Introduction
Angioimmunoblastic T-cell lymphoma (AITL) is a
subtype of peripheral T-cell lymphoma (PTCL) that accounts for 1–2%
of all cases of non-Hodgkin lymphoma (HL) and 15–20% of cases of
PTCL, and has a poor prognosis (1).
The median age at diagnosis is ~65 years, and the primary clinical
manifestations are fever, weight loss, urticaria, papules, red
nodules, and skin lesions (2).
Approximately 20–50% of patients with AITL have prodromal symptoms,
and their skin manifestations can range from urticarial lesions to
nodular tumors (3). Diagnosis is
often delayed or masked due to the atypical biochemical and
autoimmune manifestations, and it is common for the disease to have
reached an advanced stage by the time an accurate diagnosis is made
(4). As a result of the abnormal
proliferative activity of B-cells (5), AITL is often accompanied by autoimmune
disorders, such as hemolytic anemia and hypergammaglobulinemia.
Epstein-Barr virus (EBV) has been found to play an important role
in the pathogenesis of AITL (6).
EBV can stimulate the activation of helper T-cells, leading to the
development of tumors. Diagnosis of AITL is based on
histopathological examination but remains challenging given the
lack of specific pathological characteristics. This report suggests
that a combination of immunohistochemistry and gene rearrangement
can increase diagnostic accuracy. CD10 and CXCL13 are specifically
expressed in AITL and can be used as characteristic markers for
diagnostic purposes (7). Clonal
rearrangement of the IgH gene and TCRγ may also be of significance
in terms of the diagnosis (8).
Case report
The patient was a 56-year-old man who was initially
diagnosed with HL for which he received doxorubicin hydrochloride
liposome, bleomycin, vindesine, and dacarbazine (ABVD), and
doxorubicin hydrochloride liposome, vindesine, and dacarbazine
(AVD) chemotherapy in April 2020. However, a decrease in his CD21
levels and disruption of the follicular dendritic cell (FDC)
network (CD20+; PAX-5+; CD3+; Ki-67+: 20–30%; CD10-; BCL-6+;
MUM-1+; PD-1+; CXCL-13-; CD30+; CD15-) was also detected. Molecular
detection showed monoclonal rearrangement of TCRβDB+Jβ1/2 and
oligoclonal rearrangement of Vβ + Jβ2. A review of all details
concerning unsatisfactory treatment led to a diagnosis of compound
lymphoma consisting of AITL and focal classical HL (CHL).
In March 2020, the patient was admitted to Hebei
General Hospital (Shijiazhuang, China) after a 4-month history of
skin redness and itching. Physical examination revealed extensive
redness, swelling, and rough skin on the head, neck, and limbs.
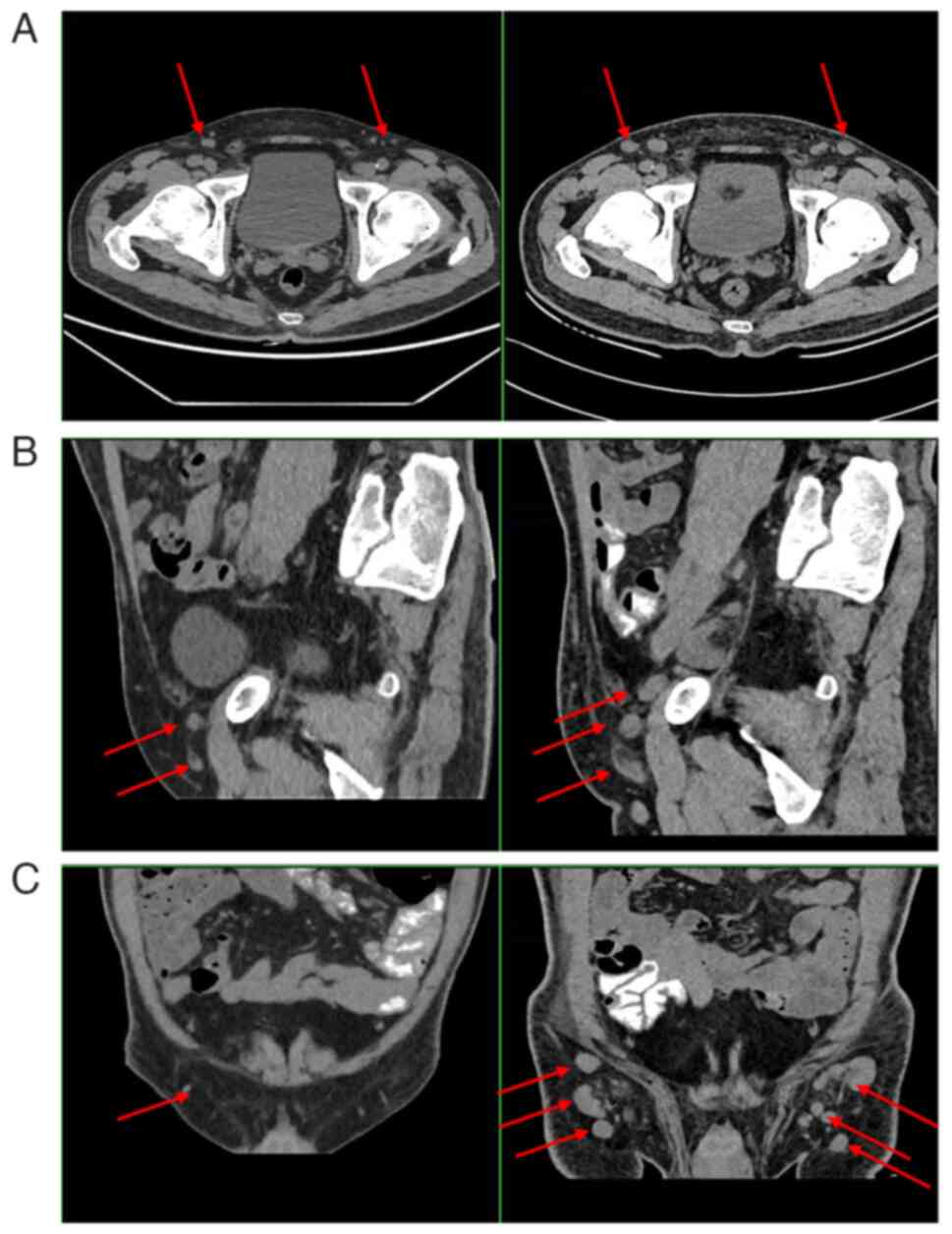
There were multiple enlarged lymph nodes in the right armpit and on
both sides of the groin. The largest node was ~4 cm in diameter
with a smooth surface and was non-tender and mobile. The laboratory
findings are presented in Table I.
A proliferative disease of the lymphatic system was suspected
initially; a punch biopsy was performed, a histological analysis of
which documented CHL, lymphocyte-rich type, with molecular
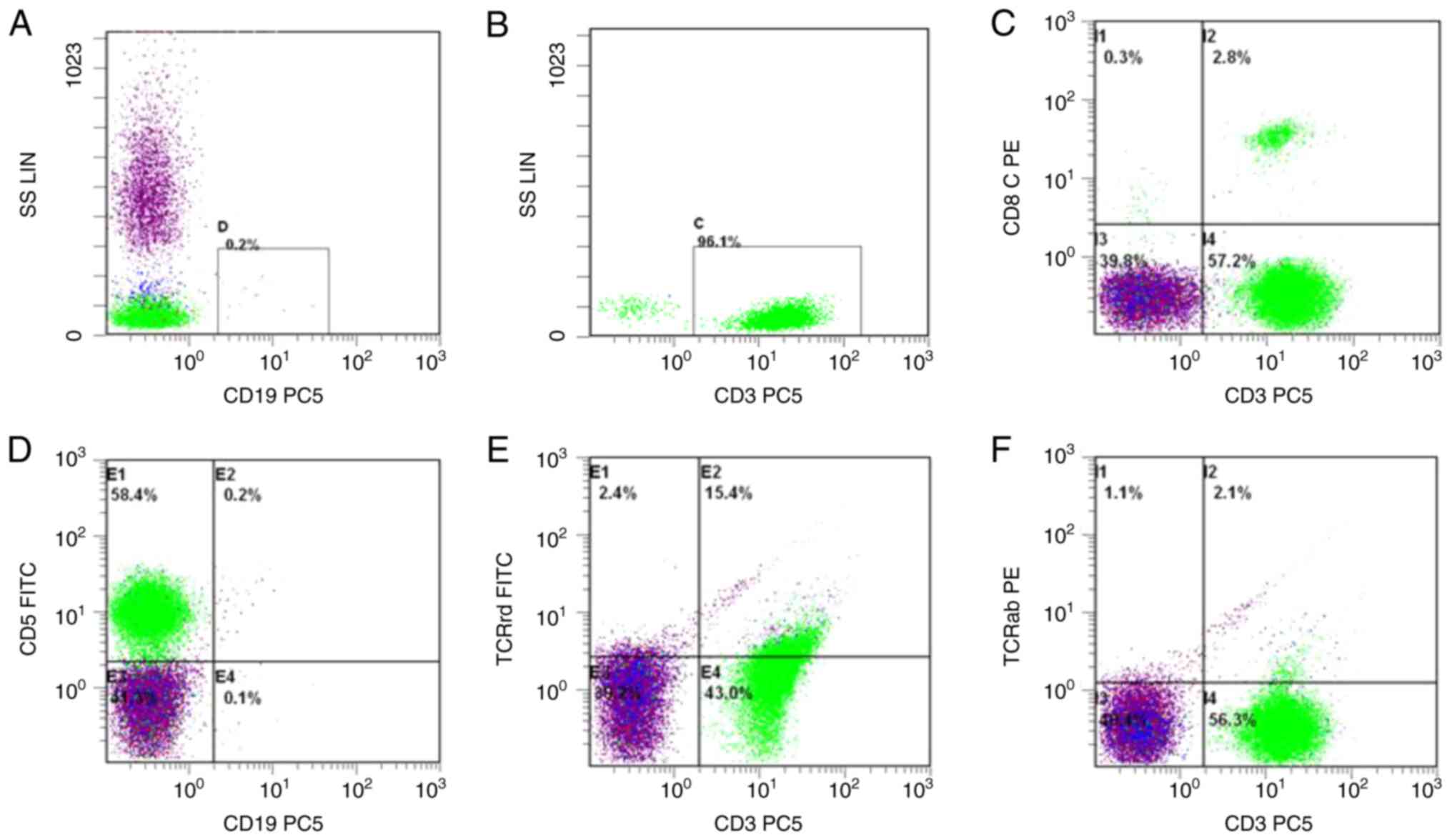
detection of EBER positivity. Flow cytometry revealed that 96.1% of
the nuclear cells in the bone marrow were CD3+ lymphocytes, with
some showing positive expression for CD3 and CD5, some expressing
TCRrd, and a small number expressing CD8 (Fig. 1). Radiological imaging detected
lymph node infiltration in multiple organs. After various
examinations, the patient was diagnosed as having
lymphocyte-predominant HL. The patient completed chemotherapy that
consisted of ABVD (doxorubicin hydrochloride liposome 40 mg IV,
bleomycin 10 mg/m2 IV, vindesine 4 mg IV, and
dacarbazine 375 mg/m2 IV on days 1 and 15), followed by
AVD as a second course.
 | Table I.Results of laboratory findings on
admission. |
Table I.
Results of laboratory findings on
admission.
| Laboratory
indicator | Normal range | Result |
|---|
| CBC |
|
|
| WBC,
×109/l | 3.5-9.5 | 15.00 |
| LY,
×109/l | 1.1-3.2 | 5.12 |
| HGB,
g/l | 115-150 | 151 |
|
PLTx109/l | 125-350 | 292 |
|
β2-microglobulin, µg/ml | 0.9-2.7 | 4.288 |
| Biochemistry |
|
|
| TP,
g/l | 65-85 | 53.5 |
| GLO,
g/l | 20-40 | 22.3 |
| LDH,
U/l | 120-250 | 323.9 |
| HBDH,
U/l | 72-182 | 222.4 |
| TG,
mmol/l | 0.1-1.7 | 1.84 |
| Coombs test |
|
|
| Direct
Coombs test |
| Negative |
| Indirect
Coombs test |
| Negative |
| Tumor Marker |
|
|
| CA 125,
U/ml | 0-35 | 125.100 |
| VEGF,
pg/ml | 0-142 | 178.4 |
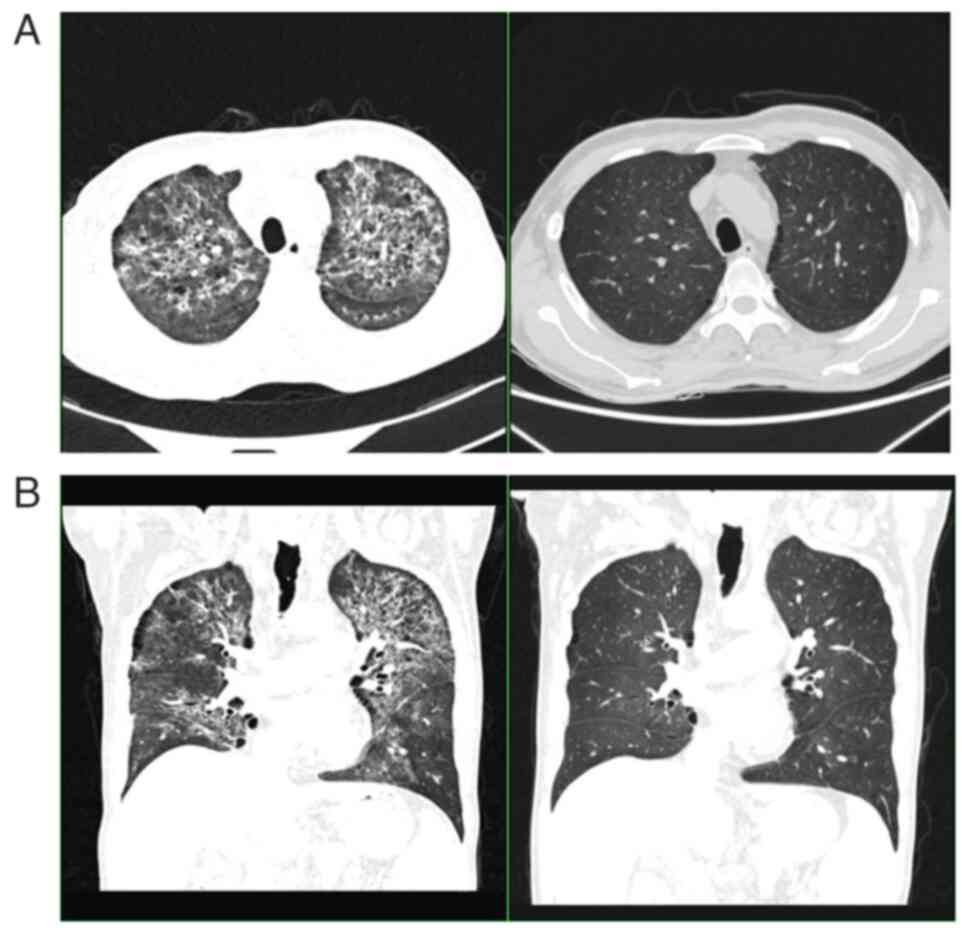
After two courses of chemotherapy, a CT showed that
the patient's lymph nodes were smaller than before (Fig. 2). However, the patient appeared to
have worsening pruritus. Immunohistochemical examination of the
lymph node specimen revealed CD20+, PAX-5+, CD3+, Ki-67+ (20–30%),
CD10-, BCL-6+, MUM-1+, PD-1+, CXCL-13-, CD30+, and CD15-; (Fig. 3). T-cell clone analysis was
performed using the BIOMED-2 polymerase chain reaction protocol. On
molecular examination, TCRβDB+Jβ1/2 showed monoclonal rearrangement
and Vβ+Jβ2 showed oligoclonal rearrangement with PTPRD gene
mutation. The pathological diagnosis was non-Hodgkin peripheral
(mature) T-cell lymphoma, prone to AITL. Thus, the patient was
diagnosed with lymphoma, which was a composite of AITL and HL
(International Prognostic Index, 3; Prognostic Index for T-cell
lymphoma, 2). Given this diagnosis, the patient was scheduled for
etoposide, prednisone, vincristine, cyclophosphamide, and
doxorubicin (EDOCH). However, the patient died of a lung infection
before starting further treatment (Fig.
4).
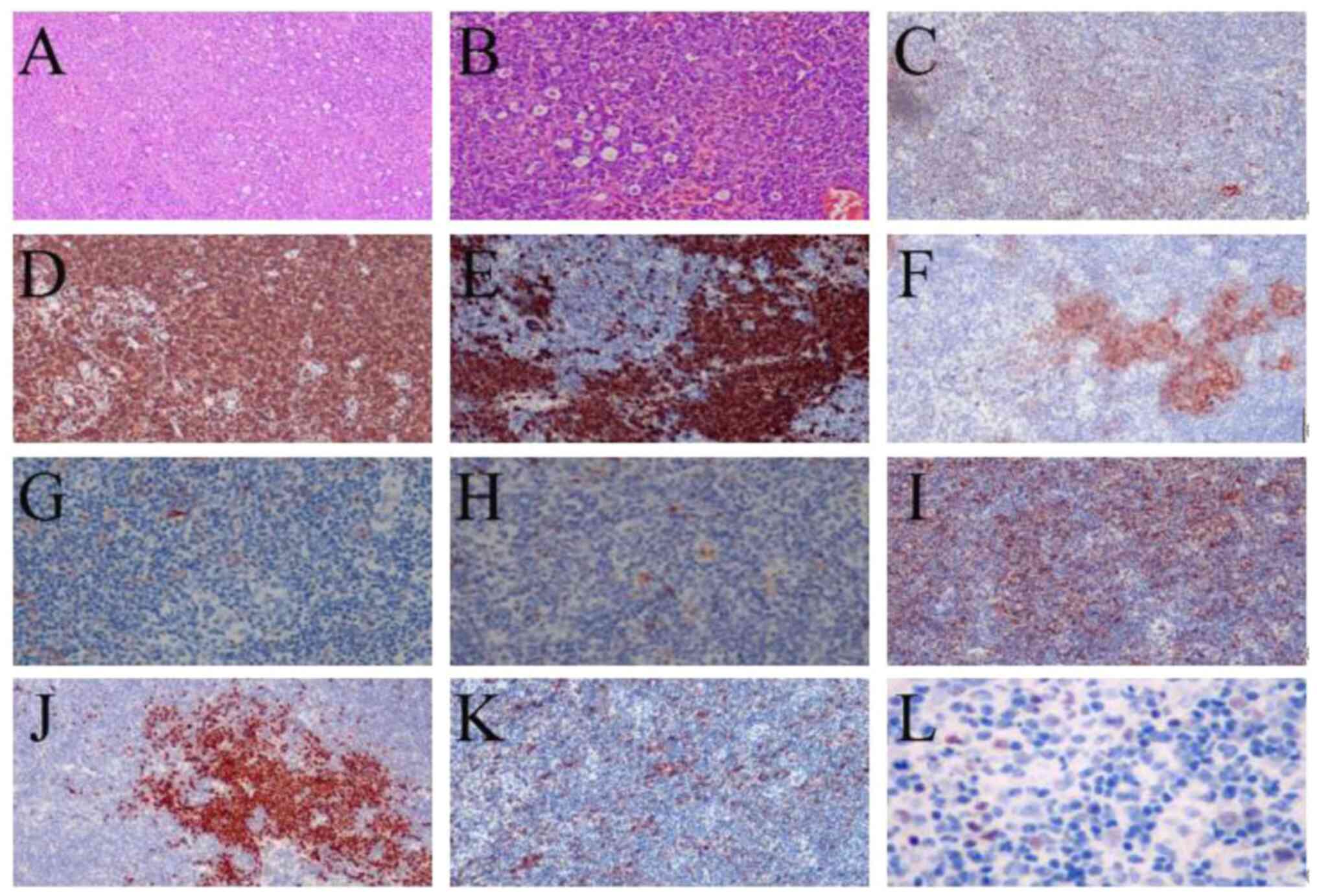 | Figure 3.Immunostaining of the right inguinal
lymph node for cytokines. (A) HE staining ×40 magnification; (B) HE
staining, ×100 magnification; (C) BCL-6 negative staining, ×100
magnification. (D) CD3 positive staining, ×100 magnification. (E)
CD20 positive staining, 100× magnification. (F) CD21 staining
negative, ×100 magnification. (G) CXCL13 negative staining, ×100
magnification. (H) Ki-67 positive staining, ×100 magnification. (I)
MUM-1 positive staining, ×100 magnification. (J) PAX-5 positive
staining, ×100 magnification. (K) PD-1 positive staining, ×100
magnification. (L) EBER (molecular diagnosis) positive staining.
Based on (A and B), the structure of the lymph node was partially
destroyed, with scattered atypical large cells, rich cytoplasm,
large nuclei, with some cells possessing some double nuclei and
larger nucleoli. There were T-cell lymphomas in multiple lymph
nodes, and certain lymph nodes also contained focal classical
Hodgkin's lymphoma. HE, hematoxylin-eosin. |
Discussion
Although this patient had obvious skin symptoms, the
pathological results and immunohistochemical analysis n his initial
admission were consistent with a diagnosis of CHL. However, the
patient's pruritic symptoms worsened after ABVD chemotherapy.
Therefore, a lymph node pathology examination was repeated.
Finally, the patient was diagnosed as having a composite of
lymphocyte-rich type HL and AITL.
AITL is a specific subtype of PTCL that originates
from T follicular helper (TFH) cells and is often accompanied by
fever, night sweats, weight loss, lymphadenopathy, skin rash, and
other clinical manifestations (9).
Skin involvement is one of the most common extranodal
manifestations of the disease (10). However, the heterogeneous
presentation of AITL means that most cases are not diagnosed until
weeks or months after the onset of symptoms (11).
Although the diagnosis of AITL relies on lymph node
biopsy, certain patients may not be diagnosed until after 2–3 lymph
node biopsies (12). The
pathological features include destruction of lymph node structure,
tumor cells primarily medium in size, lightly stained or
transparent cytoplasm, generally with round or oval nuclei, and
atypical cells. Large cells of varying numbers are scattered in the
background of inflammatory cells, including eosinophils,
lymphocytes, and plasma cells. The TFH phenotype is positive for
CD3, CD4, and CD10 (4). CD5 and CD7
expression are largely absent (13). CD30 is found in 20% of patients
(14). Cytoplasmic CXCL13 is
expressed almost uniformly and is specific for AITL (15). TFH expression of PD-1, ICOS, BCL-6,
and CD200 can be distinguished from that in benign
lymphoproliferative diseases and other subtypes of PTCL. In ~60% of
patients, TCR gene rearrangements are observed, as seen in TCβ,
TCD, and TCG, whereas some have an IgH gene rearrangement (16). In recent years, with advances in the
field of genomics, patients with AITL have been found to have a
high number of TET2, RHOA, IDH2, and DNMT3A mutations (especially
in TET2), which are associated with a poor prognosis (17). However, there is still no systematic
method for the identification of AITL, and its diagnosis remains
difficult.
There is a strong correlation between AITL and EBV
infection. EBV-infected B-cells can transmit EBV protein signals on
their surface to T-cells via major histocompatibility complex II
molecules when TFH cells interact with B-cells, which upregulate
the expression of CD ligands, provide antigen and costimulatory
signals for activation of T-cells, promote the secretion of the
chemokine CXCL13 (6), and lead to
activation of B-cells. Laforga et al (18) hypothesized that EBV promotes the
proliferation of B-cells in AITL. They suggested that as a result
of the effects of EBV, CD8-positive T-cells become
immunosuppressed, leading to immune evasion of EBV-positive
B-cells. EBV-infected B-cells exhibit abnormal proliferation and
may be polyclonal, oligoclonal, or monoclonal. If the
immunoglobulin structure is disrupted, there are three possible
outcomes for B-cells: Proliferation resembling that of
Reed-Sternberg (RS) cells; proliferation resembling that of CHL; or
the development of CHL.
The 2008 edition of the WHO Classification of Tumors
of Hematopoietic and Lymphoid Tissues mentions RS-like cells being
seen in early AITL for the first time (19). Destruction of lymph node structure
and infiltration of plasma cells, tissue cells, and other
inflammatory cells is observed in both AITL and CHL, and both
diseases are associated with EBV infection (20), and most immune phenotypes do not
have specificity, so diagnosis cannot be determined solely based on
immune phenotypes. The immunophenotype of CHL includes CD15+,
CD30+, PAX-5 weakly positive, CD3-, CD20-(mainly), CD45-, and
CD79a-, which combined with RS cells, assist in the diagnosis
(21,22). CHL and AITL are two types of
lymphoma with very different prognoses. While CHL has a good
prognosis with a 5-year overall survival rate of >80% (23), AITL has a poor prognosis. A
retrospective analysis found the complete response rate to be 25%
and the median overall survival to be only 14.9 months after CHOP
chemotherapy in elderly patients with AITL (24). At present, ABVD is the first-line
treatment for CHL, and most patients benefit from it. However, AITL
progresses rapidly with a high mortality rate. Therefore, clinical
phenotype, pathological morphology, immunohistochemistry, and gene
rearrangement studies are important for early and correct diagnoses
of AITL.
AITL is frequently misdiagnosed given its
nonspecific clinical and histologic findings. A summary of a review
of the literature on the misdiagnosis of AITL is shown in Table II. It was found that AITL can not
only be misdiagnosed as another hematological disease but also as a
disease involving another system and that more than one lymph node
biopsy is required for a definitive diagnosis.
| Table II.Literature review of misdiagnosis of
AITL. |
Table II.
Literature review of misdiagnosis of
AITL.
| First author/s,
year | Age, years | Sex | Misdiagnosis | Method leading to
misdiagnosis | Method of
diagnosis | (Ref.) |
|---|
| van den Akker and
Chen, 2021 | 62 | M | Reactive
lymphadenopathy | FNA | Excisional
biopsy | (25) |
| Ellis et al,
2018 | 72 | F | DLBCL | LN biopsy | Reexamination,
IHC | (26) |
| Keefe et al,
2022 | 65 | M | DRESS syndrome | clinical signs and
symptoms, PET-CT | LN biopsy | (27) |
| Trimech et
al, 2021 | 62 | M | Richter
syndrome | PET-CT, BM
uptake | LN biopsy | (28) |
| Papadi et
al, 2012 | 55 | F | SPBIP | LN biopsy and BM
aspirate | LN biopsy, clonal
TCR gene rearrangement | (29) |
|
| 70 | F | SPBIP | Clinical and
morphologic features | Flow cytometry and
gene rearrangement |
|
| Smithberger et
al, 2010 | 79 | F | Inflammatory
dermatosis | Skin biopsy | LN biopsy | (30) |
| Ahsanuddin et
al, 2011 | 76, 46, 60 | F, F, F | Plasma cell
leukemia | Smear morphology
and manual differential of peripheral blood | LN biopsy and BM
biopsy | (31) |
| Han et al,
2019 | 70 | M | Drug fever and
allergic purpura, septicemia | Medication history,
surgery history | LN biopsy | (32) |
| Kaffenberger et
al, 2015 | 59, 68 | M, M | MALT | Skin biopsy | LN biopsy | (33) |
| Szablewski et
al, 2019 | 41, 60, 67 | M, M, M | CHL, B cell
lymphoma | Skin biopsy | A second review of
the skin biopsy, LN biopsy | (34) |
| Suárez, 2016 | 77 | F | Marginal-zone
B-cell-lymphoma | Skin biopsy | Skin biopsy, LN
biopsy | (35) |
| Laforga, 2010 | 58 | M | HL | Autoimmune
phenomena | Touch imprints of
LN biopsy | (18) |
In conclusion, AITL is a specific subtype of
peripheral T-cell lymphoma that is challenging to diagnose.
Moreover, treatment is often delayed by misdiagnosis. The present
case underscores the importance of early and accurate diagnosis of
AITL and the potential for a poor prognosis. More than one
pathological examination should be performed to reduce the risk of
misdiagnosis. Clinical phenotype, lymph node biopsies,
immunohistochemistry, and gene rearrangement analyses should all be
considered for early and accurate diagnosis and appropriate
treatment.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
research are available from the corresponding author on reasonable
request.
Authors' contributions
YL conceived and designed the study. XG collected
the data and wrote the manuscript. LK treated the patient and
contributed to draft and revise the manuscript. JL advised on
patient treatment and participated in revising the manuscript. YL,
XG, LK and JL confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
This report was published with the written consent
of the patient's relatives.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AITL
|
angioimmunoblastic T-cell lymphoma
|
|
CHL
|
classical Hodgkin lymphoma
|
|
EBV
|
Epstein-Barr virus
|
|
HL
|
Hodgkin lymphoma
|
|
PTCL
|
peripheral T-cell lymphoma
|
|
RS
|
Reed-Sternberg
|
|
TFH
|
T follicular helper
|
References
|
1
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD and Jaffe ES: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lachenal F, Berger F, Ghesquières H, Biron
P, Hot A, Callet-Bauchu E, Chassagne C, Coiffier B, Durieu I,
Rousset H and Salles G: Angioimmunoblastic T-cell lymphoma:
Clinical and laboratory features at diagnosis in 77 patients.
Medicine (Baltimore). 86:282–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Federico M, Rudiger T, Bellei M, Nathwani
BN, Luminari S, Coiffier B, Harris NL, Jaffe ES, Pileri SA, Savage
KJ, et al: Clinicopathologic characteristics of angioimmunoblastic
T-cell lymphoma: Analysis of the international peripheral T-cell
lymphoma project. J Clin Oncol. 31:240–246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lunning MA and Vose JM: Angioimmunoblastic
T-cell lymphoma: The many-faced lymphoma. Blood. 129:1095–1102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xie Y and Jaffe ES: How I Diagnose
angioimmunoblastic T-cell lymphoma. Am J Clin Pathol. 156:1–14.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dunleavy K, Wilson WH and Jaffe ES:
Angioimmunoblastic T cell lymphoma: Pathobiological insights and
clinical implicationsl. Curr Opin Hematol. 14:348–453. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Basha BM, Bryant SC, Rech KL, Feldman AL,
Vrana JA, Shi M, Reed KA and King RL: Application of a 5 marker
panel to the routine diagnosis of peripheral T-cell lymphoma with
T-follicular helper phenotype. Am J Surg Pathol. 43:1282–1290.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu J, Tang Y, Zhao S, Zhang W, Xiu Y, Liu
T and Wu Y: Angioimmunoblastic T-cell lymphoma with coexisting
plasma cell myeloma: A case report and review of the literature.
Tohoku J Exp Med. 235:283–288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xie C, Li X, Zeng H and Qian W: Molecular
insights into pathogenesis and targeted therapy of peripheral T
cell lymphoma. Exp Hematol Oncol. 9:302020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Botros N, Cerroni L, Shawwa A, Green PJ,
Greer W, Pasternak S and Walsh NM: Cutaneous manifestations of
angioimmunoblastic T-cell lymphoma: Clinical and pathological
characteristics. Am J Dermatopathol. 37:274–283. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu WY, Yang W, Xu XH, Shen JL and Zhang
CY: An Angioimmunoblastic T cell lymphoma patient misdiagnosed as
systemic lupus erythematosus: A case report and literature review.
Clin Misdiagnosis Mistherapy. 29:14–17. 2016.(In Chinese).
|
|
12
|
Pircher A, Verdorfer I, Brunner A,
Hopfinger G and Steurer M: Paraneoplastic phenomena and diagnostic
challenges in angioimmunoblastic T-cell lymphoma (AITL): Report of
two cases and review of the literature. In Vivo. 28:327–332.
2014.PubMed/NCBI
|
|
13
|
Cortés JR and Palomero T: The curious
origins of angioimmunoblastic T-cell lymphoma. Curr Opin Hematol.
23:434–443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Leval L, Rickman DS, Thielen C, Reynies
Ad, Huang YL, Delsol G, Lamant L, Leroy K, Brière J, Molina T, et
al: The gene expression profile of nodal peripheral T-cell lymphoma
demonstrates a molecular link between angioimmunoblastic T-cell
lymphoma (AITL) and follicular helper T (TFH) cells. Blood.
109:4952–4963. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grogg KL, Attygalle AD, Macon WR, Remstein
ED, Kurtin PJ and Dogan A: Expression of CXCL13, a chemokine highly
upregulated in germinal center T-helper cells, distinguishes
angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma,
unspecified. Mod Pathol. 19:1101–1107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aung NY, Ohtake H, Iwaba A, Kato T, Ohe R,
Tajima K, Nagase T and Yamakawa M: Angioimmunoblastic T-cell
lymphoma with dual genotype of TCR and IgH genes. Pathol Res Pract.
207:317–321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hopfinger G and Staber P: Current standard
in diagnostic and therapy of peripheral T-cell lymphoma. Dtsch Med
Wochenschr. 144:1400–1404. 2019.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laforga JB, Gasent JM and Vaquero M:
Potential misdiagnosis of angioimmunoblastic T-cell lymphoma with
Hodgkin's lymphoma: A case report. Acta Cytol. 54 (5
Suppl):S840–S844. 2010.PubMed/NCBI
|
|
19
|
Gao X, Huang W, Li W, Xie J, Zheng Y and
Zhou X: Clinicopathologic analysis of angioimmunoblastic T-cell
lymphoma with Hodgkin/Reed-Sternberg-like cells. Zhonghua Bing Li
Xue Za Zhi. 44:553–558. 2015.(In Chinese). PubMed/NCBI
|
|
20
|
Parekh V and Peker D: EBV-related primary
splenic lymphocyte-depleted classical Hodgkin lymphoma. J Clin
Pathol. 68:947–950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferrarini I, Rigo A, Visco C, Krampera M
and Vinante F: The evolving knowledge on T and NK cells in classic
Hodgkin lymphoma: Insights into novel subsets populating the immune
microenvironment. Cancers (Basel). 12:37572020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Connors JM: Hodgkin lymphoma: Outsmarting
HRS cells. Blood. 136:2362–2364. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moccia AA, Aeppli S, Güsewell S, Bargetzi
M, Caspar C, Brülisauer D, Ebnöther M, Fehr M, Fischer N, Ghilardi
G, et al: Clinical characteristics and outcome of patients over 60
years with Hodgkin lymphoma treated in Switzerland. Hematol Oncol.
39:196–204. 2021. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin HN, Liu CY, Hong YC, Pai JT, Yang CF,
Yu YB, Hsiao LT, Chiou TJ, Liu JH, Gau JP, et al: Clinical features
and prognostic factors of angioimmunoblastic T-cell lymphoma in
Taiwan: A single-institution experience. Leuk Lymphoma.
51:2208–2214. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
van den Akker TA and Chen H:
Angioimmunoblastic T-cell lymphoma masquerading as granulomatous
lymphadenitis: Fine needle aspiration cytology, clinical and
radiology correlation. Diagn Cytopathol. 49:555–558. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ellis C, Ramirez J and LaFond AA:
Angioimmunoblastic T-cell lymphoma mimicking diffuse large B-cell
lymphoma. Cutis. 102:179–182. 2018.PubMed/NCBI
|
|
27
|
Keefe M, Buntinx-Krieg T and Contestable
J: Angioimmunoblastic T-cell Lymphoma mimicking DRESS syndrome.
Cutis. 109:E29–E32. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trimech M, Letourneau A, Missiaglia E, De
Prijck B, Nagy-Hulliger M, Somja J, Vivario M, Gaulard P, Lambert
F, Bisig B and de Leval L: Angioimmunoblastic T-cell lymphoma and
chronic lymphocytic Leukemia/small lymphocytic lymphoma: A novel
form of composite lymphoma potentially mimicking Richter syndrome.
Am J Surg Pathol. 45:773–786. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Papadi B, Polski JM, Clarkson DR and
Liu-Dumlao TO: Atypical angioimmunoblastic T-cell lymphomas
masquerading as systemic polyclonal B-immunoblastic proliferation.
Virchows Arch. 461:323–331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smithberger ES, Rezania D, Chavan RN, Lien
MH, Cualing HD and Messina JL: Primary cutaneous angioimmunoblastic
T-cell lymphoma histologically mimicking an inflammatory
dermatosis. J Drugs Dermatol. 9:851–855. 2010.PubMed/NCBI
|
|
31
|
Ahsanuddin AN, Brynes RK and Li S:
Peripheral blood polyclonal plasmacytosis mimicking plasma cell
leukemia in patients with angioimmunoblastic T-cell lymphoma:
Report of 3 cases and review of the literature. Int J Clin Exp
Pathol. 4:416–420. 2011.PubMed/NCBI
|
|
32
|
Han P, Yang L, Yan W and Tian D:
Angioimmunoblastic T-cell lymphoma mimicking drug fever and
infectious etiology after a thyroidectomy: A case report. Medicine.
98:e169322019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaffenberger B, Haverkos B, Tyler K, Wong
HK, Porcu P and Gru AA: Extranodal marginal zone Lymphoma-like
presentations of angioimmunoblastic T-cell lymphoma: A T-cell
lymphoma masquerading as a B-cell lymphoproliferative disorder. Am
J Dermatopathol. 37:604–613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Szablewski V, Dereure O, René C, Tempier
A, Durand L, Alame M, Cacheux V and Costes-Martineau V: Cutaneous
localization of angioimmunoblastic T-cell lymphoma may masquerade
as B-cell lymphoma or classical Hodgkin lymphoma: A histologic
diagnostic pitfall. J Cutan Pathol. 46:102–110. 2019.PubMed/NCBI
|
|
35
|
Suárez AE, Artiga MJ, Santonja C,
Montes-Moreno S, De Pablo P, Requena L, Piris MA and
Rodríguez-Pinilla SM: Angioimmunoblastic T-cell lymphoma with a
clonal plasma cell proliferation that underwent immunoglobulin
isotype switch in the skin, coinciding with cutaneous disease
progression. J Cutan Pathol. 43:1203–1210. 2016. View Article : Google Scholar : PubMed/NCBI
|