Introduction
Myelodysplastic syndrome (MDS) is a category of
malignant hematological clonal system disorders of hematopoietic
stem cells with heterogenous etiologies, characterized by
ineffective bone marrow hematopoiesis, significant morphological
dysplasia in one or more hematopoietic lineages or cytogenetic
abnormality (1). The etiology of
this disease is unknown and may be related to genetic, infectious
or immune factors (2). In
individuals aged ≥60 years, the prevalence was 7–35 cases per
100,000 individuals over the last two decades (3). MDS can be categorized into subtypes
that are associated with lower or higher risk for acute myeloid
leukemia transformation, which assists with therapy selection.
Management focuses on treating symptoms and reducing the number of
required transfusions in patients with low-risk disease. For those
with higher-risk MDS, hypomethylating agents such as azacitidine,
or decitabine, are first-line therapy. Hematopoietic cell
transplantation is considered for higher-risk patients and
represents the only potential cure (3). IgG4-related disease (IgG4-RD) is an
immune-mediated progressive inflammatory disease associated with
fibrosis (4), a specific organ
predisposition involving the submandibular glands, parotid glands,
lymph nodes, liver, and biliary tract (5). The true prevalence of IgG4-RD is
unknown, and it is likely that it is underrecognized and
underreported due to its relatively recent discovery, lack of
widespread recognition and frequently indolent presentation
(6). The separate incidence rate of
MDS and IgG4-RD is high in the elderly population, but the
coexistence of the two diseases is rare (7), and effective treatment of the
simultaneous comorbidity of both diseases is unclear (8,9).
Case report
Patient history
A 66-year-old male attended Zhejiang Provincial
Hospital of Chinese Medicine (Hangzhou, China) in September 2022
with ‘recurrent episodes of weakness and dizziness for 2 years’.
The patient felt dizzy and weak, along with leukopenia, severe
anemia and thrombocytopenia (Table
I). Bone marrow aspirations performed at the First Hospital of
Chun'an County (Chun'an Branch of Zhejiang Provincial People's
Hospital; Zhejiang, China) in June 2020 showed strong hematopoietic
function, with impaired maturation of leukocytes and
megakaryocytes, and active erythropoiesis. The patient received
oral folic acid (5 mg, 3 times a day) and methylcobalamin (0.5 mg,
3 times a day) to replenish hematopoietic raw materials and relieve
dizziness. The patient was transfused with B-type platelets when
severely lethargic. In September 2022, the patient came to Zhejiang
Provincial Hospital of Chinese Medicine with worsening fatigue and
acute pruritus of both legs. The general condition and the results
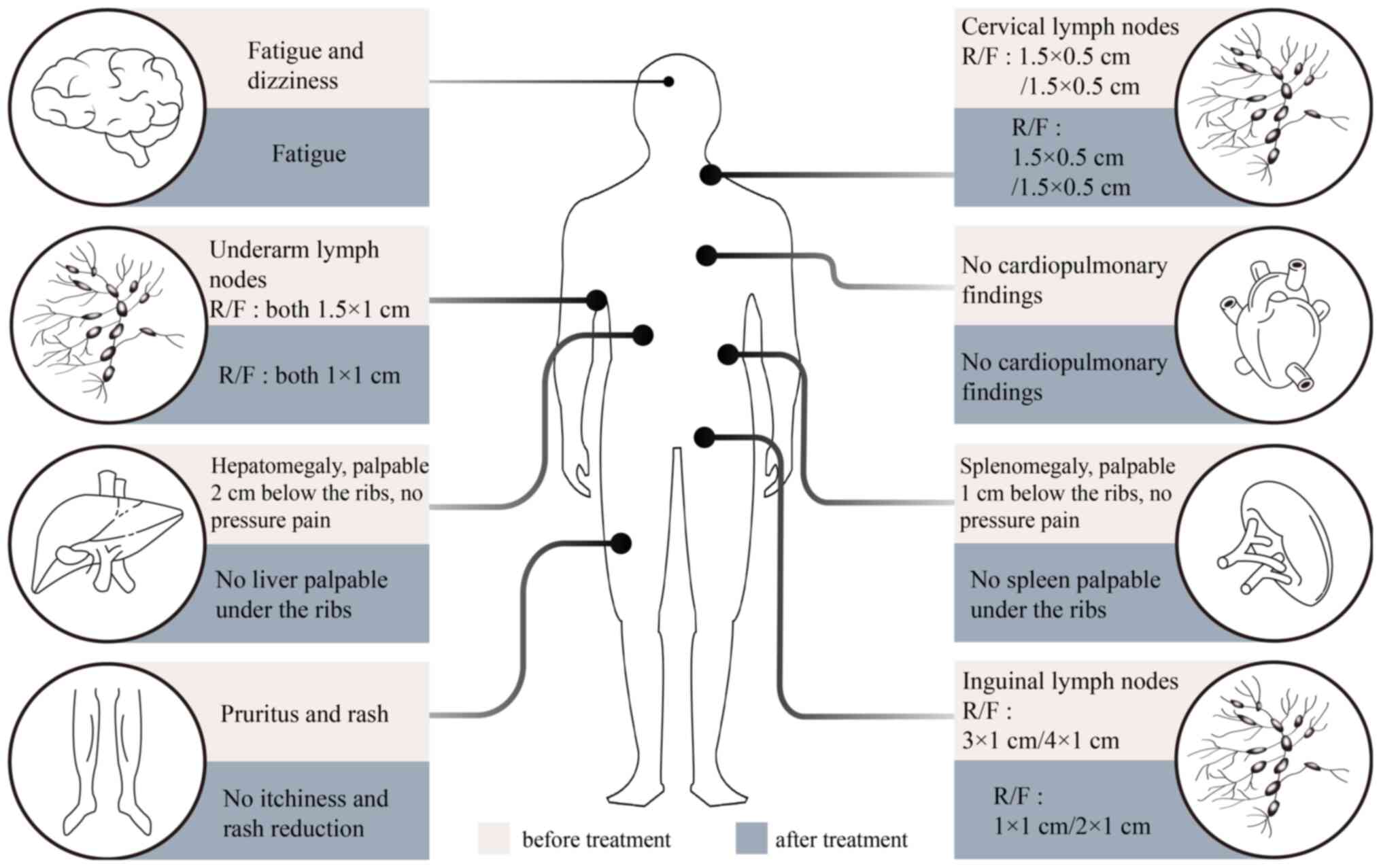
of conventional investigations are detailed in Fig. 1, Fig.
2 and the following descriptive record.
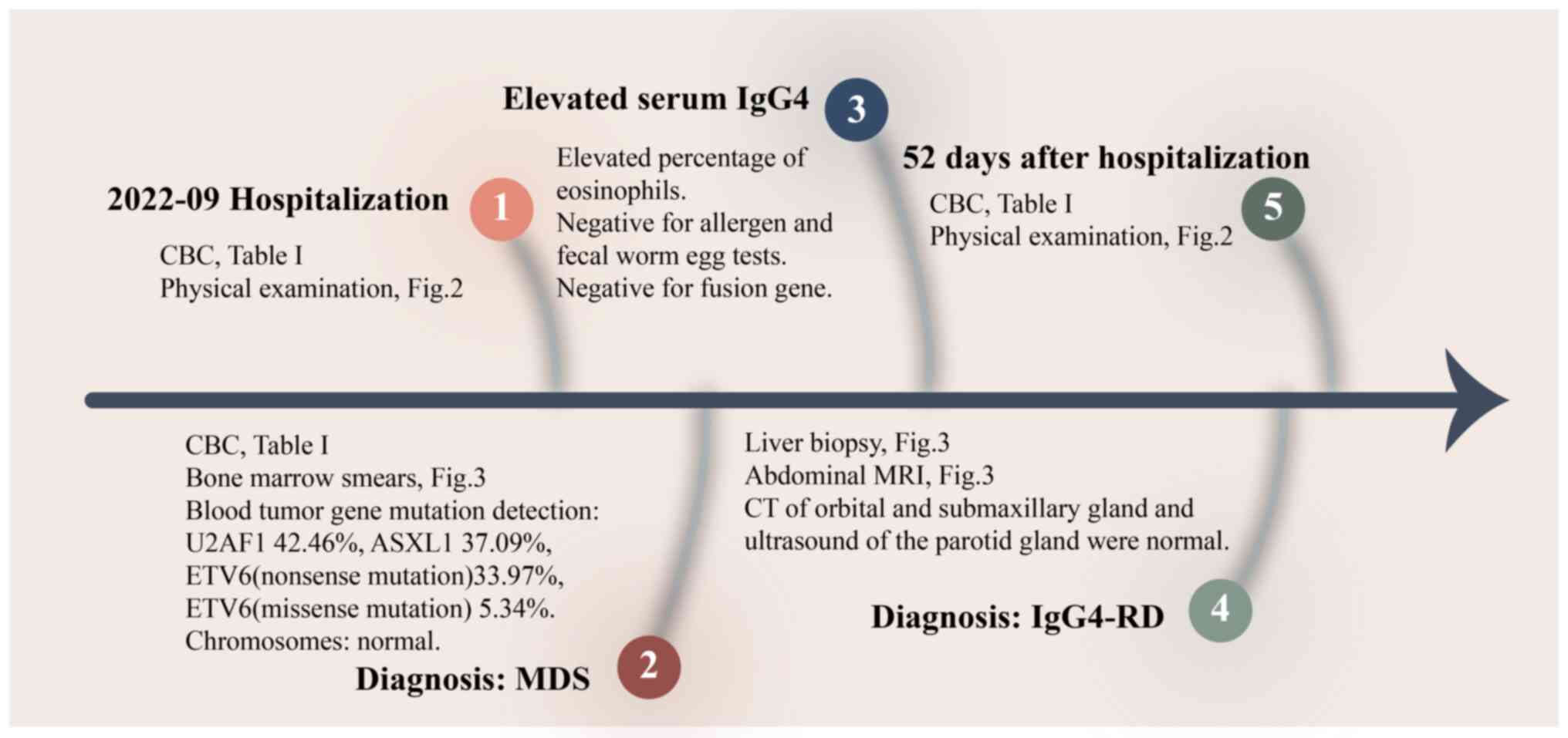 | Figure 1.Flow chart of treatment. CBC, complete
blood count; MRI, magnetic resonance imaging; CT, computerized
tomography; Ig, immunoglobulin; IgG4-RD, IgG4-related disease; MDS,
myelodysplastic syndrome; U2AF1,
U2AF1:NM_006758.2:exon2:c.C101T:p.(S34F); ASXL1,
ASXL1:NM_015338.5:exon12:c.2422delC:p.(P808Lfs*10); ETV6(nonsense
mutation), ETV6:NM_015338.5:exon3:c.C313T:p.(R105X)(nonsense
mutation); ETV6(missense mutation),
ETV6:NM_001987.4:exon6:c.G1106A:p.(R369Q)(missense mutation). |
 | Table I.Partial laboratory findings. |
Table I.
Partial laboratory findings.
| Date | Absolute leukocytes,
109/l | Absolute neutrophils,
109/l | Proportion of
eosinophil, % | Hemoglobin, g/l | Platelet,
109/l | IgG4, mg/dl |
|---|
| The First Hospital
of Chun'an County in 2020 | 1.5 | / | / | 52 | 33 | / |
| First treatment in
September 2022 | 1.8 | 0.7 | 26.3 | 40 | 41 | 493 |
| Definite diagnosis
in October 2022 | 1.4 | 0.4 | 27.2 | 44 | 42 | 510 |
| After treatment in
November 2022 | 1.4 | 1.1 | 0.0 | 56 | 49 | 398 |
Tests
Laboratory correlation examination
Immunoprogram results showed the following results:
Immunoglobulin (Ig)A, 482 mg/dl (normal range, 82–453 mg/dl); IgG,
2,050 mg/dl (normal range, 751–1,560 mg/dl), IgG4 493 mg/dl (normal
range, 3–201 mg/dl); and IgE, 12.43 IU/dl (normal level, <100
IU/dl). The patient's ferritin level was 3,917.5 ng/ml (normal
range, 16.40–293.9 ng/ml) and the erythropoietin level was
>7,500 mU/ml (normal range, 4.3–29.0 mU/ml). The normal
indicators of IgM, C3, C4, thyroid function, folic acid and vitamin
B12 ruled out some of the common diseases that cause peripheral
hypoperfusion.
Bone marrow aspirate
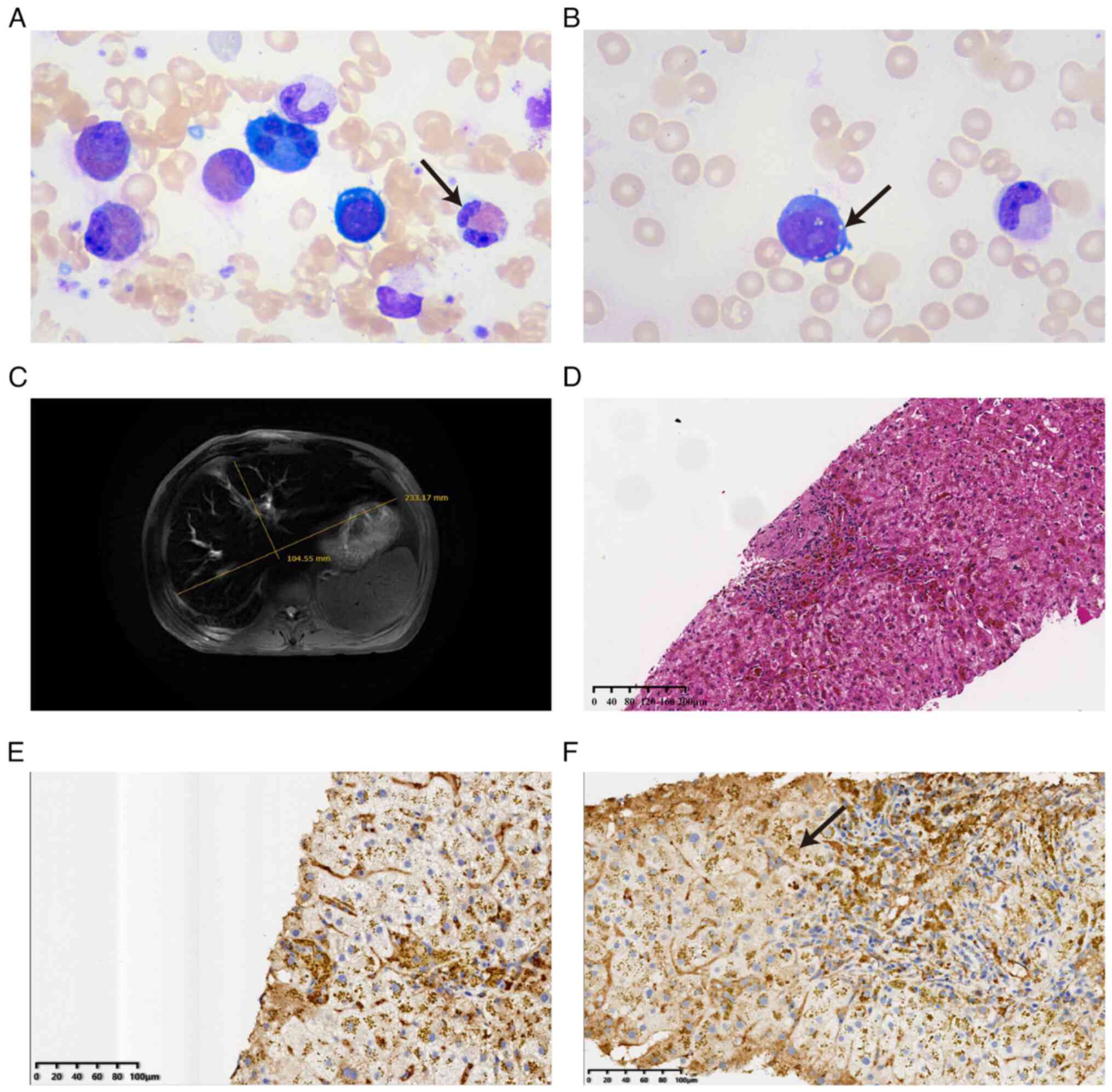
At 1 day after admission, the bone marrow smear
revealed pathological hematopoiesis and 3.5% primordial immature
granulocytes. Proliferating leukocytes accounted for 72.5% of all
cells, with a marked increase in eosinophils, accounting for 26% of
leukocytes. The erythrocyte ratio was 16% and erythrocytes showed
pathological hematopoiesis. Lymphocytes accounted for 7.5% of the
total cell count and presented normal morphology. A total of 2
megakaryocytes could be found throughout the smear, the
megakaryocyte lineage was inactive and small nucleated
megakaryocytes were easily seen. Lobulated nuclei and
multinucleated megakaryocytes were reduced (Fig. 3A and B). Bone marrow smear and
cytochemical staining analysis were performed by Department of
Hematology of Zhejiang Provincial Hospital of Chinese
Medicine(Hangzhou, China). Wright-Giemsa stain was applied for 10
sec at room temperature, then the same amount (0.5–0.8 ml) of
phosphoric acid buffer was added and mixed with the dye solution at
room temperature for 25 min. Subsequently, the smear was washed
with distilled water and the slides were air-dried. The mounted
slides were then examined and photographed using a light microscope
(magnification, ×1,000).
Flow cytometry
Flow cytometry analysis of the bone marrow revealed
1.85% medullary primitive cells with abnormal immunophenotype.
Elevated proportions of eosinophils were detected, with the overall
proportion of nucleated cells at 20.7%. The differentiation of
granulocytes was abnormal and the expression of nucleated red blood
cell CD36/CD71 expression was diminished.
Bone marrow biopsy
Bone marrow biopsy showed the fat percentage at
~65%, with 35% active nucleated cell proliferation, significantly
active granulocyte lineage proliferation and increased eosinophil
levels. A less active red lineage proliferation was shown with 2–10
megaloblasts/high power field (HPF). Multinucleated megakaryocytes
and small nucleated megakaryocytes with reduced fractionated nuclei
were easily seen. Few mature lymphocytes and plasma cells were
seen. Pathological biopsies of the bone marrow showed
immunophenotypic abnormalities, exhibiting CD34 (individual +),
CD117 (individual +), MPO, CD15, CD235a, E-cadherin (partial +),
CD61 (megakaryocyte +), CD20 (individual +); CD3 (few +), CD138, κ
and λ (few +). Histopathology was performed by the Department of
Pathology of Zhejiang Provincial Hospital of Chinese Medicine. Bone
marrow tissue was fixed in 10% neutral formalin for 30–50 min at
room temperature and rinsed under running water. The tissues were
dehydrated sequentially by ethanol gradient. An equal mixture of
pure alcohol and xylene was added for 15 min and xylene I and II
solutions were each infiltrated for 15 min. A mixture of equal
amounts of xylene and paraffin was infiltrated for 15 min, and then
put into paraffin I and paraffin II permeable wax for 50–60 min
each. Samples were slice to a thickness of 5 µm. Hematoxylin and
eosin (HE) staining was performed according to routine protocols.
Briefly, after deparaffinization and rehydration, 5-µm-thick
sections were stained with hematoxylin solution for 5 min at room
temperature, followed by five washes in 1% acid ethanol and then
rinsed in distilled water. Subsequently, the sections were stained
with eosin solution for 3 min at room temperature, followed by
dehydration in ethanol twice for 20 min each and clearing in
xylene. The mounted slides were then examined and photographed
using a light microscope (optical microscope; Leica; magnification,
×50).
Special staining
Myelofibrosis grade 0 (reticular fiber). Perls (−)
(Perls' Prussian blue Stain). Reticulated fiber staining was
performed by the Department of Pathology of Zhejiang Provincial
Hospital of Chinese Medicine. 5-µm-thick sections were
deparaffinized and fixed, oxidized with potassium permanganate
solution for 3 min and rinsed in running water. The sections were
bleached with oxalic acid solution for 1–2 min and rinsed in
running water. Sections were stained with ferric ammonium sulfate
solution for 3 min and rinsed with deionized water for 10 sec.
Staining with silver ammonia solution for 5 min, rinsing with
deionized water for 5–10 sec. Formaldehyde solution reduction for
30 sec, rinse with running water. Gold chloride solution toning 1
min and rinse. Staining with sodium thiosulfate solution for 2 min,
rinse with running water. Slices were dehydrated until transparent
and sealed. Iron staining was performed by the Department of
Pathology of Zhejiang Provincial Hospital of Chinese Medicine.
Dried bone marrow smears were placed in acidic potassium
ferricyanide solution and stained for 30 min at room temperature.
Following rinsing with distilled water, nuclear solid red stain was
restained for 10–15 min and rinsed with running water. Sections
were dried and sealed. Finally, the mounted slides were examined
and photographed using a light microscope (optical microscope;
Leica; magnification, ×50).
Cytogenetic classification
The chromosomes were normal. Hematological oncogene
mutation detection showed U2AF1:NM_006758.2:exon2:c.C101T:p.(S34F)
42.46%, ASXL1:NM_015338.5:exon12:c.2422delC:p.(P808Lfs*10) 37.09%,
ETV6:NM_015338.5:exon3:c.C313T:p.(R105X)(nonsense mutation) 33.97%
and ETV6:NM_001987.4:exon6:c.G1106A:p.(R369Q)(missense mutation)
5.34%. Fusion genes ETV6-PDGFRA, FIP1L1-PDGFRA, TEL-PDGFRB,
KIF5B-PDGFRA, STRN-PDGFRA, and PCM1-JAK2 were negative.
The patient had been leukopenic, anemic and
thrombocytopenic for 2 years. The patient bone marrow smear shows
morphologic dysplasia of erythrocytes and megakaryocytes. The 2016
WHO classification system was used to diagnose the patient with MDS
with multilineage dysplasia (10).
Three scoring methods were used to assess the MSD risk in
accordance with the ‘2019 Update on Diagnosis, Risk-stratification,
and Management’: The International Prognostic Scoring System
(IPSS), the WHO classification-based Prognostic Scoring System
(WPSS) and the Revised International Prognostic Scoring System
(IPSS-R), where the patient scored, 0.5 (intermediate risk), 2
(intermediate risk) and 5 (high risk) respectively (11). However, the patient's eosinophil
ratio (26.3% of leukocytes) and IgG4 (493 mg/dl) level were
persistently abnormally elevated, which were inconsistent with the
symptoms of MDS. The patient and his relatives requested for the
MDS-related treatment to be withheld and only be provided with
symptomatic treatment, including blood transfusion and granulocyte
colony-stimulating factor leukostimulation.
Tests at days 2–7 post-admission
Refinement of diagnostic IgG4-RD-related
auxiliary tests
Up to 6 days post-admission, the patient tested
negative on allergen and fecal worm egg tests, thus allergies and
parasitic diseases were ruled out. The parotid, orbital and
submandibular glands of the patient, in addition to the pancreas
and kidney of the patient showed no significant abnormalities,
ruling out pathology.
Abdominal ultrasound
At day 2 after admission, the ultrasound showed
regular liver morphology, the left lobe of the liver was enlarged,
with an upper and lower diameter of 11.1 cm and the anterior and
posterior diameters were 6.8 cm, while the right liver volume was
also normal, with a smooth surface. The echogenicity of the liver
area was dense and evenly distributed with well-defined blood
vessels. The spleen was full in shape, 4.6 cm thick, with uniform
echogenicity and a hyperechoic nodule of ~0.6 cm by 0.4 cm in size,
with a clear border and regular shape.
Magnetic resonance imaging (MRI) of
the upper abdomen
MRI results showed hepatic macrosomia, ferrous
deposits and decreased T2W1 signal in the liver parenchyma and a
large spleen. However, normal pancreatic morphology, no obvious
abnormal kidney morphology and no clear posterior peritoneal
structures were seen (Fig. 3C).
From the imaging results, liver lesions were considered, and a
liver tissue biopsy was completed 10 days post admission. Hepatic
puncture pathology (Fig. 3D-F)
revealed cell swelling, metachromatic deposition of iron-containing
heme and moderate chronic inflammatory cell infiltration in the
confluent area. Pathological biopsies of the liver showed
immunophenotypic abnormalities, exhibiting Ki-67 (<3% +), CK8
(+), CD10 (+), CD34 (vascular + in the confluent area), HBsAg (−),
HBcAg (−), Hep (+), IgG (partial +), IgG4 (+ >10/HPF), CD20 (−),
CD3 (lymphocytes +) and CD138 (+). The immunohistochemical staining
for IgG and IgG4 was performed by Department of Pathology of
Zhejiang Provincial Hospital of Chinese Medicine. Sections
5-µm-thick were placed in 10% neutral formalin and then stained
with 12.5% IgG (ZA-0448, Amresco) or IgG4 (ZA-0576, Amresco)
fluorescent antibody stained for 60 min at room temperature. Excess
fluorescent antibody was poured off, and the sections were washed
twice in phosphate buffered saline(PBS) at pH 7.2–7.4 with
agitation for 5 min each time, and then washed with distilled water
for 1 min to remove salt crystals. It was buffered with 50% buffer
(0.5 mol/L carbonate buffer pH 9.0–9.5) and blocked with glycerol.
The mounts were then examined and photographed using a light
microscope (magnification, ×100).
From these results IgG4-RD diagnosis was confirmed,
and the patient was prescribed 20 mg prednisone twice daily for 3
weeks. Skin pruritus of both lower extremities improved
dramatically, rashes were reduced, sizes of the liver and spleen
were decreased, the lymph node sizes in the groin were reduced,
blood cell counts were elevated compared with previous levels and
IgG4 was reduced to 398 mg/dl (Fig.
2; Table I). After 52 days of
hospitalization, the patient refused to continue treatment and
requested to be discharged. In the first telephone follow-up on in
March 2023 the patient reported a stable blood count, improved skin
pruritus and no other significant discomfort. However, in the
second follow up in September 2023 the patient reported low
platelets and hemoglobin and was transfusion-dependent due to the
lack of standardized treatment.
Discussion
MDS is a heterogeneous myeloid clonal disorder of
hematopoietic stem cell origin that presents with abnormal myeloid
cell development, ineffective hematopoiesis, refractory hematocrit
and if left untreated, possible progression to acute myeloid
leukemia (AML) (12). The life
expectancy of patients with MDS and their treatment outcomes are
highly disparate, therefore individualized treatment and prognostic
goals should be curated according to the patient's disease risk
classification, and current recommended guidelines. Thus, focusing
on hematopoietic improvement in the lower-risk group and delaying
disease progression, prolonging survival and avoiding
transformation to AML in the higher-risk group (13).
IgG4-RD, as a rare multiorgan immune-mediated
fibrotic inflammatory disease undefined until 2001 (14), is typically characterized by the
presence of a dense IgG4-positive plasma cell infiltrate, the
presence of mild to moderate eosinophilic infiltrate, extracellular
matrix fibrosis and occlusive phlebitis (15). Organs considered typical for IgG4-RD
infiltration are the lacrimal glands, major salivary glands,
orbits, lungs, paravertebral soft tissue, pancreas, biliary tree,
kidneys, retroperitoneum, aorta, meninges and thyroid gland
(6). Since IgG4-RD is highly
heterogeneous and has similar clinical manifestations, serological
and pathological findings with isolated single organ diseases such
as autoimmune pancreatitis, Mikulic disease, retroperitoneal
fibrosis, autoimmune cell syndrome (AICy) and autoimmune disease
(AID) (16), Diagnoses of these
diseases are difficult to ascertain over a long period of time
(17). Currently, hormonal shock
therapy is recommended for undiagnosed patients presenting these
symptoms, which provides good prognosis in most patients if
treatment is commenced quickly (6).
Patients with a poor survival prognosis of IgG4-RD
tend to suffer from malignancy, especially pancreatic cancer and
lymphoma, which may be associated with chronic inflammatory
stimulation and immune system dysfunction (7,18).
Immunization and biologic treatment of patients with IgG4-RD can
perturb normal immune system function and increase the possible
incidence of tumor or paraneoplastic syndromes (19), while the abnormal immune system
state of the tumor and the local inflammatory factor
microenvironment can lead to abnormal activation of immune-mediated
pathways associated with IgG4-RD (20). It has been demonstrated that
CD4+ cytotoxic T lymphocytes (CTLs) in patients with
IgG4-RD promote excessive replication of cytosine deaminase and
induce DNA mutations, while abnormal antigen replication in the
local tumor microenvironment can promote an autoimmune cascade that
ultimately leads to illnesses such as IgG4-RD and scleroderma
(21,22).
The association of IgG4-RD with malignancy has been
demonstrated in previous clinical studies, and an active IgG4-RD
state has been demonstrated to be a risk factor for the development
of malignant tumors (23,24). Patients with IgG4-RD have
significantly higher standardized incidence ratios (SIRs) (SIR,
2.57; 95% CI, 1.72–3.84) compared with the standard population of
the same sex and age, with increased overall cancer risk and a
significant decrease in patient quality of life (18). The specific SIRs for pancreatic
cancer and lymphoma were higher than those of the general
population in IgG4-RD patients (SIR, 4.07; 95% CI, 1.04–15.92; and
SIR, 69.17; 95% CI, 3.91–1223.04, respectively) (18). This correlation has been
demonstrated in a number of neoplasms, such as head and neck
neoplasms (25), pulmonary
carcinomas (26), breast carcinomas
(27), gastrointestinal (28), hepatic carcinomas and cervical
carcinomas (29).
IgG4-RD also enhances the morbidity of hematological
malignancies, decreases overall survival and accelerates disease
progression (30,31). AICy induces and complicates myeloid
malignancies such as AML and lymphoma and patients with MDS with
low- or intermediate-risk IPSS and timely diagnosis of AID or late
onset have an improved quality of survival (32). The transformation of MDS as a
premalignant stage of the hematological disease to AML is also
closely related to immune system-related diseases, but its
association with IgG4-RD has not been elucidated (33). Asano et al (34) observed a total of 34 malignancies,
including two lymphomas and one MDS, in a 12-year follow-up of 158
patients with IgG4-RD (34). This
study also confirmed that cases of IgG4-RD combined with MDS exist
but are extremely rare. Regarding treatment, AID associated with
MDS could benefit from demethylating treatments for MDS, such as
azathioprine, to reduce relapse rates and avoid hormone dependence,
as confirmed by Roupie et al (35). Current evidence demonstrates an
association between MDS and different systemic autoimmune diseases,
but the exact underlying mechanisms remain unclear (36). Therefore, the present study suggests
that there is an association between the development of these
diseases.
The present case report presents a patient with a
concurrent diagnosis of IgG4-RD and MDS who had a prolonged onset
of the diseases, and the sequence of concurrent or predisposing
events of the two diseases remained unclear. The present patient
was at substantial risk of becoming leukemic, and IgG4-RD increased
the malignancy risk. Therefore, the data suggests that the
concurrent IgG4-RD and MDS of this patient was more than a simple
coincidence, demonstrating a clinical need to raise awareness of
the development of MDS combined with IgG4-RD, as early treatment is
essential to prevent damage to multiple organs, enhance survival
quality and prolong survival time of patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Provincial Natural
Science Foundation of Zhejiang (grant no. LGF22H080005), the
National Natural Science Foundation of China (grant nos. 82274273
and 81774092) and the Innovative Talents Funding Program of
Zhejiang Health and Family Planning Commission (grant no.
1S21702).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SL, BY and ZHu contributed to the conception and
design of the study. CZ performed histopathological evaluation of
the bone marrow specimens. LW, XP and ZHo made significant
contributions to the acquisition, analysis and interpretation of
the data, wrote the manuscript, prepared the figures and revised
the article for critically important intellectual content. LW and
XP confirm the authenticity of all the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written consent for publication has been obtained
from both the patient and their daughter. All identifying
information has been removed or anonymized to ensure
confidentiality.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tanaka TN and Bejar R: MDS overlap
disorders and diagnostic boundaries. Blood. 133:1086–1095. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frumm SM, Shimony S, Stone RM, DeAngelo
DJ, Bewersdorf JP, Zeidan AM and Stahl M: Why do we not have more
drugs approved for MDS? A critical viewpoint on novel drug
development in MDS. Blood Rev. 60:1010562023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garcia-Manero G: Myelodysplastic
syndromes: 2023 update on diagnosis, risk-stratification, and
management. Am J Hematol. 98:1307–1325. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lanzillotta M, Mancuso G and Della-Torre
E: Advances in the diagnosis and management of IgG4 related
disease. BMJ. 369:m10672020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wick MR and O'Malley DP: Lymphadenopathy
associated with IgG4-related disease: Diagnosis & differential
diagnosis. Semin Diagn Pathol. 35:61–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Katz G and Stone JH: Clinical perspectives
on IgG4-related disease and its classification. Annu Rev Med.
73:545–562. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wallace ZS, Wallace CJ, Lu N, Choi HK and
Stone JH: Association of IgG4-related disease with history of
malignancy. Arthritis Rheumatol. 68:2283–2289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferry JA, Klepeis V, Sohani AR, Harris NL,
Preffer FI, Stone JH, Grove A and Deshpande V: IgG4-related orbital
disease and its mimics in a western population. Am J Surg Pathol.
39:1688–1700. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen LYC, Mattman A, Seidman MA and
Carruthers MN: IgG4-related disease: What a hematologist needs to
know. Haematologica. 104:444–455. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the world health organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patnaik MM and Tefferi A: Refractory
anemia with ring sideroblasts (RARS) and RARS with thrombocytosis:
‘2019 update on diagnosis, risk-stratification, and management’. Am
J Hematol. 94:475–488. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Estey E, Hasserjian RP and Döhner H:
Distinguishing AML from MDS: A fixed blast percentage may no longer
be optimal. Blood. 139:323–332. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Scalzulli E, Pepe S, Colafigli G and
Breccia M: Therapeutic strategies in low and high-risk MDS: What
does the future have to offer? Blood Rev. 45:1006892021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Danlos FX, Rossi GM, Blockmans D, Emmi G,
Kronbichler A, Durupt S, Maynard C, Luca L, Garrouste C, Lioger B,
et al: Antineutrophil cytoplasmic antibody-associated vasculitides
and IgG4-related disease: A new overlap syndrome. Autoimmun Rev.
16:1036–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nowak V, Agaimy A, Kristiansen G and
Gütgemann I: Increased IgG4-positive plasma cells in
nodular-sclerosing Hodgkin lymphoma: A diagnostic pitfall.
Histopathology. 76:244–250. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen LYC, Slack GW and Carruthers MN:
IgG4-related disease and Rosai-Dorfman-Destombes disease. Lancet.
398:1213–1214. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baenas DF, Miretti VS, Caeiro F and Paira
S: Differential diagnosis between pancreatic involvement in
IgG4-related disease and pancreatic cancer. Gastroenterol Hepatol.
44:144–155. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu T, Wu Y, Liu J, Zhuang Y, Jin X and
Wang L: The risk of malignancy in patients with IgG4-related
disease: A systematic review and meta-analysis. Arthritis Res Ther.
24:142022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wallace ZS, Naden RP, Chari S, Choi HK,
Della-Torre E, Dicaire JF, Hart PA, Inoue D, Kawano M, Khosroshahi
A, et al: The 2019 American college of rheumatology/european league
against rheumatism classification criteria for IgG4-related
disease. Ann Rheum Dis. 79:77–87. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martín-Nares E, Hernández-Molina G, Baenas
DF and Paira S: IgG4-related disease: Mimickers and diagnostic
pitfalls. J Clin Rheumatol. 28:e596–e604. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Furukawa S, Moriyama M, Miyake K,
Nakashima H, Tanaka A, Maehara T, Iizuka-Koga M, Tsuboi H,
Hayashida JN, Ishiguro N, et al: Interleukin-33 produced by M2
macrophages and other immune cells contributes to Th2 immune
reaction of IgG4-related disease. Sci Rep. 7:424132017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong J, Kim S and Lin PC: Interleukin-33
and ST2 signaling in tumor microenvironment. J Interferon Cytokine
Res. 39:61–71. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cush JJ and Dao KH: Malignancy risks with
biologic therapies. Rheum Dis Clin North Am. 38:761–770. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dzhus M, Ivashkivsky O, Mikukst V,
Parkishen S and Diadyk O: IgG4-related disease misdiagnosed as
neoplasm. J Clin Rheumatol. 27:e71–e72. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martínez-de-Alegría A, Baleato-González S,
García-Figueiras R, Bermúdez-Naveira A, Abdulkader-Nallib I,
Díaz-Peromingo JA and Villalba-Martín C: IgG4-related disease from
head to toe. Radiographics. 35:2007–2025. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bertoglio P, Viti A, Paiano S, Assante LR,
Bogina GS, Pomari C, Zamboni G and Terzi AC: IgG4-related disease:
A new challenging diagnosis mimicking lung cancer. Interact
Cardiovasc Thorac Surg. 28:410–412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsuda B, Kumaki N, Ishida R, Mizuno M,
Yokoyama K, Oshitanai R, Terao M, Morioka T, Okamura T, Saito Y, et
al: Distinction of IgG4-related mastitis from breast cancer: A case
report. Surg Case Rep. 5:1232019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Notohara K, Kamisawa T, Uchida K, Zen Y,
Kawano M, Kasashima S, Sato Y, Shiokawa M, Uehara T, Yoshifuji H,
et al: Gastrointestinal manifestation of immunoglobulin G4-related
disease: Clarification through a multicenter survey. J
Gastroenterol. 53:845–853. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizuno R, Yamanishi Y, Uda S, Terashima T,
Higashi T and Higuchi T: Invasive cervical cancer accompanied by
IgG4-related disease. J Obstet Gynaecol Res. 42:1198–1202. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Z, Zhang S, Zhang W, Feng J, Li M and
Zeng X: Immunoglobulin G4-related disease accompanied by primary
myelofibrosis: Case report. Front Med (Lausanne). 8:6387942021.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okamoto A, Watanabe T, Kamata K, Minaga K
and Kudo M: Recent updates on the relationship between cancer and
autoimmune pancreatitis. Intern Med. 58:1533–1539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barcellini W, Giannotta JA and Fattizzo B:
Autoimmune complications in hematologic neoplasms. Cancers (Basel).
13:15322021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiao N, He X, Niu H, Yu H, Cui N, Li H,
Yan L, Shao Z, Xing L and Wang H: Increased circulating
CD4(+)CXCR5(+) cells and IgG4 levels in patients with
myelodysplastic syndrome with autoimmune diseases. J Immunol Res.
2021:43025152021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Asano J, Watanabe T, Oguchi T, Kanai K,
Maruyama M, Ito T, Muraki T, Hamano H, Arakura N, Matsumoto A and
Kawa S: Association between immunoglobulin G4-related disease and
malignancy within 12 years after diagnosis: An analysis after
longterm followup. J Rheumatol. 42:2135–2142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roupie AL, Guedon A, Terrier B, Lahuna C,
Jachiet V, Regent A, de Boysson H, Carrat F, Seguier J, Terriou L,
et al: Vasculitis associated with myelodysplastic syndrome and
chronic myelomonocytic leukemia: French multicenter case-control
study. Semin Arthritis Rheum. 50:879–884. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gonzalez CA, Carrillo Linares JL, García
Muñoz I, Escalona García A and Valdivielso P: IgG4-Related disease
affecting testicle and myelodysplastic syndrome: Just a
coincidence? Eur J Case Rep Intern Med. 8:0029812021.PubMed/NCBI
|