Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
6th most common malignancy and the 8th cause of cancer death
worldwide (1,2). HNSCC includes carcinomas from the oral
cavity (OSCC), oropharynx (OPSCC), hypopharynx (HPSCC), larynx
(LSCC), the paranasal sinuses, and the major and minor salivary
glands. The etiology of HNSCC involves a variety of toxic,
environmental and viral agents (3).
Tobacco and alcohol exposure are the primary etiological factors
for HNSCC (4–6). Oncogenic human papillomavirus (HPV)
strains, primarily HPV-16, have been recognized as risk factor for
HNSCC, particularly for oropharyngeal cancers (7–10). Men
are more frequently diagnosed with HNSCC compared with women, and
the incidence of HNSCC has a male-to-female ratio of 3:1 in the US
(11). This incidence has been
changing as women increasingly expose themselves to HNSCC risk
factors, tobacco, alcohol and HPV-infection. Park et al
(12) showed that women with HNSCC
are at a higher risk of dying of the disease than men diagnosed
with HNSCC (HR=1.92; 95% CI, 1.07–3.43). However, HPV-associated
HNSCC is more common in men compared with women (13). Patients with HPV+ HNSCC
have a better prognosis than patients with HPV− HNSCC
(14); HPV may have a role in the
clinical manifestation of this sex disparity. HNSCC is commonly
diagnosed in patients ≥60 years old, however, an increasing number
of patients are diagnosed with HNSCC at younger ages (15). Most patients with HNSCC are
diagnosed at advanced stages of the disease (III or IV), which
leads to a poor prognosis outcome (16). HNSCC treatment is generally
multimodal including surgery, rand chemoradiation, yet the overall
survival (OS) of patients with HNSCC is relatively low, ~2.5 years
after treatment, for all HNSCC sites and stages (17).
In the US, African Americans, Hispanics/Latinos and
low-income non-Latino-White individuals are at higher risk of
developing HNSCC. In Puerto Rico, the incidence of HNSCC is 2.5
times higher than that in Hispanics/Latinos living in the US. The
HNSCC incidence of OSCC and OPSCC is 72% higher in Puerto Rico than
among Hispanics/Latinos living in the US. Similarly, the incidence
of LSCC in Puerto Rico is 51% higher than that in Hispanics/Latinos
living in the US (18). Racial and
ethnic health disparities are a serious public health concern due
to the HNSCC high mortality and morbidity rates, higher treatment
costs and the effect on quality of life. Therefore, discovery of
actionable targets for the early detection, diagnosis and prognosis
of HNSCC, and for guiding treatment would have an immediate impact
on reducing these health disparities.
Epigenetic biomarkers, such as aberrant DNA
methylation changes, have been used as molecular classifiers for
different cancer types, having a predictive capacity for patient
prognosis and treatment response (19). Aberrant changes in DNA methylation
such as global DNA hypomethylation and specific promoter DNA
hypermethylation have been associated with carcinogenesis (20). It has been proposed that aberrant
changes in DNA methylation patterns occur early in the carcinogenic
process (21).
Aberrant promoter methylation of tumor suppressor
genes (TSGs), for instance, CDH1, DAPK1, CDKN2A and
RASSF1A, have been detected in HNSCC that resulted in loss
of expression and pathway deregulation (22–24).
Several studies have demonstrated DNA methylation cancer-related
signatures (25,26), suggesting the likelihood of
differential DNA methylation patterns among HNSCC anatomical
subsites (27). Using a candidate
gene approach, the prevalence of the aberrantly methylated TSGs
CDKN2A, p14ARF and CDKN2B in HNSCC tumors
was previously evaluated. Bernabe (28) detected aberrant methylation of the
TSGs CDKN2A and CDKN2B in HNSCC tumors. A reduction
of CDKN2A expression in HNSCC tumors exhibiting methylated
(M) CDKN2A was detected with mRNA expression analysis
(28). Subsequently, the aberrant
methylation of CDH1 was evaluated in HNSCC tumors confirming
its occurrence, but hyperM CDH1 was predominantly detected
in the larynx compared with other HNSCC subsites (29). Preliminary data suggest that a
distinct pattern of aberrant DNA methylation changes may occur in
HNSCC anatomic subsites associated with HNSCC heterogeneity and its
diverse clinical manifestations.
The primary objective of the present study was to
perform a genome-wide DNA methylation analysis in HNSCC samples
from three anatomic subsites, oral cavity, oropharynx and larynx,
to identify potential DNA methylation targets with prognostic value
for HNSCC. Furthermore, a prevalence assessment of selected
candidate genes was performed, and their prognostic value was
evaluated in an independent HNSCC cohort. It was hypothesized that
a biomarker profile based on aberrant DNA methylation specific to
every anatomical site, may help clinicians to better diagnose
HNSCC, thus providing a more accurate prognosis and identify
targets for novel treatments.
Materials and methods
HNSCC discovery cohort
Demographics and clinicopathological characteristics
of the HNSCC discovery and prevalence cohorts are shown in Table I. The HNSCC discovery cohort
included 21 HNSCC tissue samples from Puerto Rican patients,
including 10 OSCC, four OPSCC and seven LSCC samples. The HNSCC
discovery cohort samples were compared with 10 healthy oral tissue
samples. The mean age of the discovery cohort was 56.62 years
(range, 24–76 years; SD, 12.62), and 90 and 10% of patients were
male and female, respectively. The HNSCC anatomical subsite
distribution included 48, 19 and 33% oral cavity, pharynx and
larynx, respectively. Most of the patients with HNSCC were at
advanced stages (III/IV; 67%) of the disease. A total of 1/3 of the
patients (33%) were HPV+. Most tumors showed moderate
differentiation (71%). Most samples were obtained from heavy
smokers (95%) and heavy drinkers (86%).
 | Table I.Clinicopathological characteristics
of the head and heck squamous cell carcinoma discovery cohort
(n=21) and the prevalence cohort (n=119). |
Table I.
Clinicopathological characteristics
of the head and heck squamous cell carcinoma discovery cohort
(n=21) and the prevalence cohort (n=119).
|
Characteristics | Discovery
cohort | Prevalence
cohort |
|---|
| Age, years | 56.62±12.62 | 61.21±12.63 |
|
| (24–76) | (24–98) |
| Sex, n (%) |
|
|
|
Male | 19 (90.5) | 107 (89.9) |
|
Female | 2 (9.5) | 12 (10.1) |
| Site of primary
tumor, n (%) |
|
|
| Oral
cavity | 10 (47.6) | 42 (35.3) |
|
Pharynx | 4 (19.1) | 25 (21.0) |
|
Larynx | 7 (33.3) | 52 (43.7) |
| Tumor stage, n
(%)a |
|
|
| Early
(I/II) | 7 (33.3) | 25 (21.0) |
|
Advanced (III/IV) | 14 (66.7) | 92 (77.3) |
| HPV-16 status, n
(%)a |
|
|
|
HPV-16+ | 7 (33.3) | 57 (47.9) |
|
HPV-16− | 12 (57.1) | 62 (52.1) |
| Differentiation, n
(%)b |
|
|
|
Poor | 2 (9.5) | 9 (7.6) |
|
Moderate | 15 (71.4) | 78 (65.5) |
|
Well | 4 (19.1) | 31 (26.1) |
| Heavy smoking, n
(%) | 20 (95.2) | 104 (87.4) |
| Heavy drinking, n
(%) | 21 (100.0) | 97 (81.5) |
HNSCC prevalence cohort
The HNSCC prevalence cohort included 119 HNSCC
tissue samples from three anatomical subsites: Oral cavity (n=42),
pharynx (n=25) and larynx (n=52). The HNSCC tissue samples of the
prevalence cohort were compared with seven healthy oral tissue
samples. The mean age of the HNSCC prevalence cohort was 61.2 years
(range, 24–98 years; SD, 12.6), and 89.9 and 10.1% were male and
female, respectively. The distribution of the HNSCC anatomical
subsites included 35.3, 21.0 and 43.7% oral cavity, pharynx and
larynx, respectively. Most of the patients with HNSCC were at
advanced stages (III/IV; 77%) of the disease. Regarding HPV
infection, 47.9% of patients were HPV+. Most tumors
showed moderate differentiation (65.5%). Most samples were obtained
from heavy smokers (87%) and heavy drinkers (84%).
The HNSCC tissue samples for both discovery and
prevalence cohorts were obtained from Puerto Rican patients with
HNSCC presenting at the School of Medicine Head and Neck Cancer
Clinic at the Puerto Rico Medical Center, a tertiary teaching
medical center. Patients that were diagnosed with HNSCC through
tissue biopsy, and whose tumors were surgically removed signed an
informed consent. The tumor tissue collected for the study was
analyzed for quality by a pathologist. Oral mucosa samples were
obtained from healthy Puerto Rican patients undergoing a routine
tooth extraction at the School of Dental Medicine, Department of
Surgery after having signed an informed consent. All procedures had
the approval of the University of Puerto Rico, Medical Sciences
Campus Institutional Review Board (IRB; approval no. MSC-IRB
protocol 2770103), and the Johns Hopkins School of Medicine IRB
(approval no. NA_00020633). The medical information of the patients
with HNSCC was obtained from medical records and pathological
reports, including date of diagnosis, site of the primary tumor,
tumor grade, date and site of tumor recurrence (if applicable), and
date and cause of death. The treatment of choice was surgery
followed in some cases by postoperative radiation or
chemoradiation. Follow-up information was prospectively collected
from either medical records or the Puerto Rican Cancer Registry.
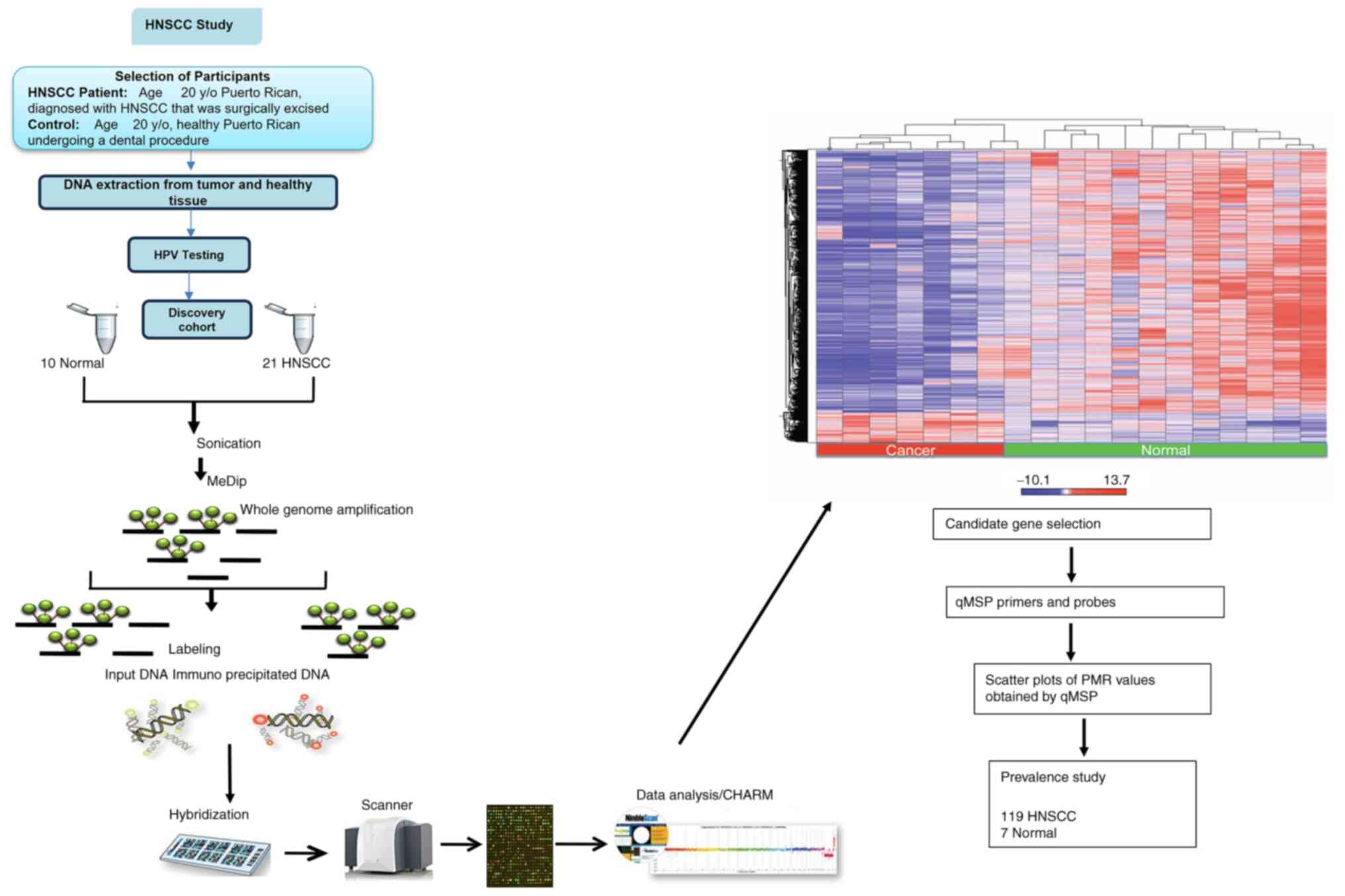
Fig. 1 shows an integrated diagram
describing the experimental study design.
DNA extraction
Genomic DNA was isolated from all HNSCC and healthy
tissues using the DNA Isolation kit for cells and tissues (catalog
no. 11814770001; Roche Diagnostics, Ltd.) following the
manufacturer's instructions. DNA concentration and quality were
measured with the NanoDrop 8000 UV–Vis Spectrophotometer (Thermo
Fisher Scientific, Inc.). DNA sample preparation and hybridization
to oligonucleotide arrays was carried out at the Head and Neck
Cancer Research laboratory, Johns Hopkins School of Medicine.
Detection of HPV-16
Genomic DNA from all HNSCC samples was analyzed for
HPV-16 infection. The HPV-16 status was previously detected by
immunohistochemistry, end-point PCR and a TaqMan-based quantitative
(q)PCR assay, targeting HPV-16 E6 and E7 viral oncogenes. All the
HNSCC samples that were classified as HPV-16+ had
amplification of E6 and E7 viral oncogenes detected through a qPCR
assay. HPV-16 E6 and E7 specific primer and probe sets, and qPCR
and thermal cycling conditions were previously described (10).
Genome-wide DNA methylation
analysis
DNA sonication
Two different genomic DNA amounts from HNSCC and
healthy samples from the discovery cohort (0.5 and 1 µg) were used
as input DNA for sonication to generate 200-800-bp long DNA
fragments. DNA sonication was performed in a Covaris E220
ultrasonicator (Covaris, LLC), and analysis of sonicated DNA was
performed on the BioAnalyzer 2100 (Agilent Technologies, Inc.) with
an Agilent High Sensitivity DNA Kit (catalog no. 5067-4626; Agilent
Technologies, Inc.) to verify DNA concentration, quality and
purity.
Methylated DNA
immunoprecipitation
DNA from HNSCC and healthy samples from the
discovery cohort was subjected to methylated DNA
immunoprecipitation (MeDIP) using the MagMeDIP kit (Diagenode SA)
following the manufacturer's instructions. Two different starting
DNA quantities were used (0.5 and 1 µg) for every sonicated sample.
A total of 10% of every sample was transferred to a 1.5-ml tube
(input DNA) and used as control of the starting DNA material. The
remaining 90% of the sonicated DNA was subjected to MeDIP and
labeled as immunoprecipitated DNA (IP DNA). IP DNA samples were
exposed to a 5-methylcytosine antibody, which recognizes methylated
cytosines in the DNA to enrich every sample with methylated DNA.
Every tumor or control (healthy) DNA sample had an IP DNA and an
input DNA. Samples were subjected to a qPCR assay to determine the
efficiency of the MeDIP assay efficiency in enriching methylated
DNA. IP DNA samples were compared with input DNA samples to
determine if enrichment of methylated DNA occurred. Both DNA
samples were tested using four primer pairs included in the
MagMeDIP kit (Diagenode SA; Table
SI).
The qPCR master mix included the following: 6.25 µl
SYBR Green Supermix, 0.5 µl primer pair (10 µM), 3.5 ng either IP
DNA or input DNA, and 3.25 µl water. The final reaction volume was
12.5 µl. The thermocycling conditions involved a denaturation step
at 95°C for 7 min, followed by 40 cycles at 95°C for 15 sec and at
60°C for 1 min, an incubation step for 95°C for 1 min to denature
the DNA, and a melting curve analysis as established by the
manufacturer instructions. The efficiency of MeDIP enrichment was
calculated using the following equation: % (meDNA-IP/total
input)=2[(Cq(10%input)-3.32)-Cq(meDNA-IP)] ×100. The
MeDIP recovery was % (meDNA-IP/total input). Samples that showed
>50% DNA methylation enrichment were subjected to hybridization
and scanning into the 3×720K CpG Island Plus RefSeq Promoter Array
(Roche Diagnostics, Ltd.; Fig.
S1).
DNA labeling and hybridization
After the MeDIP assay, all DNA samples (IP DNA and
input DNA) were subjected to a genome-wide amplification (WGA)
assay (Sigma-Aldrich; Merch KGaA) to increase the amount of DNA in
every sample. After the WGA assay, DNA samples were purified using
the QIAquick PCR Purification Kit (Qiagen Sciences, Inc.). DNA
concentration was measured with the NanoDrop 8000 UV–Vis
Spectrophotometer (Thermo Fisher Scientific, Inc.). Every DNA
sample (IP DNA and input DNA) was labeled with fluorophores using
the NimbleGen Dual-Color DNA Labeling Kits (Roche Diagnostics,
Ltd.). IP DNA was labeled with Cy5 fluorophore, and the input DNA
was labeled with the Cy3 fluorophore. Labeled IP DNA and input DNA
samples were combined and hybridized into the 3×720K CpG Island
Plus RefSeq Promoter Array. Hybridization was accomplished using
the NimbleGen Hybridization Kit (Roche Diagnostics, Ltd.) following
standard operating protocol. The 3×720K CpG Island Plus RefSeq
Promoter Array allowed hybridization of three samples
simultaneously and covered 27,728 annotated CpG islands, 22,532
RefSeq gene promoters, and regulatory elements from the HG18 build.
Each promoter array included several positive, negative and non-CpG
control regions to calculate experimental performance. Analysis of
RefSeq gene promoters involved regions 2.4 kb upstream of the
transcription start site (TSS) and 0.6 kb downstream of the TSS for
overall coverage of 3 kb of each promoter per gene. Each array was
scanned in the NimbleGen MS2 Microarray Scanner (Roche Diagnostics,
Ltd.) following the manufacturer's protocol.
Differential methylation
bioinformatics
NimbleGen's DeVa's software (Roche Diagnostics,
Ltd.) was used to create .xys files from the array's scanned
images. The images allowed a peak discovery algorithm to generate
an initial list of differentially methylated regions (DMRs) when
tumor samples were compared with control samples. The .xys files
were used as input data for the analysis using Comprehensive
high-throughput arrays for relative methylation (CHARM)
bioinformatics package within the R 4.1.2 statistical programming
(30). CHARM software is a
bioinformatics package used to discover DMRs between samples,
calculate the percentage of methylation, verify array quality and
control for batch effects. Besides, DMRs were identified with Bump
Hunting (version 1.44.0) (31), a
statistical genomics tool to identify differential peaks in
methylation data. Methylation Bump Hunting is a data analysis
pipeline that effectively models measurement error, removes batch
effects, detects regions of interest, and attaches statistical
uncertainty to regions identified as differentially methylated
(32). The bioinformatical analysis
pipeline used in the present study included analysis of TSS and CpG
Islands independently among tumor and control samples (Data S1). Frequency of genes was analyzed
for tumor and control samples. Genes with a frequency of ≥20% in
tumor samples were selected. Likewise, commonly occurring genes
between tumor and control samples were analyzed. A detailed
bioinformatical pipeline description for peak discovery algorithm
can be found in Data S1. In
summary, the raw intensity data from the array were analyzed and
the data was transformed into a log ratio of the intensities of
methylated probes vs. unmethylated probes, which represents the
M-value. An M-value ~0 represented a similar intensity between the
methylated and unmethylated probes. Positive M-values implied that
more molecules within the tested probe were methylated than
negative M-values, which represented less methylation (32). An M-value cut-off was established to
define which CpG targeted sites were aberrantly methylated in tumor
samples compared with control samples. CpG targeted sites with an
M-value ≥2.0 were classified as methylated and were further
analyzed. CpG sites with an M-value <2.0 were classified as
unmethylated. CpG targeted sites were also subjected to a
low-stringency P-value threshold (P<0.05) and ranked by
fold-change between tumor and control samples. A list of DMRs was
created using the CpG sites methylation level for every HNSCC
subsite. These lists determined the regions in the genome that were
differentially methylated between HNSCC and normal samples.
DMR validation in TCGA
DMRs identified through bioinformatical analyses
were cross-referenced with available methylation-related databases,
including publicly available HNSCC TCGA methylation database and
peer-reviewed accessible databases, as previously described
(33). Briefly, the Bump Hunting
method was used to perform an epigenome-wide analysis of the HNSCC
methylome to identify DMRs of biological interest using methylation
arrays. Two separate epigenome-wide analyses were carried out using
Bioconductor's minfi package (version 1.48.0), as previously
described (34). Briefly, an
unbiased epigenome-wide DNA methylation analysis was performed
using the minfi package in Bioconductor to identify DMRs in 274
primary chemotherapy-naïve HNSCC samples (TCGA dataset) and 32
frequency-matched uvulopalatopharyngoplasty (UPPP) controls (Johns
Hopkins Head and Neck Cancer Research laboratory). The significant
DMRs (P<0.001) were identified in a CpG island located ≤200 bp
upstream and downstream from the 5′ end of the gene. The DMRs
results obtained with MeDIP were validated with DMRs results from
HNSCC TCGA samples. Significant DMRs common to both sample sets
were subjected to Gene Ontology (GO) analysis with Database for
Annotation, Visualization and Integrated Discovery version 6.7
(https://david.ncifcrf.gov/tools.jsp).
Candidate gene selection
Based on the genome wide DMR analysis, TCGA
comparison and GO bioinformatics analysis, the 10 genes DAPK1,
PITX2, PAX5, TIMP3, SFRP1, CALCA, SOCS1, CDH1, MAGI2 and
PAX1 were selected for downstream validation as candidate
biomarker genes for HNSCC, using quantitative methylation-specific
PCR (qMSP).
DNA bisulfite modification
Bisulfite modification was used to convert
unmethylated cytosine residues of genomic DNA into uracil while
leaving the methylated cytosines unchanged. For all HNSCC and
healthy samples, including discovery and prevalence cohort, 1 µg
genomic DNA was treated with sodium bisulfite using the EZ DNA
Methylation-Gold Kit (catalog no. D5005; Zymo Research Corp.)
according to the manufacturer's protocol.
qMSP
All HNSCC and healthy DNA samples, including the
discovery and prevalence cohorts, were subjected to qMSP. Tumor and
healthy bisulfite-modified DNA samples were used as a qMSP assay
template, a fluorescence-based real-time PCR assay was previously
described (35). Primers and probe
set sequences selected had been previously described to amplify the
promoter regions of DAPK1, PITX2, PAX5, TIMP3, SFRP1, CALCA,
SOCS1, CDH1, MAGI2 and PAX1, and a reference gene,
ACTβ. Primers and probe set sequences are shown in Table SII (36–43).
All qMSPs were carried out in duplicates in a 48-well reaction
plate with a final volume of 25 µl. Each reaction contained 600 nM
forward and reverse primers, 200 nM probe (Integrated DNA
Technologies, Inc.), 1× TaqMan Universal PCR Master Mix, no UNG
(Thermo Fisher Scientific, Inc.) and 2 µl bisulfite-modified DNA.
qMSP amplifications were performed in a StepOne Real-Time PCR
System (Thermo Fisher Scientific, Inc.) using the following
conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15
sec and an annealing temperature of 58°C for 1 min. Each reaction
plate included HNSCC bisulfite-modified DNA samples, a positive,
fully methylated DNA control sample (bisulfite-converted Universal
Methylated Human DNA Standard; Zymo Research Corp.) and no-template
controls. Serial dilutions (30–0.003 ng) of bisulfite-converted
Universal Methylated Human DNA standard were used to construct a
calibration curve for each plate. After amplification, the
percentage of methylated reference (PMR) for each candidate gene in
each sample was calculated using the following equation: [(HNSCC
sample Cq value gene of interest/HNSCC sample Cq value
β-actin)/(fully methylated sample Cq value gene of interest/fully
methylated sample Cq value β-actin] ×100.
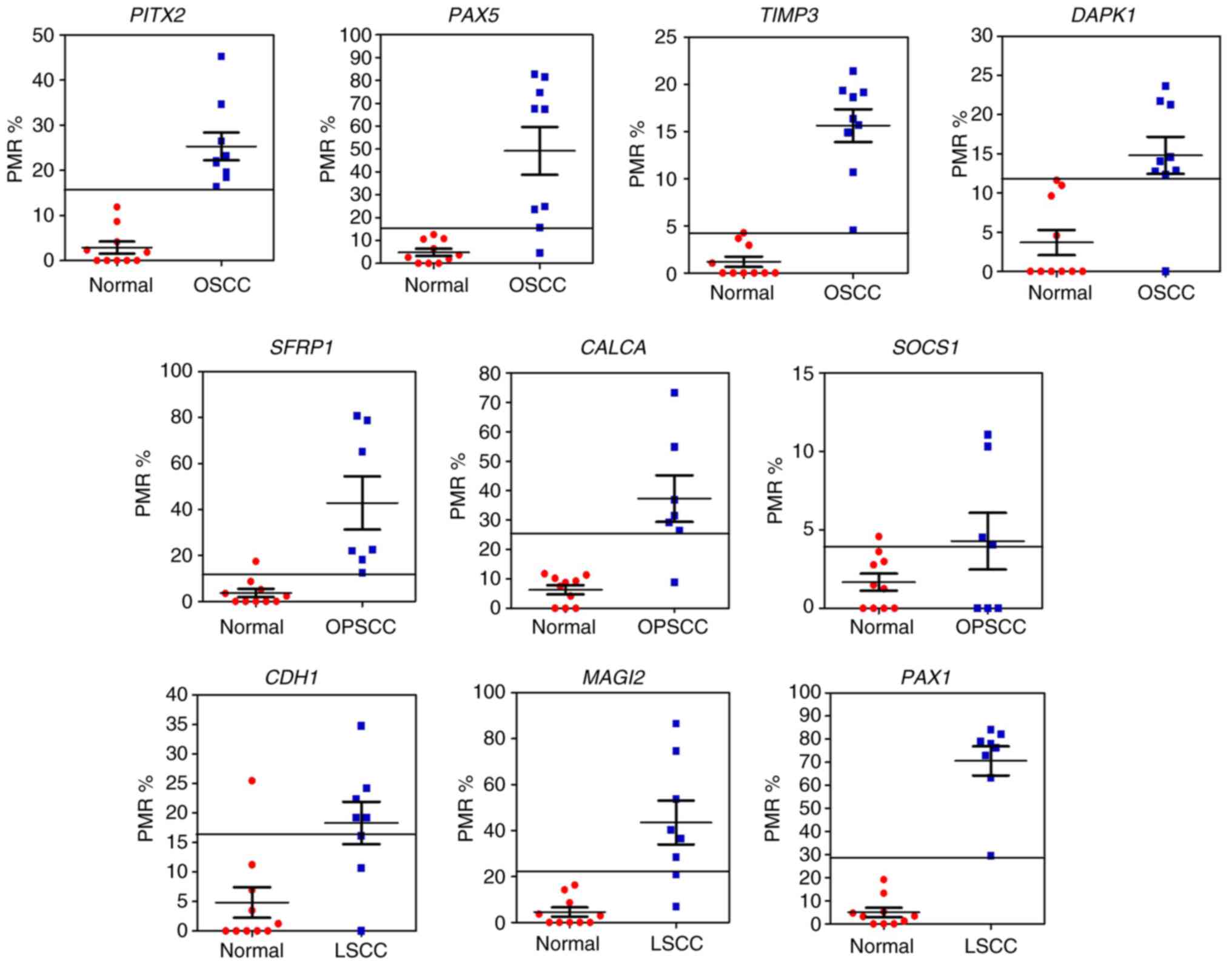
PMR values obtained from samples from the discovery
cohort were used to draw receiver operating characteristic (ROC)
curves to obtain sensitivity and specificity values for every
candidate gene. ROC curves were drawn using STATA (version 15;
StataCorp LP). Based on sensitivity and specificity values, a
suitable PMR cut-off value was chosen for every candidate gene.
Prevalence cohort samples were classified as methylated (M) or
unmethylated (UM) based on the PMR cut-off value for every
candidate gene. Promoter methylation of PITX2, PAX5 and
TIMP3 was tested in 29 OSCC samples. Promoter methylation of
SFRP1, CALCA and SOCS1 was tested in 19 OPSCC
samples. Promoter methylation of CDH1, MAGI2 and PAX1
was tested in 39 LSCC samples. Promoter methylation of DAPK1
was used as an internal control and was evaluated in all HNSCC
samples.
Statistical analysis
Data from independent groups were compared using
Fisher's exact test or χ2-test, as appropriate. Odds
ratio (OR) calculations for clinicopathological parameters were
performed using binary logistic. OS was measured in months from the
date of diagnosis until death (if applicable). Survival analyses
were performed using Kaplan-Meier curves. Log-rank Mantel-Cox and
Gehan-Breslow Wilcoxon tests were used to determine the
significance between two survival curves. Prognostic factors that
have impact on HNSCC survival were analyzed in a Cox regression
analysis. Statistical analyses were performed using SPSS (version
22; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
DMRs in HNSCC tumor samples from the
discovery cohort
Results from the discovery cohort show that the
three HNSCC subsites had in common 2,565 DMRs that included genes
previously associated with HNSCC (Table SIII). Some of the identified DMRs
corresponded to genes previously described as having a pivotal role
in HNSCC carcinogenesis, such as BRCA2, CDKN2A, CDKN1B (P27),
DAPK1,MAPK1, MAPK10, MLH1, RASSF1, HOXC6, VEGFB, WNT1 and
WNT8B (44–55). Among these genes, several of them
have roles in essential pathways for cell cycle regulation
(RASSF1 and CDKN1B), cell proliferation (MAPK1
and MAPK10) and apoptosis (DAPK1).
The genome-wide analysis also unveiled 889 DMRs
unique for OSCC, 363 DMRs for OPSCC and 738 DMRs for LSCC (Fig. S2). Results from the 450K Infinium
DNA methylation array from 274 HNSCC TCGA samples and 32
frequency-matched UPPP control samples from John Hopkins Head and
Neck Cancer Research Laboratory were used to validate
subsite-specific DMRs identified in the MeDIP experiment. A GO
analysis was used to describe the function of the most critical
DMRs. Based on the DMR and GO analyses, DAPK1, PITX2, PAX5,
TIMP3, SFRP1, CALCA, SOCS1, CDH1, MAGI2 and PAX1 were
selected as candidate genes to be further evaluated.
The promoter methylation status of the 10 candidate
genes in all HNSCC and healthy samples from the discovery cohort
was analyzed. Samples were subjected to qMSP analysis for all
candidate genes. A total of nine candidate genes showed
differential methylation between HNSCC and healthy samples. The
candidate gene SOCS1 showed no difference in the promoter
methylation status between HNSCC and healthy samples. Table II shows values obtained for
sensitivity, specificity, ROC curve and PMR cut-off value for every
candidate gene.
| Table II.Predictive accuracy of DAPK1,
PITX2, PAX5, TIMP3, SFRP1, CALCA, SOCS1, CDH1, MAGI2 and
PAX1 for head and neck squamous cell carcinoma. |
Table II.
Predictive accuracy of DAPK1,
PITX2, PAX5, TIMP3, SFRP1, CALCA, SOCS1, CDH1, MAGI2 and
PAX1 for head and neck squamous cell carcinoma.
| Target genes | ROC | P-value | Sensitivity, % | Specificity, % | Methylation cut-off
value |
|---|
| DAPK1 | 0.92 | 0.0009 | 88.89 | 100.00 | 12.36 |
| PITX2 | 1.00 | <0.0001 | 100.00 | 100.00 | 16.37 |
| PAX5 | 0.96 | <0.0001 | 88.89 | 100.00 | 15.71 |
| TIMP3 | 1.00 | <0.0001 | 100.00 | 100.00 | 4.52 |
| SFRP1 | 0.96 | 0.0005 | 100.00 | 90.00 | 12.47 |
| CALCA | 0.90 | 0.0006 | 85.71 | 100.00 | 26.37 |
| SOCS1 | 0.52 | 0.4750 | 57.14 | 90.00 | 4.06 |
| CDH1 | 0.82 | 0.0054 | 75.00 | 90.00 | 16.15 |
| MAGI2 | 0.96 | 0.0001 | 87.50 | 100.00 | 20.86 |
| PAX1 | 1.00 | <0.0001 | 100.00 | 100.00 | 29.41 |
In the OSCC samples, M PITX2 and PAX5
were detected in 58.6 and 79.3% of the samples, respectively. Also,
M TIMP3 was confirmed in 79.3% of the samples, and M
DAPK1 was detected in 51.7% of the samples. In the OPSCC
samples, M SFRP1, CALCA and SOCS1 were detected in
84.2, 78.9 and 15.8% of the samples, respectively. M DAPK1
was detected in 63.2% of the samples. As for LSCC samples, M
CDH1, MAGI1 and PAX1 were detected in 58.9, 64.1 and
74.4% of the samples, respectively. M DAPK1 was detected in
66.7% of the LSCC samples.
A total of five candidate genes were selected,
DAPK1, CDH1, PAX1, CALCA and TIMP3, for validation in
a HNSCC prevalence cohort based on predictive accuracy of the
genes. Each candidate gene's predictive accuracy to detect HNSCC
was calculated by ROC curve analysis. The sensitivity and
specificity values were used to select the PMR cut-off value for
every candidate gene. PAX1 and TIMP3 had ROC values
of 1.00, 100% sensitivity and 100% specificity; DAPK1 had a
ROC value of 0.92, 88.9% sensitivity and 100% specificity;
CALCA had a ROC value of 0.90, 85.7% sensitivity and 100%
specificity; and CDH1 had a ROC value of 0.82, 75%
sensitivity and 90% specificity.
A PMR value was calculated for every candidate gene
for all HNSCC and healthy samples in the prevalence cohort. PMR
values obtained from the prevalence cohort were compared with the
PMR cut-off value for e every candidate gene obtained from the
discovery cohort (Fig. 2).
Prevalence cohort samples with a ≥PMR value than the PMR cut-off
value for every candidate gene were classified as M. Prevalence
samples with a lower PMR than the PMR cut-off value for every
candidate gene were classified as UM. The prevalence of M candidate
genes in HNSCC and normal samples from the prevalence cohort is
shown in Table SIV. Promoter
aberrant methylation of DAPK1 was detected in 58.0% of the
HNSCC samples (P=0.005), M CDH1 was detected in 50.0% of the
HNSCC samples (P=0.112), methylation of PAX1 was confirmed
in 82.0% of the HNSCC samples (P=0.001), M CALCA was
confirmed in 44.0% of the HNSCC samples (P=0.036), while M
TIMP3 was confirmed in 76.0% of the HNSCC prevalence cohort
samples (P=0.003).
Association analysis between HNSCC
clinicopathological characteristics and aberrant methylation of
DAPK1, CDH1, PAX1, CALCA and TIMP3 (Table III) shows that the frequency of M
PAX1 was significantly different across HNSCC anatomic
subsites (P=0.029), being the highest frequency of detection
in LSCC. No significant association was found between aberrant M
genes (DAPK1, CDH1, CALCA and TIMP3) with sex, age,
smoking, alcohol abuse, HPV infection and tumor staging. Concurrent
methylation of two to five candidate genes was found in 15% of the
patients with HNSCC (Fig. S3).
| Table III.Association between
clinicopathological characteristics of the HNSCC cohort (n=50) and
aberrant promoter methylation of DAPK1, CDH1, PAX1, CALCA
and TIMP3. |
Table III.
Association between
clinicopathological characteristics of the HNSCC cohort (n=50) and
aberrant promoter methylation of DAPK1, CDH1, PAX1, CALCA
and TIMP3.
|
| DAPK1 | CDH1 | PAX1 | CALCA | TIMP3 |
|---|
|
|
|
|
|
|
|
|---|
| Clinicopathological
characteristics | Meth | Unmeth | Unknown | P-value | RR [95% CI] | Meth | Unmeth | Unknown | P-value | RR [95% CI] | Meth | Unmeth | Unknown | P-value | RR [95% CI] | Meth | Unmeth | Unknown | P-value | RR [95% CI] | Meth | Unmeth | Unknown | P-value | RR [95% CI] |
|---|
| Age, years |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
<60 | 10 | 7 |
|
| 1.02 | 9 | 8 |
|
| 1.09 | 15 | 2 |
|
| 1.12 | 6 | 11 |
|
| 0.72 | 11 | 6 |
|
| 0.79 |
|
≥60 | 19 | 14 |
| 1.000 | [0.62- | 16 | 17 |
| 1.000 | [0.62- | 26 | 7 |
| 0.699 | [0.87- | 16 | 17 |
| 0.548 | [0.35- | 27 | 6 |
| 0.294 | [0.54- |
|
|
|
|
|
| 1.67] |
|
|
|
| 1.93] |
|
|
|
| 1.44] |
|
|
|
| 1.51] |
|
|
|
| 1.16] |
| Sex, n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Male | 27 | 18 |
|
| 1.50 | 23 | 22 |
|
| 1.28 | 38 | 7 |
|
| 1.41 | 19 | 26 |
|
| 0.70 | 33 | 12 |
|
| 0.73 |
|
Female | 2 | 3 |
| 0.638 | [0.50- | 2 | 3 |
| 1.000 | [0.42- | 3 | 2 |
| 0.216 | [0.68- | 3 | 2 |
| 0.642 | [0.32- | 5 | 0 |
| 0.319 | [0.61- |
|
|
|
|
|
| 4.50] |
|
|
|
| 3.88] |
|
|
|
| 2.91] |
|
|
|
| 1.55] |
|
|
|
| 0.87] |
| HPV-16 status,
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HPV-16+ | 11 | 13 |
|
| 0.66 | 12 | 12 |
|
| 1.00 | 19 | 5 |
|
| 0.93 | 10 | 14 |
|
| 0.90 | 19 | 5 |
|
| 1.08 |
|
HPV-16− | 18 | 8 |
| 0.151 | [0.39- | 13 | 13 |
| 1.000 | [0.57- | 22 | 4 |
| 0.721 | [0.72- | 12 | 14 |
| 0.783 | [0.48- | 19 | 7 |
| 0.745 | [0.79- |
|
|
|
|
|
| 1.10] |
|
|
|
| 1.74] |
|
|
|
| 1.22] |
|
|
|
| 1.69] |
|
|
|
| 1.48] |
| Tumor site, n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Oral
cavity | 8 | 5 |
|
|
| 6 | 7 |
|
|
| 13 | 0 |
|
|
| 4 | 9 |
|
|
| 11 | 2 |
|
|
|
|
Pharynx | 3 | 3 |
|
|
| 1 | 5 |
|
|
| 3 | 3 |
|
|
| 4 | 2 |
|
|
| 6 | 0 |
|
|
|
|
Larynx | 18 | 13 |
| 0.894 |
| 18 | 13 |
| 0.169 |
| 25 | 6 |
| 0.029 |
| 14 | 17 |
| 0.334 |
| 21 | 10 |
| 0.167 |
|
| Disease staging,
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Early
(I/II) | 6 | 5 |
|
| 0.96 | 6 | 5 |
|
| 1.12 | 11 | 0 |
|
| 1.32 | 4 | 7 |
|
| 0.79 | 10 | 1 |
|
| 1.29 |
| Late
(III/IV) | 21 | 16 | 2 | 1.000 | [0.52- | 18 | 19 | 2 | 1.000 | [0.59- | 28 | 9 | 2 | 0.095 | [1.10- | 17 | 20 | 2 | 0.733 | [0.34- | 26 | 11 | 2 | 0.248 | [0.98- |
|
|
|
|
|
| 1.77] |
|
|
|
| 2.11] |
|
|
|
| 1.59] |
|
|
|
| 1.86] |
|
|
|
| 1.71] |
| Nodal |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| involvement, n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yes | 7 | 3 |
|
| 1.29 | 5 | 5 |
|
| 1.02 | 7 | 3 |
|
| 0.84 | 4 | 6 |
|
| 0.87 | 6 | 4 |
|
| 0.77 |
| No | 20 | 17 | 3 | 0.481 | [0.78- | 18 | 19 | 3 | 1.000 | [0.51- | 31 | 6 | 3 | 0.377 | [0.54- | 17 | 20 | 3 | 1.000 | [0.38- | 29 | 8 | 3 | 0.251 | [0.45- |
|
|
|
|
|
| 2.14] |
|
|
|
| 2.08] |
|
|
|
| 1.28] |
|
|
|
| 2.01] |
|
|
|
| 1.31] |
| Tumor |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| differentiation,
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Well | 6 | 7 |
|
|
| 4 | 9 |
|
|
| 11 | 2 |
|
|
| 3 | 10 |
|
|
| 11 | 2 |
|
|
|
|
Moderate | 19 | 10 |
|
|
| 17 | 13 |
|
|
| 25 | 5 |
|
|
| 14 | 16 |
|
|
| 22 | 8 |
|
|
|
|
Poor | 2 | 4 | 2 | 0.243 |
| 3 | 3 | 1 | 0.296 |
| 4 | 2 | 1 | 0.597 |
| 4 | 2 | 1 | 0.162 |
| 4 | 2 | 1 | 0.633 |
|
| Heavy smoking,
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yes | 25 | 18 |
|
| 1.74 | 23 | 20 |
|
| 8.02 | 35 | 8 |
|
| 1.22 | 19 | 24 |
|
| 0.66 | 32 | 10 |
|
| 1.14 |
| No | 1 | 2 | 4 | 0.572 | [0.34- | 0 | 3 | 4 | 0.233 | [0.39- | 2 | 1 | 4 | 0.488 | [0.54- | 2 | 1 | 4 | 0.585 | [0.28- | 2 | 1 | 5 | 1.000 | [0.50- |
|
|
|
|
|
| 8.82] |
|
|
|
| 164.9] |
|
|
|
| 2.75] |
|
|
|
| 1.58] |
|
|
|
| 2.59] |
| Heavy drinking,
n |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Yes | 22 | 16 |
|
| 1.16 | 21 | 17 |
|
| 2.21 | 32 | 6 |
|
| 1.35 | 16 | 22 |
|
| 0.67 | 28 | 10 |
|
| 0.98 |
| No | 4 | 4 | 4 | 0.713 | [0.55- | 2 | 6 | 4 | 0.243 | [0.64- | 5 | 3 | 4 | 0.176 | [0.77- | 5 | 3 | 4 | 0.439 | [0.35- | 6 | 2 | 4 | 1.000 | [0.63- |
|
|
|
|
|
| 2.43] |
|
|
|
| 7.59] |
|
|
|
| 2.35] |
|
|
|
| 1.30] |
|
|
|
| 1.53] |
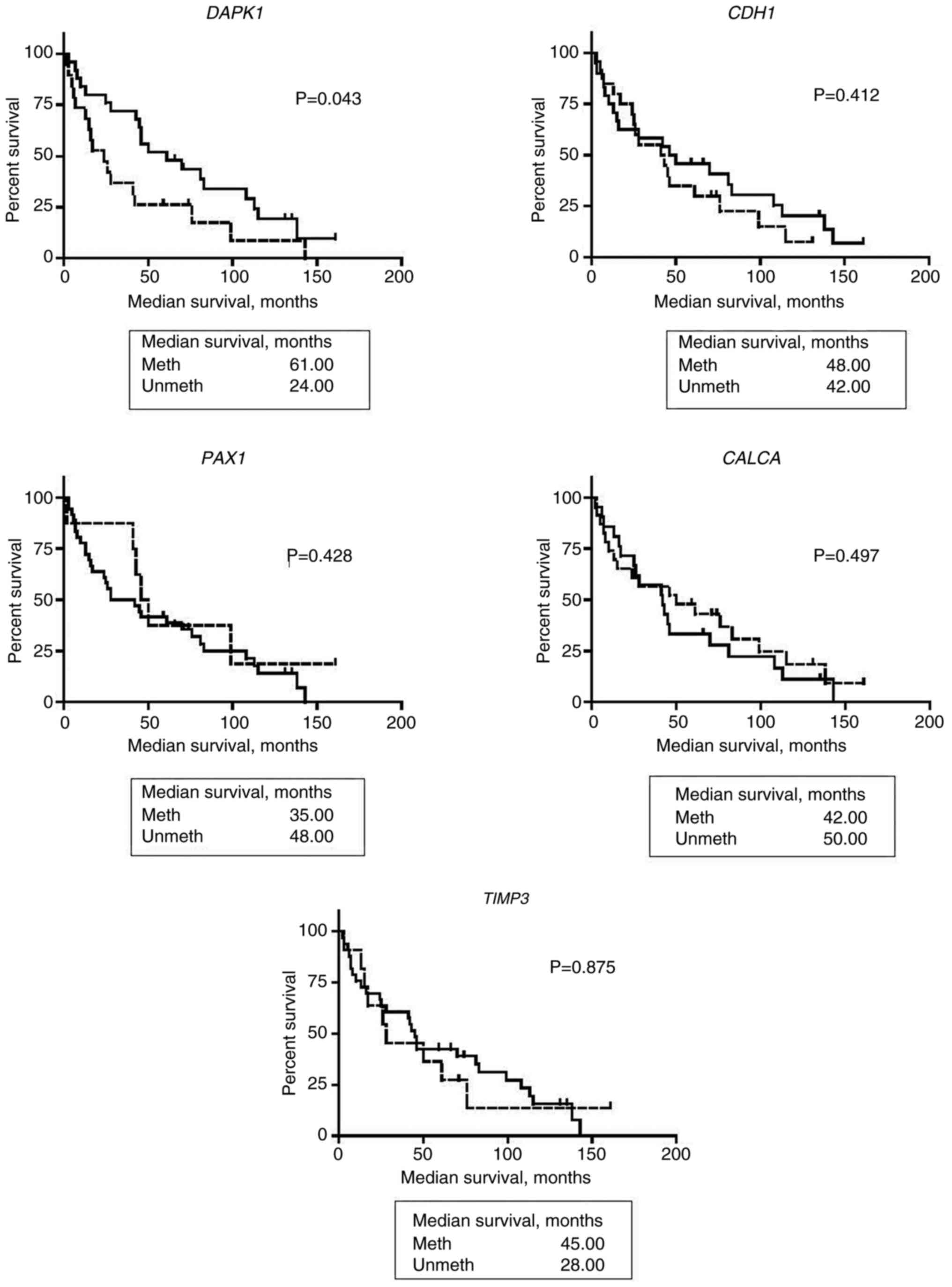
The prognostic value of DAPK1, CDH1, PAX1, CALCA
and TIMP3 was assessed using Kaplan-Meier. Kaplan Meier
survival analysis showed that patients with HNSCC and aberrant M
DAPK1 had a better OS (61.0 months) compared with UM
DAPK1 OS (24 months; P=0.043). No significant association
with the OS of patients with HNSCC and aberrant methylation of
CDH1, PAX1, CALCA and TIMP3 was found (Fig. 3).
A Cox regression analysis of OS was also performed
to evaluate the association between aberrant methylation of the
five candidate genes DAPK1, CDH1, PAX1, CALCA and
TIMP3, and the risk of death from HNSCC (Table IV). Clinicopathological indicators
such as age, tumor stage and differentiation, smoking, drinking and
HPV infection were included in the analysis to assess their effect
on HNSCC survival outcomes in this cohort. DAPK methylation
(P=0.001; HR, 0.096; 95% CI, 0.03–0.37); HPV− tumors
(P=0.006; HR, 9.72; 95% CI, 1.94–48.42); tumor site (P=0.033; HR,
0.96; 95% CI, 0.03–0.37); tumor differentiation (P=0.013; HR, 8.56;
95% CI, 1.58–46.46); and age (P=0.002; HR, 1.11; 95% CI, 1.04–1.18)
showed significant HRs.
| Table IV.Cox regression analysis of overall
survival. |
Table IV.
Cox regression analysis of overall
survival.
| Variables | P-value | HR [95% CI] |
|---|
| DAPK1
meth | 0.001a | 0.096
[0.03–0.37] |
| CDH1
meth | 0.110 | 2.990
[0.78–11.48] |
| PAX1
meth | 0.825 | 1.230
[0.20–7.55] |
| TIMP3
meth | 0.951 | 1.040
[0.30–3.66] |
| CALCA
meth | 0.259 | 0.500
[0.15–1.68] |
|
HPV-16− | 0.006a | 9.720
[1.94–48.72] |
| Tumor site |
|
|
|
Larynx | 0.765 | 1.360
[0.18–9.98] |
| Oral
Cavity | 0.920 | 0.870
[0.06–13.30] |
|
Pharynx | 0.033a | 14.170
[1.25–161.15] |
| Tumor
differentiation |
|
|
|
Well | 0.226 | 2.760
[0.53–14.27] |
|
Moderate | 0.013a | 8.560
[1.58–46.46] |
| Tumor stage |
|
|
| I | 0.997 | 1.000
[0.05–19.80] |
| II | 0.324 | 3.080
[0.03–28.90] |
|
III | 0.668 | 1.500
[0.24–9.37] |
| Positive lymph
nodes | 0.401 | 0.490
[0.09–2.61] |
| Heavy smoking | 0.969 | 0.940
[0.05–16.71] |
| Heavy drinking | 0.523 | 1.720
[0.33–9.10] |
| Age | 0.002a | 1.110
[1.04–1.18] |
Discussion
HNSCC is a heterogeneous disease comprising tumors
from multiple anatomic subsites, each differing in prognosis and
treatment strategy. Aberrant DNA methylation changes have been
shown useful as molecular classifiers in several tumor sites
because of their predictive capacity for disease detection, patient
prognosis and treatment response (56–58).
The discovery of epigenetic alterations is critical for a better
understanding of HNSCC initiation and progression. Thus, the
identification of genes epigenetically inactivated as potential
prognostic biomarkers for HNSCC is urgent.
In the current study, an epigenomic analysis of a
well-defined HNSCC cohort was shown. A genome-wide DNA methylation
analysis showed that HNSCC tumors have 2,565 DMRs common to all
HNSCC subsites. Several critical DMRs associated with a specific
HNSCC anatomical subsite were identified. A total of 889, 363 and
738 DMRs unique to OSCC, OPSCC and LSCC were identified,
respectively. These DMRs were associated with critical cellular
pathways, often deregulated in multiple cancer types, including
HNSCC (59). Among those DMRs, 10
candidate genes (DAPK1, PITX2, PAX5, TIMP3, SFRP1, CALCA, SOCS1,
CDH1, MAGI2 and PAX1) were selected and evaluated
further for their predictive and prognostic value.
Kaplan-Meier survival analysis (P=0.043) and Cox
regression analysis of OS (P=0.001) showed that DAPK1
methylation is associated with better prognosis in HNSCC.
DAPK1, a mediator of a wide range of cellular processes
including growth, apoptosis, autophagy and oxidative stress
(60,61), was identified as aberrantly
methylated in all HNSCC subsites. DAPK1 prediction analysis
suggested high levels of specificity and sensitivity for HNSCC
detection, which was later confirmed in the HNSCC prevalence study
(P=0.005). Epigenetic inactivation of DAPK1 may be a key
event in head and neck carcinogenesis (62). Aberrant methylation of DAPK1
has been confirmed in the cancer of the OSCC (63–65),
pharynx (66) and larynx (67). Aberrant methylation of DAPK1
has been shown to occur in advanced HNSCC (stages III and IV)
tumors with positive lymph node involvement, resulting in a poor
prognosis (67). Likewise,
downregulation of DAPK1 expression has also been shown in
HNSCCs (68,69). Loss of DAPK1 expression,
mediated by promoter hypermethylation, has been associated with
deregulation of autophagy in cancer cells and resistance to
radiotherapy and chemotherapy treatments (70). Thus, aberrantly methylated
DAPK1 may be associated with HNSCC carcinogenesis and
progression.
Preliminary data from our research group suggested
that aberrantly M CDH1 was associated with worse outcome
(death) in LSCC. The present study shows that CDH1 is
frequently M in LSCC. CDH1, a tumor invasion/suppressor
gene, transcribes a 120-kDa glycoprotein, E-cadherin (71), that is essential for establishing
and maintaining intercellular connections (72). Squamous carcinoma cells are
characterized by poor cellular adhesion, loss of epithelial
morphology and increased cellular motility (73). Downregulation of E-cadherin
expression, either by genetic mutation or epigenetic dysregulation,
leads to alterations in cell-to-cell adhesion and increases the
metastatic potential of squamous cell carcinoma. M CDH1 has
been associated with invasive LSCC tumors (Grade 3 and 4) and
metastasis (74). M CDH1 has
also been detected in tissue samples of surrounding mucosa of OSCC,
suggesting that M CDH1 may be a key contributor to HNSCC
carcinogenesis. Downregulation of CDH1 expression has been
observed in HNSCC, and loss of CDH1 expression was
associated with invasive HNSCC (75). Low CDH1 mRNA levels were
detected in patients with tongue cancer (76), but no statistically significant
association with clinicopathological characteristics nor patient
outcome were found. A meta-analysis of 23 studies showed that
CDH1 methylation was notably more frequent in HNSCC tissue
than healthy controls, thus, supporting the role of CDH1 as
a diagnostic biomarker (77). That
meta-analysis showed that Asians display a higher frequency of
MCDH1 than Caucasian or African subgroups and suggested that
ethnicity may account for the differences in CDH1
methylation frequency (77). In the
present study, it was confirmed that CDH1 is methylated at
high levels in LSCC tumors and could act as a promising prognostic
biomarker of LSCC.
The findings of the current study suggest that M
PAX1 is also a promising biomarker for LSCC. M PAX1
was mostly detected in LSCC and was significantly different than
control samples (P≤0.001). The frequency of M PAX1 was
significantly different across HNSCC subsites, showing a higher
frequency in LSCC (P=0.029) corroborating the genome-wide analysis.
The results of the present study show that aberrant methylation of
PAX1 had predictive accuracy for identifying HNSCC samples
(ROC, 1.00; 100% specificity and sensitivity). PAX1 belongs
to the highly conserved PAX gene family, which are developmentally
controlled and encode transcription factors regulating
embryogenesis in vertebrates (78).
The expression of PAX1 during the development process is
limited to the skeleton, thymus and parathyroid glands (79). PAX1, among other PAX
genes, has critical roles in the development of the thymus and the
parathyroid gland (80).
PAX1 dysregulation causes a hypoplastic thymus with defects
in thymocyte maturation and a delay in separation from the
oropharynx (81), suggesting that
loss of PAX1 dysregulates proliferation of the thymus
(82). Loss of PAX1
expression, mediated by aberrant promoter methylation, has been
detected in multiple cancer types, including cervical, colorectal
and esophageal cancer, and in HNSCC (83,84).
It has been shown that PAX1 is aberrantly M in HSCCC,
including PAX1 aberrant methylation association with a higher risk
of HNSCC (85,86). PAX1 methylation has been
mostly studied in OSCC and associated with larger tumor size
(87–89).
PAX1 methylation has been studied in
HPV+ cervical cancer in which HPV infection disrupts
epigenetic regulation through a series of aberrant methylation
changes in the host genome (90).
Likewise, HPV-induced aberrant methylation may affect the
carcinogenic activity and clinical manifestation of HPV+
HNSCC and distinguish it from HPV− HNSCC. Currently in
Taiwan, M PAX1 is used for cervical cancer screening due to
its association with increasing cervical dysplasia. Likewise, the
association of M PAX1 with HPV infection in cervical cancer
suggests that HPV may regulate PAX1 methylation; thus, further
analysis of the significance of M PAX1 in HPV+
patients with HNSCC is warranted.
Among other candidate genes evaluated,
CALCA, a potent vasodilator, and an essential inflammatory
response molecule (91), was
predicted as particularly M in OPSCC. Detection of aberrantly M
CALCA in HNSCC showed to be highly accurate with 100%
specificity and 85% sensitivity (P=0.0006). M CALCA was
detected in 79% of the 19 OPSCC tumor samples, corroborating the
predictive analysis. Additional assessment in 50 HNSCC samples
showed that M CALCA was predominant in 48% of HNSCC samples
(P=0.036). M CALCA has been detected in leukemia,
testicular, bladder, non-small cell lung, thyroid, and head and
neck cancer (92–97). In leukemia, M CALCA has been
associated with disease relapse and poor prognosis (98,99).
Likewise, studies in non-small lung carcinoma reveal that M
CALCA was more common in squamous cell carcinomas than
adenocarcinomas (100).
Furthermore, M CALCA was associated with a poor prognosis in
non-small lung carcinoma independent of the tumor stage (101). Aberrant methylation of
CALCA in HNSCC has been studied mostly in oral cancer
samples. Guerrero-Preston et al (102) showed a higher frequency of M
CALCA in OSCC, although the prognostic value for HNSCC was
not discussed. An additional study showed that M CALCA was
associated with metastasis and a poorer prognosis in OSCC (103). Thus, the prognostic value of
CALCA in HNSCC demands further study, particularly in
OPSCC.
M TIMP3, a tissue inhibitor of
metalloproteinase 3, was significantly detected in HNSCC (76%;
P=0.003). The frequency of M TIMP3 was higher in OSCC (85%)
compared with other subsites and corroborated the genome-wide
analysis. Previous studies have shown M TIMP3 in HNSCC
(104) in addition to various
tumor types, including kidney, brain and esophageal cancer
(105,106). TIMP3 is a critical
regulator of inflammation (107,108), and loss of TIMP3 expression
has been associated with an increase in cell proliferation, tumor
growth, angiogenesis and metastasis (109–112). No significant association was
identified between M TIMP3 and HNSCC clinical features in
the cohort of the present study. Previous studies have detected M
TIMP3 more frequently in tumor tissue of patients with
early-stage (I/II) HNSCC compared with healthy saliva samples
(113,114) but they did not find a clinical
association with cancer. A study characterizing the epigenome in
HPV+ patients with HNSCC showed that aberrant
TIMP3 was predominant in HPV+ patients (115). The authors showed that M
TIMP3 was associated with positive lymph node spread, and
consequently, with a poorer prognosis. Therefore, further studies
of M TIMP3 in HPV+ and HPV− patients
with HNSCC are warranted.
The current study also assessed the aberrant
methylation of SFRP1, MAGI2, PITX2 and PAX5 in HNSCC.
In OSCC, M PAX5 was detected in 79.3% of tumor samples, and
M PITX2 was detected in 58.6% of OSCC samples. M
MAGI2 was detected in 64.1% of LSCCs, and aberrant M
SFRP1 was detected in 84.2% of oroOPSCC samples. The
predictive accuracy of SFRP1, MAGI2, PITX2 and PAX5
was significant, and it should be further explored in a larger
HNSCC cohort to assess their predictive value accurately.
Cox regression analysis of OS revealed that
HPV− patients with HNSCC were at a higher risk of dying
of cancer (P=0.006; HR, 9.72; 95% CI, 1.94–48.42) compared to
HPV+ patients with HNSCC. Previous studies have shown
that HPV+ tumors have higher methylation levels due to
an overexpression of DNMT3a (116–118). HPV+ tumors commonly
inactivate CDKN2A by hypermethylation, whereas
HPV− tumors mostly inactivate CDKN2A by deletions
or mutations (119). Likewise,
studies have shown that HPV− tumors have a global
hypomethylation state and higher genomic instability compared with
HPV+ tumors (120–122). It has been proposed that the
machinery of HPV+ cells re-establish developmental
methylation patterns as a defense mechanism to abolish
transcription of the virus (122),
which results in increased DNA methylation. Differences in DNA
methylation patterns between HPV+ and HPV−
HNSCC must be substantiated and analyzed for their potential
clinical translation into targeted treatment options.
In summary, the findings of the current study
suggest that distinctive aberrant DNA methylation profiles arise
within every head and neck cancer anatomic subsite, thus explaining
the disease heterogeneity and possible association with disease
progression or response to treatment. It was also shown that
DAPK1, CDH1, PAX1, CALCA and TIMP3 are frequently
aberrantly M in patients with head and neck cancer and influence
survival. PAX1 hypermethylation was different across HNSCC
anatomic subsites (P=0.029), and predominantly detected in
LSCC. Kaplan-Meier survival analysis (P=0.043) and Cox regression
analysis of OS (P=0.001) showed that DAPK1 methylation is
associated with better prognosis in HNSCC.
The main limitation of the current study was the
small sample size. Further studies with larger cohorts are needed
to validate the results obtained. Determination of the global
epigenetic landscape of HNSCC will require large cohorts of samples
and coordinated research efforts to predict better biomarkers for
disease outcome and targeted therapeutic interventions.
Furthermore, epigenomic studies in which HNSCC tumors are
stratified by anatomic site and HPV positivity are needed to better
understand the modulation of the host epigenome by HPV modifying,
and subsequent impact in disease progression and severity.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the University of
Puerto Rico School of Medicine Department of Surgery Otolaryngology
Section; NIH/National Cancer Institute (grant nos. P20CA91402,
U54CA96297, K01CA164092, R44CA254690, R44CA281719 and U01CA84986),
the NIH/National Institute of General Medical Sciences (grant no.
S06GM8224), the NIH/National Institute of Dental and Craniofacial
Research Award (grant no. RC2DE20957) and the NIH/National
Institute of Minority Health and Disparities (grant no.
R44MD014911). Bianca Rivera Peña's research was supported in part
by the NIGMS-RISE award (grant no. R25 GM061838). This research
used core facilities supported by NIH/NCRR and NIH/NIMHDD
awards.
Availability of data and materials
The data that support the findings of this study
are available from LifeGeneBiomarks, but restrictions apply to the
availability of these data, which were used under license for the
current study, and so are not publicly available. Data are however
available from the authors upon reasonable request and with
permission of LifeGeneBiomarks.
Authors' contributions
BRP, RGP, DS and AB performed the study design. AB
and JAAO obtained informed consent from patients with HNSCC and
healthy individuals. BRP and AB collected all HNSCC tumor and
healthy oral samples. BRP, RV, JAAO and RL carried out DNA
extraction from HNSCC and healthy samples. BRP, OF and FP carried
out the MeDIP assay, hybridization and scanning of samples to the
microarray. BRP, NT and RGP performed bioinformatics analysis. RJRB
and MEF analyzed the data. BRP, NT, SRT and RGP performed GO
analysis, TCGA and Cancer Genome Browser analysis. DS financially
supported this study and revised and approved final manuscript.
BRP, DS and RGP confirm the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures described in the present study were
approved by the University of Puerto Rico-Medical Sciences Campus
IRB (approval no. MSC-IRB Protocol 2770103). Written informed
consent was obtained from all study participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
HPV16
|
human papillomavirus 16
|
|
LSCC
|
larynx squamous cell carcinoma
|
|
OSCC
|
oral cavity squamous cell
carcinoma
|
|
OPSCC
|
oropharynx squamous cell
carcinoma
|
|
DMRs
|
differentially methylated regions
|
|
OS
|
overall survival
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
TSGs
|
tumor suppressor genes
|
|
MeDIP
|
methylated DNA
immunoprecipitation
|
|
TSS
|
transcription starting site
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GO
|
Gene Ontology
|
|
IP DNA
|
immunoprecipitated DNA
|
|
WGA
|
whole genome amplification
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
O'Rorke MA, Ellison MV, Murray LJ, Moran
M, James J and Anderson LA: Human papillomavirus related head and
neck cancer survival: A systematic review and meta-analysis. Oral
Oncol. 48:1191–1201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Báez A: Genetic and environmental factors
in head and neck cancer genesis. J Environ Sci Health C Environ
Carcinog Ecotoxicol Rev. 26:174–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saracci R: The interactions of tobacco
smoking and other agents in cancer etiology. Epidemiol Rev.
9:175–193. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blot WJ, McLaughlin JK, Winn DM, Austin
DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB,
Stemhagen A and Fraumeni JF Jr: Smoking and drinking in relation to
oral and pharyngeal cancer. Cancer Res. 48:3282–3287.
1988.PubMed/NCBI
|
|
6
|
Hashibe M, Brennan P, Benhamou S,
Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova
E, Fernandez L, et al: Alcohol drinking in never users of tobacco,
cigarette smoking in never drinkers, and the risk of head and neck
cancer: Pooled analysis in the International Head and Neck Cancer
Epidemiology Consortium. J Natl Cancer Inst. 99:777–789. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gillison ML, Koch WM, Capone RB, Spafford
M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, et
al: Evidence for a causal association between human papillomavirus
and a subset of head and neck cancers. J Natl Cancer Inst.
92:709–720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mork J, Lie AK, Glattre E, Hallmans G,
Jellum E, Koskela P, Møller B, Pukkala E, Schiller JT, Youngman L,
et al: Human papillomavirus infection as a risk factor for
squamous-cell carcinoma of the head and neck. N Engl J Med.
344:1125–1131. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Báez A, Almodóvar JI, Cantor A, Celestin
F, Cruz-Cruz L, Fonseca S, Trinidad-Pinedo J and Vega W: High
frequency of HPV16-associated head and neck squamous cell carcinoma
in the Puerto Rican population. Head Neck. 26:778–784. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rivera-Peña B, Ruíz-Fullana FJ,
Vélez-Reyes GL, Rodriguez-Benitez RJ, Marcos-Martínez MJ,
Trinidad-Pinedo J and Báez A: HPV-16 infection modifies overall
survival of Puerto Rican HNSCC patients. Infect Agent Cancer.
11:472004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park A, Alabaster A, Shen H, Mell LK and
Katzel JA: Undertreatment of women with locoregionally advanced
head and neck cancer. Cancer. 125:3033–3039. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Katzel JA, Merchant M, Chaturvedi AK and
Silverberg MJ: Contribution of demographic and behavioral factors
on the changing incidence rates of oropharyngeal and oral cavity
cancers in northern California. Cancer Epidemiol Biomarkers Prev.
24:978–984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dayyani F, Etzel CJ, Liu M, Ho CH, Lippman
SM and Tsao AS: Meta-analysis of the impact of human papillomavirus
(HPV) on cancer risk and overall survival in head and neck squamous
cell carcinomas (HNSCC). Head Neck Oncol. 2:152010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shiboski CH, Schmidt BL and Jordan RC:
Tongue and tonsil carcinoma: Increasing trends in the U.S.
population ages 20–44 years. Cancer. 103:1843–1849. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
American Joint Committee on Cancer, . AJCC
Cancer Staging Manual. 8. New York; Springer: 2016
|
|
17
|
Baliga S, Yildiz VO, Bazan J, Palmer JD,
Jhawar SR, Konieczkowski DJ, Grecula J, Blakaj DM, Mitchell D,
Henson C, et al: Disparities in survival outcomes among
Racial/Ethnic minorities with head and neck squamous cell cancer in
the united states. Cancers (Basel). 15:17812023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suárez E, Calo WA, Hernández EY, Diaz EC,
Figueroa NR and Ortiz AP: Age-standardized incidence and mortality
rates of oral and pharyngeal cancer in Puerto Rico and among
Non-Hispanics Whites, Non-Hispanic Blacks, and Hispanics in the
USA. BMC Cancer. 9:1292009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fernandez AF, Assenov Y, Martin-Subero JI,
Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC,
Galm O, et al: A DNA methylation fingerprint of 1628 human samples.
Genome Res. 22:407–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Taby R and Issa JP: Cancer epigenetics. CA
Cancer J Clin. 60:376–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ibáñez de Cáceres I and Cairns P:
Methylated DNA sequences for early cancer detection, molecular
classification and chemotherapy response prediction. Clin Transl
Oncol. 9:429–437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Calmon MF, Colombo J, Carvalho F, Souza
FP, Filho JF, Fukuyama EE, Camargo AA, Caballero OL, Tajara EH,
Cordeiro JA and Rahal P: Methylation profile of genes CDKN2A (p14
and p16), DAPK1, CDH1, and ADAM23 in head and neck cancer. Cancer
Genet Cytogenet. 173:31–37. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Demokan S and Dalay N: Role of DNA
methylation in head and neck cancer. Clin Epigenetics. 2:123–150.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ovchinnikov DA, Cooper MA, Pandit P, Coman
WB, Cooper-White JJ, Keith P, Wolvetang EJ, Slowey PD and
Punyadeera C: Tumor-suppressor gene promoter hypermethylation in
saliva of head and neck cancer patients. Transl Oncol. 5:321–326.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lleras RA, Smith RV, Adrien LR, Schlecht
NF, Burk RD, Harris TM, Childs G, Prystowsky MB and Belbin TJ:
Unique DNA methylation loci distinguish anatomic site and HPV
status in head and neck squamous cell carcinoma. Clin Cancer Res.
19:5444–5455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bernabe RD: INK4a/ARF/INK4b tumor
suppressor locus: Its potential role in head and neck cancer
tumorigenesis (Doctoral dissertation) (order no. 3221909). ProQuest
Dissertations and Theses Global. 2006.Available from. https://search.proquest.com/docview/304984294?accountid=44820
|
|
29
|
Rivera-Peña B and Báez A: Abstract 4793:
Aberrant methylation of CDH1 correlates with poor prognosis in
patients with head and neck squamous cell carcinoma (abstract). In:
Proceedings of the 102nd Annual Meeting of the American Association
for Cancer Research. Cancer Res. 71 (Suppl 8):4793. 2011.
View Article : Google Scholar
|
|
30
|
Irizarry RA, Ladd-Acosta C, Carvalho B, Wu
H, Brandenburg SA, Jeddeloh JA, Wen B and Feinberg AP:
Comprehensive high-throughput arrays for relative methylation
(CHARM). Genome Res. 18:780–790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jaffe AE, Murakami P, Lee H, Leek JT,
Fallin MD, Feinberg AP and Irizarry RA: Bump hunting to identify
differentially methylated regions in epigenetic epidemiology
studies. Int J Epidemiol. 41:200–209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du P, Zhang X, Huang CC, Jafari N, Kibbe
WA, Hou L and Lin SM: Comparison of Beta-value and M-value methods
for quantifying methylation levels by microarray analysis. BMC
Bioinformatics. 11:5872010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
The Cancer Genome Atlas Network, .
Comprehensive genomic characterization of head and neck squamous
cell carcinomas. Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aryee MJ, Jaffe AE, Corrada-Bravo H,
Ladd-Acosta C, Feinberg AP, Hansen KD and Irizarry RA: Minfi: A
flexible and comprehensive Bioconductor package for the analysis of
Infinium DNA methylation microarrays. Bioinformatics. 30:1363–1369.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eads CA, Danenberg KD, Kawakami K, Saltz
LB, Blake C, Shibata D, Danenberg PV and Laird PW: MethyLight: A
high-throughput assay to measure DNA methylation. Nucleic Acids
Res. 28:E322000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rettori MM, de Carvalho AC, Longo AL, de
Oliveira CZ, Kowalski LP, Carvalho AL and Vettore AL: TIMP3 and
CCNA1 hypermethylation in HNSCC is associated with an increased
incidence of second primary tumors. J Transl Med. 11:3162013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Valle B, Rodriguez-Torres S, Kuhn E,
Díaz-Montes T, Parrilla-Castellar E, Lawson F, Folawiyo O,
Ili-Gangas C, Brebi-Mieville P, Eshleman J, et al: HIST1H2BB and
MAGI2 methylation and somatic mutations as precision medicine
biomarkers for diagnosis and prognosis of high-grade serous ovarian
cancer. Cancer Prev Res (Phila). 13:783–794. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Guerrero-Preston R, Valle BL, Jedlicka A,
Turaga N, Folawiyo O, Pirini F, Lawson F, Vergura A, Noordhuis M,
Dziedzic A, et al: Molecular triage of premalignant lesions in
Liquid-Based cervical cytology and circulating Cell-Free DNA from
urine, using a panel of methylated human papilloma virus and host
genes. Cancer Prev Res (Phila). 9:915–924. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guerrero-Preston R, Michailidi C,
Marchionni L, Pickering CR, Frederick MJ, Myers JN,
Yegnasubramanian S, Hadar T, Noordhuis MG, Zizkova V, et al: Key
tumor suppressor genes inactivated by ‘greater promoter’
methylation and somatic mutations in head and neck cancer.
Epigenetics. 9:1031–1046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Anglim PP, Galler JS, Koss MN, Hagen JA,
Turla S, Campan M, Weisenberger DJ, Laird PW, Siegmund KD and
Laird-Offringa IA: Identification of a panel of sensitive and
specific DNA methylation markers for squamous cell lung cancer. Mol
Cancer. 7:622008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Müller HM, Widschwendter A, Fiegl H,
Ivarsson L, Goebel G, Perkmann E, Marth C and Widschwendter M: DNA
methylation in serum of breast cancer patients: An independent
prognostic marker. Cancer Res. 63:7641–7665. 2003.PubMed/NCBI
|
|
43
|
Eads CA, Lord RV, Wickramasinghe K, Long
TI, Kurumboor SK, Bernstein L, Peters JH, DeMeester SR, DeMeester
TR, Skinner KA and Laird PW: Epigenetic patterns in the progression
of esophageal adenocarcinoma. Cancer Res. 61:3410–3418.
2001.PubMed/NCBI
|
|
44
|
van Asperen CJ, Brohet RM,
Meijers-Heijboer EJ, Hoogerbrugge N, Verhoef S, Vasen HF, Ausems
MG, Menko FH, Gomez Garcia EB, Klijn JG, et al: Cancer risks in
BRCA2 families: Estimates for sites other than breast and ovary. J
Med Genet. 42:711–719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Seiwert TY, Zuo Z, Keck MK, Khattri A,
Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, et
al: Integrative and comparative genomic analysis of HPV-positive
and HPV-negative head and neck squamous cell carcinomas. Clin
Cancer Res. 21:632–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Califano J, Van Der Riet P, Westra W,
Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B,
Koch W and Sidransky D: Genetic progression model for head and neck
cancer: Implications for field cancerization. Cancer Res.
56:2488–2492. 1996.PubMed/NCBI
|
|
47
|
Cai F, Xiao X, Niu X and Zhong Y:
Association between promoter methylation of DAPK gene and HNSCC: A
meta-analysis. PLoS One. 12:e01731942017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ngan HL, Liu Y, Fong AY, Poon PHY, Yeung
CK, Chan SSM, Lau A, Piao W, Li H, Tse J, et al: MAPK pathway
mutations in head and neck cancer affect immune microenvironments
and ErbB3 signaling. Life Sci Alliance. 3:e2019005452020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cilona M, Locatello LG, Novelli L and
Gallo O: The mismatch repair system (MMR) in head and neck
carcinogenesis and its role in modulating the response to
immunotherapy: A critical review. Cancers (Basel). 12:30062020.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Meng RW, Li YC, Chen X, Huang YX, Shi H,
Du DD, Niu X, Lu C and Lu MX: Aberrant Methylation of RASSF1A
closely associated with HNSCC, a Meta-Analysis. Sci Rep.
6:207562016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moon SM, Kim SA, Yoon JH and Ahn SG: HOXC6
is deregulated in human head and neck squamous cell carcinoma and
modulates Bcl-2 expression. J Biol Chem. 287:35678–35688. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Carla C, Daris F, Cecilia B, Francesca B,
Francesca C and Paolo F: Angiogenesis in head and neck cancer: A
review of the literature. J Oncol. 2012:3584722012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mineta H, Miura K, Ogino T, Takebayashi S,
Misawa K, Ueda Y, Suzuki I, Dictor M, Borg A and Wennerberg J:
Prognostic value of vascular endothelial growth factor (VEGF) in
head and neck squamous cell carcinomas. Br J Cancer. 83:775–781.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Iwai S, Katagiri W, Kong C, Amekawa S,
Nakazawa M and Yura Y: Mutations of the APC, beta-catenin, and axin
1 genes and cytoplasmic accumulation of beta-catenin in oral
squamous cell carcinoma. J Cancer Res Clin Oncol. 131:773–782.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Leethanakul C, Patel V, Gillespie J,
Pallente M, Ensley JF, Koontongkaew S, Liotta LA, Emmert-Buck M and
Gutkind JS: Distinct pattern of expression of differentiation and
growthrelated genes in squamous cell carcinomas of the head and
neck revealed by the use of laser capture microdissection and cDNA
arrays. Oncogene. 19:3220–3224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Paska AV and Hudler P: Aberrant
methylation patterns in cancer: A clinical view. Biochem Med
(Zagreb). 25:161–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hao X, Luo H, Krawczyk M, Wei W, Wang W,
Wang J, Flagg K, Hou J, Zhang H, Yi S, et al: DNA methylation
markers for diagnosis and prognosis of common cancers. Proc Natl
Acad Sci USA. 114:7414–7419. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen D, Wang M, Guo Y, Wu W, Ji X, Dou X,
Tang H, Zong Z, Zhang X and Xiong D: An aberrant DNA methylation
signature for predicting the prognosis of head and neck squamous
cell carcinoma. Cancer Med. 10:5936–5947. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stadler ME, Patel MR, Couch ME and Hayes
DN: Molecular biology of head and neck cancer: Risks and pathways.
Hematol Oncol Clin North Am. 22:1099–1124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bialik S and Kimchi A: The
death-associated protein kinases: Structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gade P, Manjegowda SB, Nallar SC, Maachani
UB, Cross AS and Kalvakolanu DV: Regulation of the death-associated
protein kinase 1 expression and autophagy via ATF6 requires
apoptosis signal-regulating kinase 1. Mol Cell Biol. 34:4033–448.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li C, Wang L, Su J, Zhang R, Fu L and Zhou
Y: mRNA expression and hypermethylation of tumor suppressor genes
apoptosis protease activating factor-1 and death-associated protein
kinase in oral squamous cell carcinoma. Oncol Lett. 6:280–286.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jayaprakash C, Varghese VK, Bellampalli R,
Radhakrishnan R, Ray S, Kabekkodu SP and Satyamoorthy K:
Hypermethylation of Death-associated protein kinase (DAPK1) and its
association with oral carcinogenesis-An experimental and
meta-analysis study. Arch Oral Biol. 80:117–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Melchers LJ, Clausen MJ, Mastik MF,
Slagter-Menkema L, van der Wal JE, Wisman GB, Roodenburg JL and
Schuuring E: Identification of methylation markers for the
prediction of nodal metastasis in oral and oropharyngeal squamous
cell carcinoma. Epigenetics. 10:850–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wei DM, Liu DY, Lei DP, Jin T, Wang J and
Pan XL: Aberrant methylation and expression of DAPk1 in human
hypopharyngeal squamous cell carcinoma. Acta Otolaryngol.
135:70–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
van Kempen PM, van Bockel L, Braunius WW,
Moelans CB, van Olst M, de Jong R, Stegeman I, van Diest PJ,
Grolman W and Willems SM: HPV-positive oropharyngeal squamous cell
carcinoma is associated with TIMP3 and CADM1 promoter
hypermethylation. Cancer Med. 3:1185–1196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Stephen JK, Chen KM, Shah V, Havard S,
Kapke A, Lu M, Benninger MS and Worsham MJ: DNA hypermethylation
markers of poor outcome in laryngeal cancer. Clin Epigenetics.
1:61–69. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Strzelczyk JK, Krakowczyk Ł and Owczarek
AJ: Aberrant DNA methylation of the p16, APC, MGMT, TIMP3 and CDH1
gene promoters in tumours and the surgical margins of patients with
oral cavity cancer. J Cancer. 9:1896–1904. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Schmezer P and Plass C: Epigenetic aspects
in carcinomas of the head and neck. HNO. 56:594–602. 2008.(In
German). View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Singh P, Ravanan P and Talwar P: Death
associated protein kinase 1 (DAPK1): A regulator of apoptosis and
autophagy. Front Mol Neurosci. 9:462016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Humphries MJ and Newham P: The structure
of cell-adhesion molecules. Trends Cell Biol. 8:78–83. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Riethmacher D, Brinkmann V and Birchmeier
C: A targeted mutation in the mouse E-cadherin gene results in
defective preimplantation development. Proc Natl Acad Sci USA.
92:855–859. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pećina-Slaus N: Tumor suppressor gene
E-cadherin and its role in normal and malignant cells. Cancer Cell
Int. 3:172003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Starska K, Forma E, Lewy-Trenda I, Papież
P, Woś J and Bryś M: Diagnostic impact of promoter methylation and
E-cadherin gene and protein expression levels in laryngeal
carcinoma. Contemp Oncol (Pozn). 17:263–271. 2013.PubMed/NCBI
|
|
75
|
Fan CC, Wang TY, Cheng YA, Jiang SS, Cheng
CW, Lee AY and Kao TY: Expression of E-cadherin, Twist, and p53 and
their prognostic value in patients with oral squamous cell
carcinoma. J Cancer Res Clin Oncol. 139:1735–1744. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Fujii R, Imanishi Y, Shibata K, Sakai N,
Sakamoto K, Shigetomi S, Habu N, Otsuka K, Sato Y, Watanabe Y, et
al: Restoration of E-cadherin expression by selective Cox-2
inhibition and the clinical relevance of the
epithelial-to-mesenchymal transition in head and neck squamous cell
carcinoma. J Exp Clin Cancer Res. 33:402014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shen Z, Zhou C, Li J, Deng H, Li Q and
Wang J: The association, clinicopathological significance, and
diagnostic value of CDH1 promoter methylation in head and neck
squamous cell carcinoma: A meta-analysis of 23 studies. Onco
Targets Ther. 9:6763–6773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
McGaughran JM, Oates A, Donnai D, Read AP
and Tassabehji M: Mutations in PAX1 may be associated with
Klippel-Feil syndrome. Eur J Hum Genet. 11:468–474. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wallin J, Eibel H, Neubüser A, Wilting J,
Koseki H and Balling R: Pax1 is expressed during development of the
thymus epithelium and is required for normal T-cell maturation.
Development. 122:23–30. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Xu J, Xu L, Yang B, Wang L, Lin X and Tu
H: Assessing methylation status of PAX1 in cervical scrapings, as a
novel diagnostic and predictive biomarker, was closely related to
screen cervical cancer. Int J Clin Exp Pathol. 8:1674–1681.
2015.PubMed/NCBI
|
|
81
|
Dietrich S and Gruss P: Undulated
phenotypes suggest a role of Pax-1 for the development of vertebral
and extravertebral structures. Dev Biol. 167:529–548. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Su D, Ellis S, Napier A, Lee K and Manley
NR: Hoxa3 and pax1 regulate epithelial cell death and proliferation
during thymus and parathyroid organogenesis. Dev Biol. 236:316–329.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lorincz AT: Virtues and Weaknesses of DNA
methylation as a test for cervical cancer prevention. Acta Cytol.
60:501–512. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Juodzbalys G, Kasradze D, Cicciù M,
Sudeikis A, Banys L, Galindo-Moreno P and Guobis Z: Modern
molecular biomarkers of head and neck cancer. Part I. Epigenetic
diagnostics and prognostics: Systematic review. Cancer Biomark.
17:487–502. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Huang YK, Peng BY, Wu CY, Su CT, Wang HC
and Lai HC: DNA methylation of PAX1 as a biomarker for oral
squamous cell carcinoma. Clin Oral Investig. 18:801–808. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sun R, Juan YC, Su YF, Zhang WB, Yu Y,
Yang HY, Yu GY and Peng X: Hypermethylated PAX1 and ZNF582 genes in
the tissue sample are associated with aggressive progression of
oral squamous cell carcinoma. J Oral Pathol Med. 49:751–760. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cheng SJ, Chang CF, Ko HH, Lee JJ, Chen
HM, Wang HJ, Lin HS and Chiang CP: Hypermethylated ZNF582 and PAX1
genes in mouth rinse samples as biomarkers for oral dysplasia and
oral cancer detection. Head Neck. 40:355–368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Morandi L, Gissi D, Tarsitano A, Asioli S,
Gabusi A, Marchetti C, Montebugnoli L and Foschini MP: CpG location
and methylation level are crucial factors for the early detection
of oral squamous cell carcinoma in brushing samples using bisulfite
sequencing of a 13-gene panel. Clin Epigenetics. 9:852017.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cheng SJ, Chang CF, Ko HH, Liu YC, Peng
HH, Wang HJ, Lin HS and Chiang CP: Hypermethylated ZNF582 and PAX1
genes in oral scrapings collected from cancer-adjacent normal oral
mucosal sites are associated with aggressive progression and poor
prognosis of oral cancer. Oral Oncol. 75:169–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hsu YW, Huang RL, Su PH, Chen YC, Wang HC,
Liao CC and Lai HC: Genotype-specific methylation of HPV in
cervical intraepithelial neoplasia. J Gynecol Oncol. 28:e562017.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Brain SD, Williams TJ, Tippins JR, Morris
HR and MacIntyre I: Calcitonin gene-related peptide is a potent
vasodilator. Nature. 313:54–56. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Vidal DO, Paixão VA, Brait M, Souto EX,
Caballero OL, Lopes LF and Vettore AL: Aberrant methylation in
pediatric myelodysplastic syndrome. Leuk Res. 31:175–181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Martinelli CMDS, Lengert AVH, Cárcano FM,
Silva ECA, Brait M, Lopes LF and Vidal DO: MGMT and CALCA promoter
methylation are associated with poor prognosis in testicular germ
cell tumor patients. Oncotarget. 8:50608–50617. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Brait M, Begum S, Carvalho AL, Dasgupta S,
Vettore AL, Czerniak B, Caballero OL, Westra WH, Sidransky D and
Hoque MO: Aberrant promoter methylation of multiple genes during
pathogenesis of bladder cancer. Cancer Epidemiol Biomarkers Prev.
17:2786–2794. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang Y, Zhang D, Zheng W, Luo J, Bai Y and
Lu Z: Multiple gene methylation of nonsmall cell lung cancers
evaluated with 3-dimensional microarray. Cancer. 112:1325–1336.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang B, Liu S, Zhang Z, Wei J, Qu Y, Wu
K, Yang Q, Hou P and Shi B: Analysis of BRAF(V600E) mutation and
DNA methylation improves the diagnostics of thyroid fine needle
aspiration biopsies. Diagn Pathol. 9:452014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Loyo M, Brait M, Kim MS, Ostrow KL, Jie
CC, Chuang AY, Califano JA, Liégeois NJ, Begum S, Westra WH, et al:
Asurvey of methylated candidate tumor suppressor genes in
nasopharyngeal carcinoma. Int J Cancer. 128:1393–1403. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ismail EA, El-Mogy MI, Mohamed DS and
El-Farrash RA: Methylation pattern of calcitonin (CALCA) gene in
pediatric acute leukemia. J Pediatr Hematol Oncol. 33:534–542.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Paixão VA, Vidal DO, Caballero OL, Vettore
AL, Tone LG, Ribeiro KB and Lopes LF: Hypermethylation of CpG
island in the promoter region of CALCA in acute lymphoblastic
leukemia with central nervous system (CNS) infiltration correlates
with poorer prognosis. Leuk Res. 30:891–894. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ji M, Guan H, Gao C, Shi B and Hou P:
Highly frequent promoter methylation and PIK3CA amplification in
non-small cell lung cancer (NSCLC). BMC Cancer. 11:1472011.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Morán A, Fernández-Marcelo T, Carro J, De
Juan C, Pascua I, Head J, Gómez A, Hernando F, Torres AJ, Benito M
and Iniesta P: Methylation profiling in non-small cell lung cancer:
Clinical implications. Int J Oncol. 40:739–746. 2012.PubMed/NCBI
|
|
102
|
Guerrero-Preston R, Soudry E, Acero J,
Orera M, Moreno-López L, Macía-Colón G, Jaffe A, Berdasco M,
Ili-Gangas C, Brebi-Mieville P, et al: NID2 and HOXA9 promoter
hypermethylation as biomarkers for prevention and early detection
in oral cavity squamous cell carcinoma tissues and saliva. Cancer
Prev Res (Phila). 4:1061–1072. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jithesh PV, Risk JM, Schache AG, Dhanda J,
Lane B, Liloglou T and Shaw RJ: The epigenetic landscape of oral
squamous cell carcinoma. Br J Cancer. 108:370–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
105
|
Bachman KE, Herman JG, Corn PG, Merlo A,
Costello JF, Cavenee WK, Baylin SB and Graff JR:
Methylation-associated silencing of the tissue inhibitor of
metalloproteinase-3 gene suggest a suppressor role in kidney,
brain, and other human cancers. Cancer Res. 59:798–802.
1999.PubMed/NCBI
|
|
106
|
Darnton SJ, Hardie LJ, Muc RS, Wild CP and
Casson AG: Tissue inhibitor of metalloproteinase-3 (TIMP-3) gene is
methylated in the development of esophageal adenocarcinoma: Loss of
expression correlates with poor prognosis. Int J Cancer.
115:351–358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Smookler DS, Mohammed FF, Kassiri Z,
Duncan GS, Mak TW and Khokha R: Tissue inhibitor of
metalloproteinase 3 regulates TNF-dependent systemic inflammation.
J Immunol. 176:721–725. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Mohammed FF, Smookler DS, Taylor SE,
Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B,
Yeh WC and Khokha R: Abnormal TNF activity in Timp3-/-mice leads to
chronic hepatic inflammation and failure of liver regeneration. Nat
Genet. 36:969–977. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhang L, Zhao L, Zhao D, Lin G, Guo B, Li
Y, Liang Z, Zhao XJ and Fang X: Inhibition of tumor growth and
induction of apoptosis in prostate cancer cell lines by
overexpression of tissue inhibitor of matrix metalloproteinase-3.
Cancer Gene Ther. 17:171–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ahonen M, Ala-Aho R, Baker AH, George SJ,
Grénman R, Saarialho-Kere U and Kähäri VM: Antitumor activity and
bystander effect of adenovirally delivered tissue inhibitor of
metalloproteinases-3. Mol Ther. 5:705–715. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Spurbeck WW, Ng CY, Strom TS, Vanin EF and
Davidoff AM: Enforced expression of tissue inhibitor of matrix
metalloproteinase-3 affects functional capillary morphogenesis and
inhibits tumor growth in a murine tumor model. Blood.
100:3361–3368. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Bian J, Wang Y, Smith MR, Kim H, Jacobs C,
Jackman J, Kung HF, Colburn NH and Sun Y: Suppression of in vivo
tumor growth and induction of suspension cell death by tissue
inhibitor of metalloproteinases (TIMP)-3. Carcinogenesis.
17:1805–1811. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Arantes LM, de Carvalho AC, Melendez ME,
Centrone CC, Góis-Filho JF, Toporcov TN, Caly DN, Tajara EH,
Goloni-Bertollo EM and Carvalho AL; GENCAPO: Validation of
methylation markers for diagnosis of oral cavity cancer. Eur J
Cancer. 51:632–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Righini CA, de Fraipont F, Timsit JF,
Faure C, Brambilla E, Reyt E and Favrot MC: Tumor-specific
methylation in saliva: A promising biomarker for early detection of
head and neck cancer recurrence. Clin Cancer Res. 13:1179–1185.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhang HZ, Shan CG, Huang AP and Wang JM:
Characterization of gene methylation in human papillomavirus
associated-head and neck squamous cell carcinoma. Genet Mol Res.
152016.doi: 10.4238/gmr.15038206.
|
|
116
|
Sartor MA, Dolinoy DC, Jones TR, Colacino
JA, Prince ME, Carey TE and Rozek LS: Genome-wide methylation and
expression differences in HPV(+) and HPV(−) squamous cell carcinoma
cell lines are consistent with divergent mechanisms of
carcinogenesis. Epigenetics. 6:777–787. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Nakagawa T, Matsusaka K, Misawa K, Ota S,
Takane K, Fukuyo M, Rahmutulla B, Shinohara KI, Kunii N, Sakurai D,
et al: Frequent promoter hypermethylation associated with human
papillomavirus infection in pharyngeal cancer. Cancer Lett.
407:21–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Nakagawa T, Kurokawa T, Mima M, Imamoto S,
Mizokami H, Kondo S, Okamoto Y, Misawa K, Hanazawa T and Kaneda A:
DNA Methylation and HPV-Associated Head and Neck Cancer.
Microorganisms. 9:8012021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nakahara Y, Shintani S, Mihara M, Ueyama Y
and Matsumura T: High frequency of homozygous deletion and
methylation of p16(INK4A) gene in oral squamous cell carcinomas.
Cancer Lett. 163:221–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Richards KL, Zhang B, Baggerly KA, Colella
S, Lang JC, Schuller DE and Krahe R: Genome-wide hypomethylation in
head and neck cancer is more pronounced in HPV-negative tumors and
is associated with genomic instability. PLoS One. 4:e49412009.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
van Kempen PM, Noorlag R, Braunius WW,
Stegeman I, Willems SM and Grolman W: Differences in methylation
profiles between HPV-positive and HPV-negative oropharynx squamous
cell carcinoma: A systematic review. Epigenetics. 9:194–203. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Camuzi D, Buexm LA, Lourenço SQC, Esposti
DD, Cuenin C, Lopes MdSA, Manara F, Talukdar FR, Herceg Z, Ribeiro
Pinto LF, et al: HPV infection leaves a DNA methylation signature
in oropharyngeal cancer affecting both coding genes and
transposable elements. Cancers. 13:36212021. View Article : Google Scholar : PubMed/NCBI
|