14 lung tissue samples. A
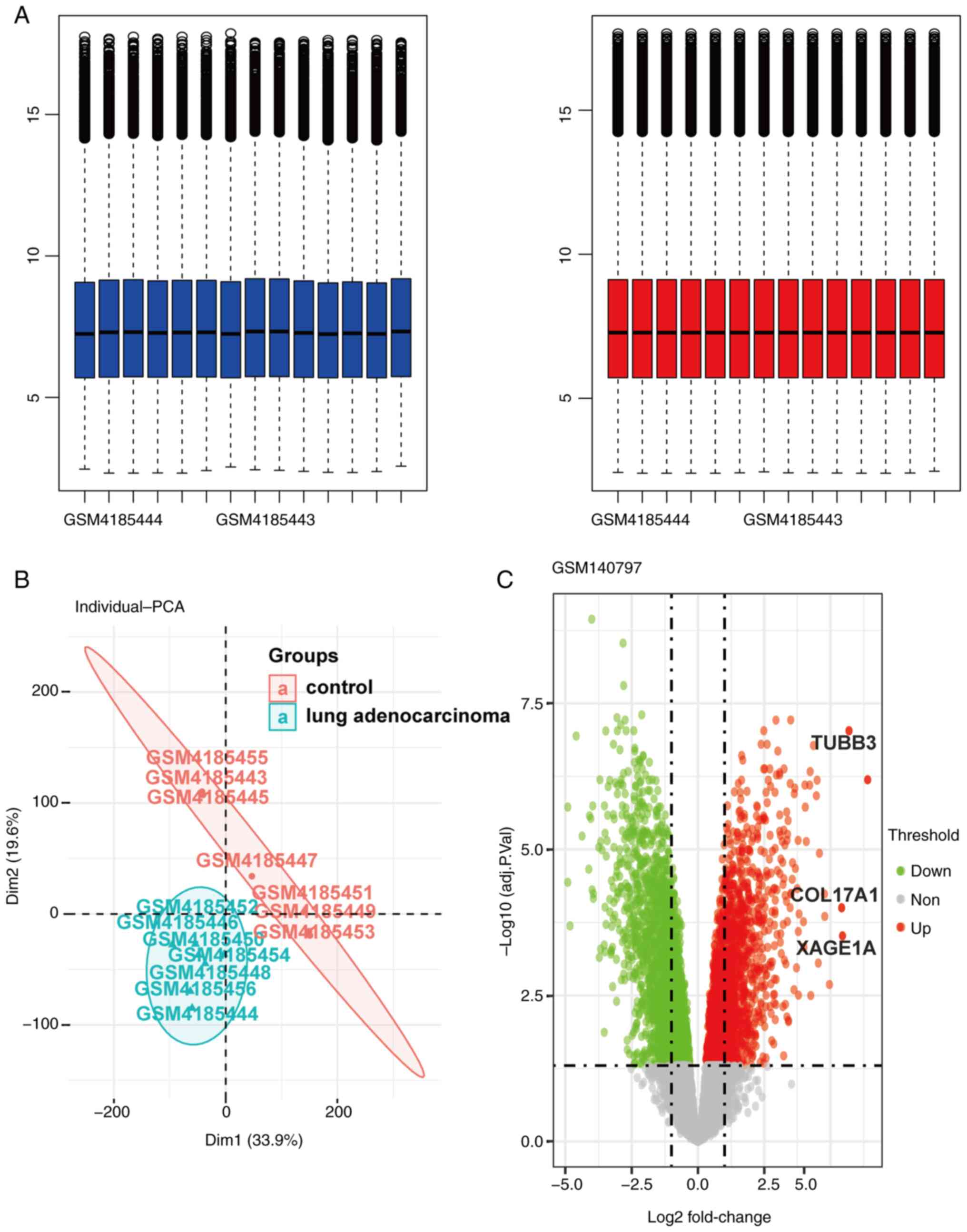
total number of 580 DEGs were identified in the dataset (Fig. 1), among which 254 genes were
downregulated and 326 genes were upregulated.
Construction of the gene co-expression
network module
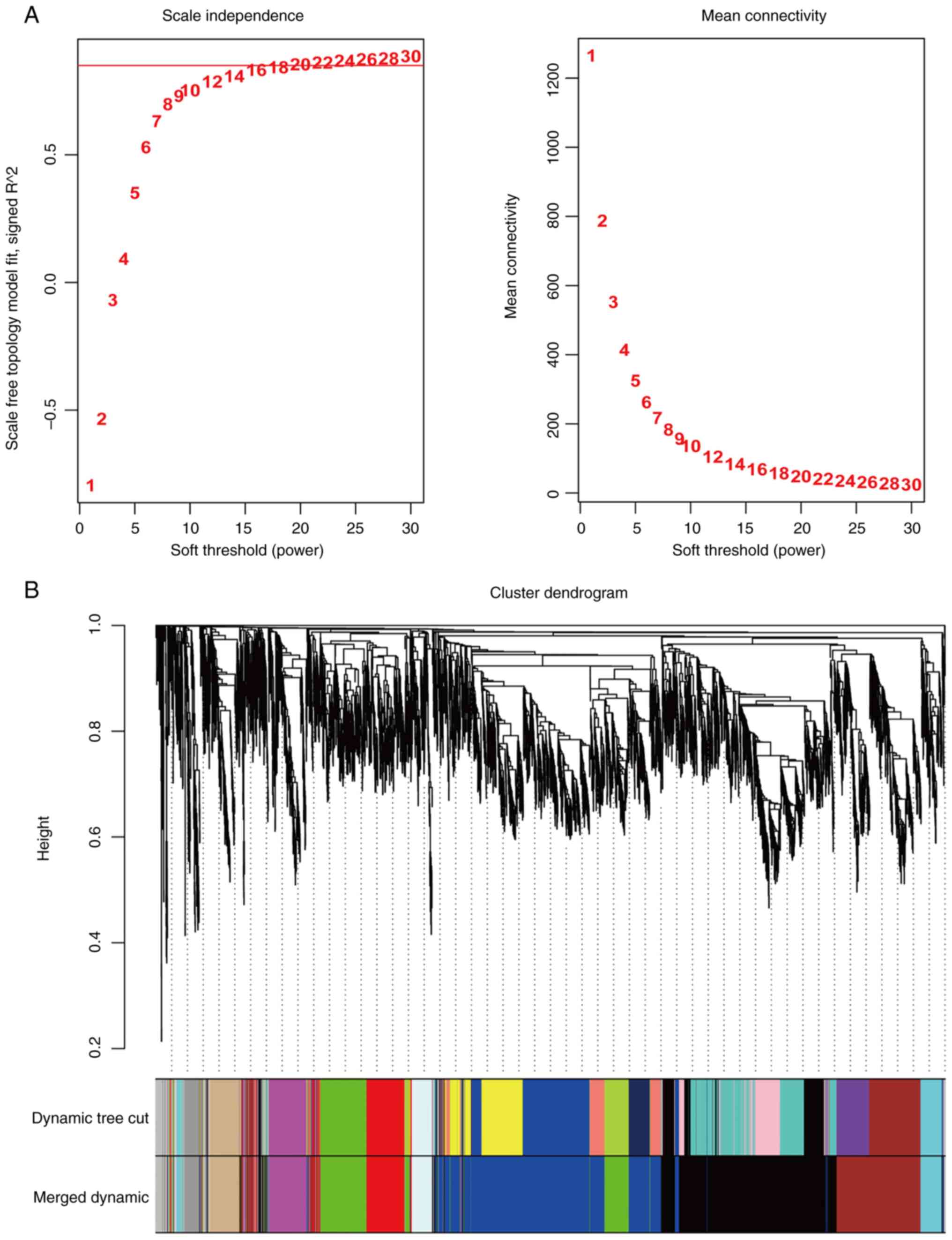
According to the non-scale network distribution
fitting, a value of 20 was selected as the soft threshold (β value)
for this dataset and a co-expression network was constructed
(Fig. 2) for module identification
using the dynamic cut tree method, finally acquiring 10 modules
(Fig. 3A).
Correlation analysis of modules and
clinical characteristics
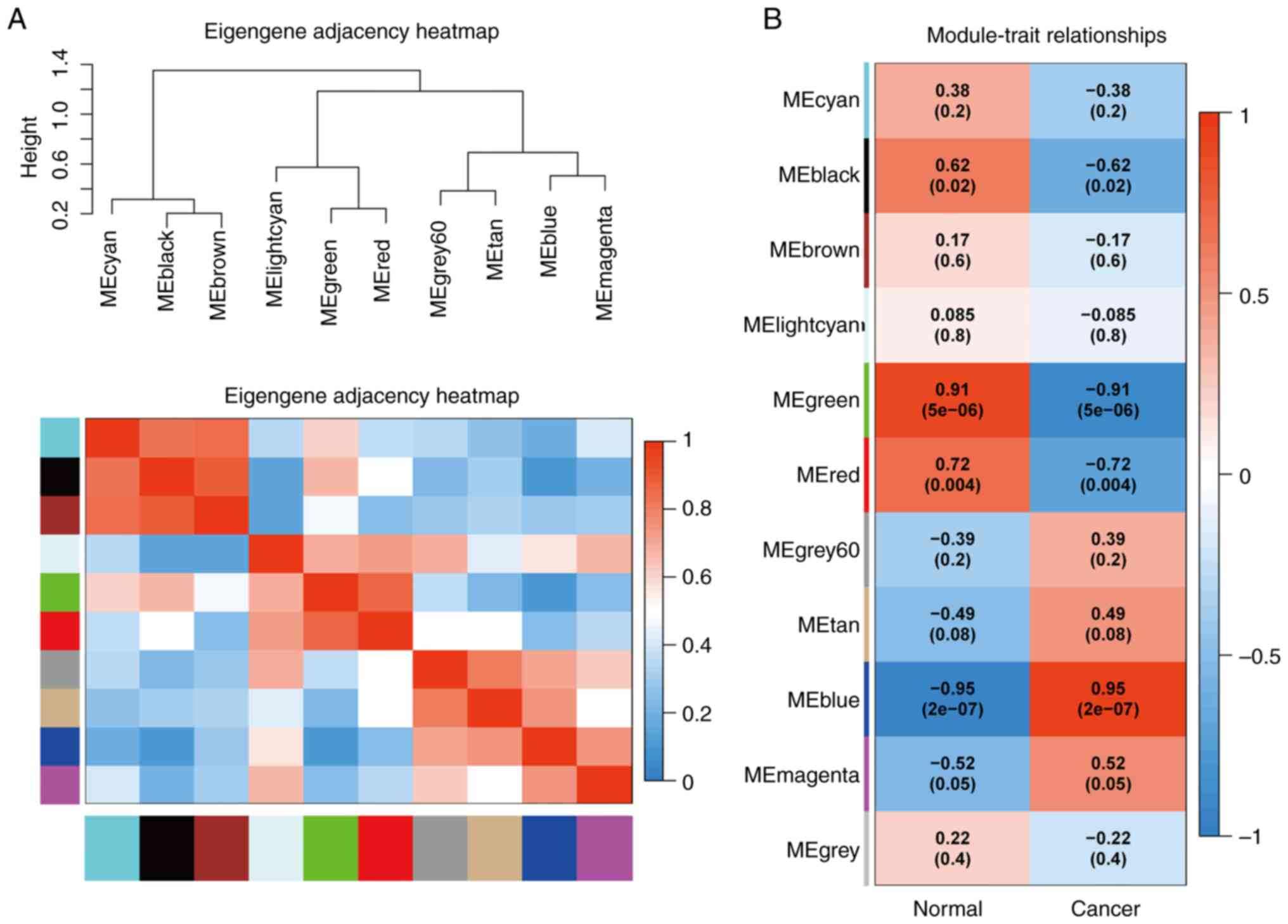
By applying the correlation analysis of each module
using sample clinical information, the green module presented with
the highest positive correlation, and the blue module the highest
degree of negative correlation with LUAD (Fig. 3B).
Identification and analysis of pivotal
genes
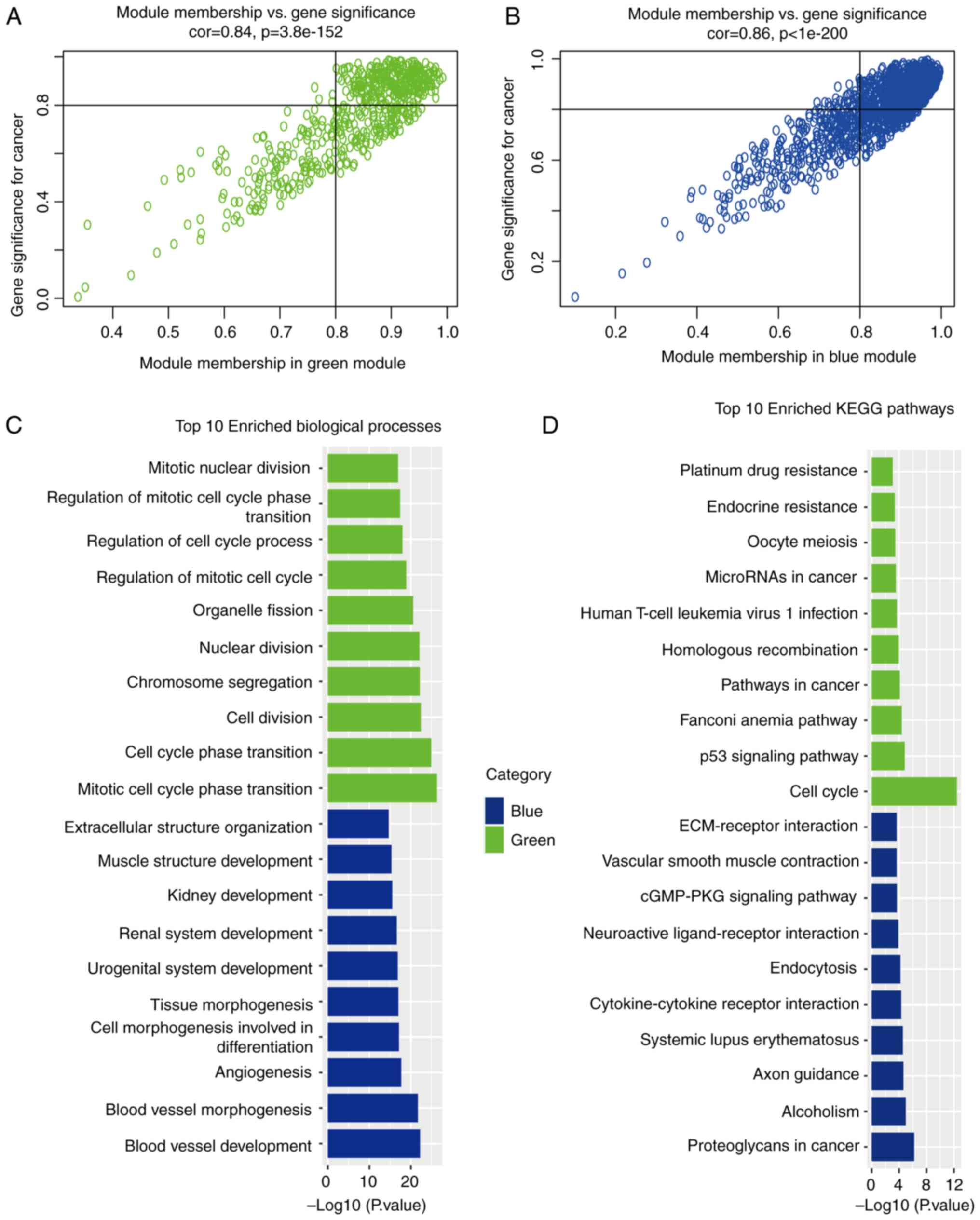
According to the criteria of GS >0.8 and MM
>0.8 to screen the key genes in the blue module and the green
module for the following research stage, 845 and 285 key genes were
selected from the blue and green modules, respectively.
Subsequently, GO function enrichment analysis and KEGG enrichment
analysis were performed on the 845 genes selected from blue module
and the 285 genes selected from green module (Fig. 4A and B). As regards the green
module, GO functional enrichment analysis revealed that common
pathogenic genes were mainly enriched in mitotic cell cycle phase
transition, cell cycle phase transition and cytoplasmic division,
whereas in the blue module, the common pathogenic genes were mainly
enriched in blood vessel development, blood vessel morphogenesis
and angiogenesis (Fig. 4C). KEGG
pathway analysis mainly demonstrated enrichment in the cell cycle,
p53 signaling pathway and Fanconi anemia pathway in the green
module, and proteoglycans in cancer, alcoholism and axon guidance
in the blue module (Fig. 4D).
PPI network construction and
analysis
The 845 genes from the blue module and 580
differentially expressed genes were intersected, in order to obtain
324 genes. Similarly, the 285 genes from the green module and 580
differential genes were intersected to obtain 107 genes. The two
PPI networks for the aforementioned 324 and 107 genes were then
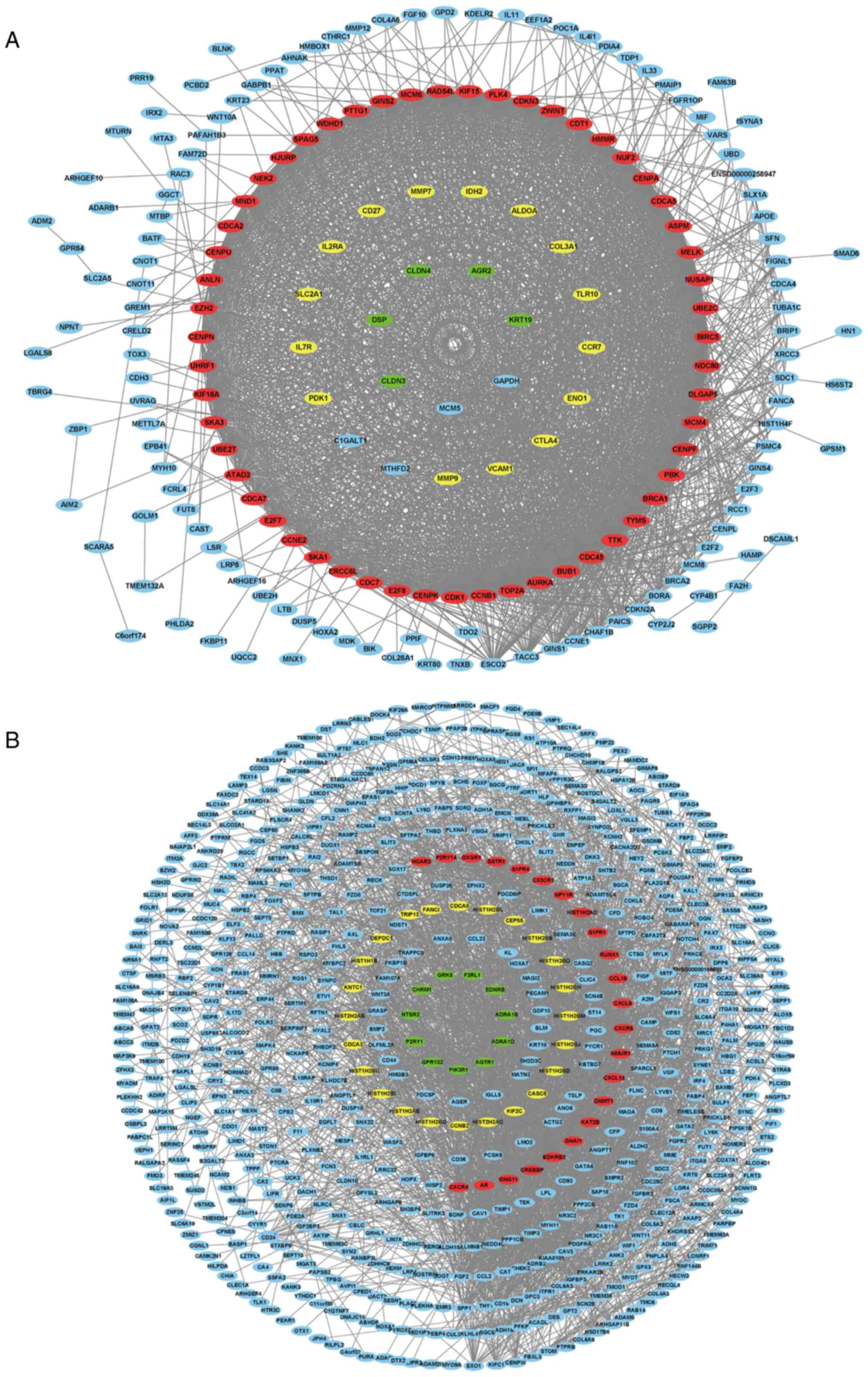
respectively established using Cytoscape software (Fig. 5), and 10 key genes were selected
from the two PPI networks, respectively according to the degree of
connectivity, including AURKA, BUB1, CCNB1, CDC45, CDK1, MELK,
NUSAP1, PBK, TOP2A, TTK, BDKRB2, CCL19, CX3CR1, CXCL13, CXCL9,
CXCR4, CXCR5, GNAI1, GNG11 and NMUR1. Among the genes,
BDKRB2, CCL19, CX3CR1, CXCL13, CXCL9, CXCR4, CXCR5, GNAI1,
GNG11 and NMUR1 were selected from the blue module, with
AURKA, BUB1, CCNB1, CDC45, CDK1, MELK, NUSAP1, PBK, TOP2A
and TTK selected from the green module.
Verification of the expression of the
20 selected genes in TCGA database
Subsequently, the expression profiles of 59 normal
lung tissues and 515 LUAD tissues were acquired from TCGA database
to verify the expression of the aforementioned 20 key genes. With
the exception of the expression of CXCR4 among the 20 genes,
the expression of the remaining 19 genes differed significantly
between normal lung tissue and LUAD tissues (Fig. 6).
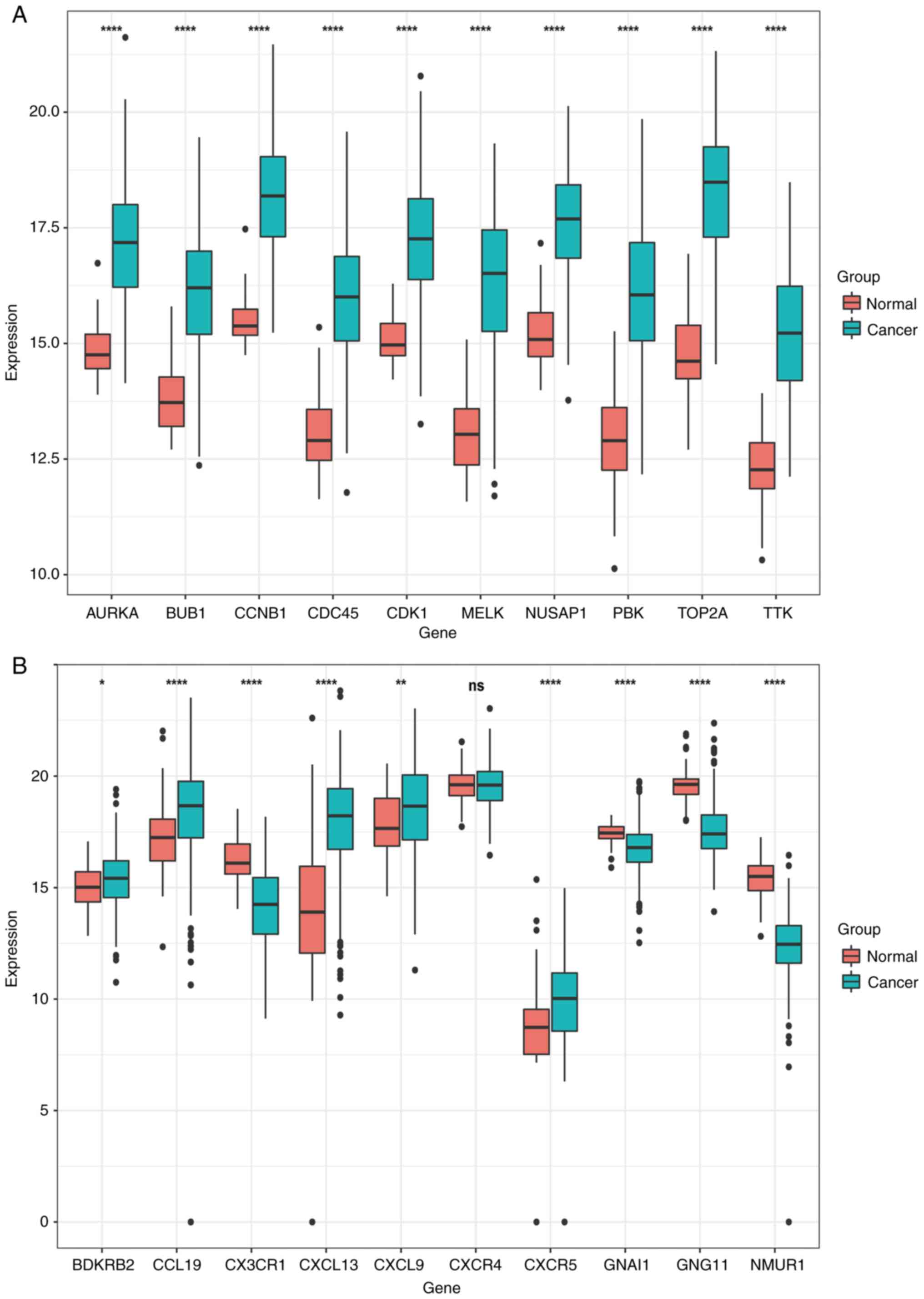 | Figure 6.In total, 59 control samples and 504
cancer samples were derived from TCGA database for verification.
From the 10 genes selected from the two modules, a total of 20
genes were verified. A total of 19 of these changes were verified,
and the other one (CXCR4) was excluded. (A) The expression
of AURKA, BUB1, CCNB1, CDC45, CDK1, MELK, NUSAP1, PBK, TOP2A
and TTK genes in normal lung tissues and lung adenocarcinoma
tissues, where red indicates normal lung tissues and green
indicates lung adenocarcinoma tissues. (B) The expression of
BDKRB2, CCL19, CX3CR1, CXCL13, CXCL9, CXCR4, CXCR5, GNAI1,
GNG11 and NMUR1 genes in normal lung tissues and lung
adenocarcinoma tissues, with red indicating normal lung tissues and
green indicating lung adenocarcinoma tissues. t-test of normal lung
tissue and lung adenocarcinoma tissue. *P<0.05, **P<0.01 and
****P<0.0001. |
ROC curve analysis
Subsequently, ROC curve analysis was performed on
the 19 genes verified in TCGA database, and it was observed that
apart from BDKRB2, CCL19, CXCR5, CXCL9, GNAL1 and
CX3CR1, and the other 13 genes had AUCs >0.9 (Fig. 7) and were considered in the
following stages of the analysis.
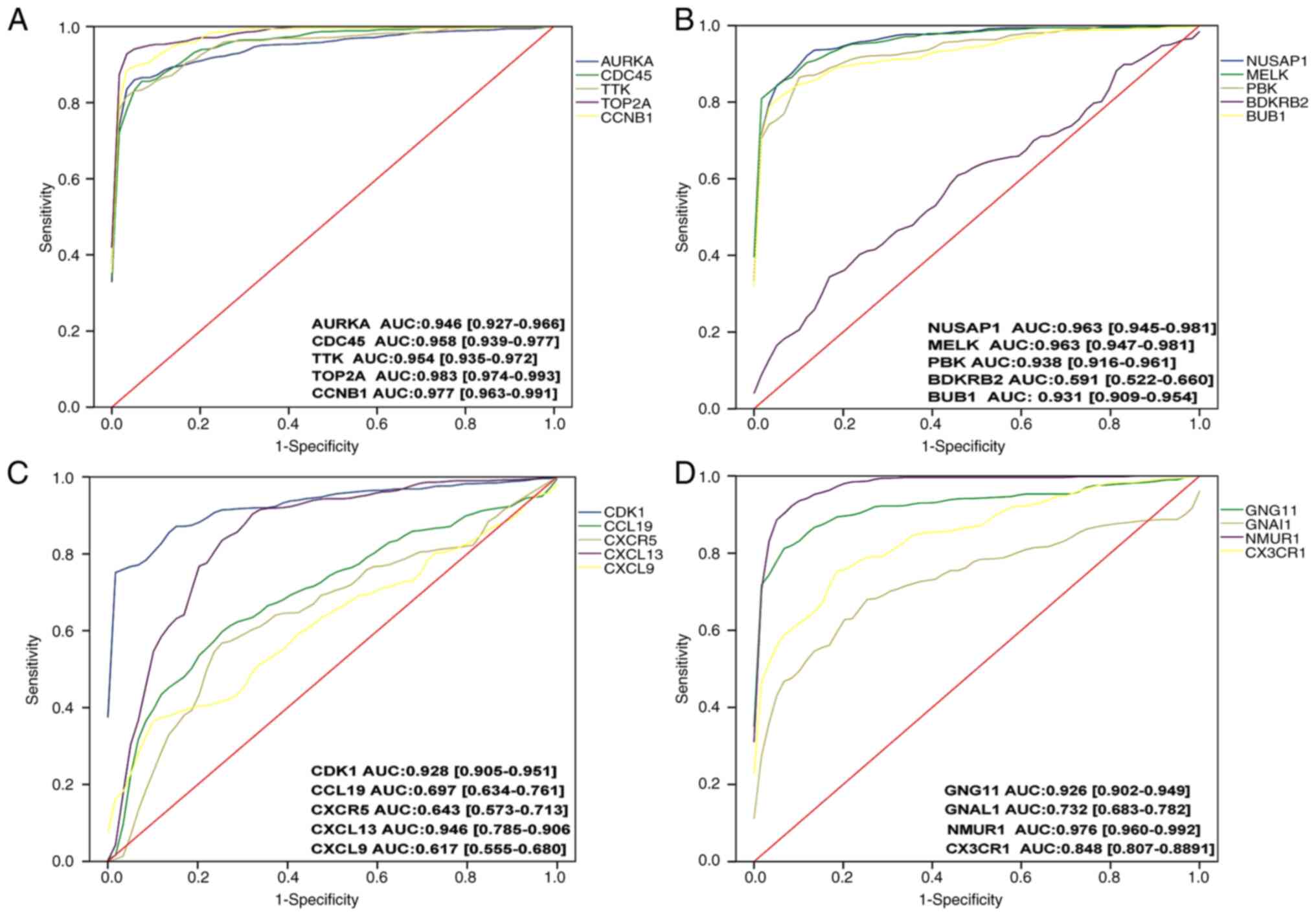 | Figure 7.ROC curve analysis of 20 genes. AUCs
>0.9 were included in the subsequent analysis. A total of 13
genes (AURKA, CDC45, TTK, TOP2A, CCNB1, NUSAP1, MELK, PBK, BUB1,
CDK1, CXCL13, GNG11 and NMUR1) were included, and seven
genes were excluded. (A) ROC curve analysis was performed for the
AURKA, CDC45, TTK, TOP2A and CCNB1 genes
sequentially. (B) ROC curve analysis was performed for the
NUSAP1, MELK, PBK, BPKPB2 and BUB1 genes
sequentially. (C) ROC curve analysis was performed for the CDK1,
CCL19, CXCR5, CXCL13 and CXCL9 genes sequentially. (D)
ROC curve analysis was performed for the CXCR4, GNG11, GNAL1
and NMUR1, CX3CR1 genes sequentially. ROC, receiver
operating characteristic; AUC, area under the curve. |
Survival analysis
Subsequently, survival analysis using the 13 genes
was performed by GEPIA and it was determined that the P-value of
eight genes was <0.05, including AURKA, BUB1, CCNB1, CDK1,
MELK, NUSAP1, PBK and TOP2A (Fig. 8), indicating that they may be key
genes that reduce lung adenocarcinoma survival and affect prognosis
and were included in the following analysis.
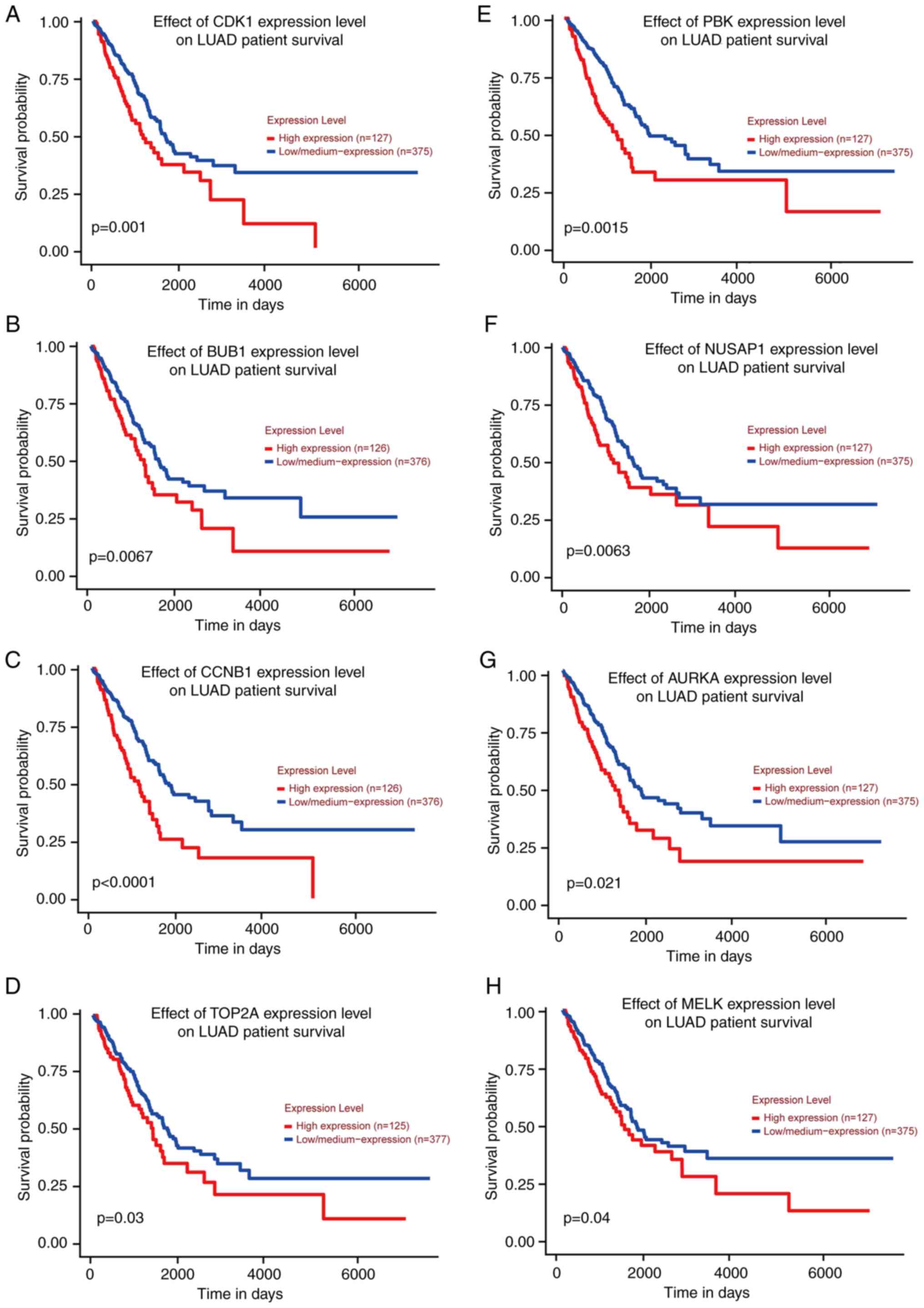 | Figure 8.Survival analysis was performed and
survival curves were obtained using GEPIA database. According to
the P-values, there were eight genes with P<0.05 (AURKA,
TOP2A, CCNB1, NUSAP1, MELK, PBK, BUB1 and CDK1). (A-H)
The survival curves of CDK1, BUB1, CCNB1, TOP2A, PBK, NUSAP1,
AURKA and MELK genes in TCGA database are presented in
order. TCGA, The Cancer Genome Atlas. |
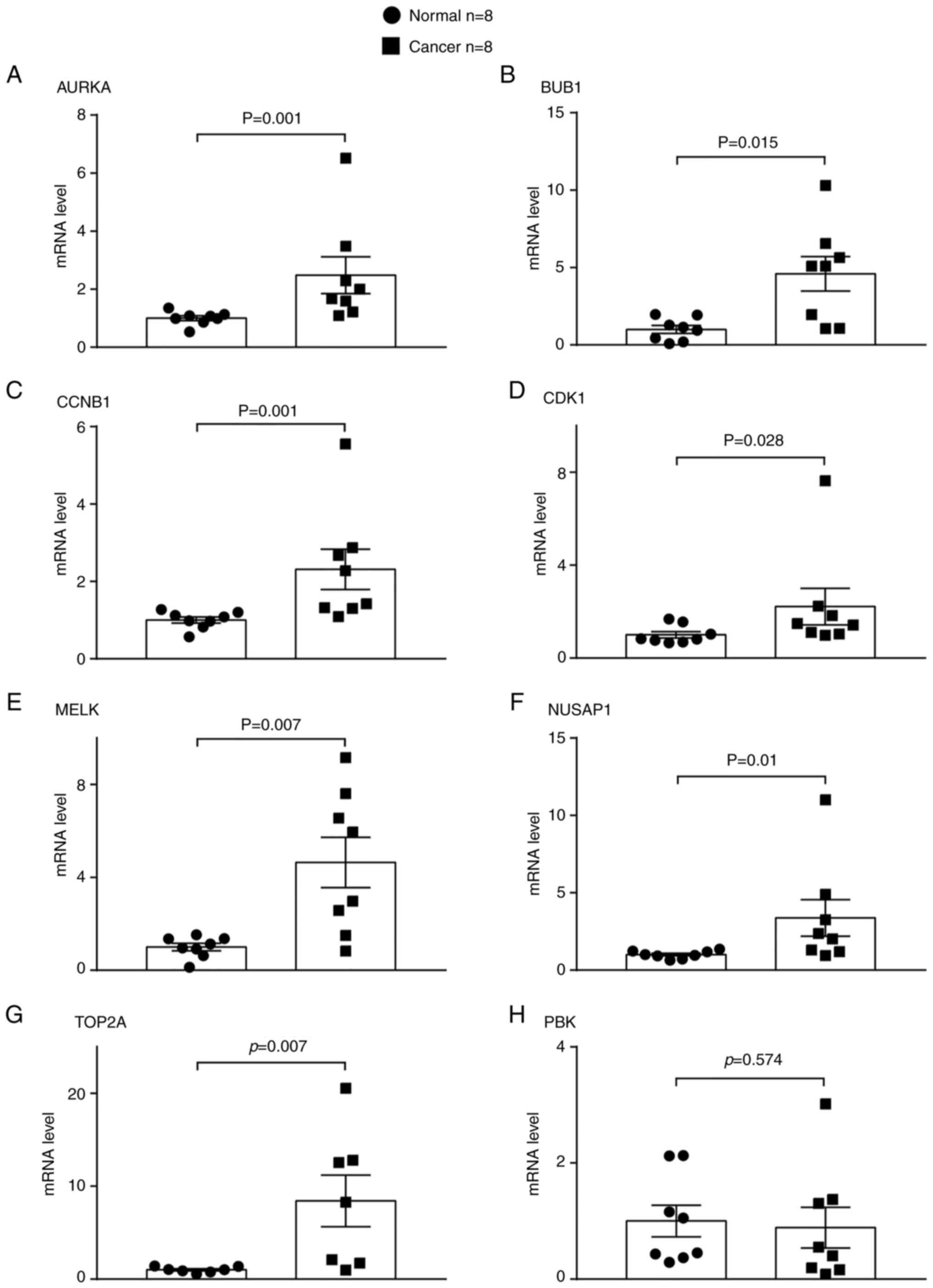
Gene expression in human LUAD and
normal paracancerous tissues
To validate the results of bioinformatics analysis,
the expression levels of the aforementioned eight genes were
verified in human LUAD tissues and paired lung paracancerous
tissues using RT-qPCR and western blot analysis. The relative mRNA
expression levels of seven out of eight genes, namely AURKA,
BUB1, CCNB1, CDK1, MELK, NUSAP1 and TOP2A, were
significantly higher in the LUAD than in the adjacent normal lung
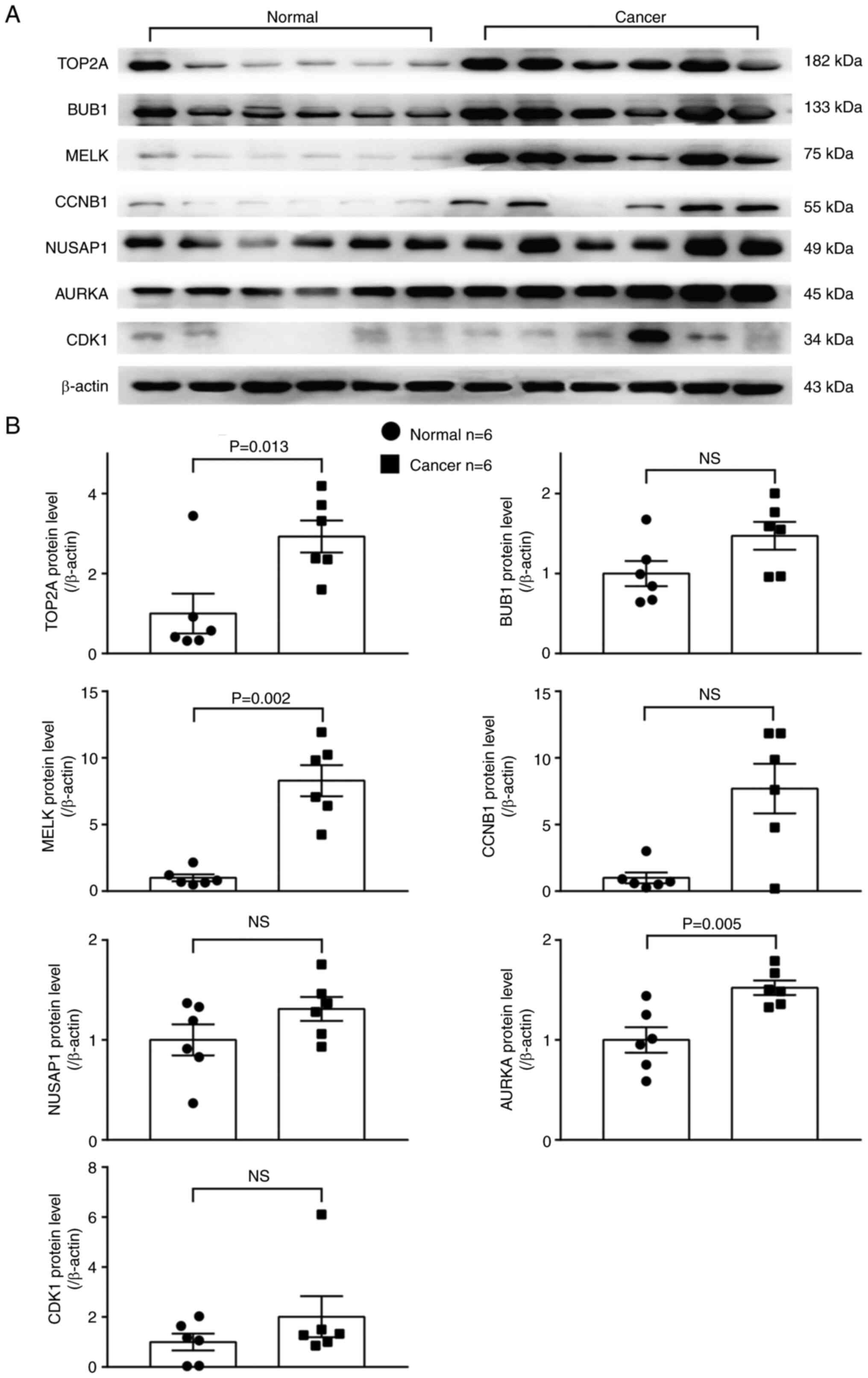
tissues (Fig. 9). The protein
levels of three out of these seven overexpressed genes, including
AURKA, MELK and TOP2A, were significantly higher in
the LUAD than in adjacent normal lung tissues (Fig. 10).
Discussion
Lung cancer is one of the most prevalent types of
cancer and currently presents with the highest mortality rate.
Among patients recently diagnosed with lung cancer, the 5-year
survival rate following diagnosis has been observed to be extremely
reduced in the majority of countries, with a survival rate of only
1/10 to 1/5 (35). Ηowever, the
molecular mechanisms underlying LUAD remain poorly understood.
Without early diagnosis, the majority of patients are not treated
promptly, resulting in a very poor prognosis. Therefore, there is
an urgent need for the identification of efficient biomarkers for
the early detection and treatment of lung cancer. The screening of
early biomarkers and key genes for malignant and benign diseases
using bioinformatics analysis has been proven a very efficient
method (36–39). However, the procedure of data
analysis in a scientifically sound and efficient manner is
currently a serious hindrance. In the present study, the
information extracted from a high-throughput gene expression
dataset was analyzed, firstly sorting the differentially expressed
genes, and WGCNA was then used to obtain the genes in the modules
with the highest correlation with the clinical phenotype.
Subsequently, PPI and correlation analyses were performed on the
common pathogenic genes of the two analyses.
Several inhibitors with high specificity for
AURKA have been developed with clinical efficacy, including
MLN8237 and ENMD-2076 (40).
Moreover, cell cycle inhibition by regulating the AURKA/ polo-like
kinase 1 (PLK1) pathway has been reported to induce apoptosis in
LUAD (41), with AURKA not
only being a potential biomarker for predicting the poor prognosis
of smoking-related LUAD. Furthermore, the AURKA rs1047972
variant has been found to be significantly associated with EGFR
mutation in patients with LUAD, particularly in women and
non-smoking patients. The AURKA variant may contribute to
the pathologic development of LUAD (42–44).
The AURKA-induced amplification or activation of liver
kinase B1 (LKB1)/AMPK signaling pathway impairment contributes to
the initiation and progression of NSCLC, suggesting that
AURKA may be a potential therapeutic target against
AURKA-driven overactive LUAD (45).
Chemotherapy resistance research has emerged as a
major challenge in cancer treatment. Currently, resistance to
radiation therapy in LUAD has been attributed to elevated levels of
autophagy and thus resistance, and AURKA is critical for the
reduction chemotherapy resistance in LUAD, as evidenced by high
levels of AURKA expression associated with chemoresistance
and proliferation in LUAD. Genetic resistance in response to
chronic EGFR inhibition attenuates drug-induced apoptosis, and
silencing AURKA reduces drug resistance in EGFR-mutant LUAD
(46,47).
It has been reported that TOP2A expression
levels are upregulated in both surgically resected lung cancer
tissues and lung cancer cell lines. As previously demonstrated, the
knockdown of TOP2A in human lung cancer cell lines inhibited
cell proliferation, migration and invasion, while the inhibition of
TOP2A reduced the expression levels of CCNB1 and
CCNB2. High expression of TOP2A has been reported to
significantly increase the risk of mortality in patients with
NSCLC, a risk that is particularly pronounced in patients with
LUAD, and its molecular mechanism is associated with activation of
PI3K/AKT and Wnt/β-catenin signaling pathways, which promote
apoptosis. Etoposide, which targets TOP2A, has been approved
for the treatment of small cell lung cancer, but there are
currently no drugs for LUAD (48,49).
Through various bioinformatics approaches, TOP2A has been
identified as an independent factor affecting the prognosis of
patients with LUAD (50–53), whereas an increased TOP2A
expression has also been identified as a potential risk factor for
pathological stage I LUAD (54).
Ciclopirox olamine and quercetin have also been demonstrated to
exert tumor-suppressive effects via TOP2A in LUAD (55,56).
MELK is highly expressed in LUAD, and the
increased expression of MELK has been associated with a poor
prognosis; MELK may serve as a potential diagnostic marker
and therapeutic target for LUAD. The molecular mechanisms by which
MELK affects cancer include the possibility of the kinase
activity of MELK affecting lung adenocarcinogenesis by
inhibiting the pro-apoptotic function of Bcl-GL. High levels of
MELK expression have been associated with high-grade tumors,
an increased aggressiveness, a poorer patient prognosis and
radioresistance, and an increased expression of MELK is
associated with TOP2A, CDK1 and AURKB (57). Various MELK inhibitors have
been developed as potential cancer therapeutic agents, molecules,
including OTS and MELK-T1 have demonstrated efficacy in
experimental animals to delay the proliferation of cancer cells
(58).
It has been reported that TOP2A interacts
directly with MELK, CDC20, CCNB2, UBE2T, KIAA0101 and
TK1 through a PPI network (11). However, this cannot systematically
reflect the interaction pattern between key pathogenic genes in
LUAD. In the present study, bioinformatics analysis of LUAD using
WGCNA and validation by human tissue samples yielded three key
genes, AURKA, MELK and TOP2A, whose co-expression may
be important for early diagnosis and prognosis as well as further
elucidation of the pathogenesis of LUAD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the seventh batch of young
and middle-aged medical backbone talent training projects in Wuhan
in 2019 [Wu Weitong (2019); Grant no. 87]; National Natural Science
Foundation Youth Project (Grant no. 82170106).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and YX contributed significantly to the concept
and design of the study. XZ and SW conducted bioinformatics
experiments and obtained data. HL, RH and MZ conducted confirmatory
experiments and obtained data. XZ and JR and LC analyzed the data.
XZ, YX and MZ drafted the manuscript. YX, SW and RH critically
modify the important intellectual content of the study. XZ and YX
confirm the authenticity of all the raw data. BX and NZ and WS
contributed to the collection and collation of clinical samples
from lung adenocarcinoma patients. All authors have read and
approved the final manuscript and have agreed to take
responsibility for all aspects of the work.
Ethics approval and consent to
participate
The present study was approved (approval no.
WDRY2022-K231) by Renmin Hospital of Wuhan University (Wuhan,
China) and written informed consent was obtained from patients in
all cases.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Denisenko TV, Budkevich IN and Zhivotovsky
B: Cell death-based treatment of lung adenocarcinoma. Cell Death
Dis. 9:1172018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu Y, Wang Z, Zheng Q and Li J: FAM72
serves as a biomarker of poor prognosis in human lung
adenocarcinoma. Aging (Albany NY). 13:8155–8176. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang C, Tan S, Liu WR, Lei Q, Qiao W, Wu
Y, Liu X, Cheng W, Wei YQ, Peng Y and Li W: RNA-Seq profiling of
circular RNA in human lung adenocarcinoma and squamous cell
carcinoma. Mol Cancer. 18:1342019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Yu X, Zhang Y, Wang X, Zhao L, Liu
D, Zhao G, Gao X, Fu J, Zang A and Jia Y: Identification of a novel
prognosis-associated ceRNA network in lung adenocarcinoma via
bioinformatics analysis. Biomed Eng Online. 20:1172021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li H, Guo L and Cai Z: TCN1 is a potential
prognostic biomarker and correlates with immune infiltrates in lung
adenocarcinoma. World J Surg Oncol. 20:832022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuanhua L, Pudong Q, Wei Z, Yuan W, Delin
L, Yan Z, Geyu L and Bo S: TFAP2A Induced KRT16 as an Oncogene in
Lung Adenocarcinoma via EMT. Int J Biol Sci. 15:1419–1428. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu ZP: Reverse engineering of genome-wide
gene regulatory networks from gene expression data. Curr Genomics.
16:3–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Z, Li M, Fang X, Shen L, Yao W, Fang
Z, Chen J, Feng X, Hu L, Zeng Z, et al: Identification of surrogate
prognostic biomarkers for allergic asthma in nasal epithelial
brushing samples by WGCNA. J Cell Biochemi. 120:5137–5150. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong S, Men W, Yang S and Xu S:
Identification of lung adenocarcinoma biomarkers based on
bioinformatic analysis and human samples. Oncol Rep. 43:1437–1450.
2020.PubMed/NCBI
|
|
12
|
Zhang L, He M, Zhu W, Lv X, Zhao Y, Yan Y,
Li X, Jiang L, Zhao L, Fan Y, et al: Identification of a panel of
mitotic spindle-related genes as a signature predicting survival in
lung adenocarcinoma. J Cell Physiol. 235:4361–4375. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang L, Li S, Wang Y, Tang Z, Liu C, Jiao
W and Liu J: Identification of differentially expressed
protein-coding genes in lung adenocarcinomas. Exp Ther Med.
19:1103–1111. 2020.PubMed/NCBI
|
|
14
|
Li J, Liu X, Cui Z and Han G:
Comprehensive analysis of candidate diagnostic and prognostic
biomarkers associated with lung adenocarcinoma. Med Sci Monit.
26:e9220702020.PubMed/NCBI
|
|
15
|
Wang Y, Zhou Z, Chen L, Li Y, Zhou Z and
Chu X: Identification of key genes and biological pathways in lung
adenocarcinoma via bioinformatics analysis. Mol Cell Biochem.
476:931–939. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen C, Tang Y, Qu WD, Han X, Zuo JB, Cai
QY, Xu G, Song YX and Ke XX: Evaluation of clinical value and
potential mechanism of MTFR2 in lung adenocarcinoma via
bioinformatics. BMC Cancer. 21:6192021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fan X, Wang Y and Tang XQ: Extracting
predictors for lung adenocarcinoma based on Granger causality test
and stepwise character selection. BMC Bioinformatics. 20 (Suppl
7):S1972019. View Article : Google Scholar
|
|
18
|
Guo W, Sun S, Guo L, Song P, Xue X, Zhang
H, Zhang G, Wang Z, Qiu B, Tan F, et al: Elevated TOP2A and UBE2C
expressions correlate with poor prognosis in patients with
surgically resected lung adenocarcinoma: A study based on
immunohistochemical analysis and bioinformatics. J Cancer Res Clin
Oncol. 146:821–841. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du R, Huang C, Liu K, Li X and Dong Z:
Targeting AURKA in Cancer: Molecular mechanisms and opportunities
for Cancer therapy. Mol Cancer. 20:152021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng J, Wei Y, Deng Q, Li L and Li X:
Study on the expression of TOP2A in hepatocellular carcinoma and
its relationship with patient prognosis. Cancer Cell Int.
22:292022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Yang H, Wang L, Zhou H, Zhang G,
Xiao Z and Xue X: TOP2A correlates with poor prognosis and affects
radioresistance of medulloblastoma. Front Oncol. 12:9189592022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang F and Wu H: MiR-599 targeting TOP2A
inhibits the malignancy of bladder cancer cells. Biochem Biophys
Res Commun. 570:154–161. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao Y, Zhao H, Ren M, Chen Q, Li J, Li Z,
Yin C and Yue W: TOP2A promotes tumorigenesis of high-grade serous
ovarian cancer by regulating the TGF-β/Smad pathway. J Cancer.
11:4181–4192. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang B, Shen Y, Zou Y, Qi Z, Huang G, Xia
S, Gao R, Li F and Huang Z: TOP2A promotes cell migration, invasion
and epithelial-mesenchymal transition in cervical cancer via
activating the PI3K/AKT signaling. Cancer Manag Res. 12:3807–3814.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pei YF, Yin XM and Liu XQ: TOP2A induces
malignant character of pancreatic cancer through activating
β-catenin signaling pathway. Biochim Biophys Acta Mol Basis Dis.
1864:197–207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen YU, Yu Y, Lv M, Shi Q and Li X:
E2F1-mediated up-regulation of TOP2A promotes viability, migration,
and invasion, and inhibits apoptosis of gastric cancer cells. J
Biosci. 47:842022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du X, Xue Z, Lv J and Wang H: Expression
of the topoisomerase II alpha (TOP2A) gene in lung adenocarcinoma
cells and the association with patient outcomes. Med Sci Monit.
26:e9291202020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen C, Guo Q, Song Y, Xu G and Liu L:
SKA1/2/3 serves as a biomarker for poor prognosis in human lung
adenocarcinoma. Transl Lung Cancer Res. 9:218–231. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gray D, Jubb AM, Hogue D, Dowd P, Kljavin
N, Yi S, Bai W, Frantz G, Zhang Z, Koeppen H, et al: Maternal
embryonic leucine zipper kinase/murine protein serine-threonine
kinase 38 is a promising therapeutic target for multiple cancers.
Cancer Res. 65:9751–9761. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Li Y, Chen Y, Xie Q, Dong N, Gao Y,
Deng H, Lu C and Wang S: MicroRNA-214-3p inhibits proliferation and
cell cycle progression by targeting MELK in hepatocellular
carcinoma and correlates cancer prognosis. Cancer Cell Int.
17:1022017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen S, Zhou Q, Guo Z, Wang Y, Wang L, Liu
X, Lu M, Ju L, Xiao Y and Wang X: Inhibition of MELK produces
potential anti-tumour effects in bladder cancer by inducing G1/S
cell cycle arrest via the ATM/CHK2/p53 pathway. J Cell Mol Med.
24:1804–1821. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The Cancer Genome
Atlas Pan-Cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Allemani C, Matsuda T, Di Carlo V,
Harewood R, Matz M, Nikšić M, Bonaventure A, Valkov M, Johnson CJ,
Estève J, et al: Global surveillance of trends in cancer survival
2000–14 (CONCORD-3): Analysis of individual records for 37 513 025
patients diagnosed with one of 18 cancers from 322 population-based
registries in 71 countries. Lancet. 391:1023–1075. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao X, Zhang L, Wang J, Zhang M, Song Z,
Ni B and You Y: Identification of key biomarkers and immune
infiltration in systemic lupus erythematosus by integrated
bioinformatics analysis. J Transl Med. 19:352021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu X, Bao M, Huang J, Zhou L and Zheng S:
Identification and validation of novel biomarkers for diagnosis and
prognosis of hepatocellular carcinoma. Front Oncol. 10:5414792020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang Q, Wang R, Wei B, Peng C, Wang L, Hu
G, Kong D and Du C: Candidate biomarkers and molecular mechanism
investigation for glioblastoma multiforme utilizing WGCNA. Biomed
Res Int. 2018:42467032018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vernocchi P, Gili T, Conte F, Del Chierico
F, Conta G, Miccheli A, Botticelli A, Paci P, Caldarelli G, Nuti M,
et al: Network analysis of gut microbiome and metabolome to
discover microbiota-linked biomarkers in patients affected by
non-small cell lung cancer. Int J Mol Sci. 21:87302020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Otto T and Sicinski P: Cell cycle proteins
as promising targets in cancer therapy. Nat Rev Cancer. 17:93–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Z, Zhang Y, Zhou Y, Wang F, Yin C, Ding
L and Zhang S: Tanshinone IIA suppresses the progression of lung
adenocarcinoma through regulating CCNA2-CDK2 complex and AURKA/PLK1
pathway. Sci Rep. 11:236812021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhong N, Shi S, Wang H, Wu G, Wang Y, Ma
Q, Wang H, Liu Y and Wang J: Silencing Aurora-A with siRNA inhibits
cell proliferation in human lung adenocarcinoma cells. Int J Oncol.
49:1028–1038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang MY, Liu XX, Li H, Li R, Liu X and Qu
YQ: Elevated mRNA Levels of AURKA, CDC20 and TPX2 are associated
with poor prognosis of smoking related lung adenocarcinoma using
bioinformatics analysis. Int J Med Sci. 15:1676–1685. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang PJ, Hsieh MJ, Lee CI, Yen CH, Wang
HL, Chiang WL, Liu TC, Tsao TC, Lee CY and Yang SF: Impact of
aurora kinase a polymorphism and epithelial growth factor receptor
mutations on the clinicopathological characteristics of lung
adenocarcinoma. Int J Environ Res Public Health. 17:73502020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zheng X, Chi J, Zhi J, Zhang H, Yue D,
Zhao J, Li D, Li Y, Gao M and Guo J: Aurora-A-mediated
phosphorylation of LKB1 compromises LKB1/AMPK signaling axis to
facilitate NSCLC growth and migration. Oncogene. 37:502–511. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shah KN, Bhatt R, Rotow J, Rohrberg J,
Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al:
Aurora kinase A drives the evolution of resistance to
third-generation EGFR inhibitors in lung cancer. Nat Med.
25:111–118. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao J, Lu F, Yan J, Wang R, Xia Y, Wang L,
Li L, Chang L and Li W: The role of radiotherapy-related autophagy
genes in the prognosis and immune infiltration in lung
adenocarcinoma. Front Immunol. 13:9926262022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grenda A, Błach J, Szczyrek M, Krawczyk P,
Nicoś M, Kuźnar Kamińska B, Jakimiec M, Balicka G, Chmielewska I,
Batura-Gun H, et al: Promoter polymorphisms of TOP2A and ERCC1
genes as predictive factors for chemotherapy in non-small cell lung
cancer patients. Cancer Med. 9:605–614. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kou F, Sun H, Wu L, Li B, Zhang B, Wang X
and Yang L: TOP2A promotes lung adenocarcinoma cells' malignant
progression and predicts poor prognosis in lung adenocarcinoma. J
Cancer. 11:2496–2508. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zeng H, Ji J, Song X, Huang Y, Li H, Huang
J and Ma X: Stemness related genes revealed by network analysis
associated with tumor immune microenvironment and the clinical
outcome in lung adenocarcinoma. Front Genet. 11:5492132020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Song Y, Tang W and Li H: Identification of
KIF4A and its effect on the progression of lung adenocarcinoma
based on the bioinformatics analysis. Biosci Rep.
41:BSR202039732021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dai JJ, Zhou WB and Wang B: Identification
of crucial genes associated with lung adenocarcinoma by
bioinformatic analysis. Medicine (Baltimore). 99:e230522020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang S, Pang K, Feng X and Zeng Y:
Transcriptomic data exploration of consensus genes and molecular
mechanisms between chronic obstructive pulmonary disease and lung
adenocarcinoma. Sci Rep. 12:132142022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Deng Y, Chen X, Huang C, Song J, Feng S,
Chen X and Zhou R: Screening and validation of significant genes
with poor prognosis in pathologic Stage-I lung adenocarcinoma. J
Oncol. 2022:37940212022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yin J, Che G, Jiang K, Zhou Z, Wu L, Xu M,
Liu J and Yan S: Ciclopirox olamine exerts tumor-suppressor effects
via topoisomerase II alpha in lung adenocarcinoma. Front Oncol.
12:7919162022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang YQ, Li K, Guo Q and Li D: A new risk
model based on 7 quercetin-related target genes for predicting the
prognosis of patients with lung adenocarcinoma. Front Genet.
13:8900792022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Du T, Qu Y, Li J, Li H, Su L, Zhou Q, Yan
M, Li C, Zhu Z and Liu B: Maternal embryonic leucine zipper kinase
enhances gastric cancer progression via the FAK/Paxillin pathway.
Mol Cancer. 13:1002014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
McDonald IM and Graves LM: Enigmatic MELK:
The controversy surrounding its complex role in cancer. J Biol
Chem. 295:8195–8203. 2020. View Article : Google Scholar : PubMed/NCBI
|